

Abstract
Whenever neurons in the CNS are injured, microglia become activated. In addition to local activation, microglia remote from the primary lesion site are stimulated. Because this so-called secondary activation of microglia is instrumental for long-term changes after neuronal injury, it is important to understand how microglia activity is controlled. The remote activation of microglia implies that the activating signals are transported along neuronal projections. However, the identity of these signals has not yet been identified. It is shown here that glutamate-treated neurons rapidly express and release the chemokine CCL21. We also provide evidence that neuronal CCL21 is packed in vesicles and transported throughout neuronal processes to reach presynaptic structures. Chemotaxis assays show that functional CCL21 is released from endangered neurons and activate microglia via the chemokine receptor CXCR3. Based on these findings, we suggest that neuronal CCL21 is important in directed neuron-microglia signaling and that this communication could account for the remote activation of microglia, far distant from a primary lesion.
Keywords: neuron-microglia signaling, chemokines, CCL21, vesicle release, apoptosis, neuroinflammation
Introduction
Brain lesions can lead to profound changes in the neuronal architecture and influence plasticity phenomena in neurons. Recent results show a significant impact of activated microglia on these lesion-induced long-term changes (Banati, 2002a; Beattie et al., 2002; Parish et al., 2002; Monje et al., 2003). Activated microglia can either inhibit neurogenesis (Monje et al., 2003) or support neuronal sprouting (Parish et al., 2002), showing that microglia activation is a double-edged sword. It is thus of particular interest to understand how microglia activity is controlled. Whenever neurons are injured, microglia are among the first cells in the CNS that become activated (Kreutzberg, 1996). Microglia activation frequently occurs not only at the primary lesion site but also remote from the primary damage in sites in which the damaged neurons project to (secondary activation of microglia) (Banati, 2002a). Thus, microglial activity is likely to be controlled by endangered neurons, and the secondary activation of microglia requires the transport of microglia activating signals. However, the identity of the molecules involved in this neuron-microglia signaling is not yet known.
Chemokines and their receptors constitute an elaborate signaling system that plays an important role in cell-cell communication not only in the peripheral immune system (Murphy et al., 2000; Rossi and Zlotnik, 2000; Mackay, 2001; Moser et al., 2004; Rot and von Andrian, 2004) but also in the CNS (for review, see Ransohoff and Tani, 1998; Bacon and Harrison, 2000; Bajetto et al., 2002; Biber et al., 2002a; Ambrosini and Aloisi, 2004). The expression of most chemokines in the CNS is induced by inflammatory and neurodegenerative events in which they mediate leukocyte infiltration (Fife et al., 2000; Izikson et al., 2000; Siebert et al., 2000; Huang et al., 2001). Conversely, the presence of chemokine receptors in neurons and glia moreover suggests that chemokines also participate in local signaling during CNS inflammation (Hesselgesser and Horuk, 1999; Biber et al., 2002b; Ambrosini and Aloisi, 2004).
We have recently provided evidence that damaged neurons upregulate the expression of CCL21 mRNA. Furthermore, we were able to show that CCL21 activates microglia via the chemokine receptor CXCR3 (Biber et al., 2001, 2002; Rappert et al., 2002; Dijkstra et al., 2004). Subsequent studies showed that mice deficient for CXCR3 displayed no secondary activation of microglia in response to enthorinal cortex lesion. Importantly, this lack of secondary microglia activation had profound effects on the neuronal reorganization after the lesion, illustrating the impact of chemokine signaling in the brain (Rappert et al., 2004).
We here further elucidated the mechanism of neuron-microglia signaling with respect to neuronal synthesis and release of CCL21 protein. It is shown here that CCL21 is specifically induced and released from neurons that were treated with high concentrations of glutamate (excitotoxicity). Several lines of evidence furthermore indicate that CCL21 in neurons is sorted into vesicles that are transported via axons and reach presynaptic structures. These results thus offer the first explanation how endangered neurons could activate microglia remote from a primary lesion.
Materials and Methods
Chemicals
DMEM, Neurobasal medium, and B27 supplement were from Invitrogen (Breda, The Netherlands). TA vectors pCRII were from Invitrogen (Leek, The Netherlands). Taq-polymerase was from InViTek (Berlin, Germany). Phosphorylated enhanced green fluorescent protein (pEGFP)-N2 vector was from Clontech (Alphen aan den Rijn, The Netherlands). Fugene was from Roche (Mannheim, Germany). Restriction enzymes were from Promega (Leiden, The Netherlands). Recombinant chemokines were from Pepro Tech (London, UK), and all other chemicals were from Sigma-Aldrich (Bornhem, Belgium). CCL21 antibody was from Pepro Tech. Neuronal-specific nuclear protein (NeuN) antibody was from Chemicon (Temecula, CA). β-Actin antibody was from Abcam (Cambridge, MA). Mitogen-activated protein 2 (MAP-2) antibody, Hoechst dye, brefeldin-A (BFA), secondary antibodies, and goat anti-rabbit cyanine 3 (Cy3)-labeled antibodies were from Jackson Immuno-Research (West Grove, PA). Goat anti-mouse FITC-labeled antibodies were from Chemicon.
Cell cultures
NG108-15 and HEK293 cells. NG108-15 (mouse glioma-rat neuroblastoma) (NG108) cells and HEK293 cells were maintained in DMEM containing 10% fetal calf serum with 0.01% penicillin and 0.01% streptomycin in a humidified atmosphere (5% CO2) at 37°C. The differentiation of NG108 cells was induced by changing the medium to DMEM containing 1% fetal calf serum supplemented with 50 μm IBMX.
Cultured cortical neurons. Cultures of cortical neurons were established as described previously (Biber et al., 2001). In brief, pregnant mice (NMRI) were anesthetized with halothane and killed by cervical dislocation, and embryonic day 17 embryos were removed. Cortices were dissected in ice-cold HBSS supplemented with 30% glucose. Meninges were removed, and brain tissues were gently dissociated by trituration in Neurobasal/B27 medium (supplemented with 0.4% glucose, 2 mm l-glutamine, 0.01% penicillin, and 0.01% streptomycin) and filtered through a cell strainer (70 μm pore size; Falcon, Franklin Lakes, NJ). After one washing step (100 × g for 10 min), cells were seeded on poly-d-lysine (10 μg/ml)-coated glass cover slides and maintained in Neurobasal/B27 medium for at least 7 d in a humidified atmosphere (5% CO2) at 37°C. To induce CCL21 expression, cells were treated with glutamate (100 μm, 30 min) as described previously (Biber et al., 2001). To increase CCL21 immune reactivity, cells were treated in some experiments with 10 μg/ml BFA 1 h before fixation.
Cultured microglia. Microglia cultures from wild-type animals and CXCR3-deficient mice were established as described previously (Biber et al., 2001; Rappert et al., 2002). In brief, mouse cortex was dissected from newborn mouse pups (<1 d). Brain tissue was gently dissociated by trituration in PBS and filtered through a cell strainer (70 μm pore size; Falcon) into DMEM. After two washing steps (200 × g for 10 min), cells were seeded in culture dishes (10 cm diameter; Nunc, Roskilde, Denmark) (8 × 106 cells per dish). Cultures were maintained 6 weeks in DMEM containing 10% fetal calf serum with 0.01% penicillin and 0.01% streptomycin in a humidified atmosphere (5% CO2) at 37°C. Culture medium was changed the second day after preparation and every 6 d thereafter. Floating microglia were harvested from confluent mixed astrocyte cultures, counted, and used for chemotaxis assays. CXCR3-deficient mice were obtained from Bao Lu and Craig Gerard (Children's Hospital, Harvard Medical School, Boston, MA).
Preparation of organotypic slice cultures
Organotypic hippocampal slices were prepared from brains of 1- to 3-d-old mouse pups under sterile conditions. After decapitation, brains were quickly removed and further dissected in ice-cold HBSS. Hippocampi from both hemispheres were dissected and sectioned perpendicular to their septotemporal axis at 400 μm thickness by a tissue chopper (McIlwain). Slices were separated and transferred onto humidified membranes of cell culture inserts (Millicell-CM; Millipore, Bedford, MA). Slices from left and right hemispheres of one animal were put on separate membranes. These membranes, containing four to six slices, were placed in a six-well plate containing 1 ml of culture medium (0.5× MEM, 25% basal medium Eagle without glutamine, 25% heat-inactivated horse serum, 2 mm glutamax, 0.65% glucose, 0.01% penicillin, and 0.01% streptomycin) per well. Slices were incubated in a humidified atmosphere (5% CO2) at 35°C. Medium was refreshed the next day after preparation and subsequently every 2 d. After 8 d in vitro, one cell culture insert containing slices from one hemisphere of an animal was placed in medium containing 100 μm glutamate, whereas the other insert remained in normal culture medium. After 30 min of glutamate exposure, medium was aspirated and replaced with fresh culture medium. Slices were kept in culture for another 8 h and subsequently fixed in 4% paraformaldehyde before immunohistochemistry.
Transfection of cells
NG108 and HEK293 cells. A total of 1 μg of the plasmid was transfected with 3 μl of Fugene (Roche) in NG108 or HEK293 cells according to the instructions of the manufacturer. The expression plasmid for mCXCR3 (pcDNA3.1-mCXCR3) was described recently (Dijkstra et al., 2004), and the expression plasmid for mCCR7 (pRC/CMV-mCCR7) was a kind gift from Dr. U. Hoekpen and Dr. M. Lipp (Max-Delbrück-Centrumfür Molekulare Medizin, Berlin, Germany). Mock transfections were performed with empty pEGFP-N2 vector.
Cortical neurons. Primary cortical neurons were transfected using the nucleofector technology (Amaxa, Cologne, Germany). The 5 × 106 cells were diluted directly after preparation in 100 μl of mouse neuronal stem cell nucleofector solution supplemented with 5 μg of plasmid DNA (CCL21-EGFP or control) and transfected with program O-05 according to the instructions of the supplier. Cells were immediately transferred to prewarmed culture medium after transfection and seeded on coated coverslips. Nonattached cells were washed away 2 h after transfection, and neurons were further cultivated as described above. This transfection procedure yielded efficiency up to 40%, and transfected neurons remained healthy and developed comparably to nontransfected cells. Mock transfections were performed with empty pEGFP-N2 vector.
Real-time PCR analysis of CCL21 mRNA levels in cultured neurons
Cells and brain material were Lysol in guanidinium isothiocyanate/mer-captoethanol buffer, and total RNA was extracted and transcribed into cDNA as described previously (Biber et al., 2001). Primers were designed using Primer Designer version 3.0. CCL21 primer sequences were as follows: forward, 5′-CAGGACTGCTGCCTTAAGTA-3; reverse, 5′-GCACATAGCTCAGGCTTAGA-3′. Hydroxymethylbilane synthase (HBMS) primer sequences were as follows: forward, 5′-CCGAGCCAAGGACCAGGATA-3′; reverse, 5′-CTCCTTCCAGGTGCCTCAGA-3′. HMBS was used as a normalizing gene because its expression remained stable under various experimental conditions. Experiments were done in 25 μl, using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA) and primer concentrations of 250 nmol in 96-well plates in an i-cycler (Bio-Rad). The reaction conditions were as follows: 3 min at 95°C, followed by 50 cycles of 10 s at 95°C and 45 s at 58°C, followed by 1 min at 95°C and by 1 min at 55°C. The comparative cycle threshold (Ct) method [amount of target amplicon X in sample S, normalized to a reference R and related to a control sample C, calculated by 2-{(CtX,S-CtR,S)-(CtX,C-CtR,C)} was used to determine the relative CCL21 mRNA expression levels between cultured neurons under control conditions and treated with glutamate for various time points (Livak and Schmittgen, 2001)].
Construction of the CCL21-EGFP fusion protein
CCL21 was coupled to EGFP by cloning into pEGFP-N2 (Clontech). Full-length CCL21 was amplified using cDNA from mouse spleen with the following primers: forward, 5′-ATA CTC GAG ATG GCT CAG ATG ATG ACT CTG AGC CTC; reverse, 5′-ATA GGA TCC ATC CTC TTG AGG GCT GTG TCT GTT C. These primers introduced a XhoI restriction site at the 5′ end and a BamHI restriction site at the 3′ end of CCL21 and omitted the stop codon in the chemokine sequence. The resulting PCR product was cloned into pCRII (Clontech) by TA cloning, and the insert was verified by sequencing. A total of 5 μg of the resulting vector, pCRII (CCL21-stop), was restricted with BamHI (Promega) and XhoI (Promega), and the excised band was purified using gel electrophoresis and gel purification (MinElute Gel Extraction kit; Qiagen, Hilden, Germany) and subsequently cloned into the BamHI/XhoI sites of pEGFP-N2 (Clontech). Again, sequence and the orientation of the insert were verified by sequencing. Transfection of HEK293 cells was performed as described above. Mock transfections were performed with empty pEFGP-N2. A Zeiss (Oberkochen, Germany) Axioscope 2 or a Leica (Nussloch, Germany) confocal laser microscope were used to analyze the expression of the CCL21-EGFP construct in transfected cells.
Immunocytochemistry
Immunocytochemistry was performed as described previously (Biber et al., 2001). In brief, before immunocytochemical processing, cells or cultured brain slices were fixed in 4% paraformaldehyde for 30 min and washed in 0.9% saline dissolved in 0.05 m Tris, pH 7.4 (TBS). All antisera were diluted in TBS containing 0.3% Triton X-100, 1% bovine serum albumin (BSA), and heparin (5 mg/ml). Cells were preincubated in 5% BSA in TBS for 30 min and incubated overnight with CCL21, caspase-3, NeuN, or MAP-2 antibodies. Antibody-antigen reactions were either detected using the biotin-streptavidin method and the complex was visualized with diaminobenzidine (DAB)/H2O2 or visualized with a Cy3- or Cy5-labeled goat anti-rabbit secondary antibody for fluorescence microscopy. Control experiments for all immunocytochemical stainings were done by incubating cells or cultured brain slices with an isotype-matched primary antibody against GFAP (Chemicon) or in the absence of primary antibodies.
Western blot analysis
Western blot was performed as described previously (Baron et al., 2000). Briefly, neuronal cultures were harvested in PBS, solubilized in sample buffer, and subjected to SDS-PAGE. Equal amounts of proteins (40 μg) were loaded onto 12.5% SDS-polyacrylamide gels and transferred to Hybond-ECL nitrocellulose membrane (Amersham Biosciences, Arlington Heights, IL). The membranes were blocked with 10% nonfat dry milk in PBS and incubated with the primary antibody (CCL21 at 1:2000; β-actin at 1:2000) overnight, followed by 2 h incubation with the appropriate horseradish peroxidase-conjugated antibody (Amersham Biosciences). Specific bands were visualized by ECL for β-actin or ECLplus Western blotting Detection System for CCL21. A total of 125 pg of recombinant CCL21 was used as a positive control.
Gold-silver-substituted peroxidase intensification and electron microscopy
EM immunocytochemistry for the ultrastructural detection of CCL21 was performed on cultured cortical neurons using a biotinylated secondary antibody and the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) with DAB as the chromogen.
Subsequently, intensification of the DAB reaction product was effectuated using the gold-silver-substituted peroxidase (GSSP) method (Liem et al., 2001). In brief, after completing the DAB reaction, the cells were rinsed twice in 2% sodium acetate solution. After destaining in 10% thioglycolic acid, the cells were immersed for 8 min in a freshly prepared physical developer containing 2.5% sodium carbonate, 0.1% ammonium nitrate, 0.1% silver nitrate, 0.5% phosphosilico-tungstic acid, and 0.9% paraformaldehyde. After a brief wash with 1% acetic acid, the deposited silver particles were replaced by gold by immersing the cells in 0.02% gold chloride for 8 min at room temperature. Subsequently, they were fixed for two times at 10 min with 3% sodium thiosulphate, rinsed briefly in 0.1 m sodium acetate cacodylate buffer, pH 7.6, and were then osmicated in 1% OsO4 (w/v) and 1.5% potassium hexacyanoferrate in the same buffer, dehydrated in a graded series of ethanols, and embedded in Epon. Semithin sections (1 μm) were cut on a Reichert Ultratome and stained with toluidine blue to check the presence of relevant areas. Ultrathin sections (60 nm) were then cut, counterstained with uranyl acetate and lead citrate, and examined using a Philips (Aachen, Germany) CM-100 transmission electron microscope.
Chemotaxis assay
Cell migration in response to neuronal supernatants or chemokines was assessed using a 48-well chemotaxis microchamber (Neuro Probe, Gaithersburg, MD) as described previously (Dijkstra et al., 2004). Chemokine stock solutions were prepared in PBS and further diluted in neuronal medium for use in the assay. Culture medium without chemokines served as a control in the assay. A total of 28 μl of neuronal supernatants, the chemoattractant solution, or control medium was added to the lower wells, lower and upper wells were separated by a polyvinylpyrrolidone-free polycarbonate filter (8 μm pore size), and 1. 5 × 104 microglial cells or 3 × 104 transfected HEK293 cells (mock, mCXCR3 or mCCR7) were applied per well (50 μl) in the upper chamber. Determinations were done six times. The chamber was incubated at 37°C, 5% CO2 in a humidified atmosphere for 120 min in case of microglia and 240 min in case of HEK293 cells. At the end of incubation, the filter was washed, fixed in methanol, and stained with toluidine blue. Migrated cells were counted with a scored eyepiece, and data are presented as percentages of control migration. To prevent bias, chemotaxis filters were occasionally counted by blinded investigators.
Analysis of CCL21-positive axons in organotypic brain cultures
Only slices that showed an intact hippocampal structure [recognizable dentate gyrus (DG) and CA region] were analyzed. Thus, 14 control slices and 17 slices from six animals were evaluated. CCL21-positive processes were counted in the molecular layers of the dentate gyrus and the CA regions in three microscopic fields (10 × 20). For each animal, the amount of CCL21-positive axons after glutamate treatment was calculated as the percentage of CCL21-positive processes in the nonstimulated slices obtained from the same animal.
Statistical analysis
Statistical analysis and comparison of the different groups was performed using with Students' t test for CCL21 mRNA expression and CCL21-positive axons in organotypic brain cultures and with the Tukey's ANOVA test for multiple comparisons for the chemotaxis data (SPSS, Chicago, IL). A significance level of 0.05 was used.
Results
Time- and concentration-dependent induction of CCL21 expression in neurons after treatment with glutamate
Previous studies have shown that glutamate treatment rapidly induces expression of CCL21 mRNA in cultured cortical neurons (Biber et al., 2001). Here we analyzed the expression of CCL21 protein in neurons exposed to glutamate. Glutamate treatment is a well known inducer of excitotoxicity and subsequent neuronal death (Chapman et al., 2000). This was also found in our cultures as investigated by nuclear stainings, vital stainings, and caspase-3 immunocytochemistry. These data indicated that glutamate treatment caused gradual neuronal death, leading to 80% neuronal death after 24 h in response to 30 min glutamate treatment (100 μm) (data not shown).
Using immunocytochemistry, CCL21 protein expression was undetectable in neurons under control culture conditions (Fig. 1A). Weak CCL21 immunoreactivity (visualized by DAB staining) was induced in cortical neurons 16 h after treatment with glutamate (100 μm, 30 min) (Fig. 1B, arrows). Similar results were obtained using fluorescent labeling (data not shown). However, compared with spleen tissue, used as a positive control to validate the CCL21 antibody (data not shown), the staining intensity in cultured neurons was rather weak. Previous studies in dendritic cells showed an increase in chemokine staining intensity that was achieved by BFA treatment. BFA leads to intracellular accumulation of proteins that are released through the Golgi endoplasmatic route (Dinter and Berger, 1998; Lore et al., 2001). Similar effects were observed for accumulation of CCL21 in glutamate-treated neurons. Brief BFA treatment (30 min) was already sufficient to increase CCL21 staining, and a maximal effect was found after 1 h of BFA treatment (Fig. 1C). Longer treatment with BFA (up to 3 h) did not further increase neuronal CCL21 staining (data not shown). Another option to increase the sensitivity of immunostaining is GSSP intensification. Because of the intensification of the signal, CCL21 expression was now detectable by light microscopy in semithin sections (1 μm) without BFA incubation and was already evident in untreated healthy neurons (Fig. 1D). Again, it was found that glutamate treatment strongly increased the expression of neuronal CCL21 (Fig. 1E,F). At lower magnification, CCL21 expression was visible in the majority of the cells, in which CCL21 immunoreactivity was located in the soma and the processes (Fig. 1F). Thus, CCL21 expression was detectable in untreated neurons (albeit in very low expression levels) and is induced by treatment with glutamate.
Figure 1.
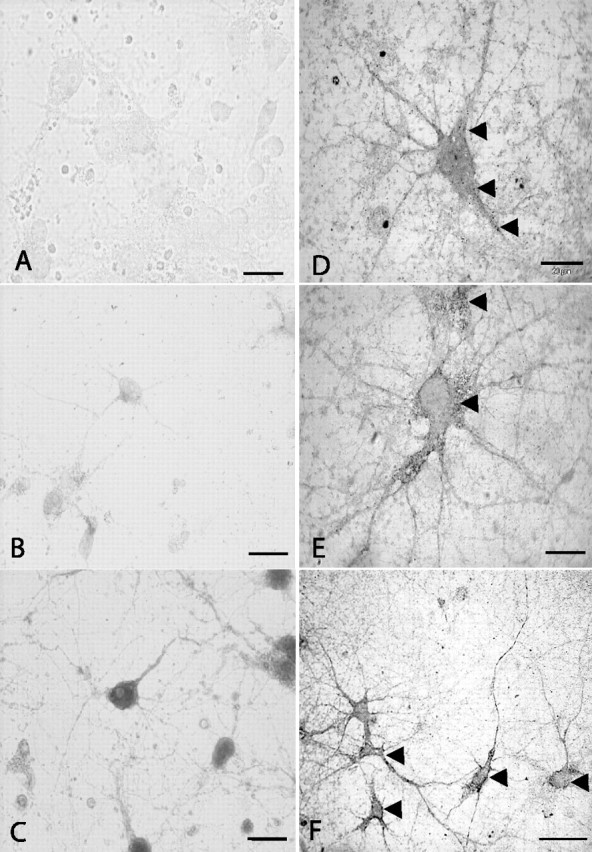
Immunohistochemical analysis revealed the expression of CCL21 protein in cultured cortical neurons. Standard immunohistochemical staining did not show immunoreactivity for CCL21 in untreated cortical cultured neurons (A), whereas weak staining for CCL21 was observed 12 h after the treatment with glutamate (100 μm, 30 min) (B). The staining intensity for CCL21 in glutamate-treated neurons was increased after pretreatment with BFA (10 μg/ml), a compound that blocks the release of proteins from the Golgi-TGN (C). Another method to increase the staining sensitivity is GSSP intensification. Semithin sections (1 μm) of cultured neurons were analyzed for CCL21 expression after GSSP intensification by light microscopy. In contrast to our standard method, CCL21 expression was now detectable in untreated cultures (arrowheads, D). Again, treatment with glutamate (100 μm, 30 min) pronouncedly induced the expression of CCL21 in these cells (arrowheads, E). Lower magnification showed the overall expression of neuronal CCL21 including the processes of the cells (arrowheads, F). Microphotographs in B, C, E, and F were taken 16 h after treatment with glutamate. Scale bars: A-C, F, 50 μm; D, E, 20 μm. Similar results have been obtained in at least three independent experiments, performed on at least three separate experimental days.
To further investigate the effect of glutamate treatment in detail, subsequent experiments have been performed in the presence of BFA (30 min), and CCL21 expression has been determined by immunofluorescence (Figs. 2, 3B). A low level of CCL21 staining was detectable in untreated cortical neurons (Fig. 2A,B). A time-dependent increase in neuronal CCL21 expression was induced by 30 min treatment with 100 μm glutamate (Fig. 2C-F). Low levels of CCL21 were detected already 3 h after glutamate treatment (Fig. 2C), and the level of CCL21 increased up to 12 h (Fig. 2D,E). At later time points, no additional increase of neuronal CCL21 expression was detected (Fig. 2F). The expression of CCL21 in glutamate-treated neurons was further investigated by Western blotting. Again, BFA treatment was used to increase the intracellular concentration of CCL21 in glutamate-treated neurons. Thus, using BFA, we detected a band for CCL21 in neurons 12 h after the treatment with glutamate (Fig. 2G). At earlier time points, we never obtained a band for CCL21 on the Western blot even with BFA treatment (data not shown). As a positive control for the Western blot, recombinant CCL21 was used (Fig. 2G). Subsequent real-time PCR analysis revealed an induction of CCL21 mRNA 2-6 h after glutamate treatment (Fig. 2H), which was not detectable at later time points (data not shown). Real-time PCR occasionally revealed the expression of CCL21 mRNA even in untreated cultures, which was found in three of seven independent experiments (data not shown).
Figure 2.
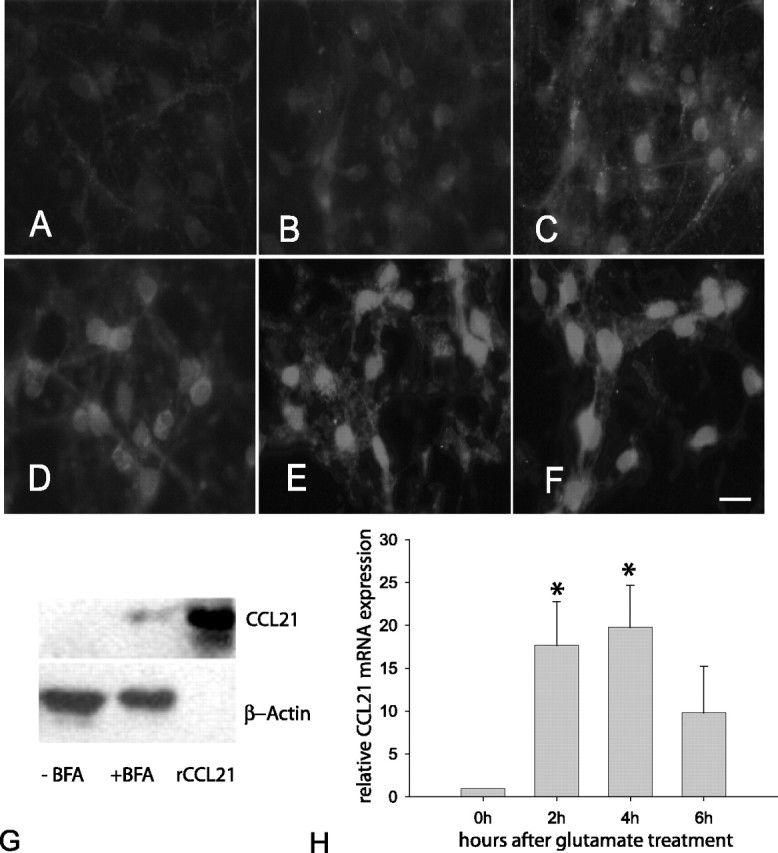
Time-dependent induction of CCL21 protein and mRNA expression in cultured cortical neurons after treatment with glutamate (100 μm, 30 min). Untreated cultured cortical neurons did not show pronounced CCL21 expression (B) compared with the negative control (A). Treatment with glutamate induced expression of CCL21, which was already observed 3 h after treatment (C), increased further at 6 h after treatment (D), and reached its maximum 12 h (E) and 16 h (F) after treatment with glutamate. Western blot analysis further confirmed the presence of CCL21 in glutamate-treated neurons 12 h after the treatment with glutamate (G). BFA treatment, however, was necessary to increase CCL21 levels for Western blotting, and no CCL21 band was observed in neurons from the same culture without BFA (G). Recombinant mouse CCL21 was used as a positive control and to confirm the identity of the CCL21 band obtained from neuronal material (G). Real-time PCR revealed the significant induction of CCL21 mRNA as early as 2 h after glutamate treatment (H). *Significantly different (p < 0.05). Scale bar (in F): A-F, 50 μm. Similar results have been obtained in five independent experiments. Data for mRNA expression are shown as mean ± SEM (n = 3).
Figure 3.

Glutamate treatment (100 μm, 30 min) induced CCL21 expression in the majority of cultured neurons. Pronounced CCL21 expression after glutamate treatment (B) (green fluorescence, exemplified by arrowheads in A and B) was found in ∼80% of the neurons costained with a nuclear dye (A) (compare A and B). Only a minority of neurons with healthy nuclei did not express CCL21 (exemplified by thin arrows in A and B). Confocal fluorescence microscopy revealed numerous CCL21-postive vesicle-like structures (green fluorescence, arrowheads) at the axonal hillock and axon-like processes of glutamate-treated neurons (C). Microphotographs (A-C) were taken 12 h after the treatment with glutamate, and microphotographs A and B were taken after BFA treatment. Scales bars: A, B, 50 μm; C, 10 μm. Similar results have been obtained in three independent experiments, performed on three separate experimental days.
The level of CCL21 expression was dependent on the concentration of glutamate. No or little CCL21 induction was observed after exposure to 1 or 10 μm glutamate (data not shown), but a pronounced CCL21 induction was found using 100 μm glutamate (Fig. 2). Higher glutamate concentrations (up to 1 mm) did not further increase neuronal CCL21 expression (data not shown). Glutamate treatment induced expression of CCL21 in ∼80% of the neurons [Fig. 3, compare nuclear staining (A, blue) and CCL21 immunoreactivity (B, green)]. Most cells were positive for CC21 (exemplified by arrowheads), whereas only few cells did not show CCL21 expression (exemplified by small arrow). As can be estimated from Figure 2, glutamate treatment did not induce a significant increase in the number of CCL21-positive cells; thus, at 2 h after the treatment, ∼70-80% of the neurons showed CCL21 immunoreactivity (Fig. 2). CCL21 immunoreactivity was often observed at the axonal hillock (Fig. 3B, arrowheads). To investigate whether this observation might be attributable to an artifact induced by BFA, CCL21 immunoreactivity was analyzed in more detail using confocal microscopy without BFA incubation. Here, numerous CCL21-positive vesicle-like structures at the axonal hillock and in axon-like processes of glutamate-treated neurons were observed (Fig. 3C).
CCL21 in neurons is sorted in vesicle-like structures
Our observations indicated that CCL21 in neurons might be sorted into vesicles and transported into axons: first, treatment with BFA enhanced the staining for CCL21, indicating that this chemokine is released via the Golgi and trans-Golgi network (TGN) route (Fig. 1). Second, expression of CCL21 in cultured neurons was frequently detected in axon-like structures (Fig. 1). Third, accumulation of CCL21 immunoreactivity was often observed at the axonal hillock, which is the site at which axonal transport starts (Fig. 3, arrowheads). Finally, confocal analysis revealed the presence of CCL21-positive vesicle-like structures in the soma and the processes of neurons (Fig. 3).
These data prompted us to further investigate the sorting of CCL21 using an expression vector for a CCL21-EGFP fusion protein. Cytoplasmic expression was observed when EGFP alone was transfected into HEK293 cells (data not shown). Expression of CCL21-EGFP in HEK293 cells revealed typical staining of the Golgi-TGN (Fig. 4A, arrowheads), indicating that CCL21 is a protein that is released via this organelle. Similar results were obtained in the mouse neuroblastoma-rat glioma cell line NG-108-15. Again, staining of the Golgi and TGN was observed when CCL21-EGPF was expressed in undifferentiated NG-108-15 cells (Fig. 4B, arrowheads). This pattern, however, changed when the cells were differentiated into neuron-like cells. In these cells, CCL21-EGFP fluorescence was still observed in the Golgi and TGN (small arrow) but could furthermore be visualized in the developing processes (Fig. 4C, arrowheads). At later time points of differentiation, CCL21-EGFP-positive vesicle-like structures were found at the most distant sites of the cellular processes (Fig. 4D).
Figure 4.
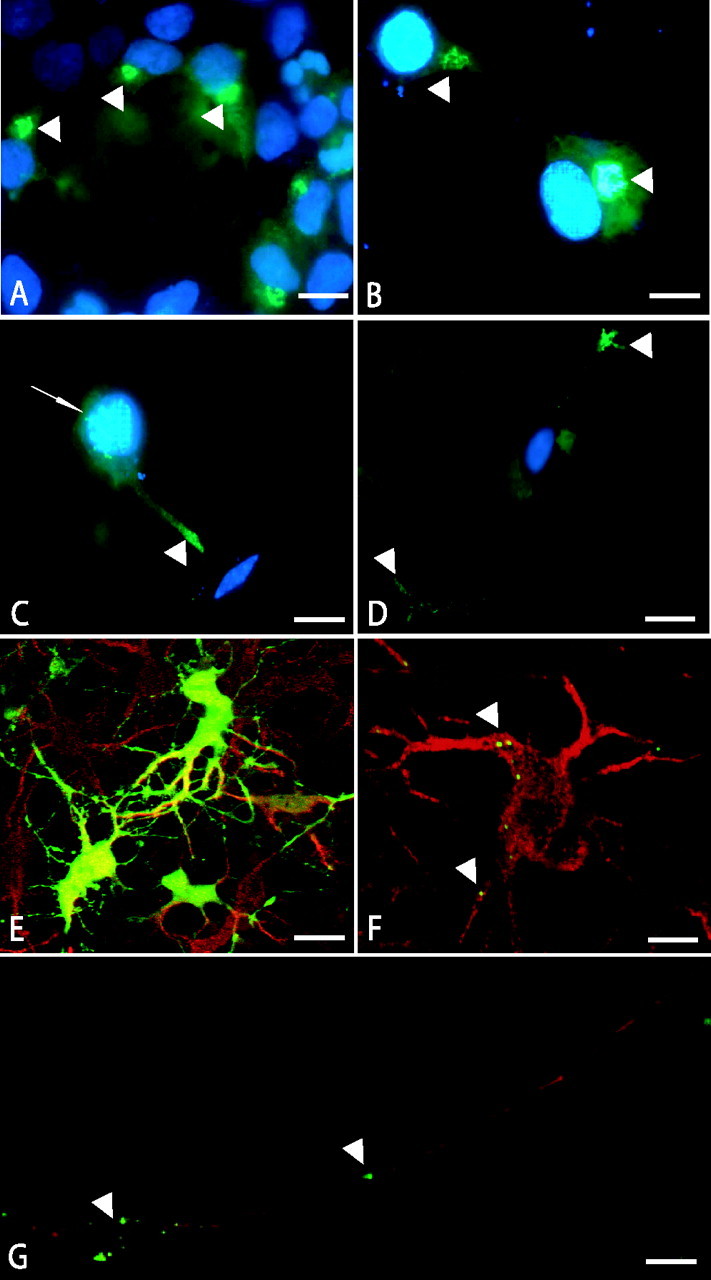
Expression of CCL21-EGFP fusion protein in HEK293, NG108, and primary cortical neurons. Typical Golgi-TGN staining (green fluorescence, arrowheads) was observed in HEK293 cells that expressed CCL21-EGFP (A). Similar results were found in undifferentiated NG108 cells (green fluorescence, arrowheads) (B). Two days after differentiation of these cells, CCL21-EGFP expression was still observable in the Golgi-TGN (green fluorescence, arrow) but also detectable in the developing processes of these cells (green fluorescence, arrowhead) (C). In more differentiated NG108 cells (4-5 d after differentiation), CCL21-EGFP expression was predominantly observed at the ends of the processes of the cells (green fluorescence, arrowheads) (D). Expression of EGFP in primary neurons (red fluorescence, MAP-2 staining) revealed overall cytoplasmic expression (green fluorescence) (E). Expression of CCL21-EGFP fusion protein in primary neurons (red fluorescence represents MAP-2 staining) revealed a punctuate staining (green fluorescence, arrowheads), indicating the expression of CCL21 in vesicle-like structures (F). These CCL21-EGFP-positive vesicle-like structures were found in the soma of the neurons (green fluorescence, arrowheads, F) and also in long processes remote from the soma (green fluorescence, arrowheads, G). The expression of the CCL21-EGFP fusion protein was analyzed by confocal fluorescence microscopy, 2 d after transfection in HEK293 and undifferentiated NG 108 and 5 d after transfection in primary neurons. Scale bars: A-C, F, G, 10 μm; D, E, 50 μm. Similar results with HEK293 and NG108 cells have been obtained in three independent experiments. At least five independent experiments have been performed with primary neurons that showed similar results.
Experiments with primary cultured neurons confirmed these findings. Control transfections with an EGFP-containing plasmid showed that ∼40% of the MAP-2-positive neurons (red fluorescence) were transfected (green fluorescence) (Fig. 4E). These control transfections showed a general, cytoplasmic expression of EFGP in the soma and processes of primary neurons (Fig. 4E). Expression of the CCL21-EGFP fusion protein resulted in a different pattern. Numerous small CCL21-EGFP-positive vesicle-like structures (green fluorescence; arrowheads) were observed in the soma and in the processes of the MAP-2-positive neurons (Fig. 4F, red fluorescence). The CCL21-EGFP-positive vesicle-like structures were also detected in long axon-like processes far away from the soma of the neurons (Fig. 4G, arrowheads).
Ultrastructural evidence for neuronal CCL21-positive vesicles
The use of an expression plasmid might create artifacts because of the overexpression of the protein of interest. It was therefore decided to further investigate the cellular localization of endogenous CCL21 in neurons by electron microscopy and GSSP intensification. At the ultrastructural level, CCL21 was detected in small vesicles in the Golgi and TGN (arrowheads) (Fig. 5A). CCL21-positive vesicles were moreover found in axon-like processes of the cells (data not shown) and in presynaptic structures (Fig. 5B, arrowheads). CCL21 immunoreactivity was also detected in the synaptic cleft (Fig. 5C, arrowheads).
Figure 5.

GSSP intensification and analysis of ultrathin sections of cultured neurons by electron microscopy showed the presence of CCL21-positive vesicles in the Golgi or TGN near the axonal hillock (arrowheads, A), in presynaptic structures (arrowheads, B), and in the synaptic cleft (arrowheads, C). These microphotographs were taken from untreated neurons because treatment with glutamate had a major impact on the ultrastructural architecture of the cells. However, no major differences were found between the CCL21 staining patterns in treated versus untreated neurons (data not shown). Scale bars: A, 500 nm; B, 200 nm; C, 100 nm. Similar results have been obtained in two independent experiments.
Neuronal expression of CCL21 in organotypic slice cultures
The pattern of neuronal CCL21 expression was further investigated in organotypic slices of the hippocampus that maintain their natural structure and neuronal circuitry (Gaehwiler et al., 1997). To verify the organotypic structure, hippocampal cultures were stained after 8 d in culture for nuclei (Hoechst dye, blue signal) and axons (MAP-2 antibody, green signal) (Fig. 6A). The overlay of both stainings clearly demonstrated that the typical hippocampal formation with CA1, CA3, and DG was still present in the slice (Fig. 6A). After 8 d in culture under control conditions, some CCL21-positive cells were present in the slices (data not shown). Like in cultured neurons, treatment with glutamate (100 μm, 30 min) significantly increased the number of CCL21-positive cells (Fig. 6B). Counting of the number of CCL21-positive cells (as described in Materials and Methods) revealed an induction by 170 ± 13% in glutamate-treated slices (n = 17) compared with untreated slices (n = 14). Strikingly, CCL21 expression was mainly observed in long processes (Fig. 6B, arrowhead). Double-labeling experiments and confocal microscopy corroborated these data. Costaining for NeuN (green fluorescence) and CCL21 (red fluorescence) revealed that numerous neuronal cell bodies were also positive for CCL21 (Fig. 6C, yellow signal, thin arrows). Solely CCL21 signal (red fluorescence) was only present in long processes (Fig. 6C, thick arrow, red staining) and was never observed in cell body-like structures. Serial pictures taken in the Z-direction clearly showed that these CCL21-positive processes always belonged to NeuN-positive neurons (data not shown). Conversely, when neurons were stained for the axonal marker MAP-2 (green fluorescence) together with CCL21 (red fluorescence), it was found that most axons showed double labeling (Fig. 6D, thin arrows, yellow signal). Only few axons were found solely positive for MAP-2 (Fig. 6D, arrowheads, green staining). Thus, CCL21 expression in organotypic brain slice cultures was clearly located in neuronal cell bodies and axon-like processes.
Figure 6.

Expression of neuronal CCL21 in organotypic brain slices. Nuclear staining (Hoechst dye, blue signal) and immunohistochemical staining of axons (MAP-2 antibody, green signal) showed the intact structure of the hippocampal organization (CA1, CA3, and DG) of an organotypic brain slice culture. The inset represents the area in which the microphotographs (B-D) were taken (A). Similar to cultured neurons, treatment with glutamate (100 μm, 30 min) induced expression of CCL21 (B). Strikingly, CCL21 staining (red fluorescence) was mostly observed in long processes (B). Double-labeling experiments with anti-NeuN antibody (green fluorescence) to stain neuronal somata and anti-CCL21 antibody (red fluorescence) showed that both CCL21-negative cell somata were found (solely green staining, arrowheads, C) and neurons that were positive for both NeuN and CCL21 (yellow signal, thin arrows, C). However, solely CCL21 signal (red fluorescence) was also observed (thick arrow, red staining, C). CCL21 immunoreactivity was also observed in axons when neurons were stained with MAP-2 antibody (green fluorescence) together with CCL21 (red fluorescence) because most axons showed double labeling (thin arrows, yellow signal) (D). Only few axons were found solely positive for MAP-2 (arrowheads, green staining, D). Scale bars: A, 500 μm; B-D, 50 μm. Similar results have been obtained in three independent experiments, performed on three separate experimental days.
Microglia and recombinant cells expressing CXCR3 or CCR7 migrate in response to supernatant from glutamate-treated neuronal cultures
In previous publications, it was shown that microglia respond to recombinant CCL21 via the chemokine receptor CXCR3 (Biber et al., 2001; Rappert et al., 2002). Because our data indicate that CCL21 is expressed in glutamate-treated neurons and most likely released, we investigated whether microglia would respond to supernatant of glutamate-treated neurons. Chemotaxis experiments confirmed previous data that microglia significantly migrated in response to CCL21 (1 nm) (Fig. 7A). Significant migration of microglia was also observed with supernatant (harvested 16 h after the treatment) from neurons that were treated with 1, 10, and 100 μm glutamate (Fig. 7A). The most pronounced effect was observed with supernatant from neurons that were treated with 100 μm glutamate (Fig. 7A). Thus, 100 μm glutamate was used in additional experiments. Microglia already migrated significantly toward supernatants from cultured neurons that were harvested 2 h after the treatment (Fig. 7B). Supernatants taken from neuronal cultures at longer time points after the glutamate treatment induced even higher microglial migration (Fig. 7B). Importantly, a significant reduction of the migration toward neuronal supernatants was observed in microglia cultured from CXCR3-deficient mice, indicating the importance of CCL21-CXCR3 signaling for this microglial behavior (Fig. 7B). Because CXCR3-deficient microglia still showed migration, it is indicated that glutamate treatment induced also the release of other neuronal factors that attracted microglia, like, for example, CX3CL1, as shown previously (Chapman et al., 2000). It was furthermore shown by Chapman et al. (2000) that pretreatment with MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] inhibited the glutamate-induced CX3CL1 cleavage, and they concluded that the release of CX3CL1 was attributable to glutamate-induced neurotoxicity. We describe here similar effects for CCL21 because our findings showed that blocking the effect of glutamate by the NMDA receptor antagonist MK-801 completely abolished the migration of cultured microglia toward neuronal supernatants (Fig. 7C). Thus, inhibiting neurotoxicity abolished the release of microglia attractants by neurons.
Figure 7.

Neuronal supernatant of glutamate-treated neurons attracts microglia as well as recombinant (CXCR3 or CCR7 expressing) HEK293 cells. Application of CCL21 (1 nm) served as a positive control and induced significant chemotaxis of cultured mouse microglia (A). Supernatants taken from neurons 16 h after glutamate treatment significantly attracted microglia cells. This effect was dependent on the concentration of glutamate and was absent when supernatant of untreated neurons was used (A). Migration of microglia was clearly dependent on the time point (0-24 h) of sampling of supernatant from glutamate-treated (100 μm) neurons (B). This time-dependent chemoattraction was not observed in microglia isolated from CXCR3-/- mice (B). The chemotactic effect of supernatant sampled from neurons after treatment with glutamate (100 μm, 6 h) was completely abolished when neurons, before glutamate treatment, were pretreated with the NMDA receptor antagonist MK-801 (C). HEK293 cells transfected with either CXCR3 or CCR7 also showed significant migration toward supernatants from glutamate-treated (100 μm, 24 h) neurons (D). Mock transfection did not influence cellular migration (D). Results have been obtained by performing the experiments six times and on three separate experimental days. Data are presented as mean ± SD (n = 6). *p < 0.05. WT, Wild type.
ELISA analyses were performed to further verify the presence of CCL21 in the supernatants of glutamate-treated neurons. Low concentrations of CCL21 have been obtained in neuronal supernatants 24 h after the treatment with glutamate. The measured values, however, were not significantly above the detection limit (0.1 nm) of the used ELISA assay and were therefore not reliable (data not shown). Thus, to further verify the specific activity of CCL21 in neuronal supernatant, a pharmacological approach was performed using HEK293 cells expressing the two currently known chemokine receptors for CCL21, CXCR3, and CCR7. We demonstrated previously that CXCR3-transfected HEK293 cells migrate in response to recombinant CCL21, making CCL21 the only yet known chemokine that activates CXCR3 and CCR7 (Dijkstra et al., 2004). A significant and comparable migration of both CXCR3- and CCR7-expressing HEK293 cells was observed in response to supernatants from glutamate-treated neurons, whereas mock-transfected HEK293 cells did not show any migratory response (Fig. 7D). Expression of the other CCR7 ligand CCL19 in neuronal cultures was never detected (reverse transcription-PCR experiments, data not shown). Together, from these data, it is strongly suggested that the migration of microglia and chemokine receptor-transfected HEK293 cells is evoked by the presence of CCL21 in neuronal supernatants.
Discussion
Although the CNS has long been considered an immune-privileged site, it is very clear today that the brain is capable of mounting an inflammatory response. There is furthermore substantial evidence that brain inflammation is a double-edged sword. Controlled and well balanced, it significantly contributes to protection and repair strategies. Excessive or chronic inflammation, conversely, can lead to additional damage of the surrounding tissue and is now considered to be a hallmark in all neurodegenerative diseases (Morganti-Kossmann et al., 2002; Olsson et al., 2003).
Microglia, the sentinels of the CNS, are key players in brain inflammation because these cells are activated in response to any type of neuronal damage (Kreutzberg, 1996; Raivich et al., 1999). It is known that activation of these cells may lead to either a neuroprotective or neurodetrimental type of inflammation (Bruce-Keller, 1999; Streit et al., 1999; Streit, 2002). Furthermore, activated microglia have been shown to be instrumental for long-term changes in response to brain lesions (Banati, 2002a,b; Beattie et al., 2002; Parish et al., 2002; Monje et al., 2003).
Healthy neurons most likely suppress microglia activity
In turn, microglia activity is most likely controlled by neurons (Bruce-Keller, 1999; Streit et al., 1999; Neumann, 2001; Streit, 2002). It has been demonstrated that myosin heavy chain II expression in microglia is suppressed by neuronal activity (Neumann et al., 1996) or by stimulation with the neurotrophins NGF or neurotrophin 4 (Neumann et al., 1998). Molecules that are constitutively expressed in the membranes of healthy neurons, such as CD200 or CX3CL1, are moreover known to inhibit microglia activity (Hoek et al., 2000; Zujovic et al., 2000), and, recently, a soluble neuronal protein (CD22) has been described that inhibits cytokine release in microglia (Mott et al., 2004). Thus, these findings suggest that healthy, normal functioning neurons suppress microglia activation.
CCL21, a signal from endangered neurons to activate microglia
In contrast to the neuronal signals that keep microglia quiescent, currently not much is known about how endangered neurons activate surrounding microglia. We have recently described that various neurotoxic treatments lead to rapid induction of CCL21 mRNA in damaged neurons in vitro and in vivo (Biber et al., 2001). Furthermore, we were able to demonstrate that CXCR3 is the primary receptor for CCL21 in microglia (Biber et al., 2001; Rappert et al., 2002; Dijkstra et al., 2004). Accordingly, a function of CCL21 in the signaling between endangered neurons and microglia was proposed (Biber et al., 2002a).
Our new findings on CCL21 expression and processing in neurons corroborate this assumption. Weak CCL21 expression in neuronal cultures under control conditions was most likely attributable to the rather stressful culture environment that cannot be compared with the situation in healthy brain. Even higher expression of neuronal CCL21 protein was rapidly inducible by glutamate treatment, a well known neurotoxic condition (Chapman et al., 2000). It is therefore assumed that the expression of CCL21 is a marker for neuronal stress.
If CCL21 acts as a signaling molecule between damaged neurons and microglia, one has to assume that it is released from neurons. CCL21 was found in the Golgi and TGN when expressed in HEK293 or NG108 cells. In neurons, BFA treatment increased the intensity of CCL21 staining, and CCL21-positive vesicles were observed in the Golgi apparatus of these cells, supporting the involvement of the Golgi and TGN in the release of neuronal CCL21. Whereas in hematopoietic cells chemokines are most likely released via the specialized secretory lysosome (Griffith, 1996; Lacy et al., 1999; Andrews, 2000), this is not likely to occur in neuronal cells. Furthermore, our observation of Golgi and TGN involvement shows that CCL21 does not belong to the family of leaderless secretory cytokines, such as interleukin-1β, that do not use the classical release pathway (Rubartelli et al., 1990). In neurons, two different release pathways, the constitutive and the regulated secretory pathways, have been described (Gerber and Sudhof, 2002). The TGN is discussed to be the organelle in which it is decided through which pathway a protein is released (for review, see Tekirian, 2002; Ponnambalam and Baldwin, 2003). However, it is not known yet which of the two release pathways is used for the secretion of CCL21 from neurons. Still, our results clearly show that the supernatants of glutamate-treated neurons attracted cultured microglia and that this response is primarily dependent on the expression of CXCR3 in these cells. Moreover, HEK293 cells expressing either CXCR3 or CCR7 responded to neuronal supernatants with migration. Because CCL21 is the only yet known chemokine that can activate these two receptors, it is indicated that CCL21 released from stressed neurons is functional and can induce significant cellular responses. However, it is not clear yet why we did not find reliable concentrations of CCL21 by ELISA analysis, although neuronal supernatants induced a comparable migration because it was found with 1 nm recombinant CCL21. One explanation might be that CCL21 released from neurons is processed in a way that it is not longer recognized by the antibodies in the ELISA assay. Alternatively, neuronal CCL21 might act in synergism with other factors, thereby increasing its potency and enabling cellular responses at very low concentrations. Both processing of chemokines and heterodimerization has already been described for chemokines (for review, see Fernandez and Lolis, 2002). Experiments to investigate the structure of neuronal CCL21 by mass spectrometry are therefore in preparation in our laboratory.
A role of CCL21 for neuroinflammatory signaling distant from a neuronal lesion
CCL21-positive vesicle-like structures were detected in neuronal processes, and they were also found at the endings of these processes. Ultrastructural evidence furthermore indicated that CCL21-positive vesicles were transported into presynaptic structures, and we even detected CCL21 immunoreactivity in the synaptic cleft. These findings are interesting in view of the fact that microglia activation can be observed at distant sites from a primary neuronal lesion (Kreutzberg, 1996; Bechmann and Nitsch, 2000; Cagnin et al., 2001; Banati, 2002a,b). Injury of the substantia nigra pars compacta (SNpc), for example, leads to secondary activation of microglia in the dorsal striatum (caudate-putamen), in places in which the neurons coming from the SNpc project to. Recent results clearly show that this glial activity is beneficial and promotes sprouting and regeneration in response to the lesion (Parish et al., 2002). Although it has long been known that secondary microglia activity can be observed remote from the primary lesion site, it could not be explained how the signal that activates microglia reaches the distant site. Our observations concerning the expression, distribution, and release by endangered neurons of CCL21 would offer an explanation for this yet unsolved problem.
Is CCL21 specific for neuron-microglia communication?
We and others have never been able to demonstrate expression of CCL21 in any glial cell type in vitro or in vivo (Biber et al., 2001; Alt et al., 2002; Columba-Cabezas et al., 2003). This is rather astonishing because glial cells are a major cellular source of chemokine release in brain. Thus, CCL21 is the only chemokine detected, so far, that is specifically expressed in endangered neurons.
Microglia are not the only cells in brain that express the CCL21 receptor CXCR3. It has been shown recently that CXCR3 is also expressed in astrocytes and neurons (Xia et al., 2000; Biber et al., 2002; Sui et al., 2004; Vlkolinsky et al., 2004). However, functional responses in these studies have only been elicited with CXCL10, the originally identified CXCR3 ligand. In none of these studies was CCL21 demonstrated to be a functional ligand for astrocytic or neuronal CXCR3. Furthermore, we have shown recently that activation of human CXCR3 by CCL21 is predominately achievable when this receptor is expressed in microglia but not in other cell types (Dijkstra et al., 2004). Next to this, we could demonstrate that, in the absence of CXCR3, microglial activation was prevented after brain damage, whereas astrogliosis was indistinguishable from the wild-type situation. This suggests that the lack of CXCR3 has a greater impact on the activation of microglia than on other cell types (Rappert et al., 2004). It is thus tempting to speculate that CCL21 released from neurons would only be detected by microglia, thus yielding a highly specific and effective neuron to microglia signaling.
In summary, the chemokine CCL21 is synthesized specifically in endangered, glutamate-treated neurons and is transported along axons and subsequently released. Clearly, CCL21 is involved in the signaling between endangered neurons and microglia cells, supporting the concept that neurons actively contribute to the onset of inflammation in the brain. We suggest that the activity of CCL21 serves to activate microglia at distance from the primary lesion.
Footnotes
This work was supported by Senter Project TSGE3101 (E.K.d.J.) and a Dutch Foundation for Scientific Research (NWO) Vidi grant (K.B.).
Correspondence should be addressed to Knut Biber, University Medical Center Groningen, Department of Medical Physiology, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands. E-mail: k.biber@med.umcg.nl.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257548-10$15.00/0
References
- Alt C, Laschinger M, Engelhardt B (2002) Functional expression of the lymphoid chemokines CCL19 (ELC) and CCL 21 (SLC) at the blood-brain barrier suggests their involvement in G-protein-dependent lymphocyte recruitment into the central nervous system during experimental autoimmune encephalomyelitis. Eur J Immunol 32: 2133-2144. [DOI] [PubMed] [Google Scholar]
- Ambrosini E, Aloisi F (2004) Chemokines and glial cells: a complex network in the central nervous system. Neurochem Res 29: 1017-1038. [DOI] [PubMed] [Google Scholar]
- Andrews NW (2000) Regulated secretion of conventional lysosomes. Trends Cell Biol 10: 316-320. [DOI] [PubMed] [Google Scholar]
- Bacon KB, Harrison JK (2000) Chemokines and their receptors in neurobiology: perspectives in physiology and homeostasis. J Neuroimmunol 104: 92-97. [DOI] [PubMed] [Google Scholar]
- Bajetto A, Bonavia R, Barbero S, Schettini G (2002) Characterization of chemokines and their receptors in the central nervous system: physiopathological implications. J Neurochem 82: 1311-1329. [DOI] [PubMed] [Google Scholar]
- Banati RB (2002a) Brain plasticity and microglia: is transsynaptical glial activation in the thalamus after limb denervation linked to cortical plasticity and central sensitization? J Physiol (Lond) 96: 289-299. [DOI] [PubMed] [Google Scholar]
- Banati RB (2002b) Visualising microglial activation in vivo. Glia 40: 206-217. [DOI] [PubMed] [Google Scholar]
- Baron W, de Jonge JC, Vries H, Hoekstra D (2000) Perturbation of myelination by activation of distinct signaling pathways: an in vitro study in a myelinating culture derived from fetal rat brain. J Neurosci Res 59: 74-85. [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Keun Ha B, von Zastrow M, Beattie MS, Malenka RC (2002) Control of synaptic strength by glial TNFα. Science 295: 2282-2285. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Nitsch R (2000) Involvement of non-neuronal cells in entorhinal-hippocampal reorganization following lesions Ann NY Acad Sci 911: 192-206. [DOI] [PubMed] [Google Scholar]
- Biber K, Sauter A, Brouwer N, Copray SCVM, Boddeke HWGM (2001) Ischemia-induced neuronal expression of the microglia attracting chemokine secondary lymphoid-tissue chemokine (SLC). Glia 34: 121-133. [DOI] [PubMed] [Google Scholar]
- Biber K, Zuurman MW, Dijkstra IM, Boddeke HWGM (2002a) Chemokines in the brain: neuroimmunology and beyond. Curr Opin Pharmacol 2: 63-68. [DOI] [PubMed] [Google Scholar]
- Biber K, Dijkstra I, Trebst C, De Groot CJA, Ransohoff RM, Boddeke HWGM (2002b) Functional expression of CXCR3 in cultured mouse and human astrocytes and microglia. Neuroscience 112: 487-497. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ (1999) Microglial-neuronal interactions in synaptic damage and recovery. J Neurosci Res 58: 191-201. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Myers R, Gunn RN, Lawrence AD, Stevens T, Kreutzberg GW, Jones T, Banati RB (2001) In vivo visualization of activated glia by [11C](R)-PK11195-PET following herpes encephalitis reveals projected neuronal damage beyond the primary focal lesion. Brain 124: 2014-2027. [DOI] [PubMed] [Google Scholar]
- Chapman GA, Moores K, Harrison D, Campbell CA, Steward BR, Strijbos PJLM (2000) Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci 20: RC87(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Columba-Cabezas S, Serafini B, Ambrosini E, Aloisi F (2003) Lymphoid chemokines CCL19 and CCL21 are expressed in the central nervous system during experimental autoimmune encephalomyelitis: implications for the maintenance of chronic neuroinflammation. Brain Pathol 13: 38-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra M, Hulshof S, van der Valk P, Boddeke HWGM, Biber K (2004) Functional activity of human adult microglia in response to CCL21. J Immunol 172: 2744-2747. [DOI] [PubMed] [Google Scholar]
- Dinter A, Berger EG (1998) Golgi-disturbing agents. Histochem Cell Biol 109: 571-590. [DOI] [PubMed] [Google Scholar]
- Fernandez EJ, Lolis E (2002) Structure, function and inhibition of chemokines. Annu Rev Pharmacol Toxicol 42: 469-499. [DOI] [PubMed] [Google Scholar]
- Fife BT, Huffnagel GB, Kuziel WA, Karpus WJ (2000) CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med 192: 899-905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaehwiler BH, Capogna M, Debanne D, McKinney RA, Thompson SM (1997) Organotypic slice cultures: a technique has come of age. Trends Neurosci 20: 471-477. [DOI] [PubMed] [Google Scholar]
- Gerber SH, Sudhof TC (2002) Molecular determinants of regulated exocytosis. Diabetes 51 [Suppl 1]: S1-S11. [DOI] [PubMed] [Google Scholar]
- Griffith GM (1996) Secretory lysosomes—a special mechanism of regulated secretion in haemopoietic cells. Trends Cell Biol 6: 329-332. [DOI] [PubMed] [Google Scholar]
- Hesselgesser J, Horuk R (1999) Chemokine and chemokine receptor expression in the central nervous system. J Neurovirol 5: 13-26. [DOI] [PubMed] [Google Scholar]
- Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, Blom B, Homola ME, Streit WJ, Brown MH, Barclay AN, Sedwick JD (2000) Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 290: 1768-1771. [DOI] [PubMed] [Google Scholar]
- Huang D, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM (2001) Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med 93: 713-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD (2000) Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med 192: 1075-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberg GW (1996) Microglia: a sensor for pathological event is the CNS. Trends Neurol Sci 19: 312-318. [DOI] [PubMed] [Google Scholar]
- Lacy P, Mahmudi-Azer S, Bablitz B, Hagen SC, Velazquez JR, Man SFP, Moqbel R (1999) Rapid mobilization of intracellularly stored RANTES in response to interferon-γ in human eosinophils. Blood 94: 23-32. [PubMed] [Google Scholar]
- Liem RSB, Brouwer N, Copray JCVM (2001) Ultrastructural localisation of intramuscular expression of BDNF mRNA by silver-gold intensified non-radioactive in situ hybridisation. Histochem Cell Biol 116: 545-551. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402-408. [DOI] [PubMed] [Google Scholar]
- Lore K, Spetz A-L, Fehninger TE, Sonnerborg A, Landay AL, Andersson J (2001) Quantitative single cell methods that identify cytokine and chemokine expression in dendritic cells. J Immunol Methods 249: 207-222. [DOI] [PubMed] [Google Scholar]
- Mackay CR (2001) Chemokines: immunology's high impact factors. Nat Immunol 2: 95-101. [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD (2003) Inflammatory blockade restores adult hippocampal neurogenesis. Science 302: 1760-1765. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T (2002) Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care 8: 101-105. [DOI] [PubMed] [Google Scholar]
- Moser B, Wolf M, Walz A, Loetscher P (2004) Chemokines: multiple levels of leukocyte migration control. Trends Immunol 25: 75-84. [DOI] [PubMed] [Google Scholar]
- Mott RT, Ait-Ghezala G, Town T, Mori T, Vendrame M, Zeng J, Ehrhart J, Mullan M, Tan J (2004) Neuronal expression of CD22: novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia 46: 369-379. [DOI] [PubMed] [Google Scholar]
- Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA (2000) International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev 52: 145-176. [PubMed] [Google Scholar]
- Neumann H (2001) Control of glia immune function by neurons. Glia 36: 191-199. [DOI] [PubMed] [Google Scholar]
- Neumann H, Boucraut J, Hahnel C, Misgeld T, Weckerle H (1996) Neuronal control of MHC class II inducibility in rat astrocytes and microglia. Eur J Neurosci 8: 2582-2590. [DOI] [PubMed] [Google Scholar]
- Neumann H, Misgeld T, Matsumuro K, Weckerle H (1998) Neurotrophins inhibit MHC class II inducibility of microglia: involvement of p75 neurotrophin receptor. Proc Natl Acad Sci USA 95: 5779-5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson T, Lidman O, Piehl F (2003) Harm or heal—divergent effects of autoimmune neuroinflammation? Trends Neurosci 24: 5-6. [DOI] [PubMed] [Google Scholar]
- Parish CL, Finkelstein DI, Tripanichkul W, Satoskar AR, Drago J, Horne MK (2002) The role of interleukin-1, interleukin-6 and glia in inducing growth of neuronal terminal arbors in mice. J Neurosci 22: 8034-8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnambalam S, Baldwin SA (2003) Constitutive protein secretion from the trans-Golgi network to the plasma membrane. Mol Mem Biol 20: 129-139. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Kloss CUA, Werner A, Jones LL, Kreutzberg GW (1999) Neuroglial activation repertoire in the injured brain: graded response molecular mechanisms and cues to physiological function. Brain Res Rev 30: 77-105. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Tani M (1998) Do chemokines mediate leukocyte recruitment in post-traumatic CNS inflammation? Trends Neurosci 4: 154-159. [DOI] [PubMed] [Google Scholar]
- Rappert A, Biber K, Nolte C, Lipp M, Schubel A, Lu B, Gerard NP, Gerard C, Boddeke HWMG, Kettenmann H (2002) Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl- current and chemotaxis in murine microglia. J Immunol 169: 3221-3226. [DOI] [PubMed] [Google Scholar]
- Rappert A, Bechmann I, Pivneva T, Mahlo J, Biber K, Nolte C, Kovac AD, Gerard C, Boddeke HWGM, Nitsch R, Kettenmann K (2004) CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J Neurosci 24: 8500-8509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D, Zlotnik A (2000) The biology of chemokines and their receptors. Annu Rev Immunol 18: 217-242. [DOI] [PubMed] [Google Scholar]
- Rot A, von Andrian UH (2004) Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol 22: 891-928. [DOI] [PubMed] [Google Scholar]
- Rubartelli A, Cozzolino F, Talio M, Sitia RA (1990) A novel secretory pathway for interleukin 1b, a protein lacking a signal sequence. EMBO J 9: 1503-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert H, Sachse A, Kuziel WA, Maeda N, Brueck W (2000) The chemokine receptor CCR2 is involved in macrophage recruitment to the injured peripheral nervous system. J Neuroimmunol 110: 177-185. [DOI] [PubMed] [Google Scholar]
- Streit WJ (2002) Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 40: 133-139. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA (1999) Reactive microgliosis. Prog Neurobiol 57: 563-581. [DOI] [PubMed] [Google Scholar]
- Sui Y, Potula R, Dhillon N, Pinson D, Li S, Nath A, Anderson C, Turchan J, Kolson D, Narayan O, Buch S (2004) Neuronal apoptosis is mediated by CXCL10 overexpression in simian human immunodeficiency virus encephalitis. Am J Pathol 164: 1557-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekirian TL (2002) The central role of the trans-Golgi network as a gateway of the early secretory pathway: physiologic vs nonphysiologic protein transit. Exp Cell Res 281: 9-18. [DOI] [PubMed] [Google Scholar]
- Vlkolinsky R, Siggins GR, Campbell IL, Krucker T (2004) Acute exposure to CXC chemokine ligand 10, but not its chronic astroglial production, alters synaptic plasticity in mouse hippocampal slices. J Neuroimmunol 150: 37-47. [DOI] [PubMed] [Google Scholar]
- Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT (2000) Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimers disease. J Neuroimmunol 108: 227-235. [DOI] [PubMed] [Google Scholar]
- Zujovic V, Benavides J, Vige X, Carter C, Taupin V (2000) Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia 29: 305-315. [PubMed] [Google Scholar]