

Abstract
Parkinson's disease (PD) is characterized by a loss of ventral midbrain dopaminergic neurons, which can be modeled by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Inflammatory oxidants have emerged as key contributors to PD- and MPTP-related neurodegeneration. Here, we show that myeloperoxidase (MPO), a key oxidant-producing enzyme during inflammation, is upregulated in the ventral midbrain of human PD and MPTP mice. We also show that ventral midbrain dopaminergic neurons of mutant mice deficient in MPO are more resistant to MPTP-induced cytotoxicity than their wild-type littermates. Supporting the oxidative damaging role of MPO in this PD model are the demonstrations that MPO-specific biomarkers 3-chlorotyrosine and hypochlorous acid-modified proteins increase in the brains of MPTP-injected mice. This study demonstrates that MPO participates in the MPTP neurotoxic process and suggests that inhibitors of MPO may provide a protective benefit in PD.
Keywords: MPTP, Parkinson's disease, oxidative stress, inflammation, neuroprotection, nitrotyrosine
Introduction
Parkinson's disease (PD) is a common neurodegenerative disorder characterized by disabling motor abnormalities, which include tremor, muscle stiffness, paucity of voluntary movements, and postural instability (Dauer and Przedborski, 2003). Its main neuropathological feature is the loss of the nigrostriatal dopaminergic neurons, the cell bodies of which reside in the substantia nigra pars compacta (SNpc) and nerve terminals of which extend to the striatum (Dauer and Przedborski, 2003). Except for a handful of inherited cases related to known gene defects (Vila and Przedborski, 2004), PD is a sporadic condition of unknown pathogenesis (Dauer and Przedborski, 2003). However, epidemiological studies suggest that inflammation increases the risk of developing PD (Chen et al., 2003), and experimental models of PD show that inflammatory oxidants modulate SNpc dopaminergic neuronal death (Liberatore et al., 1999; Gao et al., 2002; Wu et al., 2002,2003). For instance, NADPH oxidase and inducible nitric oxide synthase (iNOS), which are major sources of inflammatory oxidants, are upregulated in damaged areas in both PD and the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of PD (Hunot et al., 1996; Liberatore et al., 1999; Wu et al., 2003). Studies of mice deficient in NADPH oxidase or iNOS indicate that superoxide radical (O -2) and NO contribute to the MPTP-induced neurodegenerative process (Liberatore et al., 1999; Wu et al., 2003). However, both O -2 and NO are relatively unreactive, and a variety of secondary oxidants, such as peroxynitrite (ONOO-), are more likely to account for the injurious capacity of inflammation in PD. Supporting this view are the demonstrations that levels of 3-nitrotyrosine, a major product of ONOO- oxidation of proteins, are elevated in affected brain areas after MPTP injections to mice (Pennathur et al., 1999), for the most part in an iNOS-dependent manner (Liberatore et al., 1999).
Levels of O,O′-dityrosine also increase markedly in the SNpc of MPTP-intoxicated animals (Pennathur et al., 1999). This is an intriguing finding because O,O′-dityrosine is a relatively minor product of ONOO- (Pennathur et al., 1999). Conversely, myeloperoxidase (MPO), and not ONOO-, seems to promote O,O′-dityrosine formation in this model of PD (Pennathur et al., 1999). Moreover, MPO can use the NO degradation product NO -2 to generate reactive nitrogen species (RNS) (van der Vliet et al., 1997), and studies of mice deficient in MPO demonstrate that this enzyme is one of the major sources of 3-nitrotyrosine during acute inflammation (Gaut et al., 2002). Thus, these results raise the unanticipated possibility that MPO, a heme enzyme expressed in abundance in a variety of phagocytic cells (Hampton et al., 1998), would contribute to the MPTP-induced neurodegen erative process and would represent a previously unrecognized culprit in the inflammatory-mediated oxidative insult associated with diseases such as PD. Consistent with this hypothesis, we show here not only that MPO is detected in affected brain areas of MPTP-injected mice and PD patients, specifically in glial cells, but also that mutant mice deficient in MPO are more resistant to MPTP-induced dopaminergic neurotoxicity. These findings indicate that MPO does participate in the MPTP neurotoxic process and suggest that inhibitors of MPO may provide protective benefit in PD.
Materials and Methods
Animals and treatment. Procedures using laboratory animals were in accordance with the National Institutes of Health (NIH) guidelines for the use of live animals and approved by the Institutional Animal Care and Use Committee of Columbia University. The mice used in this study were 10-week-old male C57BL/6J mice (Charles River Laboratories, Wilmington, MA) and MPO-deficient mice that had been backcrossed >10 times into the C57BL/6J background (Brennan et al., 1985) and their wild-type (WT) littermates, all weighing 22-25 g. For MPTP intoxication, 4-10 mice per group received four intraperitoneal injections every 2 h of MPTP-HCl (18-20 mg/kg of free base; Sigma-Aldrich, St. Louis, MO) dissolved in saline. Mice were killed from 0-7 d after the last injection, and their brains were used for morphological and biochemical analyses. Control mice received saline only. MPTP handling and safety measures were in accordance with published guidelines (Przedborski et al., 2001).
RNA extraction and reverse transcription-PCR. Total RNA was extracted from selected brain regions and at selected time points after MPTP and used for reverse transcription-PCR analysis as described previously (Wu et al., 2003). The primers used for mouse MPO and β-actin were as follows: MPO, 5′-AGGATAGGACTGGATTGCCTG-3′ (forward) and 5′-GTGGTGATGCCAGTGTTGTCA-3′ (reverse); β-actin, 5′-CTTTGATGTCACGCACGATTTC-3′ (forward) and 5′-GGGCCGCTCTAGGCACCAA-3′ (reverse). The thermal cycling conditions of the PCR were 94°C for 3 min, followed by 23-35 cycles for 20 s at 94°C, 1 min at 60°C, 1 min at 72°C, and a final extension at 72°C for 5 min. After amplification, products were separated on a 5% PAGE and quantified by a FluorChem 8800 digital image system (Alpha Innotech, San Leandro, CA). PCR products were of expected sizes, and sequences were confirmed by direct cycle sequencing.
Immunoblots. Mouse brain protein extracts from selected regions were prepared and used for Western blot analysis as described previously (Wu et al., 2003). The primary antibodies used were as follows: a rabbit polyclonal antibody raised against a 14 aa peptide representing the C terminus of the mouse MPO (NTLPKLNLTSWKET; 1:1000 dilution; generated by J.W.H.'s laboratory) and a mouse monoclonal anti-β-actin antibody (1:10,000; Sigma, St. Louis, MO). A horseradish-conjugated secondary antibody (1:500-1:25,000; Amersham Biosciences, Piscataway, NJ) and a chemiluminescent substrate (SuperSignal Ultra; Pierce, Rockford, IL) were used for detection. Bands were quantified using the FluorChem 8800.
MPO isolation and activity. The methods used to prepare brain samples and to measure MPO activity are slight modifications of those described previously by Daugherty et al. (1994). In brief, fresh mouse tissues from selected brain regions were homogenized in a 100 mm sodium phosphate buffer, pH 7.0, containing 1% (wt/vol) cetyltrimethylammonium bromide (CTAB) and centrifuged (6000 × g, 4°C, 10 min). Then 1 mm CaCl2, MnCl2, and MgCl2 (final concentration) were added to each sample before being incubated overnight at 4°C with 0.3 ml of concanavalin A-Sepharose B (Sigma). The gel was then pelleted by centrifugation and washed three times with a 0.1 m sodium acetate buffer, pH 6.0, containing 0.1 m NaCl and 0.05% CTAB. Then samples were centrifuged (6000 × g, 5 min) to remove residual washing buffer. The glycoprotein bound to the lectin gel was then eluted by incubation with 0.15 ml elution buffer (0.5 m methyl α-d-mannoside in washing buffer) for 30 min. After the last centrifugation, final supernatants were collected and used immediately to assess MPO activity by monitoring the oxidation of tetramethylbenzidine as described previously (Andrews and Krinsky, 1982). The absorbance was read at 655 nm with a microplate reader (Bio-Rad, Hercules, CA).
Mouse MPO, glial fibrillary acidic protein, β2 integrin MAC-1 (CD116/CD18), neutrophil, and tyrosine hydroxylase immunohistochemistry. At selected time points after MPTP, mice were killed, and their brains were processed for immunohistochemical studies following our standard protocol for single or double immunostaining (Wu et al., 2003). The primary antibodies used were rabbit polyclonal anti-MPO (1:500; Lab Vision, Fremont, CA), rabbit polyclonal anti-glial fibrillary acidic protein (GFAP; 1:500; Chemicon, Temecula, CA), mouse monoclonal anti-MAC-1 (1:1000; Serotec, Raleigh, NC), and the monoclonal rat anti-mouse neutrophil antibody MCA771GA (1:100; Serotec). Immunostaining was visualized by 3,3′-diaminobenzidine (DAB) or fluorescein and Texas Red (Vector Laboratories, Burlingame, CA) and examined by regular or confocal microscopy. Colocalization studies were performed on doubly immunofluorescent stained sections, which were analyzed with an LSM 510 META laser-scanning microscope (Zeiss, Thornwood, NY).
For quantitative tyrosine hydroxylase (TH) immunostaining, mice were killed 7 d after MPTP. Both striatal and nigral sections (30 μm), spanning the entire extent of the structures, were incubated with a polyclonal anti-TH antibody (1:1000; Calbiochem, San Diego, CA) for 48 h at 4°C. Immunoreactivity was visualized by incubation in DAB, glucose, and glucose oxidase, and sections were counterstained with thionin. The total numbers of TH- and Nissl-positive neurons in the SNpc were counted stereologically using the optical fractionator method (West, 1993) as used previously (Tieu et al., 2003). Striatal OD of TH immunostaining, determined by the Scion (Frederick, MD) Image program, was used as an index of striatal density of TH innervation (Tieu et al., 2003). The concentration of anti-TH antibody and DAB used here and the length of time striatal sections were incubated in DAB were the same as reported previously (Tieu et al., 2003).
Human samples. All human samples were obtained from the New York Brain Bank at Columbia University (http://cumc.columbia.edu/research/equip/eq-tb_bb.htm). Procedures using this autopsy material were in accordance with the NIH guidelines for human studies and approved by the Institutional Review Board of Columbia University. Samples used in this work included the cerebellum, striatum, and ventral midbrain (for PD and controls); the caudate nucleus [for Huntington's disease (HD) and controls]; and the frontal motor cortex [for amyotrophic lateral sclerosis (ALS) or motor neuron disease and controls]. All of these cases were selected on the basis of neuropathological diagnoses using the criteria for definite PD, HD, and ALS outlined in the supplemental material (available at www.jneurosci.org). Relevant clinical and neuropathological information regarding all of the cases used here are presented in supplemental Table 2 (available at www.jneurosci.org as supplemental material). The procedures for Western blot analysis and immunohistochemistry in human tissues were identical to those described above in mouse tissues; the primary anti-MPO antibody was a rabbit anti-human MPO antibody (DakoCytomation, Carpinteria, CA) used at 1:1000 for Western blot and 1:200 for immunohistochemistry, as well as a rabbit polyclonal anti-GFAP antibody (1:10,000; DAKO, Carpinteria, CA). Visualization of the bound antibody was achieved using chromogenes SG (blue/gray) and 3-amino-9-ethylcarbazole (red) from Vector Laboratories.
MPTP metabolism. Striatal 1-methyl-4-phenylpyridinium (MPP +) levels were determined by HPLC with UV detection (λ = 295 nm) in WT and MPO-deficient mice at 90 min after the last injection of 20 mg/kg MPTP. Striatal tissue lactate production induced by MPP + and synaptosomal uptake of [3H]MPP + were performed as described previously (Wu et al., 2003). The assays were repeated three times, each time using duplicate samples.
Mass spectrometric analysis. At selected time points, anesthetized mice were perfused with ice-cold 50 mm sodium phosphate, pH 7.4, containing an antioxidant mixture made of 100 μm diethylenetriaminepentaacetic acid, 1 mm butylated hydroxytoluene, 10 mm 3-amino-1,2,4-triazole, and 1% ethanol (v/v) to minimize ex vivo oxidation. The ventral midbrain and cerebellum were then dissected and pulverized in liquid N2, delipidated, dialyzed to remove low-molecular weight compounds, and hydrolyzed using HBr instead of HCl to prevent artifactual chlorination. [13C6]-Ring-labeled internal standards were added before hydrolysis. The amino acids were isolated using a C-18 solid-phase extraction column and subjected to derivatization and analysis by isotope dilution gas chromatography/mass spectroscopy (GC/MS) (Heinecke et al., 1999).
Detection of hypochlorous acid-modified protein. Immunochemical detection of hypochlorous (HOCl)-modified proteins was performed with the antibody HOP-1 (clone 2D10G9; dilution 1:500; provided by E. Malle, Medical University of Graz, Graz, Austria). HOP-1 is specific for HOCl-modified epitopes/proteins and does not cross-react with other oxidative modifications (Malle et al., 1995; Hazell et al., 1996). Immunostaining was visualized by using DAB, and sections were counterstained with methylgreen (Vector Laboratories).
Statistical analysis. All values are expressed as mean ± SEM. Differences among means were analyzed using one- or two-way ANOVA with time, treatment, or genotype as the independent factors. When ANOVA showed significant differences, pairwise comparisons between means were tested by Newman-Keuls post hoc testing. In all analyses, the null hypothesis was rejected at the p ≤ 0.05 level.
Results
MPO is induced in the mouse ventral midbrain during MPTP-induced dopaminergic neurodegeneration
To examine the possibility that MPO is a component of the inflammatory response seen in the MPTP model of PD (Liberatore et al., 1999; Wu et al., 2002), we first assessed MPO mRNA and protein content in the ventral midbrain (i.e., brain region containing the SNpc dopaminergic neurons) over the entire active phase of neurodegeneration and gliosis provoked by this neurotoxin (Liberatore et al., 1999; Przedborski and Vila, 2001). In saline-injected control mice, the ventral midbrain contained low levels of MPO mRNA and protein (Fig. 1A-C). In contrast, in MPTP-injected mice, ventral midbrain levels of both MPO mRNA and protein increased in a time-dependent manner (Fig. 1A-C). Ventral midbrain MPO mRNA and protein expression levels peaked at 1 and 2 d after MPTP exposure, respectively (Fig. 1C), which is contemporaneous to the most-intense phase of SNpc dopaminergic neuronal death in this PD model (Przedborski and Vila, 2001). We next asked whether the observed changes in MPO ventral midbrain content in MPTP-injected animals paralleled an alteration of MPO enzymatic activity by monitoring oxidation of tetramethylbenzidine. Consistent with the protein data, we found that ventral midbrain MPO activity also rose during MPTP neurotoxicity in a time-dependent manner (Fig. 1D). In contrast, in mutant mice deficient in MPO (MPO-/-; n = 2), the ventral midbrain did not show higher oxidation of tetramethylbenzidine after MPTP administration (data not shown). Unlike in the ventral midbrain, levels of MPO mRNA, proteins, and catalytic activity in the cerebellum (brain region resistant to MPTP) were unaffected by MPTP administration. However, more unexpected was the finding that no MPO alteration could be detected in the striatum (where dopaminergic fibers degenerate after MPTP administration), as illustrated by the lack of change in striatal MPO activity: saline, 14.0 ± 4.1 (n = 7), versus MPTP (at 2 d), 16.2 ± 1.5 (n = 11; p > 0.05). Thus, both protein levels and activity of MPO increase in the MPTP mouse model of PD, specifically in ventral midbrain where the demise of the nigrostriatal dopaminergic neurons is taking place.
Figure 1.
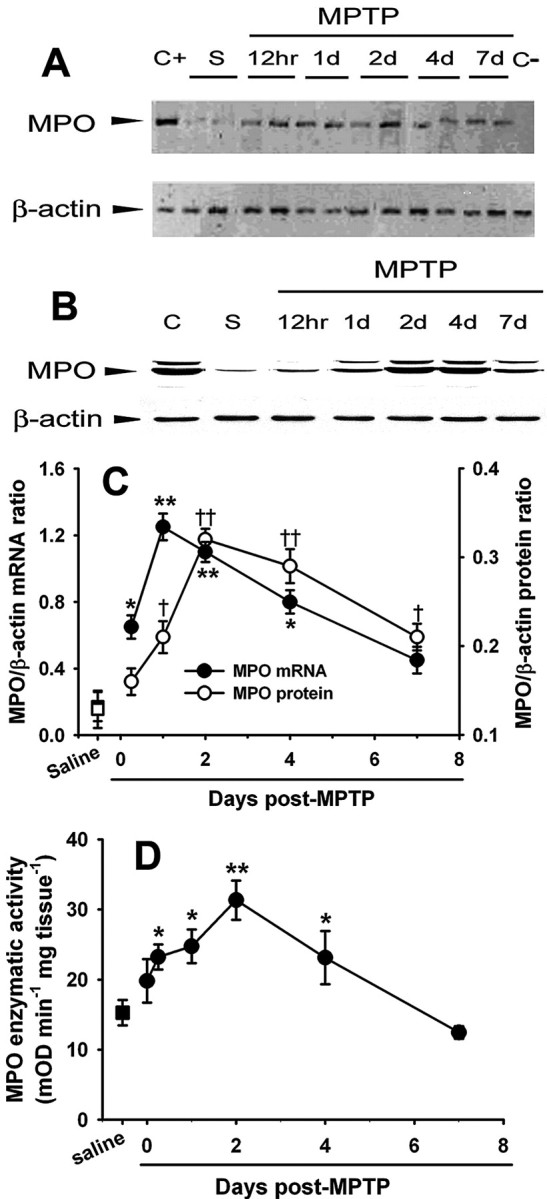
MPTP injections are associated with a time-dependent increase in ventral midbrain MPO mRNA (A, C), protein expression (B, C), and enzymatic activity (D) relative to saline injections. Data are means ± SEM for 3-11 mice per group. *,†p < 0.05, **,††p < 0.01 compared (Newman-Keuls post hoc test) with saline-injected control animals. S, Saline; C+, positive control (bone marrow); C-, negative control (absence of reverse transcriptase); mOD, millioptical density.
MPO is expressed in reactive astrocytes after MPTP injection
To elucidate the cellular origin of MPO in the ventral midbrain of MPTP-treated mice, immunohistochemical studies were performed. In saline controls, diffuse MPO immunoreactivity was seen in the neuropil (Fig. 2A,C). In MPTP-treated mice 2 d after the last injection, ventral midbrain MPO immunostaining was stronger, especially at the level of the substantia nigra, and cells with a glial morphology appeared labeled (Fig. 2B,D). These MPO-positive cells showed punctate immunoreactivity over both the cell bodies and proximal processes (Fig. 2D). To corroborate the bright-field microscopy results, we performed double immunofluorescence confocal microscopy on ventral midbrain sections from mice 2 d after MPTP. This analysis confirmed that MPO colocalized with the astrocytic marker GFAP as shown by the merged image from the two fluorochromes (Fig. 2E-G) and the computed mask of the colocalized pixels (Fig. 2H). Conversely, no evidence of MPO expression in microglial cells could be documented by using the same techniques (Fig. 2I-L). Although abundant neutrophils were seen in our mouse bone marrow preparations (positive controls) using the anti-mouse neutrophil antibody MCA771GA, none were detected within the brain parenchyma (data not shown). No noticeable cellular MPO immunoreactivity was observed in the striatum or cerebellum of either saline- or MPTP-treated mice (data not shown). These results demonstrate that MPO is primarily expressed in ventral midbrain astrocytes during the demise of dopaminergic neurons caused by MPTP.
Figure 2.
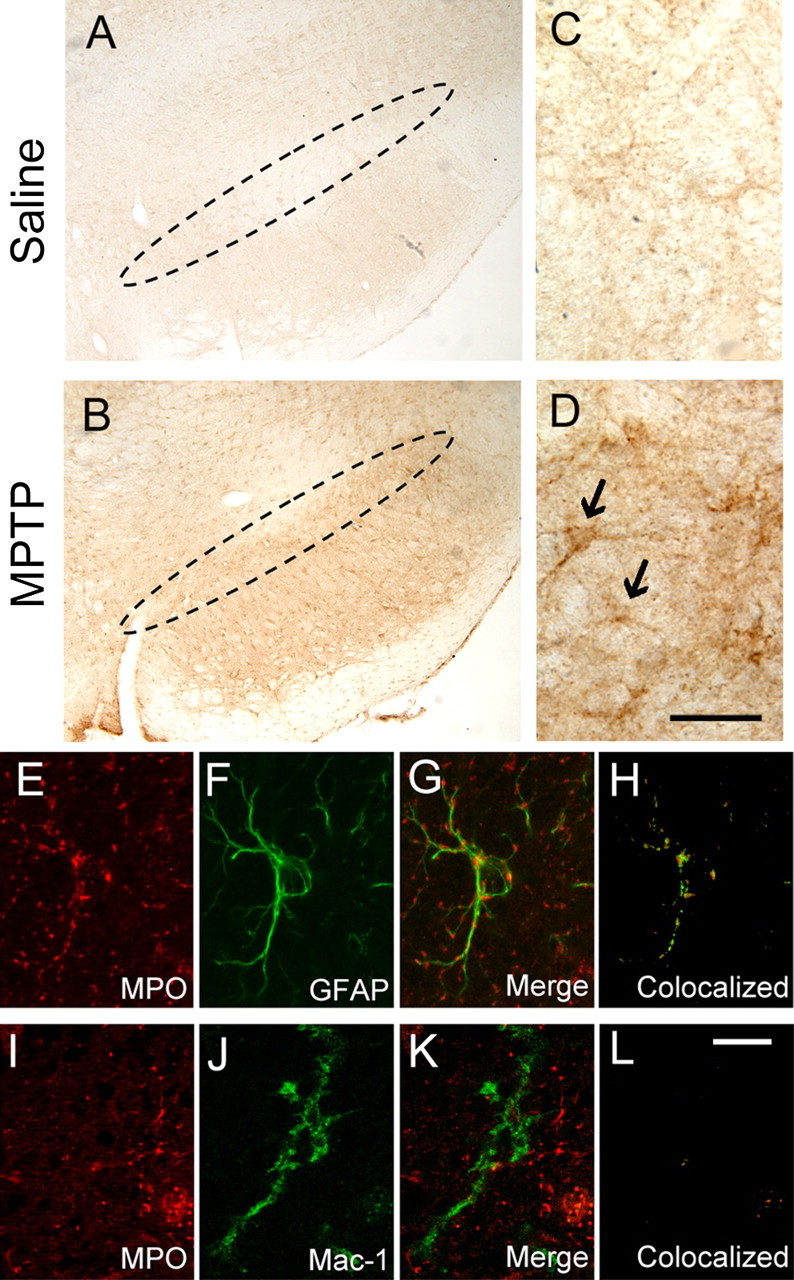
A, C, Immunochemical studies revealed no specific MPO immunoreactivity in the ventral midbrain of saline-injected control mice. The dashed oval delineates the SNpc. B, D, However, a dense network of fibers and scattered cell bodies positive for MPO are seen at the level of the SNpc after MPTP injections. Black arrows in D show the MPO-positive cellular elements. E-H, Confocal microscopy demonstrates that ventral midbrain MPO-positive structures (E, red) are also GFAP positive (F, green), as evidenced by the overlay of the two fluorochromes (G) and by the computed mask of the colocalized pixels (H). J-L, In contrast, ventral midbrain MPO-positive structures (I, red) are not MAC-1 positive (J, green), as evidenced by the overlay (K) and the mask of colocalized pixels (L). Tissue sections are from mice at 24 and 48 h after saline or MPTP injections. Scale bars: (in D) A, B, 250 μm; C, D, 25 μm; (in L) E-L,10 μm.
Expression of MPO is increased in PD midbrain
To determine whether the changes in MPO observed in the MPTP mouse model of PD were present in the human condition, we assessed MPO protein levels in postmortem ventral midbrain samples from sporadic PD patients. Consistent with the mouse data, PD samples had significantly higher MPO protein contents compared with controls (Fig. 3A,B). Like in mice, there was no significant difference in MPO to β-actin ratios in the striatum (PD, 1.1 ± 0.8, vs controls, 1.4 ± 0.8; p > 0.05; n = 7) or cerebellum (PD, 0.8 ± 0.2, vs controls, 1.0 ± 0.3; p > 0.05; n = 7) between the PD and control samples. Histologically, cellular MPO immunoreactivity was not detected in the control ventral midbrain parenchyma per se (Fig. 3C) but only in small cells within blood vessels. However, MPO immunoreactivity was seen in ventral midbrain sections from PD patients (Fig. 3D), where it was identified in SNpc glial cells in the vicinity of neuromelanin-containing neurons (Fig. 3D). The similarity of the MPO alterations between the MPTP mice and the PD postmortem specimens strengthens the relevance of using this experimental model to study the role of MPO in the PD neurodegenerative process.
Figure 3.
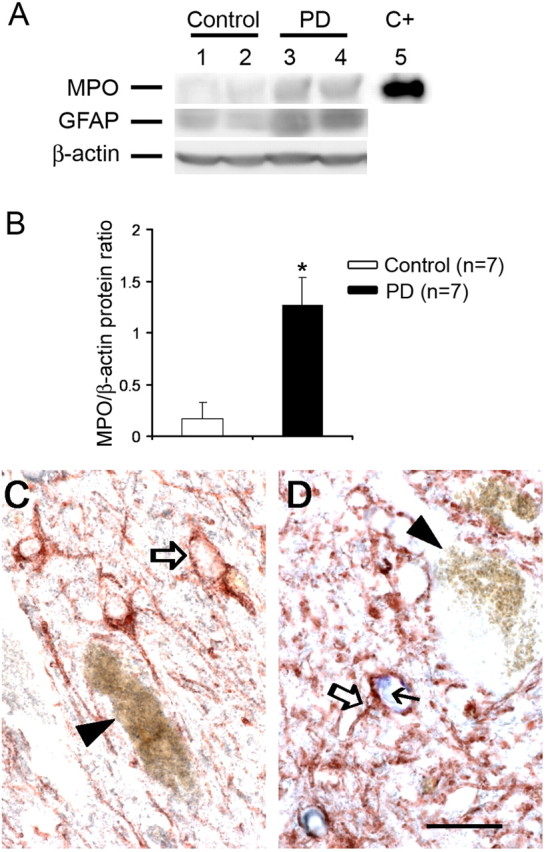
A, B, Ventral midbrain MPO tissue content is increased in postmortem tissue from PD patients compared with controls, as well as GFAP tissue content. C+, Positive control (purified MPO). C, Inventral midbrain sections, MPO (blue) is not detected in control tissues, neither in GFAP-positive cells (open arrow) nor in or around neuromelanized dopaminergic neurons (arrowhead). D, Conversely, MPO immunoreactivity (blue, small black arrow) is found in GFAP-positive cells (open arrow) in PD tissue but not in the rare remaining neuromelanized dopaminergic neurons (arrowhead). Scale bar, 20 μm. Data are means ± SEM for seven samples per group. *p < 0.05 compared with normal controls (Newman-Keuls post hoc test).
Because gliosis is a common pathological feature of many neurodegenerative diseases, we wondered whether increases in the expression of MPO within areas of neurodegeneration can be found in neurodegenerative disorders other than PD. Compared with controls, the motor cortex from ALS patients did not exhibit higher GFAP or MPO values (data not shown). Conversely, we found that caudate nucleus tissues from stage 4 HD patients had higher GFAP to β-actin ratios (HD, 0.7 ± 0.1, vs controls, 0.1 ± 0.1; p < 0.01; n = 3-4) as well as MPO to β-actin ratios (HD, 0.8 ± 0.2, vs controls, 0.2 ± 0.1; p < 0.05; n = 5-6). This suggests that brain MPO expression is not specific to PD but rather generic to neurodegenerative diseases in which areas of neuronal loss are accompanied with gliosis.
MPO deficiency protects against MPTP-induced neurodegeneration
Next we compared the effects of MPTP on the nigrostriatal pathway of mutant mice deficient in MPO (MPO-/-) and their WT littermates (MPO+/+). Seven days after the last injection of saline or MPTP, the brains of these animals were processed for quantification of dopaminergic cell bodies in the SNpc and of projecting dopaminergic fibers in the striatum using TH immunostaining. In saline-injected MPO-/- and MPO+/+ mice, stereological counts of SNpc dopaminergic neurons and striatal TH-positive OD (Fig. 4A,B,E,F) were comparable. In MPTP-injected MPO+/+ mice, there was a ∼70% loss of SNpc TH-positive neurons (Fig. 4C,E) and ∼92% reduction of striatal TH OD (Fig. 4C,F) compared with saline-injected controls (Fig. 4A,E,F). In contrast, in MPTP-injected MPO-/- mice, there was only ∼50% loss of SNpc TH-positive neurons (Fig. 4D,E) and ∼70% reduction of striatal TH OD (Fig. 4D,F) compared with saline-injected controls (Fig. 4B,E,F). The TH/Nissl ratio of neuronal counts did not differ between saline- and MPTP-injected WT mice (saline, 1.68 ± 0.15, vs MPTP, 1.46 ± 0.20; n = 5 per group), supporting the assertion that the reduction in dopaminergic neuron numbers corresponds to an actual cell loss and not to a downregulation of TH.
Figure 4.
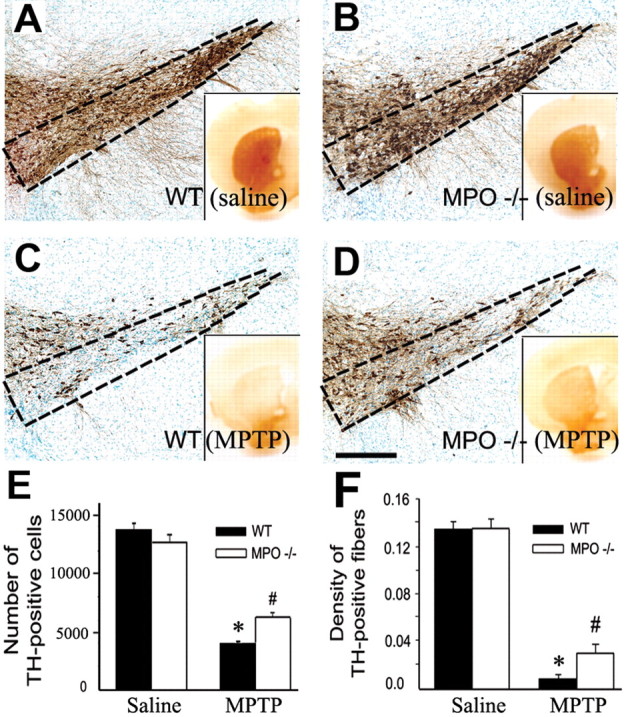
A-D, Ablation of MPO in mutant mice attenuates MPTP-induced striatal TH fibers and SNpc TH neuronal loss, as assessed 7 d after either saline or MPTP injections. E, F, Quantification of neuronal (E) and fiber (F) loss. Data are means ± SEM for four to six mice per group. *p < 0.05 compared with saline-injected animals; #p < 0.05 compared with saline- and MPTP-injected MPO +/+ mice.
To examine whether MPO ablation protects not only against structural damage but also against functional deficits caused by MPTP, we compared the levels of dopamine and its metabolites, dihydroxyphenylacetic acid and homovanillic acid, in the striatum as well as locomotor activity between MPO-/- and MPO+/+ mice, after MPTP injections. Contrasting with the protection afforded by the lack of MPO on the nigrostriatal dopaminergic neurons, the loss of striatal dopamine and the deficit in motor performance caused by MPTP were as severe in MPO-/- as in MPO+/+ mice (supplemental material, available at www.jneurosci.org).
MPTP metabolism
Major factors controlling MPTP neurotoxicity are its conversion in the brain to MPP+, followed by MPP+ entry into dopaminergic neurons and its subsequent blockade of mitochondrial respiration (Przedborski and Vila, 2001). To ascertain that the resistance of MPO-/- mice was not attributable to alterations in MPTP toxicokinetics, we assessed its three key neurotoxic steps (Tieu et al., 2003). Results show that striatal levels of MPP+, striatal uptake of [3H]MPP+, and MPP+-induced lactate production (a measure of mitochondrial function) did not differ between MPO-/- mice and their WT littermates (Table 1).
Table 1.
Striatal MPTP metabolism in MPO-deficient mice
|
|
MPP+ level (μg/g striatum) |
MPP+ uptake (IC50′ nm) |
MPP+-induced lactate (μm/100 mg protein) |
|---|---|---|---|
| MPO+/+ mice | 4.46 ± 0.24 | 113.7 ± 1.2 | 57.6 ± 7.5 |
| MPO−/− mice |
5.54 ± 0.71 |
114.3 ± 1.7 |
66.8 ± 4.4 |
Striatal MPP+ levels in WT (MPO+/+) and MPO-deficient mice (MPO−/−) were determined 90 min after the last injection of MPTP (20 mg/kg). Values are means ± SEM of either six mice per group (MPP+ level) or three independent experiments each performed in duplicate ([3H]MPP+ uptake and lactate level). None of the presented values differ significantly (p > 0.05) between MPO+/+ and MPO−/− mice.
MPO damages ventral midbrain proteins
MPO is the only known mammalian source of HOCl at plasma concentrations of halide ion (Gaut et al., 2001). HOCl reacts with tyrosine to form 3-chlorotyrosine, a specific and stable biomarker of protein damage by MPO (Heinecke et al., 1999). To determine whether MPTP promotes oxidative damage to brain proteins, we used isotope dilution GC/MS (Heinecke et al., 1999), a sensitive and specific method, to quantify 3-chlorotyrosine levels in samples from eight saline-injected controls and eight MPTP-injected mice 24 h after injection. We compared levels of 3-chlorotyrosine in the ventral midbrain and cerebellum. In MPTP-treated mice, 3-chlorotyrosine levels in the ventral midbrain were markedly increased (p < 0.05) compared with saline-injected controls: MPTP, 30.8 ± 5.7 nmol of 3-chlorotyrosine per molar of tyrosine (n = 8) versus saline controls, 4.8 ± 2.1 nmol of 3-chlorotyrosine per molar of tyrosine (n = 8). 3-Chlorotyrosine was undetectable in the cerebellum of mice injected with either saline or MPTP. In contrast, in MPTP-treated MPO-/- mice (n = 3), ventral midbrain 3-chlorotyrosine was undetectable. The identification of chlorinated tyrosine in tissues therefore supports the hypothesis that reactive intermediates produced by MPO damage brain proteins in MPTP-intoxicated mice.
To localize MPO-damaged proteins, tissue sections were immunostained with HOP-1, a mouse antibody that specifically recognizes HOCl-modified proteins (Malle et al., 1995); the chlorotyrosine antibody was not available to us. Intense HOP-1 immunoreactivity was observed in the SNpc of MPTP-injected mice (Fig. 5A-C). HOP-1-positive material was seen in the neuropil within beaded-appearing fibers and in cells with both neuronal and non-neuronal morphology within vesicular elements (Fig. 5A-C). No HOP-1 immunostaining was detected in the SNpc of saline-injected mice or MPTP-injected MPO-/- mice (data not shown).
Figure 5.

Immunohistochemical localization of HOCl-modified proteins with the HOP-1 antibody in ventral midbrain sections. Twenty-four hours after MPTP injections, HOP-1-positive immunoreactive material is seen mainly at the level of the SNpc (A) and within or around cellular elements (B, C). Scale bars: A, 250 μm; B, C, 25 μm.
Discussion
The present study shows that the level of MPO expression increases markedly in diseased SNpc from both mice exposed to MPTP (Figs. 1, 2) and human PD (Fig. 3). This work also demonstrates that changes of MPO protein content and enzymatic activity in MPTP-intoxicated mice parallel (Fig. 1) the degeneration of SNpc dopaminergic neurons (Przedborski and Vila, 2001). Moreover, MPO is found primarily in SNpc-reactive astrocytes (Figs. 2, 3), which are major cellular components of the PD- and MPTP-associated inflammatory response (Przedborski and Goldman, 2004). Conversely, we failed to detect any of the well established cellular sources of MPO (neutrophils, monocytes, or macrophages) (Hampton et al., 1998) within the ventral midbrain parenchyma of PD patients and MPTP-injected mice. The presence of MPO in damaged SNpc thus appears to derive essentially from a resident, not a blood-borne, inflammatory response associated with the degeneration of dopaminergic neurons. Based on assessments performed in two other neurodegenerative diseases, namely HD and ALS, it appears that MPO upregulation in the brain is not pathognomonic of PD. Instead, we believe that the occurrence of MPO in diseased brains is likely indicative of a disease process associated with chronic gliosis rather than a particular etiology. That said, our results are surprising because phagocytic white blood cells are generally believed to be the only cellular sources of MPO. However, neuronal expression of MPO is also increased in Alzheimer's disease (Green et al., 2004), raising the possibility that this enzyme may contribute to oxidative damage in a variety of chronic neurodegenerative disorders.
Contrary to ventral midbrain, the striatum, which is also a site of a strong inflammatory reaction after MPTP administration and sometimes in PD, did not show any alteration in MPO expression or enzymatic activity as illustrated in Figure 3. Remarkably, detectable changes in iNOS expression and enzymatic activity are also confined to ventral midbrains of MPTP-injected mice (Liberatore et al., 1999), whereas activation of NADPH oxidase is observed in both ventral midbrains and striata of these animals (Wu et al., 2003). Collectively, these observations suggest that the molecular composition of the inflammatory response to injury may be, to a certain extent, regionally specific. Supporting this view is the finding that a stereotaxic injection of 5 μg/μl bacterial endotoxin lipopolysaccharide into the hippocampus or cortex of adult rats produces no apparent neuronal loss, whereas an identical administration into the substantia nigra dramatically reduces the number of neurons (Kim et al., 2000). Although this distinct regional susceptibility has been linked to differences in microglial densities, its molecular basis might well be related to differences in the quantity or variety of the inflammatory mediators produced.
After MPTP injections, mutant mice deficient in MPO showed more spared SNpc dopaminergic neurons and striatal dopaminergic fibers than their WT littermates (Fig. 4). We also found that the lack of MPO did not alter key aspects of MPTP toxicokinetics (Table 1). Together, these findings indicate that MPO contributes to the pathogenic cascade of deleterious events responsible for the demise of dopaminergic neurons in the MPTP model and perhaps in PD as well. Surprisingly, although alterations in MPO protein and enzymatic activity were only detected in the ventral midbrain (Fig. 1), both cell bodies and fibers of nigrostriatal dopaminergic neurons were preserved in MPTP-injected MPO-/- mice (Fig. 4). This observation implies that an entire neuron may be salvaged by mitigating deleterious factors that specifically injure cell bodies and that nigrostriatal dopaminergic neurons are not degenerating solely via a dying-back process, as one may have thought based on previous observations (Herkenham et al., 1991; Wu et al., 2003).
The relative resistance of dopaminergic neurons to MPTP-induced neurotoxicity in MPO-/- mice was, however, not accompanied by a preservation of striatal dopamine levels or attenuation of motor deficits caused by this parkinsonian neurotoxin (supplemental material, available at www.jneurosci.org). This discrepancy may be explained by the fact that TH (the rate-limiting enzyme in the synthesis of dopamine) can be inactivated by injury, such as that inflicted by MPTP (Ara et al., 1998). It is thus conceivable that although ablation of MPO attenuates the loss of TH protein (as evidenced by immunostaining), this beneficial effect may not be enough to prevent the loss of TH catalytic activity (as evidenced by the dopamine levels). Targeting MPO alone may thus suffice to provide observable structural, but not functional, neuroprotection in this experimental model of PD. Accordingly, optimal therapeutic interventions for PD may rely on the combination of strategies capable of providing structural protection such as MPO inhibition, with other strategies capable of protecting/stimulating dopaminergic function. Yet, given the relentless nature of PD, it can be surmised that the death signal in this illness may not be as harsh as that provoked by MPTP. Therefore, whether MPO inhibition in PD can succeed, not only in slowing neuronal death but also in sustaining dopamine synthesis, is a possibility that should not readily be excluded.
As to how MPO neurotoxic actions on dopaminergic neurons are mediated, two distinct and not mutually exclusive mechanisms may be invoked. First and foremost, MPO is known for its production of cytotoxic reactive oxygen species and RNS (Harrison and Schultz, 1976; Eiserich et al., 1996; Hampton et al., 1998). Therefore, neurons located in the vicinity of MPO-containing cells may have their plasma membrane proteins and lipids subjected to the deleterious effects of MPO-derived oxidants such as HOCl. In keeping with this scenario, we found high levels of 3-chlorotyrosine, a specific oxidative modification of tyrosine residues mediated by HOCl in the MPTP-susceptible brain region, the ventral midbrain. Also supporting the oxidative role of MPO in the MPTP model is our immunohistochemical demonstration of HOCl-modified protein in the ventral midbrain of intoxicated mice (Fig. 5A-C). Aside from this oxidative effect, MPO can be secreted and bind CD11b/CD18 integrins to the cell surface (Lau et al., 2005). In the case of neutrophils, ligation of CD11b/CD18 by MPO stimulates signaling pathways implicated in the activation of these cells (Lau et al., 2005). Because brain microglia do express CD11b/CD18 integrins and seem to participate in the neurodegenerative process in the MPTP model and in PD, this cytokine-like effect of MPO may represent an additional mechanism by which dopaminergic neurons are affected by this enzyme.
As raised previously (Wu et al., 2003), a key issue is the selective damage to dopaminergic neurons observed during inflammation in MPTP-treated mice and humans suffering from PD. Many lines of evidence suggest that dopaminergic neurons are particularly vulnerable to oxidative stress compared with the other cells in the brain (Dauer and Przedborski, 2003). Alternatively, it is likely that in the MPTP model and in PD, the magnitude of the inflammatory response and resulting oxidative stress is mild and only inflicts sublethal lesions. Thus, inflammation-mediated oxidative stress would succeed in killing only neurons already compromised, as dopaminergic neurons probably are in PD and after MPTP injections.
Footnotes
This work was supported by grants from the National Institutes of Health-National Institute of Neurological Disorders and Stroke (NS38586, NS42269, NS38370, NS11766-27A2) and National Institute of Aging (AG021191), the United States Department of Defense Grant (DAMD17-99-1-9474 and DAMD17-03-1), the Lowenstein Foundation, the Lillian Goldman Charitable Trust, the National Institute of Environmental Health Sciences (P30ES07033), the Donald W. Reynolds Foundation, and the Parkinson's Disease Foundation. We thank Matthew Lucas for assistance in preparing this manuscript, Dr. Ernst Malle (University of Graz, Graz, Austria) for providing the HOP-1 antibody, and the New York Brain Bank at Columbia University Taub Institute for providing human Parkinson's disease samples.
Correspondence should be addressed to Dr. Serge Przedborski, Columbia University, 650 West 168th Street, BB-318, New York, NY 10032. E-mail: sp30@columbia.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256594-07$15.00/0
References
- Andrews PC, Krinsky NI (1982) Quantitative determination of myeloperoxidase using tetramethylbenzidine as substrate. Anal Biochem 127: 346-350. [DOI] [PubMed] [Google Scholar]
- Ara J, Przedborski S, Naini AB, Jackson-Lewis V, Trifiletti RR, Horwitz J, Ischiropoulos H (1998) Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Proc Natl Acad Sci USA 95: 7659-7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan Jr WA, Bird ED, Aprille JR (1985) Regional mitochondrial respiratory activity in Huntington's disease brain. J Neurochem 44: 1948-1950. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhang SM, Hernan MA, Schwarzschild MA, Willett WC, Colditz GA, Speizer FE, Ascherio A (2003) Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol 60: 1059-1064. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39: 889-909. [DOI] [PubMed] [Google Scholar]
- Daugherty A, Dunn JL, Rateri DL, Heinecke JW (1994) Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest 94: 437-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiserich JP, Cross CE, Jones AD, Halliwell B, van der Vliet A (1996) Formation of nitrating and chlorinating species by reaction of nitrite with hypochlorous acid. A novel mechanism for nitric oxide-mediated protein modification. J Biol Chem 271: 19199-19208. [DOI] [PubMed] [Google Scholar]
- Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B (2002) Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J Neurochem 81: 1285-1297. [DOI] [PubMed] [Google Scholar]
- Gaut JP, Yeh GC, Tran HD, Byun J, Henderson JP, Richter GM, Brennan ML, Lusis AJ, Belaaouaj A, Hotchkiss RS, Heinecke JW (2001) Neutrophils employ the myeloperoxidase system to generate antimicrobial brominating and chlorinating oxidants during sepsis. Proc Natl Acad Sci USA 98: 11961-11966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut JP, Byun J, Tran HD, Lauber WM, Carroll JA, Hotchkiss RS, Belaaouaj A, Heinecke JW (2002) Myeloperoxidase produces nitrating oxidants in vivo. J Clin Invest 109: 1311-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green PS, Mendez AJ, Jacob JS, Crowley JR, Growdon W, Hyman BT, Heinecke JW (2004) Neuronal expression of myeloperoxidase is increased in Alzheimer's disease. J Neurochem 90: 724-733. [DOI] [PubMed] [Google Scholar]
- Hampton MB, Kettle AJ, Winterbourn CC (1998) Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 92: 3007-3017. [PubMed] [Google Scholar]
- Harrison JE, Schultz J (1976) Studies on the chlorinating activity of myeloperoxidase. J Biol Chem 251: 1371-1374. [PubMed] [Google Scholar]
- Hazell LJ, Arnold L, Flowers D, Waeg G, Malle E, Stocker R (1996) Presence of hypochlorite-modified proteins in human atherosclerotic lesions. J Clin Invest 97: 1535-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinecke JW, Hsu FF, Crowley JR, Hazen SL, Leeuwenburgh C, Mueller DM, Rasmussen JE, Turk J (1999) Detecting oxidative modification of biomolecules with isotope dilution mass spectrometry: sensitive and quantitative assays for oxidized amino acids in proteins and tissues. Meth Enzymol 300: 124-144. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Little MD, Bankiewicz K, Yang SC, Markey SP, Johannessen JN (1991) Selective retention of MPP+ within the monoaminergic systems of the primate brain following MPTP administration: an in vivo autoradiographic study. Neuroscience 40: 133-158. [DOI] [PubMed] [Google Scholar]
- Hunot S, Boissière F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y, Hirsch EC (1996) Nitric oxide synthase and neuronal vulnerability in Parkinson's disease. Neuroscience 72: 355-363. [DOI] [PubMed] [Google Scholar]
- Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS (2000) Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci 20: 6309-6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM, Brummer J, Rudolph V, Munzel T, Heitzer T, Meinertz T, Baldus S (2005) Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci USA 102: 431-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberatore G, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WJ, Dawson VL, Dawson TM, Przedborski S (1999) Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. Nat Med 5: 1403-1409. [DOI] [PubMed] [Google Scholar]
- Malle E, Hazell L, Stocker R, Sattler W, Esterbauer H, Waeg G (1995) Immunologic detection and measurement of hypochlorite-modified LDL with specific monoclonal antibodies. Arterioscler Thromb Vasc Biol 15: 982-989. [DOI] [PubMed] [Google Scholar]
- Pennathur S, Jackson-Lewis V, Przedborski S, Heinecke JW (1999) Mass spectrometric quantification of 3-nitrotyrosine, ortho-tyrosine, and O,O′-dityrosine in brain tissue of 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine-treated mice, a model of oxidative stress in Parkinson's disease. J Biol Chem 274: 34621-34628. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Goldman JE (2004) Pathogenic role of glial cells in Parkinson's disease. In: Non-neuronal cells of the nervous system: function and dysfunction (Hertz L, ed), pp 967-982. New York: Elsevier
- Przedborski S, Vila M (2001) MPTP: a review of its mechanisms of neurotoxicity. Clin Neurosci Res 1: 407-418. [Google Scholar]
- Przedborski S, Jackson-Lewis V, Naini A, Jakowec M, Petzinger G, Miller R, Akram M (2001) The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a technical review of its utility and safety. J Neurochem 76: 1265-1274. [DOI] [PubMed] [Google Scholar]
- Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, Naini A, Vila M, Jackson-Lewis V, Ramasamy R, Przedborski S (2003) d-β-Hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest 112: 892-901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vliet A, Eiserich JP, Halliwell B, Cross CE (1997) Formation of reactive nitrogen species during peroxidase-catalyzed oxidation of nitrite. A potential additional mechanism of nitric oxide-dependent toxicity. J Biol Chem 272: 7617-7625. [DOI] [PubMed] [Google Scholar]
- Vila M, Przedborski S (2004) Genetic clues to the pathogenesis of Parkinson's disease. Nat Med [Suppl] 10: S58-S62. [DOI] [PubMed] [Google Scholar]
- West MJ (1993) New stereological methods for counting neurons. Neurobiol Aging 14: 275-285. [DOI] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S (2002) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22: 1763-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S (2003) NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc Natl Acad Sci USA 100: 6145-6150. [DOI] [PMC free article] [PubMed] [Google Scholar]