

Abstract
Chronic wasting disease (CWD), a prion disease affecting free-ranging and captive cervids (deer and elk), is widespread in the United States and parts of Canada. The large cervid population, the popularity of venison consumption, and the apparent spread of the CWD epidemic are likely resulting in increased human exposure to CWD in the United States. Whether CWD is transmissible to humans, as has been shown for bovine spongiform encephalopathy (the prion disease of cattle), is unknown. We generated transgenic mice expressing the elk or human prion protein (PrP) in a PrP-null background. After intracerebral inoculation with elk CWD prion, two lines of “humanized” transgenic mice that are susceptible to human prions failed to develop the hallmarks of prion diseases after >657 and >756 d, respectively, whereas the “cervidized” transgenic mice became infected after 118–142 d. These data indicate that there is a substantial species barrier for transmission of elk CWD to humans.
Keywords: chronic wasting disease, CWD, transmissibility to humans, transgenic mice, prion, cervids, deer, elk, species barrier
Introduction
Prion diseases are neurodegenerative diseases that affect humans and animals, including cattle, sheep, cervids (deer and elk), and mink (Sigurdson and Miller, 2003; Jeffrey and Gonzalez, 2004; Kong et al., 2004). They pose a serious threat to public health because they can be transmitted between humans and from animals to humans. Animal to human transmission was dramatically exemplified by the sudden appearance of a new form of prion disease, identified as variant Creutzfeldt–Jakob disease (vCJD), in the United Kingdom, after the emergence of a large outbreak of bovine spongiform encephalopathy (BSE) (Will, 2003). Compelling evidence indicates that vCJD is acquired after the consumption of beef or beef products from BSE-infected cattle (Scott et al., 1999; Will, 2003). To date, five cases of indigenous BSE, but no case of locally acquired vCJD, have been reported in North America.
Chronic wasting disease (CWD) is the prion disease that affects free-ranging and captive cervids, including white-tail deer, mule deer, and Rocky Mountain elk (Miller and Williams, 2004). First reported in 1967, CWD was once considered a rare and geographically contained disease; however, recent data support the presence of CWD among free-ranging and captive cervids in at least 13 states in the United States and 2 provinces in Canada, with a prevalence of up to 20% in some endemic areas (Miller and Williams, 2003; Miller et al., 2004; Prusiner et al., 2004). These findings, along with other considerations such as the high cervid population in the United States (estimated at 22 million), the several million deer and elk hunters, and the widespread consumption of elk and deer meat, underscore the likely increasing risk of human exposure to CWD. These considerations, along with the recognition that the outbreak of BSE led to the emergence of vCJD, have heightened concerns about possible direct-contact and food-borne CWD transmission to humans. In fact, many people are known to have consumed venison from confirmed CWD-affected cervids. Twenty-seven patients with CJD who regularly consumed elk and deer meat were reported to the National Prion Disease Pathology Surveillance Center at Case Western Reserve University, but none of these cases appeared to have a novel form of prion disease (Belay et al., 2001, 2003, 2004; P. Gambetti, unpublished observation); however, human disease acquired from CWD might have an unusual phenotype or a phenotype that is difficult to distinguish from that of sporadic CJD (sCJD). This uncertainty is a serious public health concern in the United States.
To assess the transmissibility of CWD to humans, we generated transgenic (Tg) Friend leukemia virus B (FVB) mice expressing either the human prion protein (PrP) or Rocky Mountain elk PrP in a PrP-null background. Here we show that, after intracerebral inoculation with elk CWD prion, the two lines of “humanized” Tg mice failed to develop the hallmarks of prion diseases after >657 and >756 d, respectively, whereas the “cervidized” Tg mice became infected after 118–142 d. These data indicate that there is a substantial species barrier for the transmission of elk CWD to humans.
Materials and Methods
Construction of transgenes expressing human PrP-129M or elk PrP-132M. The transgene constructs are based on the murine half-genomic PrP clone in plasmid pHGPRP (Fischer et al., 1996). The HuPrP-129M open reading frame (ORF) was amplified from the human genomic DNA PAC (P1-derived artificial chromosome) clone RP5–1068H6 (obtained from the Sanger Center, Cambridge, UK) with primers HRM-F (TATGTGGACTGATGTCGGCCTCTGCAAGAAGCGC) and HRM-R (CCACCTCAATTGAAAGGGCTGCAGGTGGATAC). The PCR product was digested with PshAI and MfeI and used to replace the corresponding 0.97 kb PshAI–MfeI fragment in pHGPRP to create pHGHuPrP-129M. In the resulting pHGHuPrP-129M clones, the signal-peptide sequence is still from mouse, but the rest of the PrP ORF and the first 76 bp after the stop codon are from human PRNP (prion protein) genomic DNA. The inserted 0.97 kb PshAI–MfeI fragment in pHGHuPrP-129M was then sequenced with the primers HRM-R, HRM-F, and HP306R (CATGTTGGTTTTTGGCTTACTC). One error-free clone was chosen for the creation of transgenic mice. To create the transgene construct expressing elk PrP-132M, the mouse PrP ORF in pHGPRP was first replaced with the restriction sites for ClaI and NruIby using conventional recombinant DNA techniques to create pHGD3. The ElPrP-132M ORF (eGMSE allele) was selected to make cervidized transgenic mice because it was reported that all CWD-affected elk and some deer carried this allele (Raymond et al., 2000). The ElPrP-132M ORF was amplified from the genomic DNA of an American elk with primers DePrP-F (CAGTCTAGACCGCGGCATGGTGAAAAGCCACATAGG) and DePrP-R (ACCTCTAGACCTATCCTACTATGAGAAAAATGAG), and cloned into pSTBlue 1 (Novagen, Madison, WI). The 0.78 kb ElPrP ORF thus cloned was released by SacII–XbaI double digestion, blunted with T4 DNA polymerase, and inserted into the NruI site of pHGD3 to create pHGDePrP-132M. The final pHGElPrP-132M construct was confirmed by sequencing. One error-free clone was chosen for the creation of transgenic mice.
Generation, screening, and characterization of transgenic Tg(HuPrP-129M)Prnp0/0 and Tg(ElPrP-132M)Prnp0/0 mice. The 12.2 kb HuPrP-129M and elk PrP-132M transgene constructs were microinjected into fertilized FVB/NJ eggs, and planted into the oviducts of pseudopregnant CD-1 mice at the transgenic mouse facility of Albert Einstein College of Medicine (Bronx, NY). Founder pups were screened by tail DNA PCR. All founder mice that carry the transgene were bred with FVB/Prnp0/0 mice (Fischer et al., 1996) (kindly provided by the Prusiner laboratory, University of California, San Francisco) to obtain Tg mice in PrP-null background. Transgenic PrP expression in the brain and other tissues of the Tg mice were examined by Western blot analysis with monoclonal antibodies (mAbs) 3F4 and 8H4 for humanized and cervidized Tg mice, respectively. All animal experiments in this study were approved by the Institutional Animal Use and Care Committee and the Institutional Bio-safety Committee, and the use of human brain tissues was authorized by the Institutional Review Board.
Western blot analysis. Mouse tissues were homogenized in 10 vol of lysis buffer (10 mm Tris, 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 5 mm EDTA, pH 8.0) with or without 1 mm phenylmethylsulfonyl fluoride (PMSF). The immunoblotting was performed as described previously (Pan et al., 2001), with minor modifications. The homogenate was cleared at 12,000 rpm for 10 min, and the supernatant was then stored at –80°C. To detect Proteinase K (PK)-resistant PrP fragments (PrPSc), brain extracts without PMSF were incubated with 100 μg/ml PK for 1 h at 37°C, and PMSF was added to a final concentration of 3 mm to terminate the digestion. The extracts were mixed with PAGE loading buffer (160 mm Tris, 4% SDS, 4% 2-ME, 20% glycerol, 0.04% bromophenol blue, pH 6.8), loaded onto 12% Tris-glycine SDS-PAGE or 10–20% Tris-tricine SDS-PAGE gels, transferred to polyvinylidene difluoride membrane, and probed with 8H4 or 3F4 in conjunction with horseradish peroxidase-conjugated goat anti-mouse IgG Fc antibody.
Inoculation of transgenic mice. Brain tissues from humans with sCJD or from elk with CWD were homogenized and inoculated into the brains of Tg(HuPrP-129M)Prnp0/0 or Tg(ElPrP-132M)Prnp0/0 mice. Brain tissues were homogenized in cold PBS, and the homogenate was centrifuged at 1000 × g for 10 min at 4°C. The supernatant was diluted to 10-fold of the brain tissue volume in cold PBS to obtain 10% brain homogenate, frozen at –80°C for storage, and diluted to 1% with PBS just before inoculation. After anesthetization with isoflurane, 30 μl of the 1% brain homogenate was injected into each mouse brain with a 26 gauge needle inserted to a depth of ∼2 mm at the left parietal region of the cranium.
Monitoring of symptoms and examination of PrPSc. After intracerebral inoculations, the animals were visually examined daily for symptoms such as coarse coat, waddling gait, tail plasticity, and bradykinesia. Within 2–3 d after the appearance of these symptoms or at death, the brain was removed; one-half was frozen for biochemical studies, and the other half was stored in formalin for histology and immunohistochemistry analysis as described previously (Taraboulos et al., 1992). Total PrP as well as PrPSc (PK-resistant PrP) were examined by Western blotting in Tris-glycine and/or Tris-Tricine SDS-PAGE gels, as described above. Sodium phosphotungstate precipitation of PrPSc was performed as described previously (Safar et al., 1998).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining of paraffin-embedded brain sections was performed as described previously (Shi et al., 1990) with the In Situ Cell Death Detection Kit-peroxidase (POD) (Roche Applied Science, Indianapolis, IN) according to the manufacturer's instructions. Paraffin-embedded sections were dried at 60°C. After the paraffin was removed with xylene, tissues were rehydrated in serial solutions of ethanol (100, 95, and 75%), washed in PBS, and subjected to microwave treatment for 20 min in 60 mm HCl. The slides were then air dried at room temperature, treated with 5% Triton X-100 at room temperature, washed in PBS several times, and incubated with TUNEL reagent for 2 h at 37°C in a humid chamber. The slides were then washed in PBS, incubated with converter-POD for 30 min at 37°C, developed with diaminobenzidine substrate according to the manufacturer's protocol, counter-stained with hematoxylin QS (Vector Laboratories, Burlingame, CA), and mounted. The TUNEL-positive cells were stained brown.
Results
We used two lines of humanized Tg mice, Tg40 and Tg1, and one line of cervidized Tg mice, Tg12, that express the transgene PrP in brain approximately onefold, twofold, and twofold, respectively, the level of brain PrP in wild-type FVB mice. Both humanized and cervidized Tg mice were inoculated intracerebrally with brain homogenates from two CWD-affected elk. The humanized Tg1 and Tg40 mice were also similarly inoculated with brain homogenates from human subjects with a type of sporadic CJD identified as sCJDMM1 (Parchi et al., 1996).
Thirteen of 14 cervidized Tg12 mice inoculated intracerebrally with CWD elk 1 brain homogenates developed ataxia after an average of 118 ± 6 d postinoculation (dpi) (range, 83–142 dpi) (Table 1, Fig. 1). All seven cervidized Tg12 mice inoculated intracerebrally with CWD elk 2 brain homogenates developed ataxia after an average of 142 ± 7 dpi (range, 124–178 dpi) (Table 1, Fig. 1). Histologically, all ataxic mice contained severe and wide-spread spongiform degeneration throughout the cerebral cortex and basal ganglia, as well as neuronal loss in the hippocampus and granule cell layer of the cerebellum (Fig. 2a,b). Neuronal apoptosis was detected by TUNEL staining in the hippocampus (Fig. 2f), other cortical regions, and the cerebellum (data not shown). Amyloid plaques were not present; however, immunohistochemical staining for PrP revealed spotted, round PrP deposits, the so-called plaque-like PrP deposits, similar to those found in CWD-affected elk, in the cerebral cortex (Fig. 2d) and the molecular layer of the cerebellum (Fig. 2e). Symptomatic mice also accumulated large amounts of PK-resistant PrP with gel mobility and glycoform ratios matching those of the original CWD inoculum (Fig. 3); therefore, the cervidized Tg12 mice replicated the main features of the elk CWD PrPSc. Furthermore, secondary transmission of the elk 1 CWD prion passaged once in Tg12 mice required an incubation period of 125 ± 4 d (range, 115–138 d) (Table 1), indicating that there is no species barrier for elk CWD transmission to the Tg12 mice.
Table 1.
Prion transmission in transgenic mice
|
Prion inoculum |
Mice |
PrP (level) |
Incubation time (SEM) |
Transmission ratea |
|---|---|---|---|---|
| CWD (elk 1) | Tg12 | Elk PrP-132M (2×) | 118 ± 6 d | 13/14 |
| CWD (elk 2) | Tg12 | Elk PrP-132M (2×) | 142 ± 7 d | 7/7 |
| CWD (Tg12) | Tg12 | Elk PrP-132M (2×) | 125 ± 4 d | 5/5 |
| CWD (elk 1/2) | Tg40 | HuPrP-129M (1×) | >756 d | 0/29 |
| CWD (elk 1/2) | Tg1 | HuPrP-129M (2×) | >657 d | 0/22 |
| sCJDMM1 | Tg40 | HuPrP-129M (1×) | 263 ± 13 d | 9/10 |
| sCJDMM1 |
Tg1 |
HuPrP-129M (2×) |
226 ± 5 d |
7/7 |
Number of affected/total inoculated animals.
Figure 1.

Survival curves of humanized and cervidized transgenic mice. Six- to eight-week-old cervidized Tg12 and humanized Tg1 and Tg40 mice were inoculated intracerebrally with 30 μl of 1% brain homogenate from two CWD-affected elk or a subject with sCJDMM1. The Tg12 mice were also inoculated with brain homogenates from elk 1 CWD-affected Tg12 mice to evaluate the species barrier of elk CWD transmission to the Tg12 mice. The average incubation times were as follows: 118±6 d for elk 1 CWD-inoculated Tg 12 mice (opensquares); 125±4 d for secondary transmission of elk 1 CWD in Tg12 mice (filled squares); 142 ± 7 d for elk 2 CWD-inoculated Tg12 mice (open rectangles); 263 ± 13 d for the sCJDMM1-inoculated Tg40 mice (open circles); and 226 ± 5 d for the sCJDMM1-inoculated Tg1 mice (open ovals). None of the 29 CWD-inoculated Tg40 mice (filled circle) or the 22 CWD-inoculated Tg1 mice (filled oval) had detectable PK-resistant PrPSc or substantial histopathology after >756 and >657 dpi, respectively. The asterisk indicates that three Tg40 mice became ataxic between 420 and 509 dpi but were free of prion infection. Parentheses indicate prion-positive mice per total inoculated mice.
Figure 2.
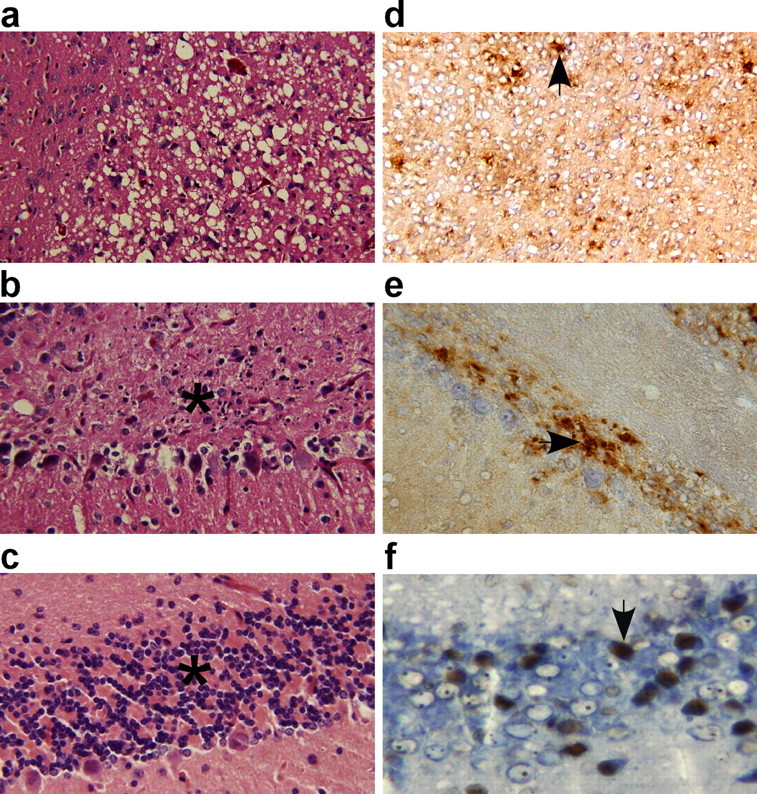
Histopathology, PrP immunohistochemistry, and apoptosis examination in CWD-affected Tg12-cervidized mice. Prominent spongiform degeneration was present in the cerebral cortex (a) along with marked neuronal loss in the granule cell layer of the cerebellum (b) when compared with age-matched control Tg12 mice (c) (hematoxylin and eosin staining; 20× magnification). PrP deposits formed a fine granular pattern interspersed with plaque-like deposits (arrow) in the cerebral cortex (d) and in the granule cell layer of the cerebellum (e) (mAb 8H4; 20× magnification). Neuronal apoptosis (arrow) was present in the granule cells of the hippocampus (f) and in other brain regions (TUNEL; 40× magnification).
Figure 3.

Characterization of PK-resistant PrPSc from the donor CWD-affected elk and CWD-affected Tg12 mice. a, Immunoblot of PrP. Lanes 1 and 2, PK-untreated (lane 1) and PK-treated (lane 2) PrP from one of the donor elks with CWD. Lanes 3–14, PK-untreated (lane 3) and PK-treated (lanes 4–14) PrP from 11 CWD-affected Tg12 mice. b, Glycoform ratio analysis of PK-resistant PrPSc. The blots were probed with mAb 8H4. Error bars are based on quantitative analyses of digital chemiluminescence images of triplicate Western blots of brain homogenates from an elk with CWD (the inoculum) and duplicate Western blots of 11 Tg12 mice infected with elk CWD. kD, Kilodalton.
Brain homogenates from the two CWD-affected elk used for the cervidized mice were also inoculated intracerebrally into 29 Tg40 humanized mice and 22 Tg1 humanized mice. None of the 29 Tg40 mice or the 22 Tg1 mice showed signs of prion diseases after >756 and >657 dpi, respectively. Three Tg40 mice appeared to be mildly ataxic before being killed at 420–509 dpi. A total of 18 Tg40 mice and 12 Tg1 mice died naturally of old age or were killed because of other illnesses; however, none of the Tg40 and Tg1 mice examined, including the three ataxic Tg40 mice, had PK-resistant PrPSc as detected by immunoblotting of PrPSc-enriched preparations after precipitation with sodium phosphotungstate (Fig. 4). Histopathological and PrP immunohistochemical examinations also were negative (data not shown). Furthermore, the PrP immunoprecipitates obtained from the three ataxic mice with the mAb OCD4, which immunoreacts with both PK-resistant and PK-sensitive abnormal PrP present in human and animal prion diseases but not with normal PrPC (Zou et al., 2004), were not different from the corresponding immunoprecipitates obtained from other elk CWD-inoculated or noninoculated Tg40 mice. Therefore, all CWD-inoculated humanized mice appeared to be free of detectable prion, and the cause of the mild ataxia in these three Tg40 mice did not appear to be related to prion disease.
Figure 4.

Absence of PK-resistant PrPSc in elk CWD-inoculated humanized Tg mice. Brain homogenates were subjected to sodium phosphotungstate treatment to precipitate PrPSc, digested with 20 μg/ml PK for 30 min, and analyzed by Western blotting with the mAb 3F4. Lane 1, An uninoculated Tg40 mouse; lane 2, an sCJDMM1-infected Tg40 mouse; lanes 3–5, three ataxic CWD-inoculated Tg40 mice; lanes 6–8, three CWD-inoculated Tg40 mice killed because of other aging-related illnesses. Lanes 1, 3–8, Forty microliters of 10% brain homogenates were loaded; lane 2, a total of 2 μl was loaded. kD, Kilodalton.
As positive controls, 10 Tg40 and 7 Tg1 humanized mice were also inoculated intracerebrally with brain homogenate from a human subject with sCJDMM1. Nine of the 10 Tg40 mice and all 7 Tg1 mice became symptomatic, with an average incubation time of 263 ± 13 d (range, 213–315 d) for the Tg40 mice and 226 ± 5 d (range, 213–244 d) for the Tg1 mice. The affected mice revealed fine spongiform degeneration in the cerebral cortex (Fig. 5a) but not in the cerebellum (Fig. 5b), and the vacuoles were different in size and distribution from those of the cervidized mice inoculated with elk CWD prion. Apoptosis of neuronal cells was present but was less prominent than in the CWD-affected Tg12 cervidized mice (data not shown). After immunohistochemical staining, fine PrP deposits mimicking the “synaptic” pattern of sCJDMM1 were found in the cerebral cortex (Fig. 5c) and cerebellum (Fig. 5d) of the CJD-affected humanized mice, whereas the plaque-like deposits observed in the elk CWD-inoculated cervidized mice were not present. Abundant PK-resistant PrPSc with gel mobility matching that of the sCJDMM1 inoculum was shown in the brain of the infected mice (Fig. 6a). The glycoform ratio also was similar to that of sCJDMM1, with underrepresentation of the diglycosylated PK-resistant PrPSc fragment (Fig. 6b), but it was quite different from that of CWD.
Figure 5.

Histopathology and PrPSc deposition in the brains of sCJDMM1-infected Tg40 mice. In the brains of sCJDMM1-infected Tg40 mice, hematoxylin and eosin staining revealed moderate spongiform degeneration with fine vacuoles in the cerebral cortex (a) accompanied by even milder spongiosis in other brain areas (20×magnification), but the granule cell layer of the cerebellum (b) appears mostly intact (40× magnification). Immunohistochemistry for PrPSc with 3F4 revealed fine PrPSc deposits in the cerebral cortex (c), with more intense PrPSc deposits in the cerebellum (d) (20× magnification).
Figure 6.

Immunoblots and glycoform ratios of PK-resistant PrPSc from the sCJDMM1 donor and sCJDMM1-inoculated Tg1 mice. a, Immunoblot of PrP. PK-untreated (lane 1) PrP and PK-treated (lane 2) PrP were obtained from the sCJDMM1 donor; PK-untreated (lane 3) PrP and PK-treated (lanes 4–10) PrP were obtained from the seven sCJDMM1-inoculated Tg1 mice. b, Glycoform ratio analysis of PK-resistant PrPSc. PK-untreated and PK-treated brain homogenates were processed as in Figure 2, but blots were probed with 3F4. Error bars are based on quantitative analyses of digital chemiluminescence images of triplicate Western blots of brain homogenates from a subject with sCJDMM1 (the inoculum) and duplicate Western blots of the seven Tg1 mice infected with sCJDMM1. kD, Kilodalton.
Discussion
We have shown that CWD of elk can be transmitted to Tg12 cervidized mice with a relatively short average incubation time of 118 ± 6 d for elk 1 and 142 ± 7 d for elk 2. The PK-resistant PrPSc of the elk CWD-inoculated Tg12 mice has the same gel mobility and glycoform ratio as the PK-treated PrPSc of the CWD-affected elk. The pattern of PrP immunostaining is also similar in the two conditions. In addition, the secondary transmission of elk 1 CWD passaged in Tg 12 mice to naive Tg12 mice required an incubation time of 125 ± 4 d, which is not substantially different from that of the primary transmissions. Furthermore, it is associated with a histopathology and a PrP immunostaining pattern that is similar to that of the primary infection. These data argue that there is no species barrier in the transmission of elk CWD to the Tg12 mice and that CWD-inoculated Tg12 mice reproduced major strain characteristics of the PrPSc associated with elk CWD. Recently, Browning et al. (2004) inoculated both hemizygous and homozygous cervidized Tg(CerPrP) mice that express mule deer PrP (S2 allele) with brain homogenates from CWD-affected mule deer and elk. The inoculated mice developed a disease with histopathological and PrP immunohistochemical characteristics that appear overall to mimic the histopathology and PrP immunohistochemistry observed in our elk CWD-inoculated Tg12 mice. The gel mobility of the PK-resistant fragments of PrPSc recovered from the Tg(CerPrP) mice also is apparently similar to that observed in our Tg12 mice; however, Browning et al. (2004) reported a difference in the glycoform ratio between the PK-resistant PrPSc fragments recovered from the Tg(CerPrP) mice and that of the original elk and deer inocula. At variance with this finding, the PrPSc glycoform ratio of our affected Tg12 mice reproduced precisely that of the elk CWD inoculum (Fig. 3b). The incubation time of the CWD-inoculated Tg(CerPrP) mice varied between 220 and 270 d for the hemizygous mice and was 160 ± 3 d for the homozygous mice despite the 3- to 5-fold and 6- to 10-fold PrP expression of the hemizygous and homozygous mice, respectively. The difference in incubation time between the Tg(CerPrP) mice and the Tg12 mice could be caused by the polymorphism of codon 226 [226Q for the S2 allele used in Tg(CerPrP) mice and 226E for the eGMSE allele used in Tg12 mice], the PrP genotype difference in the cervid CWD samples, or the difference in transgene vectors. The short incubation time combined with the only slightly elevated PrP expression make the Tg12 mice suitable for bioassay and other studies on CWD. The short incubation time for elk CWD prion in Tg12 mice also compares favorably with the incubation time of 238 d reported for BSE transmission to the Tg mice expressing bovine PrP (Scott et al., 1999).
In contrast to the efficient CWD transmission in the cervidized Tg12 mice, the same CWD inocula failed to infect the humanized Tg40 and Tg1 mice after >756 and >657 dpi, respectively, the approximate lifespan of these mice, which were ∼2 months old at the time of inoculation; however, sCJD was transmitted to these humanized mice with average incubation times of 263 and 226 d, respectively. At variance with CWD, BSE has been transmitted to a similar humanized Tg mouse model that, like our humanized Tg mice, expressed human PrP-129M at a level two times that of pooled normal human brain (Asante et al., 2002). The total attack rate was 14 of 49; of the 14 infected mice, 6 had clinical signs, and the other 8 had subclinical infection only, but all had PrPSc in the brain. The incubation times were 414 d on average (range, 338–492 d) but as short as ∼340 d in several mice (Asante et al., 2002). Combined, these findings point to the presence of a robust species barrier for elk CWD transmission to humans, which is much more effective than that for BSE transmission to humans. Because the most likely route of CWD transmission to humans is through oral consumption of CWD-contaminated meat and the intracerebral route is much more effective than the oral route (Prusiner et al., 1985), the failure to detect elk CWD transmission in the humanized Tg mice after intracerebral inoculations suggests an even lower risk of elk CWD transmission to humans. This conclusion is consistent with the lack of evidence of CWD transmission to CJD patients investigated for a possible causal link of their illness with CWD and with the low efficiency of an in vitro conversion of human PrPC by cervid PrPSc (Raymond et al., 2000). Secondary transmission experiments in naive humanized Tg mice are underway to determine whether the primary CWD-inoculated humanized Tg mice are asymptomatic carriers of prion infectivity, although PrPSc is undetectable in any of these primary mice on Western blot even after sodium phosphotungstate precipitation or immunoprecipitation with the mAb OCD4 that selectively recognizes abnormal PrP associated with prion diseases (Safar et al., 1998; Zou et al., 2004). Because the CWD prions from deer and elk appear to be indistinguishable (Williams and Young, 1992; Spraker et al., 2002) and there have been no reports of different CWD prion strains, it is likely that CWD prions from mule deer and white-tail deer are, as reported here for CWD prion from elk, of low or no transmissibility in humans.
Footnotes
This work was supported by National Institutes of Health Grant 1P01 AG14359-06 (P.G.), Centers for Disease Control and Prevention (CDC) Grant UR8\CU515004 (P.G.), and funding from the Center for Food Safety and Applied Nutrition of the Food and Drug Administration through the CDC (P.G.). We thank B. Chakraborty, K. Edmonds, D. Kofskey, P. Scalzo, and F. Scalzo for technical support as well as M. E. Potter for help and encouragement.
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the funding agency.
Correspondence should be addressed to Dr. Pierluigi Gambetti, Department of Pathology, Case Western Reserve University, Cleveland, OH 44106. E-mail: pxg13@case.edu.
DOI:10.1523/JNEUROSCI.2467-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/257944-06$15.00/0
References
- Asante EA, Linehan JM, Desbruslais M, Joiner S, Gowland I, Wood AL, Welch J, Hill AF, Lloyd SE, Wadsworth JD, Collinge J (2002) BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J 21: 6358–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belay ED, Gambetti P, Schonberger LB, Parchi P, Lyon DR, Capellari S, McQuiston JH, Bradley K, Dowdle G, Crutcher JM, Nichol CR (2001) Creutzfeldt-Jakob disease in unusually young patients who consumed venison. Arch Neurol 58: 1673–1678. [DOI] [PubMed] [Google Scholar]
- Belay ED, Maddox RA, Gambetti P, Schonberger LB (2003) Monitoring the occurrence of emerging forms of Creutzfeldt-Jakob disease in the United States. Neurology 60: 176–181. [DOI] [PubMed] [Google Scholar]
- Belay ED, Maddox RA, Williams ES, Miller MW, Gambetti P, Schonberger LB (2004) Chronic wasting disease and potential transmission to humans. Emerg Infect Dis 10: 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Mason GL, Seward T, Green M, Eliason GA, Mathiason C, Miller MW, Williams ES, Hoover E, Telling GC (2004) Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J Virol 78: 13345–13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C (1996) Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15: 1255–1264. [PMC free article] [PubMed] [Google Scholar]
- Jeffrey M, Gonzalez L (2004) Pathology and pathogenesis of bovine spongiform encephalopathy and scrapie. Curr Top Microbiol Immunol 284: 65–97. [DOI] [PubMed] [Google Scholar]
- Kong Q, Surewicz WK, Petersen RB, Zou W, Chen SG, Gambetti P, Parchi P, Capellari S, Goldfarb L, Montagna P, Lugaresi E, Piccardo P, Ghetti B (2004) Inherited prion diseases. In: Prion biology and diseases, Ed 2 (Prusiner SB, ed), pp 673–776. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Miller MW, Williams ES (2003) Prion disease: horizontal prion transmission in mule deer. Nature 425: 35–36. [DOI] [PubMed] [Google Scholar]
- Miller MW, Williams ES (2004) Chronic wasting disease of cervids. Curr Top Microbiol Immunol 284: 193–214. [DOI] [PubMed] [Google Scholar]
- Miller MW, Williams ES, Hobbs NT, Wolfe LL (2004) Environmental sources of prion transmission in mule deer. Emerg Infect Dis 10: 1003–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan T, Colucci M, Wong BS, Li R, Liu T, Petersen RB, Chen S, Gambetti P, Sy MS (2001) Novel differences between two human prion strains revealed by two-dimensional gel electrophoresis. J Biol Chem 276: 37284–37288. [DOI] [PubMed] [Google Scholar]
- Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AAF, Trojanowski JQ, Petersen RB, Gambetti P (1996) Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 39: 767–778. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, Cochran SP, Alpers MP (1985) Transmission of scrapie in hamsters. J Infect Dis 152: 971–978. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, Williams E, Laplanche J-L, Shinagawa M (2004) Scrapie, chronic wasting disease, and transmissible mink encephalopathy. In: Prion biology and diseases, Ed 2 (Prusiner SB, ed), pp 545–594. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Raymond GJ, Bossers A, Raymond LD, O'Rourke KI, McHolland LE, Bryant III PK, Miller MW, Williams ES, Smits M, Caughey B (2000) Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. EMBO J 9: 4425–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB (1998) Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 4: 1157–1165. [DOI] [PubMed] [Google Scholar]
- Scott MR, Will R, Ironside J, Nguyen HO, Tremblay P, DeArmond SJ, Prusiner SB (1999) Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci USA 96: 15137–15142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi B, De Girolami U, He J, Wang S, Lorenzo A, Busciglio J, Gabuzda D (1990) Apoptosis induced by HIV-1 infection of the central nervous system. J Clin Invest 98: 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdson CJ, Miller MW (2003) Other animal prion diseases. Br Med Bull 66: 199–212. [DOI] [PubMed] [Google Scholar]
- Spraker TR, Zink RR, Cummings BA, Wild MA, Miller MW, O'Rourke KI (2002) Comparison of histological lesions and immunohistochemical staining of proteinase-resistant prion protein in a naturally occurring spongiform encephalopathy of free-ranging mule deer (Odocoileus hemionus) with those of chronic wasting disease of captive mule deer. Vet Pathol 39: 110–119. [DOI] [PubMed] [Google Scholar]
- Taraboulos A, Jendroska K, Serban D, Yang SL, DeArmond SJ, Prusiner SB (1992) Regional mapping of prion proteins in brain. Proc Natl Acad Sci USA 89: 7620–7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will RG (2003) Acquired prion disease: iatrogenic CJD, variant CJD, kuru. Br Med Bull 66: 255–265. [DOI] [PubMed] [Google Scholar]
- Williams ES, Young S (1992) Spongiform encephalopathies in Cervidae. Rev Sci Tech 11: 551–567. [DOI] [PubMed] [Google Scholar]
- Zou WQ, Zheng J, Gray DM, Gambetti P, Chen SG (2004) Antibody to DNA specifically detects scrapie but not normal prion protein. Proc Natl Acad Sci USA 101: 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]