

Abstract
Casein kinase 1 (CK1) is a highly conserved serine/threonine kinase, present in virtually all cell types, in which it phosphorylates a wide variety of substrates. So far, no role has been found for this ubiquitous protein kinase in the physiology of nerve cells. In the present study, we show that CK1 regulates fast synaptic transmission mediated by glutamate, the major excitatory neurotransmitter in the brain. Through the use of CK1 inhibitors, we present evidence that activation of CK1 decreases NMDA receptor activity in the striatum via a mechanism that involves activation by this kinase of protein phosphatase 1 and/or 2A and resultant increased dephosphorylation of NMDA receptors. Indeed, inhibition of CK1 increases NMDA-mediated EPSCs in medium spiny striatal neurons. This effect is associated with an increased phosphorylation of the NR1 and NR2B subunits of the NMDA receptor and is occluded by the phosphatase inhibitor okadaic acid. The mGluR1, but not mGluR5, subclass of metabotropic glutamate receptors uses CK1 to inhibit NMDA-mediated synaptic currents. These results provide the first evidence for a role of CK1 in the regulation of synaptic transmission in the brain.
Keywords: excitatory postsynaptic current, EPSC, NMDA receptor, synaptic transmission, phosphorylation, striatum, metabotropic glutamate receptor
Introduction
Casein kinase 1 (CK1) was one of the first serine/threonine protein kinases to be identified (Gross and Anderson, 1998). CK1 forms one of the eight major groups of protein kinases identified in the human and mouse genome (Manning et al., 2002; Caenepeel et al., 2004). The CK1 family consists of several isoforms that include, in all animal species, CK1α, CK1γ1-CK1γ3, CK1δ, and CK1ϵ. CK1 is present in different cell types and in several subcellular compartments, including the plasma membrane, cytosol, and nucleus. The widespread distribution of CK1 suggests important regulatory roles for this protein kinase. Recently, CK1 has been implicated in diverse biological functions, such as regulation of DNA repair, cellular morphology, modulation of the Wnt/β-catenin pathway during development, and regulation of circadian rhythms in adulthood (Peters et al., 1999; Whitmore et al., 2000). In addition, CK1α was shown to enhance agonist-dependent phosphorylation of the M3 muscarinic receptor and to mediate phosphorylation of rhodopsin in response to light stimulation (Tobin, 2002).
Recent evidence shows that the activity of CK1 in the brain can be regulated by glutamate, acting on group I metabotropic receptors (mGluRs) (Liu et al., 2001, 2002). Glutamate is the major excitatory neurotransmitter in the brain and plays a critical role in multiple brain functions. For example, the activity of neurons in the striatum, a brain region involved in movement- and motivation-related behaviors, is dependent on a strong glutamatergic innervation from the cortex.
The function of CK1 in the physiology of striatal neurons and in glutamate-mediated activity is unknown. In addition, a comparative examination of the distribution of CK1 isoforms in different brain regions has not been performed. In the present study, we examined the expression of the different CK1 isoforms in sections of mouse brain and we tested whether corticostriatal synaptic transmission is regulated by CK1. We also investigated a possible participation of CK1 in the regulation of ionotropic glutamatergic synaptic transmission by metabotropic glutamate receptors. Our study demonstrates that all major CK1 isoforms are expressed in the striatum and in the cortex, and that CK1 regulates glutamatergic synaptic transmission mediated by NMDA receptors via activation of protein phosphatase 1 (PP1) and PP2A.
Materials and Methods
Electrophysiology in brain slices
Experiments were approved by the Institutional Animal Care and Use Committee and were performed using methods described previously (Chergui and Lacey, 1999; Chergui et al., 2000). Briefly, coronal brain slices (400 μm thick), containing the striatum and neocortex, were prepared with a microslicer (VT1000S; Leica, Nussloch, Germany) from C57BL/6 mice aged 22-29 d. Slices were incubated at 32°C in oxygenated (95% O2/5% CO2) artificial CSF (aCSF) containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.3 MgCl2, 2.4 CaCl2, 10 glucose, and 26 NaHCO3, pH 7.4. Slices were transferred to a recording chamber (Warner Instruments, Hamden, CT) mounted on an upright microscope (Olympus Optical, Melville, NY) and were continuously perfused with oxygenated aCSF at 32°C. Medium spiny neurons in the striatum were visualized within the brain slices using Nomarski-type differential interference contrast optics combined with infrared video microscopy. Whole-cell voltage-clamp recordings of medium-sized neurons in the dorsal striatum were made with patch electrodes filled with a solution containing the following (in mm): 140 CsCl, 2 MgCl2, 1 CaCl2, 10 HEPES, 10 EGTA, 2 MgATP, and 0.3 Na3GTP, pH 7.3. Whole-cell membrane currents were recorded with an Axopatch 200B (Axon Instruments, Foster City, CA) driven by a personal computer. Neurons were voltage clamped at -80 mV. The resting membrane potential of medium spiny striatal neurons measured with a potassium gluconate-based intracellular solution was -84.5 ± 0.5 mV (n = 34), which is in accordance with previously published observations (Calabresi et al., 1991; Jiang and North, 1991). Data were acquired and analyzed with pClamp8 software (Axon Instruments). Numerical values are expressed as means ± SEM, with n indicating the number of medium spiny striatal neurons tested. For EPSCs, data are expressed as percentage of the baseline measured for each cell during the 5 min preceding application of the drug examined. All drugs were applied in the perfusion solution except when noted. EPSCs were evoked every 15 s by electrical stimulation of the slice using a patch electrode filled with aCSF positioned on the slice surface in the vicinity of the recorded neuron. EPSCs mediated by NMDA receptors were recorded in 0.1 mm MgCl2 to reduce magnesium blockade of the receptor and in the presence of CNQX (10 μm) and bicuculline (10 μm) to inhibit AMPA-EPSCs and GABAA-IPSCs, respectively. EPSCs were mediated by NMDA receptors as demonstrated by their blockade by the NMDA receptor antagonist dl-AP-5 (data not shown). AMPA-EPSCs were recorded in the presence of dl-AP-5 (100 μm) to inhibit NMDA-EPSCs and bicuculline (10 μm). We assessed the stability of our recordings by monitoring the amplitude of NMDA-EPSCs over time with normal intracellular solution in the absence of any bath-applied compound in eight striatal neurons. We found that whole-cell recording did not affect NMDA-EPSC amplitude (see Fig. 2b). The effect of the CK1 inhibitor CK1-7 on NMDA-EPSCs was not associated with a significant change in input resistance of the recorded neurons (60.3 ± 7.2 and 66.1 ± 7.3 MΩ before and during CK1-7, respectively; n = 13). In the experiments using intracellular application of the protein phosphatase inhibitor okadaic acid, this compound was allowed to diffuse from the pipette into the recorded neuron for at least 20 min before bath application of CK1-7 or (S)-3,5-dihydroxyphenylglycine (DHPG). This also allowed NMDA-EPSC amplitude to stabilize because of the effect of okadaic acid (our unpublished observations) (Lu et al., 1999). Whole-cell inward currents were activated with a 2 min bath application of either NMDA (30 μm at a holding potential of -60 mV in normal MgCl2) or kainate (5 μm), in the presence of tetrodotoxin (0.5 μm), bicuculline (10 μm), and either CNQX (10 μm) or dl-AP-5 (50 μm) for NMDA and AMPA currents, respectively. This method of application of glutamate receptor agonists was chosen, over local application such as microiontophoresis, to stimulate all of the receptors located on the recorded neuron. Bath application of these agonists allowed us to correlate biochemical and electrophysiological data and to compare our results with those published by several other groups who have used this method to activate glutamate receptors. Drugs were obtained from the following sources: CNQX, bicuculline, dl-AP-5, kainic acid, and NMDA were from Sigma (St. Louis, MO); CK1-7 was from USBiological (Swampscott, MA); IC261 (3-[(2,4,6-trimethoxyphenyl)methylidenyl]-indolin-2-one) was from Calbiochem (La Jolla, CA); okadaic acid was from Alexis Biochemicals (San Diego, CA); and (S)-3,5-DHPG, 2-methyl-6-(phenylethynyl)-pyridine (MPEP), and LY367385 [(S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid] were from Tocris Cookson (Ballwin, MO). We followed the recommendations of Davies et al. (2000) and Bain et al. (2003) for the use of protein kinase inhibitors in cell-based assays when we designed our experiments to ensure that the concentrations of the inhibitors used (CK1-7 and IC261) are selective for CK1. (1) The concentrations of CK1-7 and IC261 used in our study were less than 10 times higher than the IC50 values found by our group and by others for inhibition of CK1 in vitro (≈10-15 and 1-16 μm, respectively) (Chijiwa et al., 1989; Behrend et al., 2000; Mashhoon et al., 2000; Liu et al., 2001). (2) In a previous study from our laboratory (Liu et al., 2001), we found that 100 μm CK1-7 (the same concentration used in the present study in the same preparation, i.e., brain slices from the striatum) decreases the basal phosphorylation level of DARPP-32 at a site phosphorylated by CK1. (3) CK1-7 and IC261 are two structurally unrelated inhibitors but they produce identical effect on both NMDA receptor activity and phosphorylation.
Figure 2.
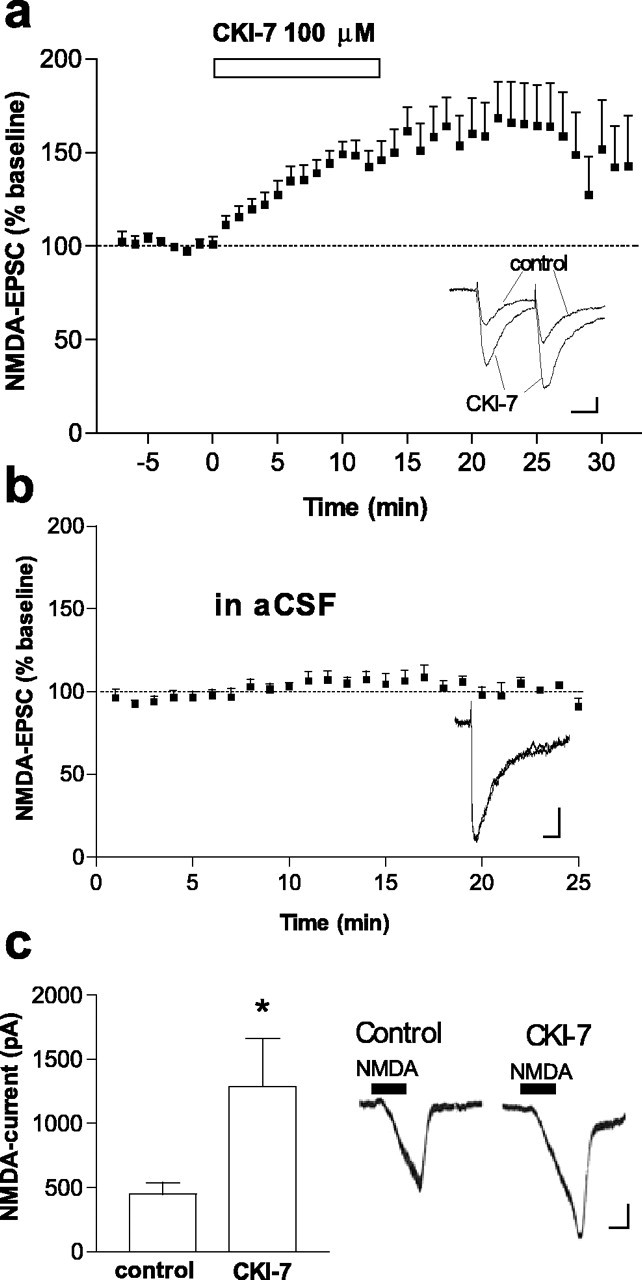
Inhibition of CK1 increases the activity of the NMDA receptor. a, Time course of the effect of the CK1 inhibitor CK1-7 (100μm), applied in the perfusion solution (as indicated by the horizontal bar), on NMDA-EPSC amplitude in striatal neurons voltage clamped at a membrane potential of -80 mV (n = 21). Inset shows superimposed NMDA-EPSCs recorded before (control) and after (CK1-7) bath application of CK1-7 (100 μm) in a representative neuron. Calibration: 20 pA, 20 ms. Inset shows no change in EPSC2/EPSC1 after CK1-7. b, Absence of effect of whole-cell recording on NMDA-EPSC amplitude in eight striatal neurons recorded in control perfusion solution. Records show two superimposed NMDA-EPSCs at the beginning of the recording and after 20 min of whole-cell recording in a representative neuron. Calibration: 20 pA, 20 ms. c, Amplitude of whole-cell inward current activated by NMDA (30 μm) before and after CK1-7 (100 μm) in six striatal neurons. Traces on the right of the graph show NMDA-activated inward currents in a representative neuron before (control) and after CK1-7. Calibration: 50 pA, 1 min. *p < 0.05 compared with control.
Anatomical studies Immunoblot analysis.
Frozen mouse cortices or striata were sonicated in 1% SDS, and proteins were resolved by 10% SDS-PAGE and transferred to a polyvinylidene difluoride membrane, followed by immunoblot analysis (Svenningsson et al., 1998). Immunoblot analysis was performed using specific antibodies against each of the CK1 isoforms α, γ, δ, and ϵ (Santa Cruz Biotechnology, Santa Cruz, CA; BD Biosciences, Franklin Lakes, NJ) and detected by enhanced chemiluminescence (Amersham Biosciences, Arlington Heights, IL).
In situ hybridization. Cryostat sections (12 μm) made from frozen C57BL/6 mouse brains were hybridized as described previously (Svenningsson et al., 1997) with [α- 35S]UTP-labeled riboprobes prepared by in vitro transcription from cDNA clones corresponding to mouse CK1 isoforms α, γ, δ, and ϵ (American Type Culture Collection, Manassas, VA). Sections were then exposed to Biomax MR films (Eastman Kodak, Rochester, NY) for 2-14 d.
Biochemical studies on NMDA receptor phosphorylation
Striatal brain slices (300 μm) were prepared from adult male C57BL/6 mice as described previously (Svenningsson et al., 1998). For studies of NR1 subunit phosphorylation, slices were incubated in Krebs' buffer at 30°C under constant oxygenation (95% O2/5% CO2) for 60 min, with a change of buffer after 30 min. The slices were thereafter treated with either CK1-7 (100 or 300 μm) or IC261 (50 μm) for 10 min. At the end of the treatment, the slices were rapidly frozen and processed for immunoblot analysis as described above. Immunoblot analysis was performed with a phospho-specific antibody against Ser897-NR1 (Upstate Biotechnology, Lake Placid, NY). Autoradiograms were quantified using NIH Image 1.62. For studies of NR2 subunit phosphorylation, 32P-labeled slices were used as described previously (Snyder et al., 1998). The slices were incubated for 60 min in phosphate-free Krebs' buffer containing 2.0 mCi of [32P]orthophosphoric acid. The labeling buffer was then removed, and the tissue sections were incubated in Krebs' buffer in the absence or presence of either CK1-7 (100 or 300 μm) or IC261 (50 μm) for 10 min. The slices were then immediately sonicated and incubated overnight at 4°C with 5-10 μg of polyclonal antibodies to immunoprecipitate 32P-NR2A, 32P-NR2B, and 32P-NR2C (Calbiochem). Immune complexes were then incubated with protein A-Sepharose, and the beads were subsequently collected, washed, eluted in SDS sample buffer, and resolved by 8% SDS-PAGE. The gels were dried, and [32P]phosphate incorporation was quantified using a PhosphorImager 400B and Image-Quant software from Molecular Dynamics (Sunnyvale, CA).
Results
CK1 isoforms are expressed in mouse striatum and neocortex
In mammals, there are seven different CK1 isoforms known as α, β, γ1-γ3, δ, and ϵ (Gross and Anderson, 1998). The β isoform is only present in bovine brain. Because there is little information on the distribution of the different isoforms of CK1 in the CNS, we examined the expression of CK1α, CK1γ, CK1δ, and CK1ϵ in the mouse brain with particular emphasis on the striatum and the cerebral cortex, the main source of glutamatergic input to the striatum. Immunoblotting performed with antibodies against each of the CK1 isoforms demonstrated the presence of CK1α, CK1γ, CK1δ, and CK1ϵ in both the striatum and the neocortex (Fig. 1a). The sizes of the bands identified in our material were similar to those calculated previously for CK1α, CK1γ1-CK1γ3, CK1δ, and CK1ϵ (37.5, 45.0-51.3, 49.1, and 47.3 kDa, respectively) (Graves et al., 1993; Fish et al., 1995; Zhai et al., 1995; Zhang et al., 1996). Using in situ hybridization, we examined the distribution of mRNAs coding for the distinct CK1 isoforms and found that all are expressed in variable amounts in many brain structures, including the neocortex and the striatum (Fig. 1b). Each of these isoforms could therefore play a role in the physiology of striatal neurons.
Figure 1.

Expression of CK1 isoforms in the brain. a, Immunoblots performed with antibodies against the CK1 isoforms α, γ, δ, and ϵ in the striatum (str; left lanes) and the neocortex (cor; right lanes). Molecular weights of 45 and 35 kDa are shown on the left of the blots to indicate relative weight of CK1 isoforms. The arrows indicate the bands that correspond to the expected molecular weight for each of the isoforms. Two clear bands of close molecular weights observed for CK1α and CK1δ could correspond to splice variants of these isoforms. The two bands observed for CK1γ could correspond to different isoforms. It is very likely that the CK1ϵ band corresponds solely to the CK1ϵ1 splice variant. b, In situ hybridization of mRNAs coding for CK1α, CK1γ, CK1δ, and CK1ϵ in parasagittal sections of mouse brain. CK1 isoforms are expressed in several brain structures, including striatum (str).
Inhibition of CK1 increases NMDA receptor but not AMPA receptor activity
To assess the possible involvement of CK1 in corticostriatal glutamatergic synaptic transmission, we evaluated the effects of two specific CK1 inhibitors, CK1-7 (Chijiwa et al., 1989) and IC261 (Behrend et al., 2000; Mashhoon et al., 2000), on EPSCs evoked by electrical stimulation of glutamatergic fibers within the striatum. EPSCs mediated by either NMDA receptors or AMPA receptors were pharmacologically isolated. CK1-7 (100 μm) applied in the perfusion solution produced a robust increase in NMDA-EPSC amplitude (percentage of baseline, 184.1 ± 10.6%; n = 21) (Fig. 2a). CK1-7 produced a small increase in the half-decay time of NMDA-EPSCs [20.4 ± 1.4 and 23.9 ± 1.5 ms, before and after CK1-7 (100 μm), respectively; n = 21]. IC261 (50 μm) also increased NMDA-EPSC amplitude (percentage of baseline, 140.2 ± 5.8%; n = 8). Whole-cell currents activated by NMDA, in the presence of tetrodotoxin, were also increased by CK1-7 (Fig. 2c). We additionally examined the effect of CK1-7 in neurons voltage clamped at +20 mV, and we found that this inhibitor also increased NMDA-EPSC amplitude (percentage of baseline, 170.1 ± 22.8%; n = 5). In contrast, AMPA-EPSCs (Fig. 3a) or whole-cell AMPA currents, activated by kainate, an AMPA/kainate receptor agonist (Fig. 3b), were not affected by bath-applied CK1-7 (100 μm). Moreover, direct application of CK1-7 into the recorded neuron by diffusion from the pipette did not affect whole-cell AMPA currents (432.8 ± 94.4 pA in seven control cells and 425.5 ± 102.2 pA in four cells loaded with CK1-7). These data indicate that CK1 specifically downregulates NMDA receptor function in the striatum. A postsynaptic rather than a presynaptic involvement of CK1 inhibition in the increased NMDA receptor function is indicated by both the absence of an effect on AMPA-EPSCs (Fig. 3a) and the observations that the EPSC2/EPSC1 ratio, a measure of presynaptic action, was not altered in either NMDA-EPSCs (0.82 ± 0.05 before CK1-7 and 0.81 ± 0.05 after CK1-7; n = 8) or AMPA-EPSCs (0.89 ± 0.04 before CK1-7 and 0.92 ± 0.06 after CK1-7; n = 5).
Figure 3.

Inhibition of CK1 does not affect the activity of the AMPA receptor. a, Absence of effect of CK1-7 (100 μm) on AMPA-EPSC amplitude (n = 10). Inset, AMPA-EPSCs recorded before and after CK1-7. Calibration: 20 pA, 20 ms. b, Amplitude of whole-cell inward current activated by kainate (5 μm) before and after CK1-7 (100 μm) in four neurons. Traces show AMPA-activated inward currents in a representative neuron before and after CK1-7. Calibration: 50 pA, 1 min.
Inhibition of CK1 increases the phosphorylation level of NR1 and NR2B
Phosphorylation of the NMDA receptor has been shown to regulate its activity (Scannevin and Huganir, 2000). We therefore examined the phosphorylation state of the NMDA receptor subunits NR1, NR2A, NR2B, and NR2C after incubation of striatal brain slices with CK1-7 (100 or 300 μm) or IC261 (50 μm). These inhibitors increased the phosphorylation levels of the two most abundant NMDA receptor subunits in the striatum, NR1 and NR2B (Fig. 4). These subunits are localized to medium spiny projection neurons in which they presumably form native heteromeric functional receptors (Landwehrmeyer et al., 1995; Ghasemzadeh et al., 1996; Götz et al., 1997). Phosphorylation levels of NR2A and NR2C, which are expressed at low levels in medium spiny striatal neurons (Landwehrmeyer et al., 1995; Ghasemzadeh et al., 1996; Götz et al., 1997), were not affected by inhibition of CK1 [percentage of control: NR2A, 100 ± 5% (control), 96 ± 31.5% (CK1-7), 117.5 ± 18% (IC261); NR2C, 100 ± 31.5% (control), 80 ± 13.3% (CK1-7)] (Fig. 4). Thus, our results show a good correlation between the phosphorylation level of functional NMDA receptors and their activity after CK1 inhibition.
Figure 4.

Inhibition of CK1 increases the phosphorylation state of the NMDA receptor. Left, Immunoblots performed with a phosphospecific antibody against Ser897 of NR1 and autoradiograms of immunoprecipitated 32P-NR2A, 32P-NR2B, and 32P-NR2C subunits in control slices and in slices treated with CK1-7 (100μm). Right, Averaged data (8-10 slices) expressed as percentage of control for P-Ser897-NR1 (top) and for 32P-NR2B (bottom) in control slices, slices treated with CK1-7 (100 and 300μm), and slices treated with IC261 (50μm). *p < 0.05 compared with control, Student's t test.
Inhibition of PP1/PP2A occludes the effect of CK1 inhibition on NMDA receptor activity
Because inhibition of CK1 increases the phosphorylation of NR1 and NR2B, it seemed likely that this kinase regulates NMDA receptor function via activation of a phosphatase. In light of previous biochemical evidence for interactions between CK1 and the protein phosphatases PP1/PP2A and between PP1/PP2A and NMDA receptors (Agostinis et al., 1987; Marin et al., 1994; Westphal et al., 1999; Chan and Sucher, 2001; Dubois et al., 2002), we hypothesized that the protein phosphatases PP1 and/or PP2A could be intermediary elements between CK1 and NMDA receptors. To examine whether inhibition of protein phosphatases might mediate the effect of CK1-7 on NMDA receptor activity, we applied the PP1/PP2A inhibitor okadaic acid in either the perfusion solution or the intracellular solution and examined the effect of CK1-7 on NMDA-EPSCs. In these neurons, CK1-7 did not increase NMDA-EPSC amplitude [percentage of baseline, 110.1 ± 12.2%, n = 7 in the presence of bath-applied okadaic acid (100 nm) (Fig. 5a) and 87.5 ± 9.2%, n = 9 neurons loaded with okadaic acid (1 μm) (Fig. 5b) compared with 184.1 ± 10.6, n = 21 in the absence of okadaic acid (Fig. 2a)]. The fact that okadaic acid occluded the effect of CK1-7 on NMDA-EPSCs indicates that increased activity of NMDA receptors after inhibition of CK1 is mediated through inhibition of PP1/PP2A. This is the first demonstration of a physiological interaction between CK1, PP1/PP2A, and an ionotropic receptor. We also found that bath-applied okadaic acid increased NMDA-EPSC amplitude [percentage of baseline, 128.0 ± 2.9 and 185.0 ± 20.5 in the presence of okadaic acid at 100 nm (n = 4) and 1 μm (n = 3), respectively]. These results are consistent with previous reports showing that protein phosphatases regulate the activity and the state of phosphorylation of NMDA receptors (Wang et al., 1994; Snyder et al., 1998; Lu et al., 1999; Westphal et al., 1999; Flores-Hernandez et al., 2002). Our observation that inhibition of CK1 did not affect AMPA receptor-mediated currents is consistent with some (but not all) observations that synaptically activated AMPA receptors were not affected by okadaic acid (1 μm) in the striatum (Colwell and Levine, 1995) or by peptides that disrupt the binding of PP1 to certain targeting proteins in the hippocampus (Morishita et al., 2001).
Figure 5.
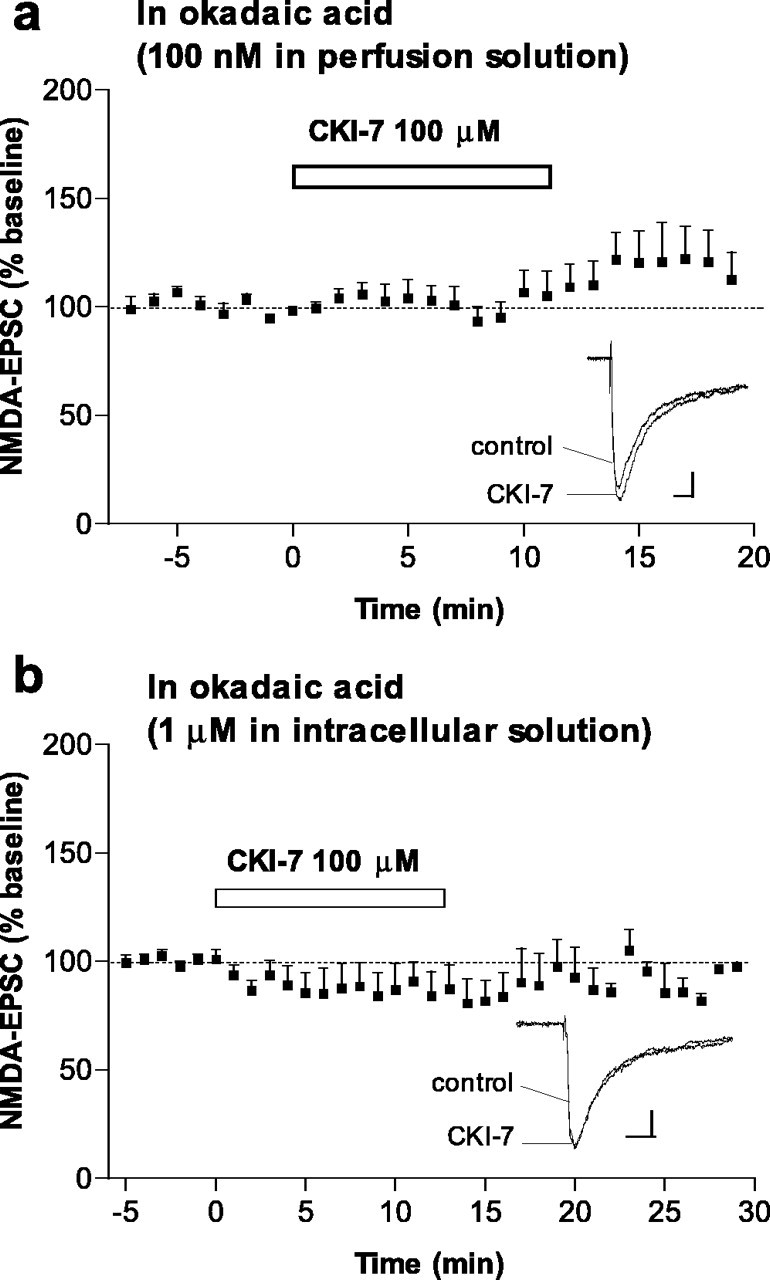
Effect of CK1-7 on NMDA-EPSCs is occluded by the PP1/PP2A inhibitor okadaic acid. a, Time course of the effect of CK1-7 (100μm) on NMDA-EPSC amplitude in neurons pretreated with bath-applied okadaic acid (100 nm; n = 7). Records show superimposed NMDA-EPSCs in a representative neuron before and after CK1-7 in the presence of okadaic acid. Calibration: 20pA, 20ms. b,Time course of the effect of CK1-7 (100 μm)in neurons loaded with okadaic acid (1 μm; n = 9). Inset shows superimposed NMDA-EPSCs recorded before and after CK1-7 in a representative neuron. Calibration: 20 pA, 20 ms.
The group I metabotropic glutamate receptor agonist DHPG depresses NMDA-EPSC amplitude via activation of CK1 and PP1/PP2A
Glutamate regulates the physiology of neuronal cells by acting on both metabotropic and ionotropic receptors. The crosstalk between these classes of glutamate receptors is very complicated and incompletely understood. The present observation that CK1 regulates NMDA-mediated synaptic transmission, together with our previous finding that group I mGluRs increase the activity of CK1 (Liu et al., 2001, 2002), raised the possibility that CK1 could act as an intermediary between metabotropic and ionotropic receptors. We therefore tested whether activation of group I mGluRs would produce an effect opposite to that seen during inhibition of CK1, namely a depression of NMDA-EPSCs. Indeed, a group I mGluR agonist, DHPG (50 μm), added to the perfusion solution, rapidly and reversibly depressed NMDA-EPSCs (Figs. 6a;7b). The observations that DHPG did not affect NMDA-EPSC2/NMDA-EPSC1 [0.83 ± 0.09% before DHPG and 0.86 ± 0.06% after DHPG; n = 6) (Fig. 6a, see EPSCs)] or AMPA-EPSCs [100.6 ± 10.1%; n = 3; as observed by others (Calabresi et al., 1999; Battaglia et al., 2001)] suggest a postsynaptic mechanism rather than a downregulation of glutamate release by group I mGluRs.
Figure 6.
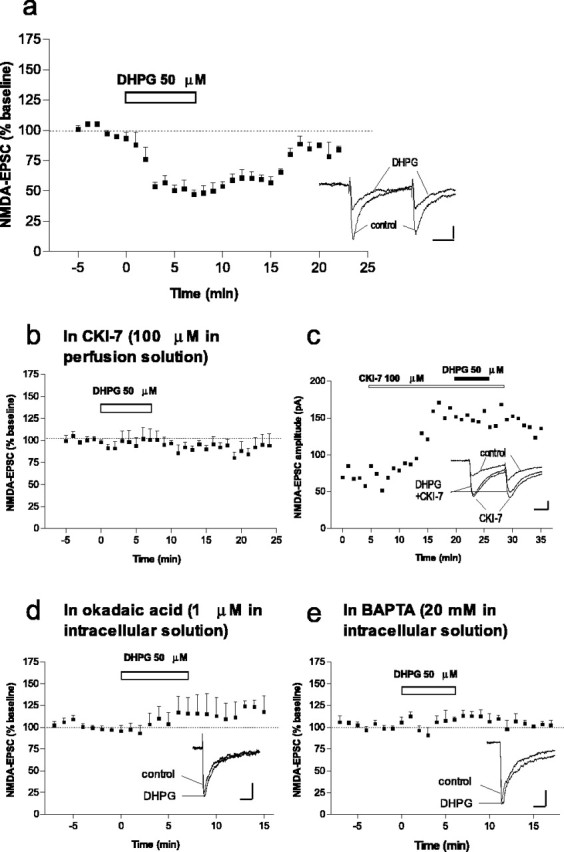
The group I metabotropic glutamate receptor agonist DHPG depresses NMDA-EPSC amplitude via activation of CK1 and PP1/PP2A. a, Time course of the effect of DHPG (50 μm) on NMDA-EPSC amplitude (n = 8). Inset shows NMDA-EPSCs recorded in a representative neuron before and during DHPG and no effect of DHPG on EPSC2/EPSC1. Calibration: 20 pA, 20 ms. b, Time course of the effect of DHPG (50 μm) on NMDA-EPSC amplitude in the presence of CK1-7 (100μm; n = 6). c, Example of a representative neuron in which CK1-7 increased NMDA-EPSCs and blocked inhibition by DHPG. Inset, NMDA-EPSCs before, during CK1-7, and during CK1-7 plus DHPG. Calibration: 20 pA, 20 ms. d, Time course of the effect of DHPG (50 μm) on NMDA-EPSC amplitude in neurons loaded with intracellular okadaic acid (1 μm; n = 7). Records show superimposed NMDA-EPSCs in a representative neuron before and during DHPG. Calibration: 20 pA, 20 ms. e, Time course of the effect of DHPG (50 μm) on NMDA-EPSC amplitude in neurons loaded with intracellular BAPTA (20 mm; n = 5). Records show superimposed NMDA-EPSCs in a representative neuron before and during DHPG. Calibration: 50 pA, 20 ms.
Figure 7.

The group I metabotropic glutamate receptor agonist DHPG depresses NMDA-EPSC amplitude via activation of mGluR1 but not mGluR5. a, Effect of DHPG (50μm) on NMDA-EPSCs in the presence of the mGluR5 antagonist MPEP (10μm; n = 6; filled squares) and in the presence of the mGluR1 antagonist LY367385 (100-300 μm; n = 6; combined data, open squares). Records show NMDA-EPSCs in two representative neurons, before and during DHPG in the presence of MPEP (top records) or LY367385 (bottom records). Calibration: 20 pA, 20 ms. b, Summary of the effect of okadaic acid (OA) (1 μm, intracellular), BAPTA (20 mm, intracellular), CK1-7 (100 μm), MPEP (10 μm), and LY367385 (100-300 μm) on DHPG-induced inhibition of NMDA-EPSCs. Data are expressed as percentage of baseline (absence of DHPG). *p < 0.05, ***p < 0.001 compared with DHPG alone (unpaired two-tailed t test).
We next tested the possible involvement of CK1 in the action of DHPG on NMDA-EPSCs. Incubation of slices with CK1-7 abolished the ability of DHPG to depress NMDA-EPSCs (Figs. 6b,c, 7b). Thus, group I mGluRs selectively downregulate synaptically activated NMDA receptors in the striatum through the activation of CK1. Because we found that CK1 modulates NMDA receptor function through activation of PP1/PP2A, we tested whether mGluR-mediated inhibition of NMDA-EPSCs also involved activation of PP1/PP2A. We found that, in neurons loaded with okadaic acid (1 μm), DHPG did not depress NMDA-EPSC amplitude (percentage of baseline, 115.6 ± 18.2%; n = 7) (Figs. 6d, 7b).
Effect of DHPG on NMDA-EPSC amplitude is calcium dependent and is mediated by mGluR1
Group I metabotropic glutamate receptors have been shown to mobilize Ca2+ from intracellular stores (Fagni et al., 2000). A biochemical study demonstrated that activation of CK1 by DPHG was reduced by the calcium chelator BAPTA (Liu et al., 2002). We therefore examined the possible involvement of intracellular calcium in DHPG-mediated effects, and we found that, in the presence of intracellular BAPTA (20 mm), activation of group I mGluRs by DHPG did not affect NMDA-EPSCs (Figs. 6e, 7b). Thus, group I mGluRs downregulate NMDA-EPSCs through a pathway that involves intracellular calcium and activation of CK1 and of PP1/PP2A.
To identify the receptor responsible for the depression in NMDA-EPSCs produced by DHPG, we treated slices with LY367385, an mGluR1 antagonist, or with MPEP, an mGluR5 antagonist, before and during application of DHPG. In the presence of LY367385 (100-300 μm), the effect of DHPG was abolished (Fig. 7). In contrast, DHPG was still able to inhibit NMDA-EPSCs in the presence of MPEP [10 μm, a concentration shown to be effective in blocking mGluR5-mediated responses in striatal slices (Nishi et al., 2003)] (Fig. 7). These results indicate that mGluR1, rather than mGluR5, mediates the ability of DHPG to depress synaptically activated NMDA currents in striatal neurons.
Discussion
Our data indicate that a cascade involving glutamate/mGluR1/CK1/PP1or PP2A plays a major role in the regulation of NMDA receptor-mediated synaptic transmission in the brain. We found that inhibition of CK1 increases NMDA receptor activity and phosphorylation levels through inhibition of PP1or PP2A and that mGluR1 downregulates NMDA-EPSCs through a Ca2+-dependent activation of CK1 and PP1or PP2A.
Expression of CK1 isoforms in the brain
Seven CK1 isoforms (α, β, γ1-γ3, δ, and ϵ), each having several splice variants, have been identified (Gross and Anderson, 1998). The expression of their mRNAs and proteins has been determined using biochemical and histochemical approaches, as well as reverse transcription-PCR analyses. CK1α has four alternatively spliced variants (CK1α, CK1αS, CK1αL, and CK1αLS) (Zhang et al., 1996; Green and Bennett, 1998) and is expressed in the human hippocampus (Ghoshal et al., 1999). CK1γ3, which has two splice variants, is abundant in rat brain extracts, but not CK1γ1orCK1γ2 (Zhai et al., 1995). CK1δ is widely expressed in the human brain, with the highest levels in hippocampus and cerebral cortex (Schwab et al., 2000; Yasojima et al., 2000). There are two variants of CK1δ: the variant 2 has a different exon at the 3′ end, which results in a shorter protein with a different C terminus compared with variant 1. A widespread distribution of CK1ϵ was found in the rat brain, with the most prominent expression in hippocampus and cerebral cortex (Takano et al., 2004). CK1ϵ has three spliced forms in the rat (Takano et al., 2000). CK1ϵ1 is expressed in the suprachiasmatic nucleus. The expression of CK1ϵ2 is restricted to cerebellum.
None of these studies specifically examined the expression of the different CK1 isoforms in the striatum. Our results revealed that all CK1 isoforms are expressed in the mouse striatum and cortex, as well as in several other brain regions such as cerebellum, hippocampus, thalamus, olfactory bulb, and midbrain (Fig. 1). Each of these isoforms could therefore regulate synaptic transmission in several brain regions. Future studies using, for example, targeted gene deletions will help identifying the isoform(s) involved in the regulation of NMDA receptor function.
Effect of CK1 inhibition on synaptic transmission and NMDA receptor activity
Although the CK1 family forms one of the eight major groups of protein kinases in the human and mouse genome (Manning et al., 2002; Caenepeel et al., 2004), no physiological role has been described for CK1 in synaptic transmission. The present study is the first that identifies a function for this major family of kinases in synaptic transmission. Our results indicate that, in medium spiny neurons in the dorsal striatum, CK1 does not regulate AMPA receptor activity or glutamate release because pharmacological inhibition of CK1 does not affect AMPA-activated whole-cell currents and does not modulate AMPA-mediated EPSC amplitude. In contrast, CK1 specifically regulates NMDA receptor-mediated glutamatergic transmission because we found that the activity of synaptic and extrasynaptic NMDA receptors is increased by inhibition of CK1. Our results show that (1) inhibition of CK1 increases NMDA receptor activity and increases phosphorylation levels of NR1 and NR2B, (2) inhibition of PP1/PP2A mimics the effect of CK1 inhibition on NMDA-EPSCs, and (3) inhibition of PP1/PP2A occludes the effect of CK1 inhibition on NMDA-EPSC amplitude. These results demonstrate that regulation of NMDA receptor function by CK1 involves dephosphorylation of NR1 and NR2B via activation of PP1 or PP2A. Although our experiments do not address the precise mechanism by which CK1 and PP1/PP2A regulate functional properties of the NMDA receptor, changes in the opening probability or desensitization of this receptor may contribute to the observed increased amplitude and slight increased decay time of NMDA-EPSCs after CK1 inhibition. Indeed, single-channel and whole-cell studies demonstrated that PP1 or PP2A depress the open probability of the NMDA receptor channel and that inhibition of PP1/PP2A increases open probability of these channels and NMDA-evoked currents in cultured hippocampal neurons (Wang et al., 1994; Lu et al., 1999; Herzig and Neumann, 2000).
We observed that CK1 inhibition slightly increased the half-decay time of NMDA-EPSCs and increased the phosphorylation of NR2B but not NR2A. These results agree with the slow deactivation kinetics of NR2B-containing NMDA receptors (Cull-Candy et al., 2001) and with the study by Lu et al. (1999) showing that, in hippocampal pyramidal neurons, activation of PKC enhances desensitization of NMDA receptors as well as NMDA-activated whole-cell currents, open probability of the channel, and amplitude of NMDA-mediated miniature EPSCs. Several other mechanisms shown previously to influence the shape of NMDA-EPSCs could also contribute to the regulation of NMDA receptor function by CK1. These mechanisms may include changes in the affinity of NMDA receptors for glutamate, in their sensitivity to magnesium, or in their channel conductance, changes in the subunit composition of NMDA receptors, in NMDA receptor desensitization mediated by dephosphorylation of NR2A by the calcium-dependent protein phosphatase PP2B (Herstrin et al., 1990; Lester and Jahr, 1992; Lieberman and Mody, 1994; Tong et al., 1995; Dingledine et al., 1999; Herzig and Neumann, 2000; Cull-Candy et al., 2001; Krupp et al., 2002). However, our observations that the phosphorylation level of NR2A is not affected by inhibition of CK1, that the concentrations of okadaic acid used in our study (0.1 and 1 μm) do probably not inhibit PP2B, and that similar effects of CK1 inhibitors are observed at hyperpolarized and depolarized membrane potentials and in low and normal concentration of extracellular Mg2+ suggest that such mechanisms may not significantly contribute to the regulation of NMDA receptor activity by CK1. Additional studies examining single-channel currents evoked by brief applications of glutamate agonists to outside-out membrane patches from striatal neurons should provide valuable information on the precise mechanisms by which CK1 and PP1/PP2A regulate NMDA receptor activity in the striatum.
Effect of DHPG on synaptic transmission and NMDA receptor activity
Previous biochemical studies showed that the group I metabotropic glutamate receptor agonist DHPG stimulates CK1 kinase activity in a calcium-dependent manner (Liu et al., 2001, 2002). We found that DHPG had an effect opposite to that produced by CK1 inhibitors, i.e., a decrease in NMDA-EPSC amplitude. This depressant action of DHPG was most probably not attributable to direct action of this compound on NMDA receptors, as shown previously (Contractor et al., 1998), because it was dependent on calcium and on activation of CK1 and PP1/PP2A. In contrast to its inhibitory effect on synaptically activated NMDA receptors, DHPG increased whole-cell-activated NMDA currents (data not shown) (Pisani et al., 1997), indicating that synaptic and extrasynaptic NMDA receptors are regulated by different signaling pathways by group I mGluRs. Indeed, the decrease in synaptically mediated NMDA-EPSCs involves CK1 and mGluR1 (present findings), whereas the increase in whole-cell NMDA currents by DHPG involves PKC and mGluR5 (Pisani et al., 1997, 2001). These previous studies by Pisani et al. (1997, 2001) found that DHPG does not modulate whole-cell-activated NMDA currents in mGluR5 knock-out mice or after pharmacological blockade of mGluR5. Given the present data, one could expect DHPG to reduce, via activation of mGluR1 and CK1, NMDA-activated whole-cell currents in the absence of functional mGluR5. Together, these results suggest that activation of the mGluR1/CK1/PP1-PP2A cascade by glutamate depresses the activity of NMDA receptors located at the synapse and does not modulate extrasynaptic NMDA receptors. These results also suggest that the contribution of synaptic NMDA receptors to the whole-cell-activated current may be significantly less than that of extrasynaptic receptors. We found that the activity of extrasynaptic NMDA receptors (whole-cell-activated NMDA currents) is increased after inhibition of CK1. These results, combined with the findings of Pisani et al. (1997, 2001), suggest that activation of receptors other than mGluRs may regulate, through a CK1 pathway, whole-cell-activated NMDA responses. For example, serotonin acting on 5-HT2A/2C receptors also activates CK1 and modulates NMDA receptor function in the striatum (our unpublished observations).
In the hippocampus, DHPG induces a long-term depression of glutamatergic synaptic transmission (Bashir, 2003). This finding contrasts with our observations in the striatum in which DHPG does not modulate AMPA receptor-mediated synaptic transmission (under our experimental conditions) and in which the depressant effect of DHPG on NMDA-EPSCs is short lasting. Furthermore, the type of mGluR and the signaling pathway activated by DHPG in the striatum (mGluR1, calcium, CK1, and PP1/PP2A) are different from those reported in the hippocampus (mGluR5, protein tyrosine kinases, and phosphatases). Together, these findings are in agreement with the suggestion that mGluR1 and mGluR5 have diverse roles and mechanisms of action in synaptic transmission, neuronal depolarization, and modulation of NMDA receptor activity in different brain areas (Pellegrini-Giampietro, 2003).
In conclusion, the present results implicate CK1 as a negative regulator of glutamate-induced neuronal excitation. Given the central role of NMDA receptors in several physiological and pathological processes in the striatum and throughout the CNS, CK1 may be an important regulatory enzyme in normal states such as movement- and motivation-related behaviors. Another important function for CK1 might be to prevent intense activation of NMDA receptors attributable to excessive glutamatergic activity, which is known to cause neuronal death (Hardingham and Bading, 2003).
Footnotes
This work was supported by Vetenskapsrådet Grant K2003-04X-14517-01A (K.C.), National Institutes of Health Grants MH-40899 and DA 10044, and the Michael Stern Foundation for Parkinson Research (P.G.). K.C. was supported by a fellowship from the Swedish Society for Medical Research and a travel grant from the Swedish Medical Research Council and was an Essel Investigator (Essel Foundation) within the Young Investigator Award program from the National Alliance for Research on Schizophrenia and Depression. P.S. was supported by a fellowship from the Stiftelsen för Internationalisering av Högre Utbilding och Forskning.
Correspondence should be addressed to Paul Greengard, Laboratory of Molecular and Cellular Neuroscience, The Rockefeller University, 1230 York Avenue, New York, NY 10021. E-mail: greengd@mail.rockefeller.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256601-09$15.00/0
References
- Agostinis P, Vandenheede JR, Goris J, Meggio F, Pinna LA, Merlevede W (1987) The ATP, Mg-dependent protein phosphatase: regulation by casein kinase-1. FEBS Lett 224: 385-390. [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P (2003) The specificities of protein kinase inhibitors: an update. Biochem J 371: 199-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir ZI (2003) On long-term depression induced by activation of G-protein coupled receptors. Neurosci Res 45: 363-367. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Bruno V, Pisani A, Centonze D, Catania MV, Calabresi P, Nicoletti F (2001) Selective blockade of type-1 metabotropic glutamate receptors induces neuroprotection by enhancing Gabaergic transmission. Mol Cell Neurosci 17: 1071-1083. [DOI] [PubMed] [Google Scholar]
- Behrend L, Milne DM, Deppert W, Campbell LE, Meek DW, Knippschild U (2000) IC261, a specific inhibitor of the protein kinases casein kinase 1-delta and -epsilon, triggers the mitotic checkpoint and induces p53-dependent postmitotic effects. Oncogene 19: 5303-5313. [DOI] [PubMed] [Google Scholar]
- Caenepeel S, Charydczak G, Sudarsanam S, Hunter T, Manning G (2004) The mouse kinome: discovery and comparative genomics of all mouse protein kinases. Proc Natl Acad Sci USA 101: 11707-11712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Mercuri NB, De Murtas M, Bernardi G (1991) Involvement of GABA systems in feedback regulation of glutamate- and GABA-mediated synaptic potentials in rat neostriatum. J Physiol (Lond) 440: 581-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Bernardi G (1999) Metabotropic glutamate receptors and cell-type-specific vulnerability in the striatum: implication for ischemia and Huntington's disease. Exp Neurol 158: 97-108. [DOI] [PubMed] [Google Scholar]
- Chan SF, Sucher NJ (2001) An NMDA receptor signaling complex with protein phosphatase 2A. J Neurosci 21: 7685-7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chergui K, Lacey MG (1999) Modulation by dopamine D1-like receptors of synaptic transmission and NMDA receptors in rat nucleus accumbens is attenuated by the protein kinase C inhibitor Ro 32-0432. Neuropharmacology 38: 223-231. [DOI] [PubMed] [Google Scholar]
- Chergui K, Bouron A, Normand E, Mulle C (2000) Functional GluR6 kainate receptors in the striatum: indirect downregulation of synaptic transmission. J Neurosci 20: 2175-2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chijiwa T, Hagiwara M, Hidaka H (1989) A newly synthesized selective casein kinase I inhibitor, N-(2-aminoethyl)-5-chloroisoquinoline-8-sulfonamide, and affinity purification of casein kinase I from bovine testis. J Biol Chem 264: 4924-4927. [PubMed] [Google Scholar]
- Colwell CS, Levine MS (1995) Excitatory synaptic transmission in neostriatal neurons: regulation by cyclic AMP-dependent mechanisms. J Neurosci 15: 1704-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Gereau IVRW, Green T, Heinemann SF (1998) Direct effects of metabotropic glutamate receptor compounds on native and recombinant N-methyl-d-aspartate receptors. Proc Natl Acad Sci USA 95: 8969-8974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M (2001) NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 11: 327-335. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7-61. [PubMed] [Google Scholar]
- Dubois T, Howell S, Zemlickova E, Aitken A (2002) Identification of casein kinase I alpha interacting protein partners. FEBS Lett 517: 167-171. [DOI] [PubMed] [Google Scholar]
- Fagni L, Chavis P, Ango F, Bockaert J (2000) Complex interactions between mGluRs, intracellular Ca2+ stores and ion channels in neurons. Trends Neurosci 23: 80-88. [DOI] [PubMed] [Google Scholar]
- Fish KJ, Cegielska A, Getman ME, Landes GM, Virshup DM (1995) Isolation and characterization of human casein kinase I ϵ (CKI), a novel member of the CKI gene family. J Biol Chem 270: 14875-14883. [DOI] [PubMed] [Google Scholar]
- Flores-Hernandez J, Cepeda C, Hernandez-Echeagaray E, Calvert CR, Jokel ES, Fienberg AA, Greengard P, Levine MS (2002) Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: role of D1 receptors and DARPP-32. J Neurophysiol 88: 3010-3020. [DOI] [PubMed] [Google Scholar]
- Ghasemzadeh MB, Sharma S, Surmeier DJ, Eberwine JH, Chesselet M-F (1996) Multiplicity of glutamate receptor subunits in single striatal neurons: an RNA amplification study. Mol Pharmacol 49: 852-859. [PubMed] [Google Scholar]
- Ghoshal N, Smiley JF, DeMaggio AJ, Hoekstra MF, Cochran EJ, Binder LI, Kuret J (1999) A new molecular link between the fibrillar and granulovacuolar lesions of Alzheimer's disease. Am J Pathol 155: 1163-1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz T, Kraushaar U, Geiger J, Lübke J, Berger T, Jonas P (1997) Functional properties of AMPA and NMDA receptors expressed in identified types of basal ganglia neurons. J Neurosci 17: 204-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves PR, Haas DW, Hagedorn CH, DePaoli-Roach AA, Roach PJ (1993) Molecular cloning, expression, and characterization of a 49-kilodalton casein kinase I isoform from rat testis. J Biol Chem 268: 6394-6401. [PubMed] [Google Scholar]
- Green CL, Bennett GS (1998) Identification of four alternatively spliced isoforms of chicken casein kinase I alpha that are all expressed in diverse cell types. Gene 216: 189-195. [DOI] [PubMed] [Google Scholar]
- Gross SD, Anderson RA (1998) Casein kinase I: spatial organization and positioning of a multifunctional protein kinase family. Cell Signal 10: 699-711. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H (2003) The yin and yang of NMDA receptor signalling. Trends Neurosci 26: 81-89. [DOI] [PubMed] [Google Scholar]
- Herstrin S, Sah P, Nicoll RA (1990) Mechanisms generating the time course of dual component excitatory synaptic currents recorded in hippocampal slices. Neuron 5: 247-253. [DOI] [PubMed] [Google Scholar]
- Herzig S, Neumann J (2000) Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev 80: 173-210. [DOI] [PubMed] [Google Scholar]
- Jiang Z-G, North RA (1991) Membrane properties and synaptic responses of rat striatal neurons in vitro. J Physiol (Lond) 443: 533-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Thomas CG, Heinemann SF, Westbrook GL (2002) Calcineurin acts via the C-terminus of NR2A to modulate desensitization of NMDA receptors. Neuropharmacology 42: 593-602. [DOI] [PubMed] [Google Scholar]
- Landwehrmeyer GB, Standaert DG, Testa CM, Penney Jr JB, Young AB (1995) NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J Neurosci 15: 5297-5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester RAJ, Jahr CE (1992) NMDA channel behavior depends on agonist affinity. J Neurosci 12: 635-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman DN, Mody I (1994) Regulation of NMDA channel function by endogenous Ca2+-dependent phosphatase. Nature 369: 235-239. [DOI] [PubMed] [Google Scholar]
- Liu F, Ma X-H, Ule J, Bibb JA, Nishi A, DeMaggio AJ, Yan Z, Nairn AC, Greengard P (2001) Regulation of cyclin-dependent kinase 5 and casein kinase 1 by metabotropic glutamate receptors. Proc Natl Acad Sci USA 98: 11062-11068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Virshup DM, Nairn AC, Greengard P (2002) Mechanism of regulation of casein kinase I activity by group I metabotropic glutamate receptors. J Biol Chem 277: 45393-45399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W-Y, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF (1999) G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci 2: 331-338. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298: 1912-1934. [DOI] [PubMed] [Google Scholar]
- Marin O, Meggio F, Sarno S, Andretta M, Pinna LA (1994) Phosphorylation of synthetic fragments of inhibitor-2 of protein phosphatase-1 by casein kinase-1 and -2. Evidence that phosphorylated residues are not strictly required for efficient targeting by casein kinase-1. Eur J Biochem 223: 647-653. [DOI] [PubMed] [Google Scholar]
- Mashhoon N, DeMaggio AJ, Tereshko V, Bergmeier SC, Egli M, Hoekstra MF, Kuret J (2000) Crystal structure of a conformation-selective casein kinase-1 inhibitor. J Biol Chem 275: 20052-20060. [DOI] [PubMed] [Google Scholar]
- Morishita W, Connor JH, Xia H, Quinlan EM, Shenolikar S, Malenka RC (2001) Regulation of synaptic strength by protein phosphatase 1. Neuron 32: 1133-1148. [DOI] [PubMed] [Google Scholar]
- Nishi A, Liu F, Matsuyama S, Hamada M, Higashi H, Nairn AC, Greengard P (2003) Metabotropic mGluR5 receptors regulate adenosine A2A receptor signalling. Proc Natl Acad Sci USA 100: 1322-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE (2003) The distinct role of mGlu1 receptors in post-ischemic neuronal death. Trends Pharmacol Sci 24: 461-470. [DOI] [PubMed] [Google Scholar]
- Peters JM, McKay RM, McKay JP, Graff JM (1999) Casein kinase I transduces Wnt signals. Nature 401: 345-350. [DOI] [PubMed] [Google Scholar]
- Pisani A, Calabresi P, Centonze D, Bernardi G (1997) Enhancement of NMDA responses by group I metabotropic glutamate receptor activation in striatal neurones. Br J Pharmacol 120: 1007-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Gubellini P, Bonsi P, Conquet F, Picconi B, Centonze D, Bernardi G, Calabresi P (2001) Metabotropic glutamate receptor 5 mediates the potentiation of N-methyl-d-aspartate responses in medium spiny striatal neurons. Neuroscience 106: 579-587. [DOI] [PubMed] [Google Scholar]
- Scannevin RH, Huganir RL (2000) Postsynaptic organization and regulation of excitatory synapses. Nat Rev Neurosci 1: 133-141. [DOI] [PubMed] [Google Scholar]
- Schwab C, DeMaggio AJ, Ghoshal N, Binder LI, Kuret J, McGeer PL (2000) Casein kinase 1 delta is associated with pathological accumulation of tau in several neurodegenerative diseases. Neurobiol Aging 21: 503-510. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Fienberg AA, Huganir RL, Greengard P (1998) A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci 18: 10297-10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Le Moine C, Kull B, Sunahara R, Bloch B, Fredholm BB (1997) Cellular expression of adenosine A2A receptor messenger RNA in the rat central nervous system with special reference to dopamine innervated areas. Neuroscience 80: 1171-1185. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Lindskog M, Rognoni F, Fredholm BB, Greengard P, Fisone G (1998) Activation of adenosine A2A and dopamine D1 receptors stimulates cyclic AMP-dependent phosphorylation of DARPP-32 in distinct populations of striatal projection neurons. Neuroscience 84: 223-228. [DOI] [PubMed] [Google Scholar]
- Takano A, Shimizu K, Kani S, Buijs RM, Okada M, Nagai K (2000) Cloning and characterization of rat casein kinase 1epsilon. FEBS Lett 477: 106-112. [DOI] [PubMed] [Google Scholar]
- Takano A, Hoe HS, Isojima Y, Nagai K (2004) Analysis of the expression, localization and activity of rat casein kinase 1epsilon-3. NeuroReport 15: 1461-1464. [DOI] [PubMed] [Google Scholar]
- Tobin AB (2002) Are we β-ARKing up the wrong tree? Casein kinase 1α provides an additional pathway for GPCR phosphorylation. Trends Pharmacol Sci 23: 337-343. [DOI] [PubMed] [Google Scholar]
- Tong G, Shepherd D, Jahr CE (1995) Synaptic desensitization of NMDA receptors by calcineurin. Science 267: 1510-1512. [DOI] [PubMed] [Google Scholar]
- Wang LY, Orser BA, Brautigan DL, MacDonald JF (1994) Regulation of NMDA receptors in cultured hippocampal neurons by protein phosphatases 1 and 2A. Nature 369: 230-232. [DOI] [PubMed] [Google Scholar]
- Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser IDC, Langeberg LK, Sheng M, Scott JD (1999) Regulation of NMDA receptors by an associated phosphatase-kinase signalling complex. Science 285: 93-96. [DOI] [PubMed] [Google Scholar]
- Whitmore D, Cermakian N, Crosio C, Foulkes NS, Pando MP, Travnickova Z, Sassone-Corsi P (2000) A clockwork organ. Biol Chem 381: 793-800. [DOI] [PubMed] [Google Scholar]
- Yasojima K, Kuret J, DeMaggio AJ, McGeer E, McGeer PL (2000) Casein kinase 1 delta mRNA is upregulated in Alzheimer disease brain. Brain Res 865: 116-120. [DOI] [PubMed] [Google Scholar]
- Zhai L, Graves PR, Robinson LC, Italiano M, Culbertson MR, Rowles J, Cobb MH, DePaoli-Roach AA, Roach PJ (1995) Casein kinase I gamma sub-family. Molecular cloning, expression, and characterization of three mammalian isoforms and complementation of defects in the Saccharomyces cerevisiae YCK genes. J Biol Chem 270: 12717-12724. [DOI] [PubMed] [Google Scholar]
- Zhang J, Gross SD, Schroeder MD, Anderson RA (1996) Casein kinase I alpha and alpha L: alternative splicing-generated kinases exhibit different catalytic properties. Biochemistry 35: 16319-16327. [DOI] [PubMed] [Google Scholar]