

Abstract
Leukocyte-common antigen related (LAR)-like phosphatase receptors are conserved cell adhesion molecules that function in multiple developmental processes. The Caenorhabditis elegans ptp-3 gene encodes two LAR family isoforms that differ in the extracellular domain. We show here that the long isoform, PTP-3A, localizes specifically at synapses and that the short isoform, PTP-3B, is extrasynaptic. Mutations in ptp-3 cause defects in axon guidance that can be rescued by PTP-3B but not by PTP-3A. Mutations that specifically affect ptp-3A do not affect axon guidance but instead cause alterations in synapse morphology. Genetic double-mutant analysis is consistent with ptp-3A acting with the extracellular matrix component nidogen, nid-1, and the intracellular adaptor α-liprin, syd-2. nid-1 and syd-2 are required for the recruitment and stability of PTP-3A at synapses, and mutations in ptp-3 or nid-1 result in aberrant localization of SYD-2. Overexpression of PTP-3A is able to bypass the requirement for nid-1 for the localization of SYD-2 and RIM. We propose that PTP-3A acts as a molecular link between the extracellular matrix and α-liprin during synaptogenesis.
Keywords: synaptogenesis, axon guidance, cell adhesion, α-liprin, syd-2, LAR receptor tyrosine phosphatase, nidogen
Introduction
The type IIa family of receptor protein tyrosine phosphatases (RPTPs), which include leukocyte-common antigen related (LAR) PTP-σ and PTP-δ, are cell adhesion molecules that regulate multiple developmental events, including neurogenesis (den Hertog et al., 1999; Johnson and Van Vactor, 2003; Paul and Lombroso, 2003; Ensslen-Craig and Brady-Kalnay, 2004). The extracellular domains of LAR-like RPTPs contain Ig-like and fibronectin type III (FNIII) repeats. Alternative splicing of RPTP genes result in isoforms that differ in the extracellular domain and can exhibit tissue-specific expression and localization, suggesting the ectodomain confers functional specificity (O'Grady et al., 1994; Pulido et al., 1995; Honkaniemi et al., 1998).
LAR-RPTPs bind laminin-nidogen via the fifth FNIII repeat, and RPTPσ binds agrin and collagen XVIII via its first Ig domain (O'Grady et al., 1998; Aricescu et al., 2002). The functions of the LAR and extracellular matrix (ECM) interaction are not clear. Mutations in human collagen XVIII result in Knobloch's syndrome and defects in neural cell migrations (Kliemann et al., 2003). Mice lacking nidogen-1 exhibit motor deficits that suggest a neurological defect (Dong et al., 2002). LAR-RPTPs may exert specific functions in neural development through distinct ligands.
The intracellular domains of LAR interact with several molecules including the TRIO guanine nucleotide exchange factor (Debant et al., 1996), Enabled (Wills et al., 1999), β-catenin (Kypta et al., 1996), and liprins (Serra-Pages et al., 1998). α-Liprins have emerged as key regulators of synaptogenesis. The presynaptic densities of neuromuscular junctions (NMJs) in Caenorhabditis elegans and Drosophila are abnormal in α-liprin mutants (Zhen and Jin, 1999; Kaufmann et al., 2002). Drosophila LAR (DLAR) null mutations cause presynaptic density defects that are similar to, but weaker than, those in Dliprin mutants (Kaufmann et al., 2002). Dliprin loss-of-function mutations are epistatic to DLAR overexpression effects, suggesting that Dliprin is required for DLAR function at synapses. In mammals, LAR and α-liprin are involved in the development and maintenance of excitatory synapses (Dunah et al., 2005).
ptp-3, the single C. elegans type IIa RPTP gene, encodes two isoforms that differ in the extracellular domain (see Fig. 1A,C) (Harrington et al., 2002). PTP-3A is most similar to vertebrate LAR, whereas the shorter isoform, PTP-3B, lacks the Ig domains and the first four FNIII repeats. PTP-3B regulates neuroblast migration during embryogenesis (Harrington et al., 2002). The function of PTP-3A has not been defined. In C. elegans mutations in collagen XVIII, cle-1 or nidogen, nid-1, causes distinct defects in cell migration, axon guidance, as well as morphological and functional defects in synapses (Kang and Kramer, 2000; Kim and Wadsworth, 2000; Ackley et al., 2001, 2003).
Figure 1.

The ptp-3 locus and protein localization. A, ptp-3 gene structure: exons are shown as shaded boxes, and introns are shown as lines. Large introns are indicated as broken lines with the size listed below. The genomic regions used to drive isoform-specific expression are indicated. The positions of restriction enzyme sites used to make minigenes and GFP fusions are also indicated. ptp-3A-specific exons are shown in red, whereas the ptp-3B-specific exon is shown in yellow. The exons common to ptp-3A and ptp-3B are in blue. The locations of the tm352, ok244, op147, and mu256 lesions are indicated. nt, Nucleotide. B, The gene structure of the PTP-3::GFP isoforms is illustrated. Exons are indicated by boxes, and introns are indicated as lines. The placement of the GFP coding sequences is indicated by the green box. C, PTP-3 proteins: Ig-like domains are illustrated as circles, FNIII domains are illustrated as hexagons, the predicted transmembrane domain is illustrated as an oval, and the phosphatase domains are illustrated as rectangles. The asterisk indicates the FNIII repeat most homologous to the FNIII repeat containing the laminin-nidogen-binding site from mouse LAR. The effects of the mutations are shown below. The X-headed line indicates the deleted portion of the protein and the introduction of a stop codon. D-F, Whole-mount immunolocalization of PTP-3 in wild-type (D), ptp-3(ok244) (E), and ptp-3(mu256) (F) animals. Staining is observed in the nerve ring (arrow) and ventral nerve cord (arrowhead) of wild-type and ptp-3(ok244) animals. All mu256 animals examined lacked reactivity in the nerve cord (F, arrowhead). Approximately 50% of ptp-3(mu256) animals exhibited weak PTP-3 staining in the nerve ring (F, arrow). Scale bar, 10 μm.
We report here the role of ptp-3 in axon guidance and synaptic morphology. PTP-3A specifically localizes to synapses, overlapping the presynaptic proteins SYD-2 and UNC-10. Isoform-specific mutants and transgenes demonstrate that ptp-3B functions in axon guidance and that ptp-3A contributes to synapse development. The synaptic defects of ptp-3 resemble those of nid-1 and syd-2. Genetic double-mutant analyses and cell biology studies support a model whereby PTP-3A acts as a link between extracellular cues and a synaptic organizer.
Materials and Methods
C. elegans strains. All C. elegans strains were maintained at 20-22.5°C as described previously (Brenner, 1974). The following strains were used in this study: N2 (var. Bristol), CH119 [nid-1(cg119)] (Kang and Kramer, 2000), RB633 [ptp-3A(ok244)], CZ333 (juIs1), CZ1200 (juIs76), CZ631 (juIs14), CZ900 [syd-2(ju37)] (Zhen and Jin, 1999), CZ1991 (mnDf90/mIn1mIs14), CZ3761 [ptp-3(mu256)], CZ2857 [ptp-3A(tm352);juIs1], CZ4660 [ptp-3A(ok244);juIs1], CZ3555 [ptp-3(mu256);juIs1], CZ4661 [ptp-3A(ok244);nid-1(cg119); juIs1], CZ3997 [ptp-3(mu256); nid-1(cg119); juIs1], CZ3139 [nid-1(cg119);syd-2(ju37);juIs1], CZ2981 [syd-2(ju37);ptp-3A(tm352);juIs1], CZ4929 [ptp-3(mu256);syd-2(ju37); juIs1], CZ4918 [ptp-3(mu256); nid-1(cg119); syd-2 (ju37); juIs1], CZ4884 (juIs194), CZ4913 [ptp-3(mu256); juIs194], CZ4919 (juIs197), CZ4914 [ptp-3(mu256); juIs197], CZ4885 [syd-2(ju37);juIs194], CZ3992 [ptp-3A(tm352);juEx584], CZ3839 (juEx563), CZ4249 (juEx686), DR2078 [mIn1 mIs14/bli-2(e678)unc-4(e120)] (Edgley and Riddle, 2001), and ZM54 (hpIs3) (Yeh et al., 2005).
The mu256 allele was isolated in a screen for mutants with defective migrations for the Q neuroblast as described previously (Ch'ng et al., 2003). Double and triple mutants were constructed by crossing nid-1(cg119)/+; mIn1 mIs14/+ or syd-2(ju37); mIn1 mIs14/+ males to homozygous ptp-3 mutant animals. The mIn1mIs14 balances the ptp-3 region and contains a green fluorescent protein (GFP) marker (Edgley and Riddle, 2001). Non-GFP offspring were selected and homozygosed. The presence of the nid-1(cg119) allele was confirmed by PCR (Kang and Kramer, 2000). Homozygotes of syd-2(ju37) were chosen by the sluggish movement and egg-laying defects (Zhen and Jin, 1999).
Molecular biology. The ptp-3 gene spans two cosmid clones, C09D8 (GenBank accession number Z46811) and F38A3 (GenBank accession number Z49938). All numbering in this report is given relative to the ATG for ptp-3A (position 7765 in C09D8). The mu256 lesion was identified by sequencing PCR-amplified regions of the ptp-3 exons (primer sequences available on request). Analysis of the sequencing results revealed a single adenosine insertion at position 34784 relative to the ATG of the ptp-3A isoform (corresponding to position 9670 in F38A3; Gen-Bank accession number Z49938). The tm352 deletion removes nucleotides 8494-9040 and the ok244 deletion removes nucleotides 8295-9639 of the ptp-3A coding region.
pCZ512 (PTP-3B::GFP) has been described previously (Harrington et al., 2002). pCZ521 (PTP-3A::GFP) was created by fusing the GFP DNA from A. Fire's vector pPD113.29 to a ptp-3A cDNA generated by reverse transcription-PCR (primer sequences available on request) at a unique PstI site in exon 27 (Fig. 1 A). A genomic fragment corresponding to nucleotide-2565 through exon 4 was generated by PCR, cut with KpnI and NsiI, and fused to the ptp-3A cDNA-GFP fragment. pCZ544 (PTP-3A::GFPΔphos) was created by removing a BglII-AvrII fragment from pCZ512 and replacing it with a BglII-SpeI fragment from pJJ471, a transmembrane::GFP fusion (J. Kramer, personal communication).
Transgenic animals were generated by germ-line transformation as described previously (Mello et al., 1991). PTP-3A::GFP (pCZ521) and PTP-3AΔphos::GFP (pCZ544) were injected into N2 animals at 75 and 68 ng/μl, respectively, with pRF4 [rol-6(SD)] (Mello et al., 1991) at 100 ng/μl to generate juEx563 and juEx584. PTP-3B::GFP (pCZ512) was injected at 25 ng/μl with Pttx-3::RFP (Altun-Gultekin et al., 2001) at 100 ng/μl to generate juEx686 and juEx687. The integrated transgenes PTP-3A::GFP (juIs194) and PTP-3B::GFP (juIs197) were generated by UV/trimethylpsoralen mutagenesis.
Whole-mount immunostaining. Anti-UNC-10 staining was done using the modified Bouin's fixation as described previously (Nonet et al., 1997). For other antibodies, animals were fixed as described previously (Finney and Ruvkun, 1990), with the following modifications. The animals were fixed for 2 h on ice, and the reduction step was performed using 1× borate buffer for 2 h at 37°C. The following primary antisera were used in this study: guinea pig anti-PTP-3 (1:20) (Harrington et al., 2002), mouse anti-GFP (1:1000; Roche, Indianapolis, IN), rabbit anti-UNC-10 (1:1000) (Koushika et al., 2001), chicken anti-UNC-10 (1:2000), rabbit anti-SNT-1 (1:2000) (Nonet et al., 1993), and rabbit anti-SYD-2 (1:2000) (Zhen and Jin, 1999). The following secondary antibodies were purchased from Molecular Probes (Eugene, OR) and were used at 1:2000: Alexa 488-labeled goat anti-guinea pig, Alexa 488-labeled anti-mouse, Alexa 594-labeled anti-rabbit, Alexa 594-labeled anti-chicken, and Alexa 647-labeled anti-rabbit. All images were collected on a Zeiss (Oberkochen, Germany) Pascal confocal microscope using multi-track parameters, with either a 63× (SNB-1::GFP) or 100× (immunofluorescence) objective.
Colocalization. Confocal stacks were projected into a single plane and analyzed using the histogram tool in the Zeiss LSM5 software (version 3.2). A region of interest was drawn around the nerve cord. Thresholding levels were set using the “determine from region of interest” function. The colocalization table was exported to Microsoft Excel (Redmond, WA) for statistical analysis. The percentage of colocalization was determined as the total area of region 3 (pixels at which both channels are above threshold) divided by the total area for that channel (total pixels above threshold).
GFP analysis. For axon morphology, animals were scored blind to genotype by examining cell type-specific GFP markers (juIs76 and juIs14) with an Axioplan 2 microscope using a 63× Plan-apochromat objective and a GFP long-pass filter set (Chroma, Battleboro, VT). A defasciculation event was counted as a region of the nerve cord where two or more processes appeared to become split from one fascicle and were visible along a distance that was greater than the length of two neuronal cell bodies, which is ∼20 μm. Synapse morphology of D type neurons was visualized by juIs1 (Punc-25-SNB-1::GFP) and hpIs3 (Punc-25-SYD-2::GFP) using a Zeiss Pascal LSM confocal microscope. Animals were anesthetized using 0.5% phenoxy-propanol (TCI America, Portland, OR) in M9 and mounted on 5% agar pads. Images were acquired using a 63× Plan-apochromat lens and a 488 argon laser line at 3% power. The microscope was set to use a single scan with a 505 long-pass filter set.
Quantification. Measurements of SNB-1::GFP and UNC-10 puncta were performed on confocal images as described previously (Ackley et al., 2003), with minor modifications and the experimenter blind to genotype. Briefly, confocal images were projected into a single plane using the maximum projection and exported as a tiff file with a scale bar. Using Scion (Frederick, MD) Image, the files were converted to a binary image using the threshold command, so that the binary image resembled the RGB image. A region of interest was drawn around the relevant region of the nerve cords. The following measurement options were selected: area, X-Y center, perimeter/length, ellipse major axis, ellipse minor axis, include interior holes, wand auto measure, and headings. Scaling was set by measuring the scale bar; for GFP, images 7 pixels equaled 1 μm, and for UNC-10 puncta, 11 pixels equaled 1 μm. The “analyze particle” command was used with a minimum size of 4 pixels and a maximum of 10,000 pixels. The following options were selected: outline particles, ignore particles touching edge, include interior holes and reset counter. The resulting measurements were exported to Microsoft Excel for statistical analysis. Significance was determined by a two-tailed Student's t test.
Results
Isolation of new mutations in ptp-3
The previously reported ptp-3 mutation, op147, is a transposon insertion in the phosphatase domain common to both isoforms and results in a strong ptp-3 loss of function during embryogenesis (Harrington et al., 2002). We isolated a new ptp-3 allele, mu256, in a screen for animals with defective Q cell migration (Williams, 2002). Sequencing of the ptp-3 locus from mu256 animals identified a single nucleotide insertion in exon 25 that would cause a frame shift after amino acid Glu1756 in the first phosphatase domain, leading to a premature stop (Fig. 1C). Immunostaining using antisera raised against the PTP-3 phosphatase domains showed that 50% of mu256 animals had no detectable protein (data not shown) and 50% exhibited only a weak reactivity in the synapse-rich nerve ring (Fig. 1F) (n = 100). mu256 animals displayed partially penetrant embryonic (Emb) and larval lethality (Lva) as well as variably abnormal (Vab) morphology phenotypes (Table 1). The lethality and morphology defects were similar to those observed for op147 and in the broods of animals injected with ptp-3 double-stranded RNA (Harrington et al., 2002). Thus, mu256 is a strong loss-of-function mutation affecting both PTP-3 isoforms.
Table 1.
Lethality and Vab in ptp-3 mutants
|
|
|
% Lethalitya |
||||
|---|---|---|---|---|---|---|
| Genotype |
n
|
Emb |
Lva |
Vab |
||
| Wild type | 823 | 0.1 ± 0.01 | 0.1 ± 0.01 | 0.1 ± 0.01 | ||
| ptp-3A(tm352) | 994 | 0.5 ± 0.02 | 0.1 ± 0.01 | 0.1 ± 0.01 | ||
| ptp-3(mu256) | 726 | 5.0 ± 0.12 | 1.1 ± 0.03 | 4.5 ± 0.03 | ||
|
ptp-3(op147)
|
501 |
3.8 ± 0.01 |
0.4 ± 0.02 |
4.8 ± 0.08 |
||
Data are expressed as mean ± SEM.
Percentage of animals that reach adult stage with Vab phenotype.
To analyze the specific requirements for the PTP-3A isoform, we obtained two deletion alleles in the ptp-3A coding region, tm352 and ok244. The tm352 and ok244 deletions remove one or three FNIII repeats, respectively, and result in frame shifts and premature terminations of PTP-3A. Both ptp-3A alleles are likely to eliminate ptp-3A function but are unlikely to affect the expression of PTP-3B because the lesions are outside of the minimal rescuing fragment for ptp-3B (Harrington et al., 2002). By whole-mount immunostaining, a reduced level of staining was observed in tm352 and ok244 animals (Fig. 1 E). Both mutations cause similar phenotypes (see below), and tm352 animals did not exhibit significant Emb or Vab phenotypes (Table 1), suggesting that PTP-3A does not function in embryonic morphogenesis.
PTP-3 expression in the postembryonic nervous system
We have reported previously that ptp-3A and ptp-3B were highly expressed in the postembryonic nervous system using promoter-driven GFP reporter transgenes (Harrington et al., 2002). By modifying the immunostaining procedure, we improved the sensitivity of detecting endogenous PTP-3. We observed that PTP-3 was concentrated at the nerve ring and along the nerve cords in larval and adult animals and showed a punctate pattern (Fig. 1D). To determine the subcellular localization of PTP-3, we performed colocalization experiments with several known synaptic proteins including UNC-49, a GABA receptor present on the postsynaptic muscle surface (Gally and Bessereau, 2003), synaptotagmin (SNT-1), a component of synaptic vesicles (Nonet et al., 1993), and two presynaptic density proteins, SYD-2 and UNC-10, the C. elegans RIM (Zhen and Jin, 1999; Koushika et al., 2001).
Along the nerve cord, PTP-3 was adjacent to but did not overlap UNC-49 (Fig. 2A), consistent with it being expressed in neurons. Within the neurons, PTP-3 partially overlapped the SNT-1-containing domain but was concentrated at the edges of the SNT-1 staining (Fig. 2B). There was a precise colocalization of PTP-3 with SYD-2 and UNC-10 (Fig. 2C,D). More specifically, we observed a smaller punctum of PTP-3 in or near the center of each of the larger SYD-2 puncta. These data indicate that PTP-3 is predominantly associated with the presynaptic density.
Figure 2.
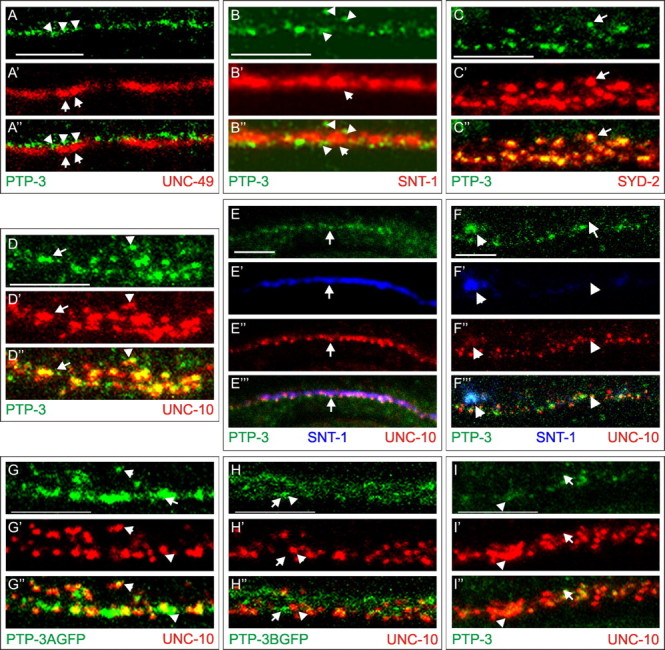
PTP-3A localizes to presynaptic regions. A-D, Costaining of PTP-3 (green) with proteins (red) that label different synaptic domains. The bottom panels are the merged images. A-A″, PTP-3 (arrowheads) does not overlap with UNC-49 (arrows). B-B″, PTP-3 (arrowheads) is concentrated at the edges of SNT-1 (arrows). C-C″, PTP-3 colocalizes with SYD-2. Note the smaller foci of PTP-3 at the center of the SYD-2 puncta (arrows). D-D″, Seventy-five percent of UNC-10 puncta are either overlapped (arrowheads) or coincident (arrows) with PTP-3. E, F, PTP-3 is trafficked to synapses independent of synaptic vesicles. A wild-type animal (E) and an unc-104 animal (F) are shown with triple labeling of anti-PTP-3 (green), anti-SNT-1 (blue), and anti-UNC-10 (red). The merged views are shown in the bottom panels. In unc-104 mutants, SNT-1 is mostly retained in cell bodies (arrow), whereas PTP-3 and UNC-10 are present along the nerve cord (arrowheads). G, H, The localization of the PTP-3 isoform-specific GFP fusion proteins is shown relative to UNC-10. G-G″, PTP-3A::GFP appeared punctate and overlapped with the UNC-10 puncta (arrows), although the protein was also observed in larger clusters than the endogenous protein (arrowheads). H-H″, PTP-3B::GFP showed diffused staining throughout the nerve cord (arrows) and rarely overlapped with UNC-10 puncta (arrowhead). I-I″, In ptp-3A(ok244) mutants, PTP-3 (arrowhead) staining is reduced, and showed little overlap with UNC-10 (arrows). Scale bars, 5 μm.
To address whether PTP-3 localization was dependent on synaptic vesicles, we analyzed the accumulation of SNT-1, PTP-3, and UNC-10 in unc-104(e1265) kinesin mutants, which causes synaptic vesicles to be retained in cell bodies (Hall and Hedgecock, 1991). We observed that PTP-3 and UNC-10 puncta showed colocalization in regions of the nerve cord lacking synaptic vesicles (Fig. 2E,F). Thus, like other presynaptic density components (Zhen and Jin, 1999; Koushika et al., 2001), PTP-3 appears to be trafficked to synapses independent of synaptic vesicles.
PTP-3A is synaptic, and PTP-3B is extrasynaptic
To address the subcellular localization of the PTP-3A and PTP-3B isoforms, we generated isoform-specific GFP fusion transgenes. GFP was inserted in-frame in the second phosphatase domain 53 aa from the stop codon, and each fusion protein was expressed under the isoform endogenous promoter (Fig. 1B). Both transgenes are functional (Harringon et al., 2002) (see below). PTP-3B::GFP showed a temporally regulated expression, with high expression throughout embryogenesis and larval development until the L1-L2 larval transition (data not shown). In the adult nervous system, PTP-3B::GFP was present in the nerve ring and along axons of the ventral and dorsal nerve cords (Fig. 2H and data not shown) and was also observed in the pharyngeal epithelium and the developing uterus throughout development (data not shown). Most of the PTP-3B::GFP was adjacent to the UNC-10 puncta, with only a small amount of GFP overlapping with UNC-10 (Fig. 2H). PTP-3A::GFP expression was first detected around the twofold stage of embryonic development (450 min after fertilization) in the nerve ring and nerve cords and continued at a constant level through larval development (data not shown). PTP-3A::GFP was seen in a punctate pattern along nerve processes (Fig. 2G) and overlapped with UNC-10 in a pattern that was similar to the endogenous protein (Fig. 2D,G) These results suggest that PTP-3A is the major isoform that is localized to presynaptic domains. Consistent with this conclusion, in ptp-3A(ok244) mutant animals, <25% of the UNC-10 puncta (n = 200) contained any PTP-3, and the amount of PTP-3 present at those UNC-10 puncta was reduced (Fig. 2I).
PTP-3B has a major role in motor axon guidance
Previous analyses using a pan-neuronal morphological marker revealed that the organization of the nervous system was grossly normal in ptp-3(op147) animals (Harrington et al., 2002). To address the effects of ptp-3 mutations on the nervous system, we focused our analysis on the ventral cord motor axons using cell type-specific markers. The DA and DB neurons extend a dendritic process along the ventral nerve cord and an axonal commissure circumferentially along the epidermis to the dorsal nerve cord (Fig. 3A). The DD and VD neurons extend a single process along the ventral nerve cord that bifurcates and extends to the dorsal nerve cord (Fig. 3F). These axon projection patterns are highly stereotyped. Using a GFP marker driven from the unc-25 promoter for D type neurons and a GFP marker driven from the acr-2 promoter for the A and B type neurons (Huang et al., 2002), we observed <10% of wild-type animals that exhibited any deviations (Table 2).
Figure 3.

Mutations in ptp-3 cause defects in axon guidance. A, Schematic diagram describing the axon outgrowth patterns for the DA and DB cholinergic motor neurons. The cell bodies are illustrated as open circles, whereas the processes are indicated as black lines. The position of the vulva is indicated by a open diamond. The shaded box is the region shown in B and C. B, In wild-type animals, the DB6 and DB7 neurons (indicated by arrows) extend processes to the right, whereas the adjacent neurons, DA6 and DA7, extend processes to the left. C, In this ptp-3(mu256) animal, the DB6 and DB7 axons (arrows) incorrectly extend on the left side of the nerve cord. D, Region of the ventral nerve cord that has become defasciculated (arrow) from the main bundle (arrowheads). E, Two axons that failed to terminate at the end of the dorsal nerve cord (arrow) are observed to turn anteriorly and continue extending. F, Schematic diagram of the DD and VD GABAergic motor neurons. G, In wild-type animals, the dorsal nerve cord (arrows) is observed as a continuous bundle. H, In ptp-3 mutants, we observed gaps along the nerve cord where the axons appear to have terminated their migration prematurely. I, In wild-type animals, the commissural processes (arrows) extend from the ventral nerve cord to the dorsal cord (arrowheads). J, Two axon commissures failed to reach the dorsal nerve cord (arrowheads): one stopped migrating prematurely (arrow), whereas a second turned prematurely (dashed arrow). DA and DB (K) and DD and VD (L) axon guidance data are displayed as the percentage of animals that exhibited a side choice error (□), targeting error ( ), or defasciculation (▪). Information on specific axon outgrowth phenotypes is in Table 2. L/R, Left/right.
), or defasciculation (▪). Information on specific axon outgrowth phenotypes is in Table 2. L/R, Left/right.
Table 2.
Axon guidance defects in ptp-3 mutant animals
|
Genotype |
n |
Left/righta |
Targetingb |
Defasciculationc |
|---|---|---|---|---|
| DA/DB cholinergic neurons | ||||
| Wild type | 60 | 8.3% | 0.0% | 1.7% |
| ptp-3A(tm352) | 15 | 13.3% | 0.0% | 6.7% |
| ptp-3A(ok244) | 20 | 5.0% | 0.0% | 0.0% |
| ptp-3(op147) | 40 | 22.5% | 10.0% | 20.0% |
| ptp-3(mu256) | 20 | 40.0% | 10.0% | 50.0% |
| ptp-3(mu256);juEx563(PTP-3A::GFP) | 50 | 36.0% | 8.0% | 40.0% |
| ptp-3(mu256);juEx686(PTP-3B::GFP) | 60 | 12.3% | 5.3% | 8.3% |
| DD/VD GABAergic neurons | ||||
| Wild type | 50 | 2.0% | 0.0% | 0.5% |
| ptp-3A(tm352) | N/D | |||
| ptp-3A(ok244) | N/D | |||
| ptp-3(op147) | 50 | 22.0% | 12.0% | 50.0% |
| ptp-3(mu256) | 50 | 30.0% | 22.0% | 52.0% |
|
ptp-3(mu256)/mnDf90
|
25 |
16.0% |
28.0% |
56.0% |
N/D, Not determined.
Percentage of animals with axons that made a side choice error in exit from the ventral nerve cord.
Percentage of animals with axons that terminated prematurely or overgrew the target.
Percentage of animals that exhibited two or more regions of defasciculated axons along the nerve cords.
Neither of the ptp-3A-specific alleles, tm352 or ok244, exhibited gross defects in motor neuron axon outgrowth and guidance (Table 2). However, the mu256 and op147 mutations caused a significant level of projection defects, such as axons that made incorrect choices in their exit from the ventral midline (Fig. 3C). The most noticeable defect was the defasciculation of motor axons along the dorsal or ventral nerve cords (Fig. 3D). In addition, some axons were observed to overgrow their appropriate termination point (Fig. 3E) or stop prematurely (Fig. 3H,J). The axon projection defects of the DD and VD neurons in mu256 in trans to mnDf90, a chromosomal deficiency for the ptp-3 locus, was comparable with that of mu256 homozygous animals, consistent with mu256 being a strong loss-of-function mutation in ptp-3 (Fig. 3L). To further address how the two PTP-3 isoforms function in axon guidance, we introduced the isoform-specific GFP transgenes into mu256 animals. Expression of the PTP-3B::GFP isoform essentially rescued the DA and DB axon guidance defects in mu256 animals (Table 2, Fig. 3K). Specific expression of PTP-3A::GFP only showed a weak partial rescue (Table 2). These data therefore argue that PTP-3B is the major isoform that functions in axon outgrowth and guidance in C. elegans.
ptp-3A contributes to synapse development
Because PTP-3A is localized to presynaptic regions, we asked whether mutations in ptp-3 caused any defects in synaptic development. We first examined the pattern of synapses by staining animals with UNC-10 antisera. In wild-type animals, UNC-10 was distributed in a set of evenly sized and spaced puncta (Fig. 4A). We measured the puncta area and the number of puncta along a 100 μm section of nerve cord (see Materials and Methods for details). In wild-type animals, the average area was 0.17 ± 0.01 μm2 (mean ± SEM), with an average of 87.3 ± 10.5 puncta per 100 μm (Fig. 4 E, Table 3). In ptp-3A mutants (tm352 and ok244), the overall UNC-10 puncta appeared to be formed in larger aggregates than in wild-type animals (Fig. 4 B, Table 3). In ptp-3A(-) animals, UNC-10 puncta showed an average area of 0.48 ± 0.03 μm2, with an average of 53.0 ± 4.0 puncta per 100 μm. In ptp-3(mu256) animals, the defects seen with UNC-10 expression were not worse (0.36 ± 0.03 μm2, 43.0 ± 10.2 puncta per 100 μm), suggesting that the loss of PTP-3A would account for the observed defects.
Figure 4.

Expression of UNC-10 in mutants. A-D, UNC-10 puncta are indicated by arrows, whereas abnormally sized gaps are indicated by arrowheads. A, In wild-type animals, UNC-10 puncta were evenly sized and spaced along the nerve cords. B, In ptp-3, UNC-10 puncta appeared misshapen. In this ptp-3A mutant, we observed smaller and larger puncta that were aberrantly spaced. C, D, Similar defects were observed in nid-1(cg119) (C) and syd-2(ju37)(D) animals. E, Area of UNC-10 puncta in wild type and in ptp-3, nid-1, and syd-2 mutants, plotted as the mean ± SEM.
Table 3.
RIM puncta measurements by genotype
|
Genotype |
Number of synapses |
Area (mm2) |
Number of animals |
Puncta/100 mm |
|---|---|---|---|---|
| Wild type | 804 | 0.17 ± 0.01 | 8 | 87.3 ± 10.5 |
| ptp-3A(−) | 613 | 0.49 ± 0.03** | 8 | 51.2 ± 4.5* |
| ptp-3(mu256) | 442 | 0.36 ± 0.03** | 4 | 43.0 ± 10.2* |
| juls194 (PTP-3A::GFP) | 131 | 0.22 ± 0.02 | 3 | 71.3 ± 2.5 |
| ptp-3(mu256);juls194 | 493 | 0.17 ± 0.01 | 3 | 96.2 ± 14.5 |
| juls197 (PTP-3B::GFP) | 501 | 0.18 ± 0.01 | 3 | 89.7 ± 2.2 |
| ptp-3(mu256);juls197 | 982 | 0.35 ± 0.05** | 3 | 43.6 ± 3.5* |
| ptp-3A(tm352);juEX584 (PTP-3ADphos::GFP) | 85 | 0.39 ± 0.08** | 3 | 39.0 ± 23.0* |
| nid-1(cg119) | 345 | 0.39 ± 0.04** | 3 | 66.8 ± 2.8 |
| nid-1(cg119);juls194 | 700 | 0.18 ± 0.01** | 3 | 37.0 ± 6.7** |
| syd-2(ju37) | 563 | 0.43 ± 0.02** | 5 | 67.03 ± 10.4 |
|
syd-2(ju37);juls194
|
583 |
0.12 ± 0.01**
|
3 |
55.0 ± 7.1*
|
Data are expressed as mean ± SEM. Values significantly different from wild type: *p < 0.05; **p < 0.001.
To evaluate the effect on the morphology of individual synapses, we used the transgene expressing synaptobrevin fused to GFP (SNB-1::GFP) driven by the unc-25 promoter (Hallam and Jin, 1998; Nonet, 1999). This SNB-1::GFP marker provides spatial resolution to detect changes in GABA presynaptic morphology. In wild type animals, SNB-1::GFP appears as a set of evenly sized and spaced fluorescent puncta, with an average area of 0.8 ± 0.0 μm2 (Fig. 5A, Table 4). In ptp-3A and ptp-3(mu256) mutants, the SNB-1::GFP puncta showed significant enlargement, with an average area of 1.5 ± 0.1 μm2. The area of SNB-1::GFP puncta in tm352/mnDf90 animals (1.7 ± 0.2 μm2) was not significantly different from the ptp-3A(tm352) homozygous animals, supporting that tm352 and ok244 represent a complete loss of function for ptp-3A, and suggesting that the defects in SNB-1::GFP puncta are mostly caused by the loss of PTP-3A. As another way of assessing the overall effect of ptp-3 mutations on SNB-1::GFP puncta, we plotted the observed areas using a box-and-whiskers plot (Fig. 6). This analysis revealed that the population of SNB-1::GFP puncta in ptp-3 mutants had a greater number of puncta that were larger than those in wild type. We calculated the values representing the 10th and 90th percentiles in wild-type animals as being 0.2-1.5 μm2, respectively, and found that 30% of the SNB-1::GFP puncta in ptp-3A(-) mutants were >1.5 μm2, whereas only 1.5% were <0.2 μm2. Overall, the defect in SNB-1::GFP was parallel to the alterations in UNC-10 staining.
Figure 5.

Pattern of SNB-1::GFP in GABAergic synapses in mutants. A, In wild-type animals, the SNB-1::GFP pattern was observed as equally sized and evenly spaced fluorescent puncta (arrow). B, In ptp-3A(tm352) mutants, SNB-1::GFP was present in larger, irregularly spaced puncta (arrows). C, In syd-2(ju37) mutants, SNB-1::GFP puncta were diffused and often appeared contiguous. D, SNB-1::GFP puncta in nid-1(cg119) mutants were elongated and aberrantly spaced. Cell bodies (arrowhead) were occasionally visible along the ventral nerve cord in all genotypes. E, The area of SNB-1::GFP puncta in the wild-type and various mutants, plotted as the mean ± SEM.
Table 4.
SNB-1::GFP area by genotype
|
|
|
|
Wild-type outliersa |
||
|---|---|---|---|---|---|
|
|
Number of synapses |
Area (mm2) |
<0.22 mm2
|
>1.31 mm2
|
|
| Wild type | 874 | 0.8 ± 0.0 | 8.8% | 10.1% | |
| ptp-3A(tm352) | 289 | 1.6 ± 0.1** | 1.5% | 30.0% | |
| ptp-3A(ok244) | 569 | 1.4 ± 0.1** | 3.7% | 28.1% | |
| ptp-3A/mmDf90 | 202 | 1.7 ± 0.2** | 9.4% | 36.1% | |
| ptp-3(mu256) | 418 | 1.5 ± 0.1** | 2.9% | 38.3% | |
| syd-2(ju37) | 852 | 1.2 ± 0.1** | 30.0% | 19.6% | |
| syd-2(ju37);ptp-3A(tm352) | 982 | 1.0 ± 0.1* | 42.3% | 18.4% | |
| syd-2(ju37);ptp-3(mu256) | 501 | 1.3 ± 0.1** | 26.1% | 29.1% | |
| nid-1(cg119) | 331 | 1.7 ± 0.1** | 10.9% | 34.1% | |
| nid-1(cg119);ptp-3A(ok244) | 493 | 1.9 ± 0.1** | 14.0% | 44.2% | |
| nid-1(cg119);ptp-3(mu256) | 717 | 1.7 ± 0.1** | 3.5% | 35.6% | |
| cle-1(cg120) | 205 | 2.3 ± 0.1** | 0.5% | 57.1% | |
| cle-1(cg120);ptp-3A(tm352) | 146 | 1.7 ± 0.2** | 26.7% | 32.9% | |
| cle-1(cg120);ptp-3(mu256) | 100 | 1.1 ± 0.2 | 41.0% | 28.0% | |
| syd-2(ju37);nid-1(cg119) | 484 | 1.5 ± 0.2** | 36.1% | 20.2% | |
| syd-2(ju37);nid-1(cg119);ptp-3A(tm352) | 366 | 1.2 ± 0.1** | 33.6% | 23.5% | |
|
syd-2(ju37);nid-1(cg119);ptp-3(mu256)
|
438 |
1.4 ± 0.1**
|
14.2% |
28.5% |
|
Data are expressed as mean ± SEM. Values significantly different from wild type:*p<0.05;**p<0.001.
Percentage of puncta below the 10th percentile or above the 90th percentile of wild type.
Figure 6.

Boxplots of SNB-1::GFP quantitation. The area of all SNB-1::GFP measurements by genotype were represented as box plots. The box represents the values within the 25th to 50th percentiles, and the white bar (or gray arrowhead) indicates the mean observed value. The ends of the whiskers show the 10th to 90th percentiles, with the 5th and 95th percentiles indicated by black circles. The dotted lines illustrate the values that we set as limits to define puncta that are outliers from wild type as defined by the 10th (0.2 μm2) and 90th (1.5 μm2) percentile values. In wild type, the box and whiskers are compact, indicating the measurements fall into a reproducible pattern. The SNB-1::GFP puncta in mutants was more variable in size, resulting in larger plots. The shape of the box and whiskers is similar in the syd-2 single, double, and triple mutants demonstrating that these mutants are phenotypically similar in their effect on SNB-1::GFP accumulation. nid-1 single and nid-1;ptp-3A double mutants exhibit a similar distribution. In contrast, cle-1 single and cle-1;ptp-3A double mutants are phenotypically distinct.
To further address the role of PTP-3A in synapse formation, we tested whether expressing PTP-3A::GFP could specifically rescue the synaptic defects in ptp-3(mu256) animals. Because of interference from the GFP tags, we were unable to use the SNB-1::GFP and instead used UNC-10 immunostaining. Neither PTP-3A::GFP nor PTP-3B::GFP caused a significant change in the size of UNC-10 puncta in wild-type animals (Fig. 4E, Table 3). ptp-3(mu256) animals that were expressing PTP-3A::GFP had an average area that was equivalent to wild type (0.17 ± 0.01 μm2), whereas UNC-10 puncta in mu256 animals expressing PTP-3B::GFP were significantly larger than wild type (0.35 ± 0.05 μm2) (Fig. 4E, Table 3). The distribution of UNC-10 puncta was also rescued in PTP-3A::GFP-expressing animals (96.0 ± 14.5 puncta per 100 μm) but not in PTP-3B::GFP-expressing animals (43.6 ± 3.5 puncta per 100 μm) (Table 3). We conclude from these analyses that PTP-3A is the major functional isoform at synapses and that loss of PTP-3A results in UNC-10 and SNB-1::GFP accumulating in larger-than-normal puncta.
Because PTP-3A and PTP-3B only differ in the extracellular domain, we asked whether the extracellular domain was sufficient for the function at synapses. Experiments from Drosophila have demonstrated that the extracellular portion of LAR may act to signal to other cells, independent of the phosphatase domains (Maurel-Zaffran et al., 2001). We generated a version of PTP-3A::GFP lacking the phosphatase domains (PTP-3AΔphos::GFP). ptp-3A(tm352) transgenic animals expressing this protein had an average UNC-10 area of 0.39 ± 0.08 μm2 (Fig. 4E, Table 3), indicating that the function of PTP-3A in synapse formation is dependent on the cytoplasmic domain.
ptp-3 genetically interacts with syd-2/α-liprin and nid-1 during synapse formation
To understand how PTP-3A could function at synapses, we examined whether ptp-3 mutants exhibited genetic interactions with other known synaptogenesis genes. LAR-RPTPs can bind α-liprins via the second phosphatase domain, and the two proteins have been reported to function together in synapse formation (Kaufmann et al., 2002; Dunah et al., 2005). We have shown that mutations in C. elegans α-liprin, syd-2, alter the morphology of synapses (Figs. 4D, 5C) (Zhen and Jin, 1999). The extracellular portion of LAR-like receptors can bind nidogen-laminin and collagen XVIII. We have shown that mutations in C. elegans nidogen, nid-1, and collagen XVIII, cle-1, result in distinct changes in synaptic morphology (Ackley et al., 2003). To assay the interactions between ptp-3 and these genes, we constructed double and triple mutants among the three genes and examined the synapse morphology using the SNB-1::GFP transgene.
The SNB-1::GFP puncta in syd-2(ju37) animals had an average area of 1.2 ± 0.1 μm2 and were not enhanced in the ptp-3A; syd-2 (1.0 ± 0.1 μm2) or ptp-3(mu256); syd-2 (1.3 ± 0.1 μm2) double mutants (Fig. 5E, 6; Table 4). The syd-2; ptp-3 double mutants were significantly different from ptp-3A(-) alone (p < 0.05) but not from syd-2(ju37) (p > 0.05). Box-and-whiskers plot analysis revealed that syd-2 mutants had more small puncta (<0.2 μm2; 30.0%) and more large puncta (>1.5 μm2; 19.6%) than the wild type. syd-2;ptp-3A double mutants resembled syd-2, having 42.3% smaller puncta and 18.4% larger puncta. Thus, our results indicate that the loss of syd-2 is epistatic to the mutations in ptp-3.
Loss-of-function mutations in nid-1 cause SNB-1::GFP puncta to appear elongated, with more severe effects in the ventral nerve cord than the dorsal cord (Ackley et al., 2003). In the ventral nerve cord, nid-1(cg119) animals had an average punctum area of 1.7 ± 0.1 μm2, which was significantly larger than in wild type (p < 0.005). The puncta size, distribution, and morphology in the ventral cords of nid-1;ptp-3A(ok244) (1.9 ± 0.1 μm2) or nid-1;ptp-3(mu256) (1.7 ± 0.1 μm2) double mutants were not significantly different from nid-1 single mutants (p > 0.05) (Figs. 5E, 6; Table 4). These results suggest that mutations in ptp-3 do not enhance the SNB-1::GFP accumulation defects associated with the loss of nid-1.
In contrast, in either ptp-3A;cle-1 or ptp-3(mu256);cle-1 double mutants, we observed more severe phenotypes than in either single mutant alone. In cle-1 mutants, SNB-1::GFP puncta are larger than in wild type, with an average area of 2.3 ± 0.1 μm2, and are separated by long distances (Ackley et al., 2003). These defects are morphologically distinct from those in the syd-2 or nid-1 mutants. In cle-1 mutants, 57.1% of the puncta were larger than the 90th percentile in wild type, and none were smaller than the 10th percentile (Fig. 6). In the double mutants, we observed an average area of 1.7 ± 0.1 μm2 for ptp-3A;cle-1 and 1.1 ± 0.1 μm2 for ptp-3(mu256);cle-1, as well as a new phenotype such that 26.7 and 40.0% of the puncta, respectively, were <0.2 μm2, a phenotype that was not seen in either of the single mutants (Table 4). The combined loss of cle-1 and ptp-3 has a more detrimental effect on SNB-1::GFP accumulation than the loss of either single gene. These data argue that cle-1 and ptp-3 likely affect synaptic development via distinct mechanisms.
Because both nid-1 and syd-2 appeared epistatic to ptp-3, we asked whether removing nid-1 from either the syd-2 single-mutant or the syd-2;ptp-3 double-mutant animals caused any further alterations in SNB-1::GFP accumulation. The nid-1; syd-2 double mutants and nid-1; syd-2; ptp-3A or nid-1;syd-2;ptp-3(mu256) triple mutants all resembled syd-2 single mutants in terms of the shape and distribution of SNB-1::GFP puncta (Figs. 5E, 6; Table 4). nid-1;syd-2 animals had an average area of 1.5 ± 0.2 μm2, and the triple mutants had average areas of 1.2 ± 0.1 and 1.3 ± 0.1 μm2, respectively. All were significantly different from wild type (p < 0.005) but not from syd-2 single mutants (p > 0.05). In addition, all of the mutant combinations with syd-2 mutants were observed to have SNB-1::GFP puncta that were of a size and morphology most similar to syd-2 single mutants (Table 4). These data show that the loss of syd-2 is epistatic to mutations in nid-1 and ptp-3.
PTP-3 localization is dependent on nid-1 and syd-2
The genetic interactions between ptp-3, nid-1, and syd-2 could be explained by several different models. NID-1 and PTP-3 could act to recruit SYD-2 to synapses, or SYD-2 and PTP-3 could function to organize NID-1 in the ECM. To differentiate between these models, we examined the amount of PTP-3 immunofluorescence that colocalized with UNC-10 in the nid-1 and syd-2 single mutants and nid-1;syd-2 double-mutant animals (Fig. 7) (see Materials and Methods). In wild-type animals, 41.7% of PTP-3 signal above threshold was colocalized with UNC-10 (n = 13). In nid-1 mutants, PTP-3 levels appeared reduced, and the protein was abnormally distributed into smaller and larger puncta with gaps in the staining pattern (Fig. 7B). However, 35.1% of the remaining PTP-3 was colocalized with UNC-10 (n = 15). This suggests that NID-1 is required to maintain PTP-3 levels but does not seem to be necessary for the association of PTP-3 with UNC-10.
Figure 7.

NID-1 and SYD-2 have distinct effects in PTP-3 localization. The localization of PTP-3 (green) and UNC-10 (red) in nid-1(cg119), syd-2(ju37), and nid-1(cg119);syd-2(ju37) double mutants is shown. The boxed regions are magnified in the right panels. A, A′, PTP-3 and UNC-10 exhibited an overlapped pattern of accumulation in wild-type animals. B, B′, In nid-1 animals, PTP levels were reduced, but the pattern remained punctate. When present, PTP-3 coincided with UNC-10 puncta (arrowhead), whereas some UNC-10 puncta lacked PTP-3(arrow). C, C′, In syd-2 mutants, PTP-3 was diffuse and had a reduced coincidence with UNC-10 (arrowhead). PTP-3 and UNC-10 had accumulated independently (arrows), an event that was rarely seen in wild type (<25%). D, D′, In nid-1;syd-2 double mutants, PTP-3 detection was moderate but present in a more punctate pattern than syd-2 single mutants. However, PTP-3 and UNC-10 only rarely coincided (arrowhead); rather, each appeared to be exclusively punctate (arrows). Scale bar, 5 μm.
In syd-2 animals, the levels of PTP-3 protein were comparable with that of wild type, but PTP-3 was distributed diffusely along the nerve cords. Only 14.7% of the PTP-3 present was colocalized with UNC-10 (Fig. 7C)(n = 8). PTP-3 was seen in some cell bodies in syd-2 animals (15 of 100). This phenotype was not observed in wild type (0 of 100) or nid-1 mutants (0 of 100). These observations are suggestive of SYD-2 acting as a molecular scaffold at synapses and being important for PTP-3 trafficking to synapses. In nid-1;syd-2 double mutants an additive effect was seen such that PTP-3 levels were reduced, and ∼19.9% of the remaining PTP-3 protein was colocalized with UNC-10 (Fig. 7D) (n = 10). Thus, NID-1 and SYD-2 have distinct roles in the synaptic accumulation of PTP-3.
The decreased PTP-3 levels in nid-1 mutants suggested that PTP-3 does not function to organize NID-1. To confirm this, we examined the accumulation of nidogen in ptp-3 mutants. In wild-type animals, NID-1 was present in the mantle of the mechanosensory neurons and between the muscle edge and nerve processes (Kang and Kramer, 2000) (Fig. 8A). NID-1 accumulation was normal in ptp-3 mutants, indicating that PTP-3A does not act to organize NID-1 in the synaptic ECM (Fig. 8B).
Figure 8.

PTP-3 and NID-1 regulate SYD-2 accumulation. A, B, NID-1 localization in wild-type (A) and ptp-3A (B) animals is comparable. NID-1 (green) is localized in a continuous pattern at the interface of the ventral nerve cord fascicles and the muscles (arrowheads) and in a punctate pattern in the mantle of the mechanosensory neurons (arrows). C-H, SYD-2 (red) localization. C, In wild-type animals, SYD-2 was present along the nerve cords in a punctate pattern (arrowheads). D, syd-2(ju37) animals lacked staining (the position of the ventral nerve cord is indicated by arrowheads). E, F, In ptp-3 mutants, SYD-2 was observed in larger puncta (arrows) and also retained in cell bodies (arrowhead). G, In nid-1 animals, SYD-2 was diffuse, and the puncta (arrows) appeared enlarged. SYD-2 was also found in cell bodies (arrowheads). H, In nid-1 mutants expressing PTP-3A::GFP, SYD-2 expression is punctate (arrows), and the amount of SYD-2 in cell bodies is reduced (arrowheads). I, SYD-2::GFP expressed in the GABAergic neurons of wild-type animals appears punctate with the clusters being evenly sized and spaced (arrowheads). GFP is observed in the cell bodies in all genotypes (arrows; I-K). This effect is likely a result of overexpression and not the genetic background. J, ptp-3A(tm352) mutants exhibit an altered pattern of SYD-2::GFP accumulation, with larger clusters of GFP (arrowheads). K, nid-1(cg119) animals have enlarged SYD-2::GFP puncta (arrowheads) that are separated by large gaps. L, The mean area of SYD-2::GFP puncta was plotted by genotype. Scale bars, 10 μm.
SYD-2 synaptic localization is altered in ptp-3 and nid-1 mutants
We subsequently examined the localization of SYD-2 in the ptp-3 and nid-1 mutant animals using SYD-2 antibodies (Zhen and Jin, 1999). In wild-type animals, SYD-2 localization was detected as puncta along the nerve cords (Fig. 8C) and in the body wall muscles (Zhen and Jin, 1999). In ptp-3 mutants, SYD-2 puncta were large and diffuse with frequent gaps along the nerve cord (Fig. 8E,F). In nid-1 mutants, SYD-2 appeared diffuse but had few gaps (Fig. 8G).
To quantitate the effect of the loss of ptp-3A and nid-1 on SYD-2, we used a SYD-2::GFP fusion protein that was expressed under the unc-25 promoter (Yeh et al., 2005). In wild-type animals, the SYD-2::GFP appears as a set of regularly sized and spaced puncta along the nerve cords, with an average of 0.7 ± 0.1 μm2 (Fig. 8I). In ptp-3A(tm352) mutants, we found the puncta were significantly enlarged and clustered together (Fig. 8J). The average area of SYD-2::GFP puncta in ptp-3A(tm352) mutants was 1.0 ± 0.1 μm2 (p < 0.05). Similarly, we found that in nid-1 mutants, SYD-2 was abnormally spaced and clustered into larger aggregates, with an average of 1.0 ± 0.1 μm2 (p < 0.05) (Fig. 8K). These observations suggest that both NID-1 and PTP-3 are required to localize or maintain SYD-2 at synapses.
Expression of PTP-3A bypasses nid-1 but not syd-2 mutants
To further test whether nid-1 solely functions in maintaining synaptic PTP-3A levels, we elevated the expression of PTP-3A in nid-1 mutants and examined UNC-10 expression. Although the average number of puncta per 100 μm (41.9 ± 8.9) was not restored to wild-type levels (87.3 ± 10.5), measurement of UNC-10 puncta showed that the average area (0.17 ± 0.01 μm2) was restored to wild-type levels (p > 0.05). The UNC-10 area was significantly different from nid-1 animals not expressing PTP-3A::GFP (0.39 ± 0.04 μm2) (Fig. 4E, Table 3). Moreover, immunostaining of anti-SYD-2 showed that SYD-2 accumulation was more punctate in nid-1; juIs194[PTP-3A::GFP] than nid-1 alone (Fig. 8H). In contrast to the effects with nid-1, PTP-3A::GFP expression in syd-2 mutant animals did not restore UNC-10 puncta to wild-type levels (Fig. 4 E, Table 3). These results support our conclusion that NID-1 is required to maintain PTP-3A protein levels at synapses and that SYD-2 is required for PTP-3A to regulate UNC-10 accumulation.
Discussion
LAR-RPTP isoforms provide functional specificity
LAR-like receptor tyrosine phosphatases have been broadly implicated in the patterning of the nervous system by regulating events such as neurite outgrowth, target recognition, and synaptogenesis (Clandinin et al., 2001; Johnson and Van Vactor, 2003; Dunah et al., 2005). One challenge in understanding RPTP function is identifying how these molecules function in multiple aspects of neurogenesis. Alternative splicing of RPTP genes generates receptor isoform diversity, suggesting that distinct isoforms could regulate specific RPTP functions (O'Grady et al., 1994; Pulido et al., 1995). We have shown here that the two PTP-3 isoforms exhibit distinct roles in axon guidance and synaptic organization. The types of axon outgrowth and guidance defects in ptp-3 mutants resemble those that have been reported for LAR homologs in Drosophila and vertebrates (Desai et al., 1996; Krueger et al., 1996; Garrity et al., 1999; Clandinin et al., 2001; Xie et al., 2001; Stepanek et al., 2005). Our observations indicate these functions of LAR are evolutionarily conserved and that receptor isoforms provide functional specificity.
PTP-3 isoforms signal via distinct pathways
In addition to axon guidance and synaptic defects, mutations that affect both ptp-3 isoforms can cause embryonic lethality resulting from defects in neuroblast migration. ptp-3 mutations synergize strongly with mutations affecting other pathways involved in neuroblast migration (VAB-1/Eph receptor, EFN-4/Ephrin) (Chin-Sang et al., 2002; Harrington et al., 2002). Both neuroblast migration and axon guidance appear to be fulfilled by the short isoform PTP-3B, because ptp-3A-specific mutations do not exhibit axon guidance or embryonic morphogenesis defects, nor do they synergize with Eph signaling mutations (our unpublished results).
PTP-3A regulates synapse formation, as does NID-1 and SYD-2. Mutations in nid-1 cause axon guidance defects that are distinct from those in ptp-3 mutant animals (Kim and Wadsworth, 2000; Ackley et al., 2003). Likewise, mutations in Eph signaling cause synaptic defects that are distinct from those in ptp-3 (B. D. Ackley and Y. Jin, unpublished data). These observations suggest that ventral neuroblast migration and axon guidance are regulated by similar mechanisms.
The PTP-3A protein contains all of the domains of PTP-3B, but the two proteins are not functionally equivalent. What might lead to the differences observed in function for PTP-3B and PTP-3A? PTP-3B is expressed earlier, and in more tissue types, than is PTP-3A, suggesting that some of the differences in PTP-3 function may be attributable to expression differences. However, within neurons, we found that PTP-3A and PTP-3B are localized to distinct subcellular compartments. An interaction between PTP-3A and a ligand that is dependent on the PTP-3A-specific domains could explain the differences in subcellular accumulation. Together, our results suggest that PTP-3A and PTP-3B likely interact with distinct molecular partners in controlling specific aspects of neuronal development.
PTP-3A functions as a molecular linker at synapses
The genetic interactions we identified between ptp-3 and syd-2 were expected given that LAR has been found to function with α-liprin to regulate synaptic morphology in vertebrates and in Drosophila (Kaufmann et al., 2002; Dunah et al., 2005). However, the cytoplasmic domains of PTP-3A and PTP-3B are identical, suggesting both should interact with SYD-2 equally. PTP-3A is sufficient to rescue the synaptic defects of ptp-3 mutants, but PTP-3B is not. Thus, an interaction with SYD-2 does not explain the specificity of PTP-3A in synapse formation.
Previous work has shown that α-liprins are capable of inducing LAR to accumulate at focal adhesion-like plaques in cultured cells (Serra-Pages et al., 1998). Focal adhesions are sites of cell-matrix interactions, suggesting that α-liprins may regulate an interaction of LAR receptors with the ECM. Specific ECM molecules are, in fact, ligands for LAR receptors, and biochemical and genetic work has shown that LAR isoforms exhibit ligand specificity (O'Grady et al., 1998). The data suggest that specific extracellular ligands could regulate the distinct functions we have observed for the two PTP-3 isoforms.
The genetic epistasis of ptp-3 and nid-1 indicates the two genes have a common output in synaptic development. PTP-3 and SYD-2 are present at presynaptic densities (Zhen and Jin, 1999; Yeh et al., 2005; present study). We found that the accumulation of SYD-2 was aberrant in both ptp-3 and nid-1 mutants. The function of NID-1 in SYD-2 localization is, at least partially, dependent on PTP-3, because we could restore a normal SYD-2 accumulation pattern in nid-1 mutants by supplying additional PTP-3A. Likewise, PTP-3 localization is altered in nid-1 mutants, suggesting the SYD-2 mislocalization may be a direct result of the PTP-3 mislocalization. These data indicate that the ECM is necessary for PTP-3 and SYD-2 to remain at specific foci. Supplying additional PTP-3A also overcomes the UNC-10 accumulation defects in nid-1 mutants, suggesting that the synaptic defects observed are attributable, at least in part, to the mislocalization of PTP-3 and SYD-2.
Our genetic analyses indicate that mutations in syd-2 are more severe than those in ptp-3, indicating that loss of SYD-2 has more detrimental effects on synaptic patterning than SYD-2 mislocalization. We also found that, in syd-2 mutants, PTP-3 is no longer efficiently colocalized with UNC-10, another protein associated with presynaptic densities. The simplest interpretation of these results is that SYD-2 acts as a molecular scaffold for synaptic proteins and that a PTP-3-ECM interaction acts as an anchor that keeps SYD-2 localized in tight foci.
Our data indicate that PTP-3A, NID-1, and SYD-2 are required for the size and spacing of synaptic proteins, rather than being required for synapse formation per se. Similarly, Drosophila DLAR and Dliprin appear to regulate the size and shape of synaptic boutons at the NMJ (Kaufmann et al., 2002), and vertebrate LAR and liprins regulate the morphology of excitatory synapses (Dunah et al., 2005). The altered synaptic shape affects synaptic transmission as measured either electrophysiologically or pharmacologically (Zhen and Jin, 1999; Kaufmann et al., 2002; Ackley et al., 2003; Dunah et al., 2005). We propose that the interactions between PTP-3A, NID-1, and SYD-2 serve to generate a spatial synaptic organization that provides the most efficient arrangement for synaptic transmission.
Footnotes
B.D.A. was supported by the Howard Hughes Medical Institute (HHMI) and by a development grant from the Muscular Dystrophy Association. Grants from the National Institutes of Health supported research in A.D.C.'s laboratory (GM54657) and in Y.J.'s laboratory (NS35546). Y.J. is an HHMI Investigator. ptp-3(ok244) and ptp-3(tm352) were generated by the C. elegans gene knock-out consortium and the National Bioresource Project for the Experimental Animal C. elegans (Tokyo, Japan), respectively. Some of the strains were provided by the Caenorhabditis Genetics Center (Minneapolis, MN). We thank M. Nonet for the anti-UNC-10 and anti-SNT-1 antibodies, J. Bessereau for anti-UNC-49 antibodies, J. Kramer for pJJ471 plasmid, M. Zhen for hpIs3 strain, and members of our laboratories for critical readings of this manuscript.
Correspondence should be addressed to Dr.Yishi Jin, 329 Sinsheimer Laboratories, University of California-Santa Cruz, 1156 High Street, Santa Cruz, CA 95064. E-mail: jin@biology.ucsc.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257517-12$15.00/0
References
- Ackley BD, Crew JR, Elamaa H, Pihlajaniemi T, Kuo CJ, Kramer JM (2001) The NC1/endostatin domain of Caenorhabditis elegans type XVIII collagen affects cell migration and axon guidance. J Cell Biol 152: 1219-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackley BD, Kang SH, Crew JR, Suh C, Jin Y, Kramer JM (2003) The basement membrane components nidogen and type XVIII collagen regulate organization of neuromuscular junctions in Caenorhabditis elegans J Neurosci 23: 3577-3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun-Gultekin Z, Andachi Y, Tsalik EL, Pilgrim D, Kohara Y, Hobert O (2001) A regulatory cascade of three homeobox genes, ceh-10, ttx-3 and ceh-23, controls cell fate specification of a defined interneuron class in C. elegans Development 128: 1951-1969. [DOI] [PubMed] [Google Scholar]
- Aricescu AR, McKinnell IW, Halfter W, Stoker AW (2002) Heparan sulfate proteoglycans are ligands for receptor protein tyrosine phosphatase sigma. Mol Cell Biol 22: 1881-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans Genetics 77: 71-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin-Sang ID, Moseley SL, Ding M, Harrington RJ, George SE, Chisholm AD (2002) The divergent C. elegans ephrin EFN-4 functions inembryonic morphogenesis in a pathway independent of the VAB-1 Eph receptor. Development 129: 5499-5510. [DOI] [PubMed] [Google Scholar]
- Ch'ng Q, Williams L, Lie YS, Sym M, Whangbo J, Kenyon C (2003) Identification of genes that regulate a left-right asymmetric neuronal migration in Caenorhabditis elegans Genetics 164: 1355-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clandinin TR, Lee CH, Herman T, Lee RC, Yang AY, Ovasapyan S, Zipursky SL (2001) Drosophila LAR regulates R1-R6 and R7 target specificity in the visual system. Neuron 32: 237-248. [DOI] [PubMed] [Google Scholar]
- Debant A, Serra-Pages C, Seipel K, O'Brien S, Tang M, Park SH, Streuli M (1996) The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc Natl Acad Sci USA 93: 5466-5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hertog J, Blanchetot C, Buist A, Overvoorde J, van der Sar A, Tertoolen LG (1999) Receptor protein-tyrosine phosphatase signalling in development. Int J Dev Biol 43: 723-733. [PubMed] [Google Scholar]
- Desai CJ, Gindhart Jr JG, Goldstein LS, Zinn K (1996) Receptor tyrosine phosphatases are required for motor axon guidance in the Drosophila embryo. Cell 84: 599-609. [DOI] [PubMed] [Google Scholar]
- Dong L, Chen Y, Lewis M, Hsieh JC, Reing J, Chaillet JR, Howell CY, Melhem M, Inoue S, Kuszak JR, DeGeest K, Chung AE (2002) Neurologic defects and selective disruption of basement membranes in mice lacking entactin-1/nidogen-1. Lab Invest 82: 1617-1630. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Hueske E, Wyszynski M, Hoogenraad CC, Jaworski J, Pak DT, Simonetta A, Liu G, Sheng M (2005) LAR receptor protein tyrosine phosphatases in the development and maintenance of excitatory synapses. Nat Neurosci 8: 458-467. [DOI] [PubMed] [Google Scholar]
- Edgley ML, Riddle DL (2001) LG II balancer chromosomes in Caenorhabditis elegans: mT1(II;III) and the mIn1 set of dominantly and recessively marked inversions. Mol Genet Genomics 266: 385-395. [DOI] [PubMed] [Google Scholar]
- Ensslen-Craig SE, Brady-Kalnay SM (2004) Receptor protein tyrosine phosphatases regulate neural development and axon guidance. Dev Biol 275: 12-22. [DOI] [PubMed] [Google Scholar]
- Finney M, Ruvkun G (1990) The unc-86 gene product couples cell lineage and cell identity in C. elegans Cell 63: 895-905. [DOI] [PubMed] [Google Scholar]
- Gally C, Bessereau JL (2003) GABA is dispensable for the formation of junctional GABA receptor clusters in Caenorhabditis elegans J Neurosci 23: 2591-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity PA, Lee CH, Salecker I, Robertson HC, Desai CJ, Zinn K, Zipursky SL (1999) Retinal axon target selection in Drosophila is regulated by a receptor protein tyrosine phosphatase. Neuron 22: 707-717. [DOI] [PubMed] [Google Scholar]
- Hall DH, Hedgecock EM (1991) Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans Cell 65: 837-847. [DOI] [PubMed] [Google Scholar]
- Hallam SJ, Jin Y (1998) lin-14 regulates the timing of synaptic remodelling in Caenorhabditis elegans Nature 395: 78-82. [DOI] [PubMed] [Google Scholar]
- Harrington RJ, Gutch MJ, Hengartner MO, Tonks NK, Chisholm AD (2002) The C. elegans LAR-like receptor tyrosine phosphatase PTP-3 and the VAB-1 Eph receptor tyrosine kinase have partly redundant functions in morphogenesis. Development 129: 2141-2153. [DOI] [PubMed] [Google Scholar]
- Honkaniemi J, Zhang JS, Yang T, Zhang C, Tisi MA, Longo FM (1998) LAR tyrosine phosphatase receptor: proximal membrane alternative splicing is coordinated with regional expression and intraneuronal localization. Brain Res Mol Brain Res 60: 1-12. [DOI] [PubMed] [Google Scholar]
- Huang X, Cheng HJ, Tessier-Lavigne M, Jin Y (2002) MAX-1, a novel PH/MyTH4/FERM domain cytoplasmic protein implicated in netrin-mediated axon repulsion. Neuron 34: 563-576. [DOI] [PubMed] [Google Scholar]
- Johnson KG, Van Vactor D (2003) Receptor protein tyrosine phosphatases in nervous system development. Physiol Rev 83: 1-24. [DOI] [PubMed] [Google Scholar]
- Kang SH, Kramer JM (2000) Nidogen is nonessential and not required for normal type IV collagen localization in Caenorhabditis elegans Mol Biol Cell 11: 3911-3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann N, DeProto J, Ranjan R, Wan H, Van Vactor D (2002) Drosophila liprin-alpha and the receptor phosphatase Dlar control synapse morphogenesis. Neuron 34: 27-38. [DOI] [PubMed] [Google Scholar]
- Kim S, Wadsworth WG (2000) Positioning of longitudinal nerves in C. elegans by nidogen. Science 288: 150-154. [DOI] [PubMed] [Google Scholar]
- Kliemann SE, Waetge RT, Suzuki OT, Passos-Bueno MR, Rosemberg S (2003) Evidence of neuronal migration disorders in Knobloch syndrome: clinical and molecular analysis of two novel families. Am J Med Genet A 119: 15-19. [DOI] [PubMed] [Google Scholar]
- Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML (2001) A post-docking role for active zone protein Rim. Nat Neurosci 4: 997-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger NX, Van Vactor D, Wan HI, Gelbart WM, Goodman CS, Saito H (1996) The transmembrane tyrosine phosphatase DLAR controls motor axon guidance in Drosophila Cell 84: 611-622. [DOI] [PubMed] [Google Scholar]
- Kypta RM, Su H, Reichardt LF (1996) Association between a transmembrane protein tyrosine phosphatase and the cadherin-catenin complex. J Cell Biol 134: 1519-1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurel-Zaffran C, Suzuki T, Gahmon G, Treisman JE, Dickson BJ (2001) Cell-autonomous and -nonautonomous functions of LAR in R7 photo-receptor axon targeting. Neuron 32: 225-235. [DOI] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V (1991) Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10: 3959-3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML (1999) Visualization of synaptic specializations in live C. elegans with synaptic vesicle protein-GFP fusions. J Neurosci Methods 89: 33-40. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Grundahl K, Meyer BJ, Rand JB (1993) Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell 73: 1291-1305. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Staunton JE, Kilgard MP, Fergestad T, Hartwieg E, Horvitz HR, Jorgensen EM, Meyer BJ (1997) Caenorhabditis elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. J Neurosci 17: 8061-8073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Grady P, Krueger NX, Streuli M, Saito H (1994) Genomic organization of the human LAR protein tyrosine phosphatase gene and alternative splicing in the extracellular fibronectin type-III domains. J Biol Chem 269: 25193-25199. [PubMed] [Google Scholar]
- O'Grady P, Thai TC, Saito H (1998) The laminin-nidogen complex is a ligand for a specific splice isoform of the transmembrane protein tyrosine phosphatase LAR. J Cell Biol 141: 1675-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Lombroso PJ (2003) Receptor and nonreceptor protein tyrosine phosphatases in the nervous system. Cell Mol Life Sci 60: 2465-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido R, Serra-Pages C, Tang M, Streuli M (1995) The LAR/PTP delta/PTP sigma subfamily of transmembrane protein-tyrosine-phosphatases: multiple human LAR, PTP delta, and PTP sigma isoforms are expressed in a tissue-specific manner and associate with the LAR-interacting protein LIP. 1. Proc Natl Acad Sci USA 92: 11686-11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra-Pages C, Medley QG, Tang M, Hart A, Streuli M (1998) Liprins, a family of LAR transmembrane protein-tyrosine phosphatase-interacting proteins. J Biol Chem 273: 15611-15620. [DOI] [PubMed] [Google Scholar]
- Stepanek L, Stoker AW, Stoeckli E, Bixby JL (2005) Receptor tyrosine phosphatases guide vertebrate motor axons during development. J Neurosci 25: 3813-3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams L (2002) A genetic analysis of the left-right asymmetric polarizations and migrations of the Q neuroblasts in Caenorhabditis elegans PhD thesis, University of California, San Francisco.
- Wills Z, Bateman J, Korey CA, Comer A, Van Vactor D (1999) The tyrosine kinase Abl and its substrate enabled collaborate with the receptor phosphatase Dlar to control motor axon guidance. Neuron 22: 301-312. [DOI] [PubMed] [Google Scholar]
- Xie Y, Yeo TT, Zhang C, Yang T, Tisi MA, Massa SM, Longo FM (2001) The leukocyte common antigen-related protein tyrosine phosphatase receptor regulates regenerative neurite outgrowth in vivo J Neurosci 21: 5130-5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Kawano T, Weimer RM, Bessereau JL, Zhen M (2005) Identification of genes involved in synaptogenesis using a fluorescent active zone marker in Caenorhabditis elegans J Neurosci 25: 3833-3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen M, Jin Y (1999) The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans Nature 401: 371-375. [DOI] [PubMed] [Google Scholar]