

Abstract
Generation of mammalian circadian rhythms involves molecular transcriptional and translational feedback loops. It is not clear how membrane events interact with the intracellular molecular clock or whether membrane activities are involved in the actual generation of the circadian rhythm. We examined the role of membrane potential and calcium (Ca2+) influx in the expression of the circadian rhythm of the clock gene Period 1 (Per1) within the rat suprachiasmatic nucleus (SCN), the master pacemaker controlling circadian rhythmicity. Membrane hyperpolarization, caused by lowering the extracellular concentration of potassium or blocking Ca2+ influx in SCN cultures by lowering [Ca2+], reversibly abolished the rhythmic expression of Per1. In addition, the amplitude of Per1 expression was markedly decreased by voltage-gated Ca2+ channel antagonists. A similar result was observed for mouse Per1 and PER2. Together, these results strongly suggest that a transmembrane Ca2+ flux is necessary for sustained molecular rhythmicity in the SCN. We propose that periodic Ca2+ influx, resulting from circadian variations in membrane potential, is a critical process for circadian pacemaker function.
Keywords: circadian rhythm, calcium, potassium, suprachiasmatic nucleus, Period 1, PERIOD 2
Introduction
The hypothalamic suprachiasmatic nucleus (SCN) plays a critical role in controlling mammalian circadian rhythmicity. Although other brain regions and non-neural tissues express circadian rhythms in molecular expression (Yamazaki et al., 2000; Abe et al., 2002), the SCN plays a pervasive role in the generation and control of physiological and behavioral rhythms (Moore, 1991). SCN rhythm generation is a cell-autonomous property (Welsh et al., 1995). Several genes, including Period (Per1, Per2, and Per3) and Cryptochromes (Cry1, Cry2), have been identified that play critical roles in generating circadian rhythms. Although models of the mammalian clock continue to evolve, periodicity is presently believed to be generated by an autoregulatory transcriptional and posttranslational feedback loop involving “clock genes” and their products (Dunlap, 1999). Membrane phenomena, such as electrical impulses and ionic currents, have not been generally recognized as part of the core clock mechanism but rather as part of the pathways by which environmental synchronizing signals reach the clock (Block et al., 1993; Meijer and Schwartz, 2003) and by which the clock regulates tissue and organ targets (Schaap et al., 2003). This view has been challenged in Drosophila, in which silencing of electrical activity leads to arrhythmic expression of the PERIOD and TIMELESS proteins, core constituents of the circadian clock in Drosophila (Nitabach et al., 2002). These data raise the issue of whether electrical activity and underlying ionic fluxes play a more central role in rhythm generation.
We now provide data suggesting that a transmembrane calcium (Ca2+) flux is critical for molecular rhythmicity within the mammalian SCN and at least one peripheral oscillator, the liver. We used a luciferase reporter to assay real-time clock gene activity in tissue cultures. Preventing Ca2+ influx by hyperpolarizing SCN neurons, removing extracellular Ca2+, or applying Ca2+ channel blockers leads to a loss of rhythmic expression of Per1 and PER2. The apparent requirement for Ca2+ influx to maintain rhythmicity reveals an unforeseen role for ionic currents in mammalian circadian rhythm generation.
Materials and Methods
Animals. A Per1-luciferase transgenic rat line [W(per1)1], a Per1-luciferase transgenic mouse line (C57x+/-) (Dr. Hajime Tei, Mitsubishi Kagaku Institute of Life Sciences) and a PER2:LUCIFERASE knock-in mouse line (provided by Dr. Joseph Takahashi, Department of Neurobiology and Physiology, Northwestern University, Evanston, IL) were raised at the University of Virginia, Department of Biology. The animals were kept on a 12 h light/dark cycle. All animal use was conducted in accordance with the recommendations of the Committee on Animal Care and Use at the University of Virginia.
Dispersed cell culture procedure. SCNs were punched out with a 440 μm diameter neural punch from selected coronal sections (275 μm) obtained from 3- to 7-d-old transgenic rat pup brains. The punches were incubated with papain enzyme (Sigma, St. Louis, MO), dissociated by trituration, dispersed on coverslips (1000-3000 cells/slip; glass or Thermanox), coated with laminin (5.8 μg/coverslip) and poly-d-lysine (0.02 mg/ml) (Sigma), and kept in 95% O2-5% CO2. Half of the medium [containing 10 g/L DMEM 2902 (Invitrogen, Grand Island, NY), 10% B27, 84 mm NaHCO3, 180.2 mm d-glucose, 0.25% penicillin-streptomycin, pH 7.2-7.3, osmolarity 290 mOsm) was exchanged three times each week.
Explant cultures. Rats and mice, 2-3 months of age, were anesthetized with CO2 and decapitated. The brains were removed, hypothalamic coronal sections (rat, 300 μm; mouse, 250 μm) were cut using a vibroslicer, and bilateral or unilateral SCNs were explanted. The explants were placed on culture membranes (Millicell-CM, PICM030-50; Millipore, Bedford, MA) in 35 mm Petri dishes with 1.2 ml of culture medium [pH 7.2; serum-free, low-sodium bicarbonate, no phenol red; manufactured at the University of Virginia according to the recipe of DMEM (13000-021; Invitrogen)] supplemented with 10 mm HEPES (Sigma), B27 (2%; 17504-010; Invitrogen), 0.1 mm luciferin (beetle luciferin, potassium salt; Promega, Madison, WI), and antibiotics (25U/ml penicillin, 25 mg/ml streptomycin; Sigma). The dishes were sealed with cover glasses and vacuum grease and transferred to the recording environment (Yamazaki et al., 2000).
Bioluminescence measurements. Bioluminescence was measured with photomultiplier tube (PMT) detector assemblies (HC135-11 MOD; Hamamatsu, Shizuoka, Japan) modified from HC135-01. The modules and cultures were maintained in light-tight incubators at 36°C and interfaced to IBM (White Plains, NY) personal computers for continuous data acquisition. The PMTs were positioned ∼1 cm above the cultures. Photon counts were made through the glass coverslip and integrated over 1 min intervals (Yamazaki et al., 2000).
Whole-cell patch-clamp recordings. The culture medium was replaced with a physiological recording solution containing the following (in mm): 0.81 MgSO4, 5.37 KCL, 1.8 CaCl2, 110 NaCl, 0.79 NaH2PO4, 10.0 HEPES, 4.19 NaHCO3, and 25.0 d-glucose (Sigma). Osmolarity and pH were adjusted to 290-300 mOsm and 7.3, respectively. Patch-clamp pipettes (resistance, 3-6 MΩ) were filled with a solution containing the following (in mm): 140 K+-gluconic acid, 4.0 NaCl, 10 HEPES, 0.38 CaCl2, 5 EGTA, and 0.49 MgCl2 (osmolarity, 280 mOsm; pH 7.3). Gigaseal formation was achieved by monitoring changes in current responses to voltage pulses. Membrane rupture was made by syringe suction. Resting potentials were recorded in current-clamp mode with an Axoclamp 2A amplifier (Molecular Devices, Foster City, CA) in conjunction with a digital interface (Digidata 1200; Molecular Devices) and read directly from the amplifier. Data acquisition was performed using pClamp 8.0 software (Molecular Devices). For each recording, the physiological solution in the dish was exchanged several times with three different solutions containing different concentrations of K+ or Ca2+. For the three different solutions, designated as 1 (control; high [ion] solution), 2 (medium [ion] solution), and 3 (low [ion] solution), the recording in each concentration and each cell was performed in the following order: 1, 2, 3, 2, 1, 2, 3. The average resting potential in each solution (1, 2, or 3) was then obtained for each cell and used as one data point.
Drugs. Drugs were solubilized in water or dimethylsulfoxide (DMSO; Sigma) to make stock solutions 10-1000× the working solutions, which were added at the time of culture, except for Ca2+ channel antagonists, which were added after 3-5 d in culture. Equivalent volumes of medium compared with the volume of the added drugs were discarded so the total volume of medium in cultures was always 1.2 ml. All medium changes and drug manipulations were done manually by manipulating volumes directly in the dish with a pipette in sterile environment outside the recording incubator. Drugs used were tetrodotoxin (TTX; sodium channel blocker; Sigma), ω-agatoxin IVA, ω-agatoxin TK (P/Q-type Ca2+ channel blockers), ω-conotoxin GVIA (N-type blocker), ω-conotoxin MVIIC (Q-type blocker), ω-grammotoxin SIA (N- and P-type blocker), and pimozide (T-type channel blocker; Alomone Labs, Jerusalem, Israel).
Data analysis. Period analysis was performed with software (written by Dr. Y. Kwak, Department of Biology, University of Virginia) calculating the 2 h running average of photon counts. Peaks (defined as maximum) were selected, and the time between two peaks was defined as the period during one cycle. For statistical comparisons of periods, the average periods of a minimum of four cycles and a maximum of eight cycles and SEM for each treatment were calculated. A Student's t test was used to calculate the p values.
Results
Decreasing [K+] causes membrane hyperpolarization and stops the Per1 oscillation
We used transgenic rats to measure rhythmic Per1 expression. In these animals, a reporter for the firefly enzyme luciferase (luc) has been linked to the mouse Per1 promoter (Yamazaki et al., 2000), enabling real-time assay of Per1 expression by recording luciferase activity. To examine the effects of membrane hyperpolarization on SCN Per1 rhythmicity, we prepared SCN slice cultures maintained in culture media containing different [K+]. In parallel experiments, we performed patch-clamp recordings to examine the effects of lowering [K+] on membrane potential (n = 9). Dispersed SCN neurons recorded in control medium (5.4 mm [K+]; n = 20) had an average resting potential of -49.8 ± 1.41 mV. In 0.5 and 2.7 mm [K+], the cells were markedly hyperpolarized (-58.2 ± 2.33 mV; -53.3 ± 1.77 mV) and depolarized in 10 mm [K+] (-38.7 ± 0.71 mV) (Fig. 1A). In SCN tissue cultures, the Per1-luc signals showed a robust circadian expression with an average period of 25.4 ± 0.11 h (n = 12). When [K+] was lowered, we found that the amplitude of the Per1-luc signal decreased. In 0 mm K+ (defined as 0 mm K+ added to the culture medium), no rhythm in Per1 expression could be detected (Fig. 1B). This effect was reversible, and the Per1 rhythm could be restored by washing with control medium.
Figure 1.

SCN recordings of membrane potential and bioluminescence of Per1-luc expression in various levels of [K+]. A, Current-clamp recordings in culture medium containing 0.5, 2.7, 5.4 (control), or 10 mm K+. Each color represents membrane potential in a single neuron in various levels of [K+]. B, Bioluminescence recordings from explants obtained from transgenic Per1-luc rats. The explants were cultured in 0, 0.5, 1, 3, or 5.4 mm K+.
To evaluate whether hyperpolarizing the SCN “stops the clock,” in three repeated experiments, we cultured three SCN explants in medium containing 0 mm K+ for 18, 24, and 30 h before replacing with control medium. We reasoned that if the clock was stopped, each rhythm would return after 18, 24, and 30 h, respectively, when 0 [K+] medium was replaced with control medium. As a control, the Per1-luc rhythms were recorded from three explants that were initially cultured in control medium, which was replaced with fresh control medium after 18, 24, and 30 h. The Per1-luc rhythms in the control slices were expressed in phase with each other and remained in phase after replacement with fresh control medium (Fig. 2A). In contrast, in the three explants started in 0 [K+], there was no evidence of Per1 rhythmicity until the tissue culture medium was exchanged with control medium. Furthermore, we found that the restored Per1-luc rhythms in the three cultures were 6 h out of phase from one another, suggesting that the motion of the clock, as measured by Per1-luc rhythmicity, was stopped in the 0 [K+] environments (Fig. 2B).
Figure 2.

Example of SCN phase delays in hyperpolarizing medium. A, Bioluminescence recordings from three explants in control medium represented by green, red, and black traces. The control medium was replaced with fresh control medium at 6 h intervals the second day in culture (indicated by arrows), and the phases were compared using the first peaks after medium replacement as reference points (indicated by dashed lines). B, Bioluminescence recordings from three bilateral explants in medium containing 0 mm K+, represented by green, red, and black traces. The 0 [K+] medium was replaced with control medium at 6 h intervals the second day in culture (indicated by arrows). Dotted lines indicate the peaks of the Per1-luc signals after medium replacement.
Decreasing [Ca2+] abolishes the Per1 rhythm in the SCN and liver
Depolarization of SCN neurons causes influx of Ca2+ (van den Pol et al., 1992; Tominaga et al., 1994; Colwell, 2001). We next addressed the question of whether preventing Ca2+ influx by removing extracellular Ca2+ has a similar effect on SCN Per1 expression as does hyperpolarizing the cells with low [K+]. We cultured SCN slices in different [Ca2+] and found that, as with [K+], the cyclic expression of Per1 was reversibly reduced or abolished in medium containing low [Ca2+] (Fig. 3A). In addition, the period was shortened (24.2 ± 0.30 h in 0.72 mm Ca2+, n = 5; 24.0 ± 0.37 h in 0.54 mm Ca2+, n = 4; p < 0.05; Student's t test) compared with controls. Reversibility was demonstrated after 12 h (n = 2), 18 h (n = 4), and 24 h (n = 4) in six separate explants cultured in 0 mm Ca2+ (Fig. S1, available at www.jneurosci.org as supplemental material). Lowering [Ca2+]depolarized the membrane (-43.8 ± 2.85 mV in 0 mm; -48.5 ± 2.53 mV in 0.33 mm), and increasing [Ca2+] slightly hyperpolarized the neurons (Fig. 3B) (-52.7 ± 1.92 mV in 3.36 mm; n = 11) compared with controls. To further demonstrate that the effect of low [Ca2+] on the Per1-luc rhythm is oscillator specific and not caused by non-specific effects, such as decreased cell viability or general transcription/translation, we added forskolin to SCN slices (n = 7) kept in 0 [Ca2+], which transiently increases Per1-luc transcription in brain slices (Yamazaki et al., 2000; Abe et al., 2002). Forskolin markedly elevated the bioluminescence baseline in slices kept in medium containing 0 [Ca2+] during 12, 18, 24, and 48 h, demonstrating that Per1 transcription was functional in the explants (Fig. S2, available at www.jneurosci.org as supplemental material).
Figure 3.
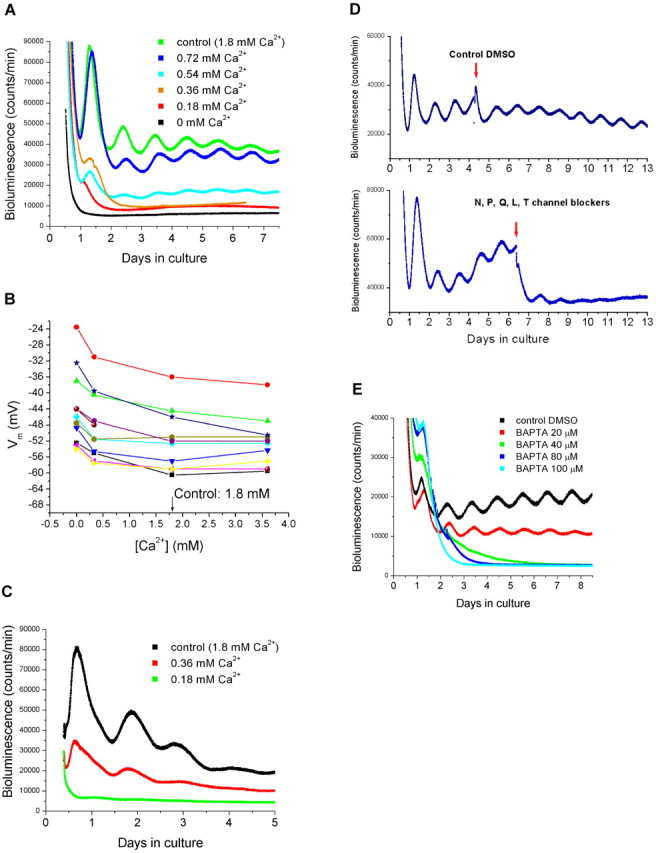
Recordings of membrane potential and bioluminescence in various concentrations of Ca2+. A, Bioluminescence recordings from SCN explants obtained from transgenic Per1-luc rats. The explants were cultured in medium containing 0, 0.18, 0.36, 0.54, 0.72, or 1.8 mm (control) Ca2+. B, Current-clamp recordings in recording solutions containing 0, 0.33, 1.8, and 3.36 mm Ca2+. Each color represents recorded membrane potentials in a single SCN neuron in various concentrations of Ca2+. C, Bioluminescence recordings from liver explants obtained from Per1-luc rats. Liver tissues were cultured in medium containing 0.18, 0.36, or 1.8 mm Ca2+. D, Effect of Ca2+ channel antagonists on the SCN Per1-luc rhythm. SCN explants were cultured in control medium. After 3-5 d in culture, a DMSO control (top trace) or a mixture of Ca2+ channel blockers specific to N-, P-, Q-, L-, and T-types (bottom trace) was added (indicated by arrows) to the explants. E, The intracellular Ca2+ chelator BAPTA-AM (20, 40, 80, and 100 μm) was added to SCN explants, and bioluminescence was recorded. DMSO was added as a control.
To examine the effects of low [Ca2+] on Per1 expression on non-neural tissues, we cultured liver tissue (n = 3) in various [Ca2+]. As with the SCN, low [Ca2+] blunted or abolished the rhythm in Per1-luc expression (Fig. 3C).
Blocking voltage-dependent Ca2+ conductances and buffering intracellular Ca2+ cease the rhythmic expression in Per1
To analyze the role of voltage-dependent Ca2+ conductances in Per1 rhythmicity, we added Ca2+ channel blockers (specific to N-, P-, Q-, L-, and T-types) to SCN slices cultured in control medium (n = 3). The channel blockers did not immediately abolish rhythmicity, but rather rhythms were lost after two to three cycles. As a control, slices (n = 4) were treated with equivalent (0.1%) and higher concentrations of DMSO (0.2, 0.3, and 0.4%), which had no effect on the Per1 rhythm (Fig. 3D). DMSO at a very high concentration (4%) did not affect the amplitude but shortened the period of the Per1 rhythm (23.75 ± 2.01 h). None of the channel blockers alone led to arrhythmicity, although the L-type channel blocker nimodipine slightly reduced the amplitude of Per1 expression culture (n = 5) (Fig. S3, available at www.jneurosci.org as supplemental material). To further explore the role of Ca2+ in regulating Per1 expression, in two different sets of experiments, we treated SCN cultures with the intracellular chelator BAPTA-AM. Buffering intracellular Ca2+ stopped Per1 rhythmicity at 40, 80, and 100 μm but not at 20 μm of original loading concentration of BAPTA-AM (Fig. 3E).
The requirement of Ca2+ for Per1 rhythmicity is not species-specific to rat
To evaluate the generality of the Ca2+ requirement for the SCN Per1 rhythm, we performed similar experiments on SCN slices from a transgenic Per1-luc mouse. Mouse slices were cultured in control medium or 0 mm Ca2+ (defined as 0 mm Ca2+ added to the culture medium; n = 3), and the rhythm in Per1-luc expression was recorded. In slices cultured in control medium, the Per1-luc expression was rhythmic (period, 24.4 ± 0.39 h). However, similar to rat slices, the mouse Per1 rhythm was immediately abolished in 0 mm Ca2+, indicating that the Ca2+ requirement of molecular rhythmicity in the SCN is not species specific (Fig. 4A).
Figure 4.

Bioluminescence recordings in mouse SCN explants. A, Recordings of Per1-luc signals. The explants were cultured in 0 or 1.8 mm (control) Ca2+. B, Recordings of PER2:LUC expression. Explants were cultured in 0 or 1.8 mm Ca2+. C, Recordings of PER2:LUC expression. Explants were cultured in DMSO control (0.3%) and BAPTA-AM (20 or 80 μm).
A Ca2+flux is required also for PER2 rhythmicity
We also evaluated whether Ca2+ is required for sustained rhythmicity of other clock genes and their protein products. We used slices from a Per2Luc knock-in mouse (provided by Dr. Joseph S. Takahashi), in which a PERIOD2:LUCIFERASE fusion protein is used as a real-time reporter. In control medium, the PER2:LUC protein showed a robust circadian expression (period, 23.9 ± 0.21 h; n = 5), as reported recently (Yoo et al., 2004). When we cultured slices in 0 mm Ca2+ (n = 4), similar to Per1, the PER2 protein rhythm was immediately attenuated and absent after two to three cycles (Fig. 4B). In addition, we treated PER2:LUC slices with 20 μm (n = 3) or 80 μm (n = 3) loading concentration of BAPTA-AM, which, as for Per1, abolished the PER2:LUC rhythm (Fig. 4C).
Discussion
Our data provide the first demonstration of an ionic flux requirement for rhythms in molecular expression in mammals. These findings are consistent with the observations in Drosophila that electrical silencing stops the free-running circadian clock (Nitabach et al., 2002). Our experiments extend this observation to mammals and suggest that a transmembrane Ca2+ flux is essential for molecular rhythmicity. A critical role of Ca2+ influx in rhythm generation is supported by several observations. First, reducing the transmembrane flux by hyperpolarizing the membranes of pacemaker cells, and thereby presumably closing voltage-controlled Ca2+ conductances, leads to arrhythmicity in Per1 expression. Second, removing extracellular Ca2+ abolishes rhythms in Per1 and PER2. Third, blocking various Ca2+ channels results in aperiodicity after two to three cycles. We are uncertain why Ca2+ channel blockers do not immediately abolish rhythmicity; however, we suspect that these compounds fail to completely block Ca2+ flux, which may explain the relatively delayed effect. Fourth, the membrane-permeable form of BAPTA blocks rhythmicity of Per1 and PER2. Finally, the phase of the mPer1 rhythm is delayed proportionally to the time spent in 0 [K+] medium. This suggests that the molecular clock is stopped rather than the rhythm being “masked” by any effects of reducing extracellular K+ or Ca2+ on the ability of the reporter gene to reliably express the state of Per1 transcription. This result also makes it unlikely that a low-amplitude rhythm persists in the absence of K+ or Ca2+, hidden within the noise level of the photomultiplier. Although we did not record the effects of manipulations with [K+] and [Ca2+] in individual pacemaker cells, we think it is unlikely that the effects observed are caused by intercellular uncoupling insomuch as the action of most of our treatments on rhythmicity was rapid, within 24 h. Although uncoupling would lead to phase scattering and apparent ensemble arrhythmicity, this typically occurs gradually over 5-7 d (Welsh et al., 2004).
The fact that a non-neural tissue, liver, also becomes arrhythmic in low [Ca2+] suggests that the requirement for molecular rhythmicity extends beyond neuronal tissues. Indeed, hepatocytes contain Ca2+ channels that are mainly receptor activated but may also include voltage-activated Ca2+ channels (Sawanobori et al., 1989; Brereton et al., 1997).
Although previous studies show conflicting conclusions regarding the role of membrane electrical events and ion fluxes in mammalian rhythm generation (Schwartz et al., 1987; Shibata and Moore, 1987; Shibata et al., 1987; Welsh et al., 1995; Shinohara et al., 1998), our data are consistent with recent findings in mice lacking vasoactive intestinal polypeptide receptor subtype 2 (VPAC2) receptors, which are activated by vasoactive intestinal polypeptide (VIP) and pituitary adenylate cyclase activating polypeptide. Mice missing the VPAC2 receptor are unable to maintain normal behavioral rhythmicity and rhythmic expression of Per1, Per2, and Cry1 (Harmar et al., 2002). These mice also fail to exhibit the midday peak in electrical activity that is characteristic of impulse rhythms from SCN brain slices (Cutler et al., 2003). Consistent with these results, VIP/peptide histidine isoleucine-deficient mice were found to exhibit profound abnormalities in locomotor rhythmicity (Colwell et al., 2003). These studies suggest an important requirement for the VPAC2 receptor in normal SCN rhythmicity. The reductions in spontaneous electrical activity in SCN neurons and the observation that these cells are capable of responding with high-impulse frequencies to stimulation suggest that loss of the VPAC2 receptor may alter the resting potential of SCN neurons (Harmar et al., 2002; Cutler et al., 2003). This situation may therefore be similar to Drosophila (Nitabach et al., 2002), in which expression of Drosophila open rectifier K+ channels in the pigment dispersing factor-expressing lateral neurons leads to a loss of behavioral and molecular rhythmicity through presumptive hyperpolarization of the membrane potential. A recent study, however, calls into question whether the lack of VIP/VPAC2 alters the SCN membrane potential. Aton et al. (2005) reported that dispersed arrhythmic SCN neurons from Vip-/- and Vipr2-/- mice exhibited normal frequencies of spontaneous impulse activity, suggesting that loss of rhythmicity may not involve hyperpolarization. The basis of the discrepancy between the different studies using VIP/VPAC2-deficient mice is presently unclear.
Another study measuring Per1-luc activity within single neurons in a slice (Yamaguchi et al., 2003) supports the view that membrane electrical activity is critical for circadian rhythmicity. When SCN slices were exposed to TTX, Per1 rhythmicity damped immediately in individual neurons. Furthermore, TTX treatment caused decreased levels of Per1 and Per2 transcripts and proteins.
In a previous study, Ikeda et al. (2003) demonstrated a TTX-resistant circadian rhythm in cytosolic, but not nuclear, Ca2+ concentration. These data are not necessarily at variance with our findings. It may indicate that there are two rhythmic systems: one membrane-related electrical rhythm and one intracellular cytosolic Ca2+ rhythm. Whether the cytosolic Ca2+ rhythm affects clock gene rhythms or vice versa has not yet been determined and additional experiments are required to investigate the relationship between molecular, electrical, and intracellular Ca2+ rhythms. Another possibility is that TTX does not block rhythmic membrane oscillations that lead to periodic Ca2+ influx. A more direct experiment would therefore be to test whether the cytosolic Ca2+ rhythm persists in hyperpolarizing or low [Ca2+] medium.
As shown in our previous studies in the invertebrate Bulla model (McMahon and Block, 1987), a transmembrane Ca2+ flux may also play a major role in entrainment of the mammalian pacemaker. This has been demonstrated recently in the SCN by Kim do et al. (2005), who showed that voltage-gated Ca2+ channels are required for glutamate-induced phase shifts.
The findings described in our study strengthen the view about the importance of membrane electrical phenomena and directly implicate Ca2+ influx in rhythm generation. Important issues remain about the nature of the required Ca2+ influx (e.g., single cell recordings; restricted to specific circadian phases) and intracellular targets of the flux required for sustained molecular rhythmicity.
Footnotes
This work was supported by National Institutes of Health Grants HL71510 and MH62517 (G.D.B.) and by funds from the Royal Swedish Academy of Sciences (G.B.L.). We thank Dr. Joseph Takahashi (Northwestern University) for supplying the PER2 transgenic mouse.
Correspondence should be addressed to Gene D. Block, Center for Biological Timing, Department of Biology, University of Virginia, Charlottesville, VA 22903. E-mail: gdb@virginia.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257682-05$15.00/0
References
- Abe M, Herzog ED, Yamazaki S, Straume M, Tei H, Sakaki Y, Menaker M, Block GD (2002) Circadian rhythms in isolated brain regions. J Neurosci 22: 350-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aton SJ, Colwell CS, Harmar AJ, Waschek J, Herzog ED (2005) Vasoactive intestinal polypeptide mediates circadian rhythmicity and synchrony in mammalian clock neurons. Nat Neurosci 8: 476-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block GD, Khalsa SB, McMahon DG, Michel S, Geusz M (1993) Biological clocks in the retina: cellular mechanism of biological timekeeping. Int Rev Cytol 146: 83-144. [DOI] [PubMed] [Google Scholar]
- Brereton HM, Harland ML, Froscio M, Petronijevic T, Barritt GJ (1997) Novel variants of voltage-operated calcium channel alpha 1-subunit transcripts in a rat liver-derived cell line: deletion in the IVS4 voltage sensing region. Cell Calcium 22: 39-52. [DOI] [PubMed] [Google Scholar]
- Colwell CS (2001) NMDA-evoked calcium transients and currents in the suprachiasmatic nucleus: gating by the circadian system. Eur J Neurosci 13: 1420-1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS, Michel S, Itri J, Rodriguez W, Tam J, Lelievre V, Hu Z, Liu X, Waschek JA (2003) Disrupted circadian rhythms in VIP and PHI deficient mice. Am J Physiol Regul Integr Comp Physiol 285: R939-R949. [DOI] [PubMed] [Google Scholar]
- Cutler DJ, Haraura M, Reed HE, Shen S, Sheward WJ, Morrison CF, Marston HM, Harmar AJ, Piggins HD (2003) The mouse VPAC2 receptor confers suprachiasmatic nuclei cellular rhythmicity and responsiveness to vasoactive intestinal polypeptide in vitro. Eur J Neurosci 17: 197-204. [DOI] [PubMed] [Google Scholar]
- Dunlap JC (1999) Molecular bases for circadian clocks. Cell 96: 271-290. [DOI] [PubMed] [Google Scholar]
- Harmar AJ, Marston HM, Shen S, Spratt C, West KM, Sheward WJ, Morrison CF, Dorin JR, Piggins HD, Reubi JC, Kelly JS, Maywood ES, Hastings MH (2002) The VPAC(2) receptor is essential for circadian function in the mouse suprachiasmatic nuclei. Cell 109: 497-508. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Sugiyama T, Wallace CS, Gompf HS, Yoshioka T, Miyawaki A, Allen CN (2003) Circadian dynamics of cytosolic and nuclear Ca2+ in single suprachiasmatic nucleus neurons. Neuron 38: 253-263. [DOI] [PubMed] [Google Scholar]
- Kim do Y, Choi HJ, Kim JS, Kim YS, Jeong do U, Shin HC, Kim MJ, Han HC, Hong SK, Kim YI (2005) Voltage-gated calcium channels play crucial roles in the glutamate-induced phase shifts of the rat suprachiasmatic circadian clock. Eur J Neurosci 21: 1215-1222. [DOI] [PubMed] [Google Scholar]
- McMahon DG, Block GD (1987) The Bulla circadian pacemaker: I. Pacemaker neuron membrane potential controls phase through a calcium-dependent mechanism. J Comp Physiol 161: 335-346. [DOI] [PubMed] [Google Scholar]
- Meijer JH, Schwartz WJ (2003) In search of the pathways for light-induced pacemaker resetting in the suprachiasmatic nucleus. J Biol Rhythms 18: 235-249. [DOI] [PubMed] [Google Scholar]
- Moore RY (1991) The suprachiasmatic nucleus and the circadian timing system. In: Suprachiasmatic nucleus-the mind's clock (Klein D, Moore R, Reppert S, eds), pp 13-15. New York: Oxford UP.
- Nitabach MN, Blau J, Holmes TC (2002) Electrical silencing of Drosophila pacemaker neurons stops the free-running circadian clock. Cell 109: 485-495. [DOI] [PubMed] [Google Scholar]
- Sawanobori T, Takanashi H, Hiraoka M, Iida Y, Kamisaka K, Maezawa H (1989) Electrophysiological properties of isolated rat liver cells. J Cell Physiol 139: 580-585. [DOI] [PubMed] [Google Scholar]
- Schaap J, Pennartz CM, Meijer JH (2003) Electrophysiology of the circadian pacemaker in mammals. Chronobiol Int 20: 171-188. [DOI] [PubMed] [Google Scholar]
- Schwartz WJ, Gross RA, Morton MT (1987) The suprachiasmatic nuclei contain a tetrodotoxin-resistant circadian pacemaker. Proc Natl Acad Sci USA 84: 1694-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S, Moore RY (1987) Development of neuronal activity in the rat suprachiasmatic nucleus. Brain Res 431: 311-315. [DOI] [PubMed] [Google Scholar]
- Shibata S, Newman GC, Moore RY (1987) Effects of calcium ions on glucose utilization in the rat suprachiasmatic nucleus in vitro. Brain Res 426: 332-338. [DOI] [PubMed] [Google Scholar]
- Shinohara K, Honma S, Katsuno Y, Abe H, Honma K (1998) Circadian release of amino acids in the suprachiasmatic nucleus in vitro. NeuroReport 9: 137-140. [DOI] [PubMed] [Google Scholar]
- Tominaga K, Geusz ME, Michel S, Inouye ST (1994) Calcium imaging in organotypic cultures of the rat suprachiasmatic nucleus. NeuroReport 5: 1901-1905. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Finkbeiner SM, Cornell-Bell AH (1992) Calcium excitability and oscillations in suprachiasmatic nucleus neurons and glia in vitro J Neurosci 12: 2648-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh DK, Logothetis DE, Meister M, Reppert SM (1995) Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron 14: 697-706. [DOI] [PubMed] [Google Scholar]
- Welsh DK, Yoo SH, Liu AC, Takahashi JS, Kay SA (2004) Bioluminescence imaging of individual fibroblasts reveals persistent, independently phased circadian rhythms of clock gene expression. Curr Biol 14: 2289-2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Isejima H, Matsuo T, Okura R, Yagita K, Kobayashi M, Okamura H (2003) Synchronization of cellular clocks in the suprachiasmatic nucleus. Science 302: 1408-1412. [DOI] [PubMed] [Google Scholar]
- Yamazaki S, Numano R, Abe M, Hida A, Takahashi R, Ueda M, Block GD, Sakaki Y, Menaker M, Tei H (2000) Resetting central and peripheral circadian oscillators in transgenic rats. Science 288: 682-685. [DOI] [PubMed] [Google Scholar]
- Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, Menaker M, Takahashi JS (2004) PERIOD2: LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci USA 101: 5339-5346. [DOI] [PMC free article] [PubMed] [Google Scholar]