

Abstract
Ethanol exposure during fetal development is a leading cause of learning disabilities. Studies suggest that it alters learning and memory by permanently damaging the hippocampus. It is generally assumed that this is mediated, in part, via alterations in glutamatergic transmission. Although NMDA receptors are presumed to be the most sensitive targets of ethanol in immature neurons, this issue has not been explored in the developing hippocampus. We performed whole-cell patch-clamp recordings in hippocampal slices from neonatal rats. Unexpectedly, we found that acute ethanol (10–50 mm) exposure depresses inward currents elicited by local application of exogenous AMPA, but not NMDA, in CA3 pyramidal neurons. These findings revealed a direct effect of ethanol on postsynaptic AMPA receptors. Ethanol significantly decreased the amplitude of both AMPA and NMDA receptor-mediated EPSCs evoked by electrical stimulation. This effect was associated with an increase in the paired-pulse ratio and a decrease in the frequency of miniature EPSCs driven by depolarization of axonal terminals. These findings demonstrate that ethanol also acts at the presynaptic level. ω-Conotoxin-GVIA occluded the effect of ethanol on NMDA EPSCs, indicating that ethanol decreases glutamate release via inhibition of N-type voltage-gated Ca2+ channels. In more mature rats, ethanol did not affect the probability of glutamate release or postsynaptic AMPA receptor-mediated currents, but it did inhibit NMDA-mediated currents. We conclude that the mechanism by which ethanol inhibits glutamatergic transmission is age dependent and challenge the view that postsynaptic NMDA receptors are the primary targets of ethanol early in development.
Keywords: development, ethanol, glutamate, presynaptic, postsynaptic, electrophysiology
Introduction
Ethanol exposure during development has long-lasting and devastating effects, ranging from full-blown fetal alcohol syndrome to cognitive deficits in information processing, learning and memory, and problem-solving skills (Warren and Foudin, 2001). The term fetal alcohol spectrum disorder (FASD) is now used to denote this array of conditions (Sokol et al., 2003). Studies suggest that the effects of ethanol in the developing CNS are, to some extent, the result of alterations in neurotransmission at glutamatergic synapses, which are critical for the maturation of neuronal circuits (Costa et al., 2000b; Ikonomidou et al., 2001; Zhang and Poo, 2001; Hua and Smith, 2004; Olney, 2004). Therefore, characterizing the mechanism of action of ethanol on excitatory transmission in developing neuronal circuits is important to further understanding of the pathophysiology of FASD.
Glutamate mediates fast excitatory transmission in the CNS and interacts with three classes of ionotropic receptors (Dingledine et al., 1999): NMDA receptors (NMDARs), AMPA receptors (AMPARs), and kainate receptors (KARs). Most studies have focused on the effect of ethanol on NMDAR-mediated synaptic transmission because of the initial demonstration of a high sensitivity of these receptors to ethanol in hippocampal neurons (Lovinger, 1997). Although it is widely accepted that ethanol acutely inhibits NMDARs, several studies suggest that their sensitivity varies among different CNS regions and as a function of development. For instance, the sensitivity to ethanol of NMDARs in the hippocampus and posterior cingulate cortex is higher in juvenile than adult rats (Swartzwelder et al., 1995; Li et al., 2002). However, the sensitivity of NMDARs during earlier developmental periods remains an open question.
In addition to modulating NMDARs, ethanol regulates the function of non-NMDARs. For instance, non-NMDAR-mediated responses are acutely inhibited by ethanol in nucleus accumbens and central amygdala rat slices (Nie et al., 1994; Roberto et al., 2004). Studies in the rat somatosensory cortex revealed an age-dependent ethanol-induced modulation of AMPAR responses (Lu and Yeh, 1999). Moreover, several studies indicate that hippocampal KARs are potently inhibited by ethanol (Weiner et al., 1999; Costa et al., 2000a; Crowder et al., 2002; Carta et al., 2003). In contrast, studies with hippocampal slices have shown that AMPARs are relatively insensitive to ethanol (Lovinger et al., 1989, 1990; Weiner et al., 1999; Carta et al., 2003; Hendricson et al., 2003). Given that hippocampal AMPAR-mediated synaptic transmission is acquired during the first 2 postnatal weeks in rats and that it contributes to network formation and synapse development (Durand et al., 1996; Molnar et al., 2002), it is important to understand the effects of ethanol on AMPARs during this developmental period.
In the present study, we addressed the hypothesis that ethanol depresses AMPAR-mediated transmission in the hippocampus of newborn rats by inducing changes at the presynaptic and postsynaptic levels. To this end, we prepared acute hippocampal slices from these rats, and, using patch-clamp electrophysiological techniques, we tested the effect of pharmacologically relevant concentrations of ethanol on glutamatergic transmission in the CA3 hippocampal region.
Materials and Methods
Electrophysiology. Unless indicated, all chemicals were from Sigma (St. Louis, MO) or Tocris Cookson (Bristol, UK). Sprague Dawley rats (Harlan Sprague Dawley, Indianapolis, IN) were used in all cases. Experiments were performed in hippocampal slices prepared from neonatal rats at postnatal day 3 (P3) to P26. Animals were anesthetized with 250 mg/kg ketamine, and 250- to 350-μm-thick slices were prepared with a vibratome, as described previously (Shuttleworth and Connor, 2001). Artificial CSF (ACSF) contained the following (in mm): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 2 CaCl2, 10 glucose, and 0.02 bicuculline methiodide (equilibrated with 95% O2 plus 5% CO2). When indicated, 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX) (Axxorra, San Diego, CA), d,l-APV, GYKI 53655 [1-(4-aminophenyl)-3-methylcarbamyl-4-methyl7,8-methylenedioxy-3,4-dihydro-5H-2,3-benzodiazepine] (custom synthesized by Tocris Cookson), tetrodotoxin (TTX) (Calbiochem, San Diego, CA), KCl, sucrose, ω-conotoxin-GVIA (Alomone Labs, Jerusalem, Israel), ω-agatoxin-IVA (Alomone Labs), or ethanol (AAPER Chemical, Shelbyville, KY) were added to the ACSF.
After a recovery period of ≥80 min, slices were transferred to a chamber perfused with ACSF at a rate of 2 ml/min. Whole-cell patch-clamp electrophysiological recordings from CA3 pyramidal neurons were performed under infrared-differential interference contrast microscopy at 32°C with an Axopatch 200B amplifier (Axon Instruments, Union City, CA).
A pneumatic picopump (World Precision Instruments, Sarasota, FL) was used to apply puffs of 5 μm AMPA in the presence of TTX (Vh of –65 mV) or 50 μm NMDA in the presence of TTX and 10 μm NBQX (Vh of –10 mV). The puffing pipette was placed ∼200 μm away from the patched neuron, and this produced a response that was ∼20–40% of the response obtained when the pipette was ∼50 μm away from the patched neuron. Thus, the effect of ethanol was tested on submaximally activated currents. The pressure was set at 7 psi, and the puff duration was 500 ms. Patch pipettes had resistances of 3–5 MΩ. Recording electrodes were filled with an internal solution containing the following (in mm): 110 Cs-gluconate, 5 NaCl, 10 tetraethylammonium-Cl, 4 Mg-ATP, 0.6 EGTA, 10 HEPES, and 4 QX-314 [2(triethylamino)-N-(2,6-dimethylphenyl) acetamine], pH 7.25. Access resistances were between 10 and 30 MΩ; if access resistance changed >20%, the recording was discarded.
AMPAR-mediated EPSCs were recorded at a holding potential of –65 mV in presence of 4 mm Mg2+ and 4 mm Ca2+ to avoid polysynaptic activity (Schmitz et al., 2001). NMDAR-mediated currents were recorded in regular ACSF at a holding potential of –10 mV to relieve the Mg2+ blockade from the receptor. Currents were evoked at a frequency of 0.05 Hz with a concentric bipolar electrode (Frederick Haer Company, Bowdoinham, ME) placed ∼200 μm away from the patched pyramidal cell. An input/output curve was measured at the start of the recording, and stimulation intensity was set at 20–30% of maximum for all of these experiments. AMPA and NMDA EPSCs in slices from juvenile rats were evoked via stimulation of the stratum lacunosum-moleculare. When indicated, NMDA EPSCs were evoked by stimulation of the mossy fibers. Paired-pulse experiments were performed at an interpulse interval of 50 ms. Recordings of AMPAR-mediated miniature EPSCs (mEPSCs) were performed at a holding potential of –65 mV in the presence of 0.5 μm TTX. When indicated, 30 mm KCl or 100 mm sucrose were added to the ACSF to increase basal mEPSC frequency. Each slice was exposed once to a single ethanol concentration, and the duration of ethanol exposure was limited to 5 min in all cases to avoid the development of rapid tolerance (Miyakawa et al., 1997). The effect of ethanol reached a stable level 3–4 min after the start of the application (see Figs. 1 A2, 2 B2). An average of the responses obtained during the stable phase of the ethanol effect was used for data analyses. Effects of ethanol were calculated with respect to the average of control and washout responses.
Figure 1.

Ethanol potently depresses postsynaptic AMPA-evoked currents in neurons from neonatal rats. A1, Effect of ethanol (EtOH) on inward currents evoked at a holding potential of –65 mV by local application of exogenous AMPA (5 μm; represented by the dots) in the presence of TTX (0.5μm) and bicuculline (20μm) in a P4 neuron. NBQX (10μm) eliminated the currents. A2, Time course of the effect of ethanol (same neuron as above). A3, Effect of increasing concentrations of ethanol on AMPA-evoked currents in slices from neonatal rats. B1, Representative traces from a P26 neuron illustrating the lack of an effect of ethanol on AMPA-evoked currents. NBQX (10μm) eliminated the currents. B2, Time course of the effect of ethanol (same neuron as above). B3, Effect of increasing concentrations of ethanol on AMPA-evoked currents in slices from juvenile rats. *p < 0.05, **p < 0.01, and ***p < 0.001 by one-sample t test versus a theoretical mean of 0 (for results of ANOVA, see Results). Event amplitude was normalized with respect to the average amplitude obtained during the first 3 min of recording.
Figure 2.

Ethanol decreases postsynaptic NMDA-evoked currents in neurons from juvenile rats. A1, Lack of an effect of 50 mm ethanol (EtOH) on inward currents evoked at a holding potential of –10 mV by local application of exogenous NMDA (50μm; represented by the dots) in the presence of TTX (0.5μm), bicuculline (20μm), and NBQX (10μm) in a P5 neuron. dl-APV (100 μm) eliminated the currents. A2, Time course of the effect of ethanol (same neuron as above). A3, Effect of increasing concentrations of ethanol on NMDA-evoked currents in slices from neonatal rats. B1, Sample traces from a P25 neuron illustrating that ethanol exposure decreases the amplitude of currents elicited by exogenous application of NMDA. dl-APV (100 μm) eliminated the currents. B2, Time course of the effect of ethanol (same neuron as above). B3, Effect of increasing concentrations of ethanol on NMDA-evoked currents in slices from juvenile rats. *p < 0.05 and **p < 0.01 by one-sample t test versus a theoretical mean of 0 (for results of ANOVA, see Results). Event amplitude was normalized with respect to the average amplitude obtained during the first 3 min of recording.
Data were acquired and analyzed with pClamp 9 (Axon Instruments); mEPSCs were analyzed with MINI ANALYSIS program (Synaptosoft, Decatur, GA), and a Kolmogorov–Smirnov test was used initially to test for significant differences between treatments in individual cells. The level of significance was set at p < 0.01. Decay time and relative τ values of responses evoked by exogenous application of AMPA and NMDA were determined with pClamp 9. Analyses of evoked EPSCs were performed after subtraction of the stimulation artifact. Statistical analysis of pooled data were performed by one-way ANOVA, followed by Bonferroni's post hoc test, t test, or one-sample t test versus a theoretical mean of 0 or 100. Data are presented as mean ± SEM, and the level of significance was set at p < 0.05. Numbers in parentheses define the number of neurons.
Western immunoblotting. Slices were prepared from animals belonging to the same litter at the indicated ages and allowed to recover as described above. This procedure was repeated with three different litters. The CA3 region was microdissected from the slices, homogenized in PBS containing a protease inhibitor cocktail (catalog #P-8340; Sigma), and stored at –80°C. Total protein concentrations were determined by the Lowry assay. Samples (3–10 μg of total protein per lane) were separated on 7.5% polyacrylamide minigels and electrotransferred to nitrocellulose membranes. Nonspecific binding of antibodies to nitrocellulose membranes was prevented by blocking with a solution containing 10% nonfat dry milk and 0.4% Tween 20. Blots were analyzed using a chemiluminescence assay kit following the instructions of the manufacturer (Roche, Indianapolis, IN). Membranes were probed with rabbit anti-NR2A (1: 500) and anti-NR2B (1:500) (generously provided by Dr. Michael Browning, Department of Pharmacology, University of Colorado Health Sciences Center, Denver, CO). Membranes were also probed with rabbit anti-NR1 (1:100) from Chemicon (Temecula, CA). Densitometric analyses of Western blot chemiluminescence x-ray films were performed using Quantity-One software (Bio-Rad, Hercules, CA). To control for differences in protein loading, each membrane was stained with Coomassie blue; the optical density of the glutamate receptor (GluR) subunits was normalized to the average optical density of two randomly chosen Coomassie bands (approximate molecular weight of 40 and 85 kDa) within each lane.
Results
Effect of ethanol on currents evoked by exogenous AMPA or NMDA
We investigated the effect of acute ethanol exposure on AMPAR function in neonatal (P3–P6) and juvenile (P21–P26) CA3 pyramidal neurons in hippocampal slices (Fig. 1). Pressure application of AMPA (5 μm) onto a P4 neuron, in the presence of TTX (500 nm) and bicuculline (20 μm), caused an inward current that was abolished by the non-NMDA antagonist NBQX (n = 12) (Fig. 1A1). Bath application of 50 mm ethanol caused a reversible reduction in the amplitude of the AMPA-evoked current (Fig. 1A1,A2). Figure 1A3 shows the effect of increasing concentrations of ethanol on currents evoked by exogenous AMPA in neonatal neurons. We detected a significant even effect at a concentration as low as 10 mm. One-way ANOVA followed by Bonferroni's post hoc test did not reveal statistically significant differences among concentrations of ethanol. Application of 50 mm ethanol onto a neuron from a P26 rat (Fig. 1B1,B2) did not affect the amplitude of AMPA-evoked currents. As shown in Figure 1B3, the AMPAR inhibitory effect of ethanol was not observed in neurons from juvenile animals at concentrations ≤50 mm; however, a significant inhibitory effect was detected with 75 mm (p < 0.05 by one-way ANOVA followed by Bonferroni's post hoc test vs 10 mm ethanol). Ethanol did not affect the decay of AMPA responses in either neonatal (control, τ = 2.9 ± 0.8 s; ethanol, τ = 3 ± 0.8 s; n = 9) or juvenile (control, τ = 2.32 ± 0.5 s; ethanol, τ = 2.35 ± 0.5 s; n = 9) rats.
We next tested the effect of ethanol on currents evoked by exogenous application of NMDA (50 μm) (Fig. 2). The experiments were performed in the presence of TTX, bicuculline, and NBQX (10 μm). Unexpectedly, ethanol, at concentrations ≤50 mm, did not affect postsynaptic NMDA currents in neurons from neonates (Fig. 2A); a small inhibitory effect of ethanol was detected with 75 mm (p < 0.05 by one-way ANOVA followed by Bonferroni's post hoc test vs 10 mm ethanol) (Fig. 2A3). The decay time of the NMDA responses also remained unchanged after 50 mm ethanol exposure (control, τ = 1.9 ± 0.3 s; ethanol, τ = 1.88 ± 0.3 s; n = 5). However, in agreement with a previous report (Weiner et al., 1999), ethanol dose dependently depressed the same currents in neurons from juvenile animals (p < 0.05 by one-way ANOVA followed by Bonferroni's post hoc test vs 10 mm ethanol) (Fig. 2B) without affecting their decay (control, τ = 2.3 ± 0.4 s; 50 mm ethanol, τ = 2.4 ± 0.5 s; n = 5). As shown in Figure 2, A and B, the selective NMDA antagonist d,l-APV (100 μm) abolished the evoked currents in neurons from neonatal and juvenile rats (n = 6), confirming the selective activation of NMDARs under our recording conditions.
We performed a control experiment to eliminate the possibility that the lack of ethanol coapplication during the pressure application of NMDA is responsible for the low ethanol sensitivity of NMDARs in neonatal neurons. We evoked NMDAR currents by bath applying homoquinolinic acid in the presence of bicuculline and NBQX and measured the effect of 50 mm ethanol on these currents (Fig. 3). Ethanol was both preapplied (∼4 min) and coapplied with homoquinolinic acid. In agreement with the results described above, we found that ethanol inhibited homoquinolinic acid-evoked currents in CA3 pyramidal neurons from juvenile but not neonatal rats (Fig. 3).
Figure 3.

Effect of ethanol (EtOH) on NMDAR currents evoked by bath application of homoquinolinic acid (HQA) (3–6 μm). Recordings were performed at a holding potential of –20 to –40 mV in Mg2+-containing ACSF in the presence of 0.5μm tetrodotoxin, 20μm bicuculline, and 10 μm NBQX. Ethanol was preapplied for 4 min and then coapplied with homoquinolinic acid. A, Ethanol does not reduce homoquinolinic acid-evoked currents in a CA3 pyramidal neuron from a P4 rat. B, Ethanol reduces homoquinolinic acid-evoked currents in a CA3 pyramidal neuron from a P27 rat. C, Summary of the effect of ethanol on the area of homoquinolinic acid-evoked currents. *p < 0.05 by unpaired t test.
As shown in Figure 4A, the effect of ethanol on AMPA-evoked currents gradually decreased with age, whereas the effect on NMDA-evoked currents gradually increased (interaction, p < 0.0001 by two-way ANOVA). AMPA and NMDA receptors displayed relatively similar sensitivities to 50 mm ethanol between P9 and P15. We next investigated whether changes in subunit composition could play a role in the age-dependent change in sensitivity to ethanol of NMDARs. In agreement with the results of a myriad of studies (for review, see van Zundert et al., 2004), we found that the subunit composition of NMDARs dramatically changes during development. Figure 4B shows that expression of the NR2B subunit predominates during P4–P10, when NMDARs are insensitive to ethanol, and that expression of the NR2A subunit gradually increases in parallel with the increase in ethanol sensitivity of these receptors. Finally, we assessed expression of NR2D subunits. We were unable to procure suitable antibodies from either commercial or academic sources for measuring expression of this subunit via Western immunoblotting. Therefore, we evaluated expression of this subunit by means of a functional assay. We measured the effect of (2S*, 3R*)-1-(phenanthrene-2-carbonyl)piperazine-2,3-dicarboxylic acid (PPDA), which has been shown to preferentially antagonize NMDARs containing NR2D subunits in oocyte and slice experiments (Feng et al., 2004; Lozovaya et al., 2004; Mameli et al., 2005). We used a low concentration of PPDA (50 nm) to minimize the possibility of effects at receptors containing NR2A or NR2B subunits (Feng et al., 2004). This submaximal concentration of PPDA significantly inhibited NMDA-evoked currents in CA3 pyramidal neurons from ≤P10 rats but not ≥P15 rats (Fig. 4C). These findings are in agreement with previous reports indicating that the NR2D subunit is transiently expressed in the hippocampus during the first week of postnatal life in rodents (Wenzel et al., 1996, 1997; Okabe et al., 1998). It is noteworthy that we did not assess NR2C subunit expression because this subunit has not been shown to be expressed in the hippocampus (Wenzel et al., 1995). Moreover, developmentally regulated changes in subunit composition could also modulate sensitivity of AMPARs to ethanol. In particular, GluR4 subunit expression predominates at early postnatal ages (Pickard et al., 2000; Zhu et al., 2000; Molnar et al., 2002), and we are planning on examining its expression in the CA3 region when we are able to procure a suitable anti-GluR4 antibody; we attempted to use commercially available anti-GluR4 antibodies (Upstate Biotechnology, Charlottesville, VA) in Western immunoblotting studies and obtained unsatisfactory results.
Figure 4.
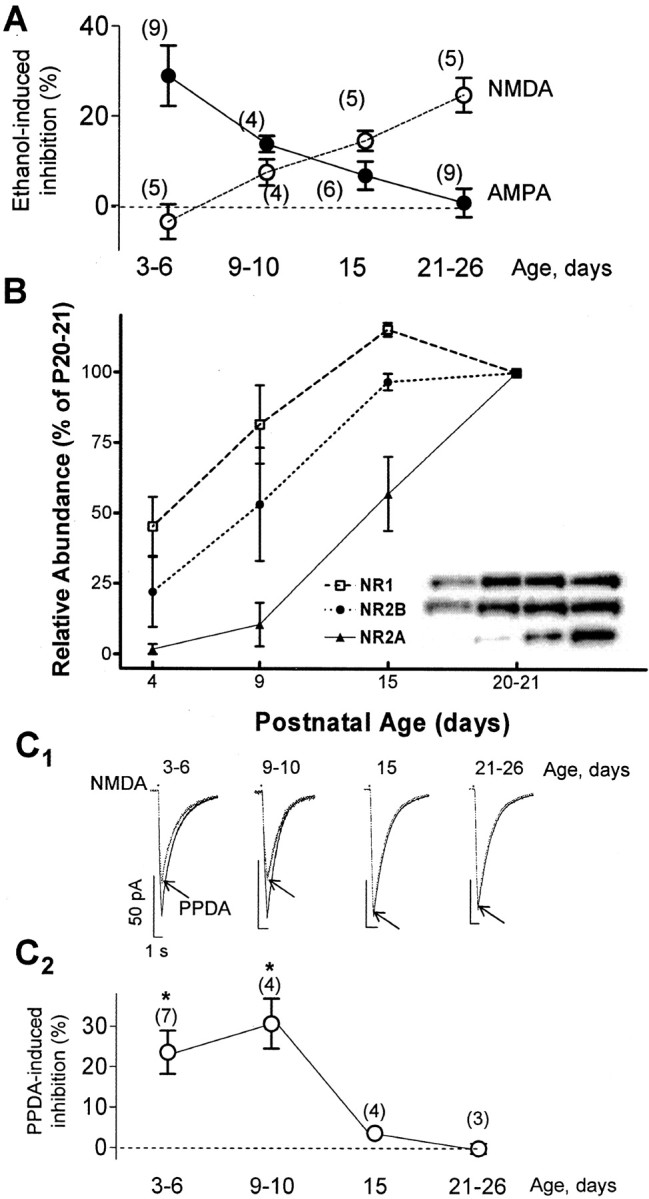
Age dependency of the effect of ethanol and changes in NMDAR subunit composition. A, The effect of 50 mm ethanol on AMPAR-mediated currents gradually decreases as age increases, and the opposite is true for NMDAR-mediated currents. Currents were evoked by pressure agonist application (see Figs. 1, 2). For results of two-way ANOVA, see Results. B, Expression profiles for NR1, NR2A, and NR2B during postnatal development plotted as the percentage of maximum (n = 3 litters). Expression was assessed by Western immunoblots using CA3 region homogenates. Inset, Sample immunoblots for the indicated subunits; each lane corresponds to the postnatal age indicated in the abscissa. C1, Sample traces illustrating the effect of 50 nm PPDA (antagonist of NR2D-containing receptors) on currents evoked at a holding potential of –10 mV by local application of exogenous NMDA (50μm; represented by the dots) in the presence of TTX (0.5 μm), bicuculline (20 μm), and NBQX (10 μm) in P3–P26 neurons. C2, Summary graph illustrating the age dependency of the effect of PPDA. *p < 0.05 by one-way ANOVA followed by Bonferroni's post hoc test (vs effect at P21–P26). In some cases, error bars are smaller than the symbols.
Effect of ethanol on EPSCs mediated by AMPA or NMDA receptors
We determined the effect of ethanol on AMPA and NMDA currents activated by synaptic glutamate release. As described previously (Schmitz et al., 2001), we included 4 mm Mg2+ and 4 mm Ca2+ in the ACSF to prevent polysynaptic activity. Events were elicited with a relatively large stimulating electrode (outer pole, 125 μm; inner pole, 5 μm). Therefore, in neonatal slices, events were likely elicited by stimulation of the mossy fibers and associational–commissural fibers (Buchhalter et al., 1990), as well as the perforant path. In juvenile slices, events were elicited by stimulation of the perforant path for the majority of the experiments; we chose to stimulate this pathway because it preferentially targets synapses abundantly expressing NMDARs (Siegel et al., 1994; Weisskopf and Nicoll, 1995; Berzhanskaya et al., 1998; Watanabe et al., 1998). As shown in Figure 5, 50 mm ethanol induced a significant decrease in the amplitude of AMPA-mediated EPSCs (Vh of –65 mV) in neurons from neonatal but not from juvenile rats. The reduction on AMPA-mediated EPSCs by 50 mm ethanol in neonatal neurons was also detectable at Vh of –10 mV (in the presence of ethanol, amplitude was 68.5 ± 18.6% of control; n = 5; data not shown) (compare with Fig. 5C). NMDA-mediated EPSCs (Vh of –10 mV) (Fig. 6) were inhibited to a similar extent by 50 mm ethanol in both neonatal and juvenile rats. In slices from juvenile rats, it was demonstrated previously that AMPA EPSCs evoked by stimulation of mossy fibers are insensitive to ethanol concentrations as high as 80 mm (Weiner et al., 1999). For comparison, we measured the effect of ethanol on NMDA EPSCs evoked by stimulation of these fibers. In agreement with previous reports, NMDA EPSCs could be evoked in CA3 neurons by stimulation of the mossy fiber pathway in slices from juvenile animals (Jonas et al., 1993; Weisskopf and Nicoll, 1995; Watanabe et al., 1998), and the amplitude of these events was 72 ± 9% of control in the presence of 50 mm ethanol (n = 4; data not shown) (compare with Fig. 6C).
Figure 5.

Ethanol inhibits synaptic AMPAR-mediated responses in an age-dependent manner. A, Representative traces and time course graph illustrating the effect of 50 mm ethanol (EtOH) on AMPAR-mediated EPSCs in a CA3 pyramidal neuron from a P4 rat. AMPA EPSCs were recorded at a holding potential of –65 mV in presence of 20μm bicuculline. B, Same as above at P23. C, Bar graph illustrating the developmental change in sensitivity of AMPA EPSCs to ethanol (**p < 0.01 by unpaired t test). One-sample t test analysis versus a theoretical mean of 100 revealed significance only for neonatal neurons (p < 0.05). Event amplitude was normalized with respect to the average amplitude obtained during the first 3 min of recording.
Figure 6.

Ethanol inhibits synaptic NMDAR-mediated responses in both neonatal and juvenile rats. A, Representative traces and time course graph illustrating the effect of 50 mm ethanol (EtOH) on NMDAR-mediated EPSCs in a CA3 pyramidal neuron from a P4 rat. Recordings were obtained at a holding potential of –10 mV in the presence of 20 μm bicuculline and 10 μm NBQX. B, Same as above at P23. C, Bar graph illustrating the lack of change in sensitivity of NMDA EPSCs to ethanol during development (n.s., not significant by unpaired t test). One-sample t test versus a theoretical mean of 100 revealed significance for both the neonate and juvenile groups (p < 0.0001). Event amplitude was normalized with respect to the average amplitude obtained during the first 3 min of recording.
To further characterize the effects of ethanol on glutamatergic transmission in the CA3 region of the hippocampus, we attempted to record kainate receptor-mediated synaptic responses. We stimulated with a train of five stimuli at 50 Hz every 20 s in the presence of bicuculline, d,l-APV, and GYKI-53655 (30 μm). However, in agreement with a recent report (Marchal and Mulle, 2004), we were unable to evoke kainate receptor-mediated responses in slices from neonatal rats (n = 3; data not shown).
Ethanol depresses excitatory transmission in neonatal slices by reducing the probability of glutamate release
The effect of ethanol on paired-pulse plasticity of AMPA-mediated currents was next investigated to assess whether it acts at a presynaptic site. Paired-pulse plasticity was induced by delivering two pulses with an interpulse interval of 50 ms every 20 s. The paired-pulse ratio was higher in slices from juvenile rats than neonatal rats (Fig. 7A,B). This finding indicates that the probability of glutamate release at CA3 pyramidal neurons is lower in juvenile rats, which is in general agreement with some studies of glutamatergic synaptic transmission at developing CA3–CA1 synapses (Bolshakov and Siegelbaum, 1995; Wasling et al., 2004) (but see Hsia et al., 1998). Figure 7A1 shows the effect of 50 mm ethanol on paired-pulse plasticity in a P5 neuron. The pooled data in Figure 7A2 indicate that, in all neurons tested (n = 7), ethanol significantly increased the ratio between EPSC2 and EPSC1. In addition, ethanol increased the paired-pulse ratio of NMDA-mediated currents in neonatal rats (control, 0.97 ± 0.05; ethanol, 1.3 ± 0.08; n = 5; data not shown). Conversely, in neurons from juvenile rats (Fig. 7B), ethanol application did not change the paired-pulse ratio of AMPA-mediated currents. The age dependency of the effect of ethanol on paired-pulse plasticity is further illustrated in Figure 7C; this effect disappears between P10 and P15.
Figure 7.

Acute ethanol (EtOH) exposure increases the paired-pulse ratio (PPR) of AMPA-mediated EPSCs in neurons from neonate but not juvenile rats. A1, Representative traces illustrating the effect of 50 mm ethanol on the paired-pulse ratio of AMPA EPSCs (50 ms interpulse interval) in neurons from a P5 rat. Holding potential was –65 mV, and bicuculline (20 μm), 4 mm Mg2+, and 4 mm Ca2+ were present in the ACSF. These events were blocked by NBQX (10 μm; data not shown). A2, Pooled data of paired-pulse ratio changes in neonatal rats after ethanol application, expressed as the second EPSC over the first (**p < 0.01 by paired t test). B1, B2, Same as above for juvenile rats (n.s., not significant by paired t test). C, Summary graph illustrating the effect of 50 mm ethanol on the paired-pulse ratio as a function of age. **p < 0.01 by one-way ANOVA followed by Bonferroni's post hoc test (vs P21–P26). ctrl, Control.
To further assess the effect of ethanol on glutamate release, we recorded AMPA receptor-mediated mEPSCs in neurons from neonatal rats. Although the frequency of these events was very low under basal conditions (0.01 ± 0.003 Hz; n = 11) (Fig. 8A, first trace), we were able to record a small number of mEPSCs and did not detect a significant effect of ethanol on mEPSC frequency (120.8 ± 19% of control; n = 4; data not shown). However, mEPSC amplitude was significantly decreased (64.5 ± 2% of control; n = 4; p < 0.001; data not shown). Analysis of the cumulative probability distribution of individual cells by means of the Kolmogorov–Smirnov test revealed a statistically significant (p < 0.01) effect of ethanol in one of four neurons on mEPSC frequency and three of four neurons on mEPSC amplitude. To further characterize the effect of ethanol, we elected to increase mEPSC frequency by depolarizing the axon terminal with bath application of 30 mm KCl (Lei and McBain, 2003). KCl, which acts by increasing Ca2+ influx via voltage-gated Ca2+ channel (VGCC) activation, increased mEPSC frequency to 5.1 ± 1.6 Hz (n = 5) (Fig. 8A, second trace). Acute application of 50 mm ethanol in the presence of TTX, bicuculline, and KCl, reversibly decreased both mEPSC frequency (Fig. 8A,B,E) and amplitude (Fig. 8A,C,E); however, ethanol had no effect on the decay time course of these events (half-width: control, 4.56 ± 0.48 ms; ethanol, 4.61 ± 0.51 ms; n = 5) (Fig. 8D). Analysis of the cumulative probability distribution of individual cells by means of the Kolmogorov–Smirnov test revealed a statistically significant (p < 0.01) effect of ethanol in four of five neurons on both mEPSC frequency and amplitude.
Figure 8.

Ethanol depresses glutamatergic transmission onto CA3 pyramidal neurons of neonatal hippocampus. A, Sample traces of AMPAR-mediated mEPSCs illustrating that 50 mm ethanol (EtOH) induces a transient decrease in mEPSC frequency and amplitude. Basal mEPSC frequency was initially increased by application of 30 mm KCl. B, C, Cumulative probability frequency and amplitude plots, respectively, corresponding to the recording shown above. D, Superimposed average mEPSC traces obtained from the recording shown in A; the bottom traces were scaled to illustrate that ethanol did not affect mEPSC decay. E, Summary graph showing the effect of ethanol on mEPSC frequency and amplitude (***p < 0.001 by one-sample t test vs a theoretical mean of 100). For results of Kolmogorov–Smirnov test, see Results. ctrl, Control.
We next tested the effect of ethanol on mEPSC frequency under conditions of increased glutamate release via a Ca2+-independent mechanism. To this end, we used 100 mm sucrose, which acts independently of Ca2+ by an osmotic effect (Stevens and Sullivan, 1998). Application of sucrose increased mEPSC frequency from 0.01 ± 0.003 Hz (n = 11) to 0.18 ± 0.01 Hz (n = 6) (Fig. 9A). Application of ethanol, in the presence of sucrose, did not affect mEPSC frequency (Fig. 9A,B,E); however, ethanol did decrease the amplitude of the events (Fig. 9A,C–E). Analysis of the cumulative probability distribution of individual cells by means of the Kolmogorov–Smirnov test revealed a statistically significant (p < 0.01) effect of ethanol in zero of six cells on mEPSC frequency and in four of six cells on mEPSC amplitude. To confirm that a decrease in mEPSC frequency could be detected in the presence of sucrose, we measured the effect of 50 μm adenosine on two of the neurons used in the ethanol experiments. Adenosine decreased mEPSC frequency by 44 and 70% in these cells, whereas it minimally affected mEPSC amplitude (i.e., it was reduced by 6 and 4%, respectively).
Figure 9.

Ethanol does not affect mEPSC frequency when it is increased via a Ca2+-independent mechanism. A, Sample traces of AMPA-mediated mEPSCs illustrating that 50 mm ethanol (EtOH) induces a transient decrease in mEPSC amplitude, but not in frequency, when basal mEPSCs frequency is initially increased by application of 100 mm sucrose. B,C, Cumulative probability frequency and amplitude plots, respectively, corresponding to the recording shown above. D, Superimposed average mEPSC traces obtained from the recording shown in A; the bottom traces were scaled to illustrate that ethanol did not affect mEPSC decay. E, Summary graph showing the lack of an effect of ethanol on mEPSC frequency and the ethanol-induced reduction in mEPSC amplitude (***p < 0.001 by one-sample t test vs a theoretical mean of 100). For results of Kolmogorov–Smirnov test, see Results. ctrl, Control.
Role of VGCCs on the mechanism of action of ethanol
Ethanol inhibition of VGCCs is well documented (for review, see Walter and Messing, 1999). Therefore, we assessed the involvement of presynaptic VGCCs on the inhibitory effect of ethanol on glutamate release. We first characterized the VGCC subtypes that mediate glutamate release early in development in the CA3 hippocampal region (Fig. 10A,B). The effect of selective VGCC blockers were tested on pharmacologically isolated NMDA EPSCs at Vh of –10 mV. We elected to study these events because NMDARs are postsynaptically insensitive to ethanol in neonatal slices (Figs. 2, 3, 4). We found that application of the N-type Ca2+ channel antagonist ω-conotoxin-GVIA (1 μm) reduced the NMDA EPSC in neonatal slices (Fig. 10A). ω-Agatoxin-IVA (100 nm), a P/Q-type Ca2+-channel blocker, was then bath applied, and it abolished the remainder of the EPSC. In slices from juvenile rats, application of ω-conotoxin-GVIA reduced the NMDA EPSC less than in neonatal slices (Fig. 10B). ω-Agatoxin-IVA was then applied, and it abolished the remainder of the EPSC.
Figure 10.

Glutamate release is regulated by the activity of N- and P/Q-type presynaptic VGCCs, and ethanol (EtOH) decreases transmitter release through inhibition of the N-type. A, Time course of the effect of N- and P/Q-type antagonists (ω-conotoxin-GVIA and ω-agatoxin-IVA, respectively) on synaptically evoked NMDA currents (Vh of –10 mV) in neurons from neonatal rats (n = 10) recorded in the presence of bicuculline (20 μm) and NBQX (10 μm). Right, Sample traces obtained from a P4 neuron. B, Same as A but for neurons from juvenile rats (n = 6). Right, Sample traces obtained from a P23 neuron. C, Time course of the effect of 50 mm ethanol on the P/Q-type-dependent residual NMDA EPSCs after blockade of the N-type-dependent component in neurons from neonatal rats (n = 6). Note that ethanol does not induce additional changes in the amplitude and that the current was abolished by the P/Q-type channel antagonistω-agatoxin-IVA. Right, Sample traces illustrating the effect of ethanol and the toxins on a P3 neuron. D, Time course of the effect of ethanol on the N-type-dependent NMDA residual EPSCs after blockade of the P/Q-type-dependent component in neurons from neonatal rats (n = 4). Note that ethanol reversibly reduces the amplitude of the residual EPSC and that, after ethanol washout, the current was abolished by the N-type channel antagonist ω-conotoxin-GVIA. Right, Sample traces illustrating the effect of ethanol and the toxins on a P4 neuron. In all cases, the bin size for the data shown on the time course graphs is 1 min (stimulation was delivered every 20 s). EPSC amplitude was normalized with respect to the average amplitude obtained during the first 3 min of recording. *p < 0.05 by one-way ANOVA followed by Bonferroni's post hoc test versus responses obtained at 12 min. ω-cTx-GVIA, ω-Conotoxin-GVIA; ω-AgaTx-IVA, ω-agatoxin-IVA.
We then tested the effect of ethanol under conditions of N-type channel blockade in neonatal slices. Application of ω-conotoxin-GVIA occluded the inhibitory effect of 50 mm ethanol on NMDA EPSCs (Fig. 10C), and there was no difference between the reduction induced by ω-conotoxin-GVIA alone or in the presence of ethanol (Fig. 10A,C). The remainder of the EPSC was blocked by application of ω-agatoxin-IVA. As shown in Figure 10D, 50 mm ethanol reversibly inhibited NMDA EPSCs in neurons pretreated with ω-agatoxin-IVA, and the reminder of the EPSC was abolished by application of ω-conotoxin-GVIA.
Discussion
We have identified two novel effects of ethanol in CA3 pyramidal neurons from neonatal rats. First, it inhibits postsynaptic AMPARs but not NMDARs at subanesthetic concentrations. Second, it reduces glutamate release via inhibition of N-type presynaptic VGCCs; this finding adds to growing evidence indicating that ethanol inhibits glutamate release in the CA1 hippocampal region (Hendricson et al., 2004; Maldve et al., 2004), central amygdala (Roberto et al., 2004), spinal cord (Ziskind-Conhaim et al., 2003), and crayfish neuromuscular junction (Strawn and Cooper, 2002).
Postsynaptic AMPA receptors are inhibited by ethanol in neonatal CA3 pyramidal neurons
In a previous study with slices from P20–P40 rats, it was demonstrated that AMPAR-mediated currents in CA3 pyramidal neurons, evoked by brief application of kainate, are insensitive to concentrations of ethanol as high as 80 mm (Weiner et al., 1999). Here, we replicated this finding with the exception that brief application of AMPA rather than kainate was used to activate the receptors. Unexpectedly, we found that AMPA-evoked currents were potently inhibited by ethanol in CA3 pyramidal neurons from P3–P6 rats. The lowest concentration we tested was 10 mm (0.045 g/dl; legal intoxication limit in most states is 0.08 g/dl), which significantly decreased AMPAR-mediated currents. Consequently, AMPARs in CA3 pyramidal neurons are one of the most ethanol-sensitive ionotropic glutamatergic receptor subtypes in the neonatal rat hippocampus. These findings are in general agreement with those of Moykkynen et al. (2003), who showed that ethanol preferentially inhibits steady-state currents activated by AMPA and currents activated by low concentrations of kainate in acutely isolated hippocampal neurons from P10–P20 mice. However, we found that ethanol did not inhibit AMPAR-mediated currents in neurons from more mature rats. This may be attributable to developmental differences in subunit composition (Pickard et al., 2000, 2001) because it is well established that it changes as a function of the developmental stage (Burnashev and Rozov, 2000; Molnar et al., 2002). Whereas GluR4 subunit expression predominates at early postnatal ages (Pickard et al., 2000; Zhu et al., 2000; Molnar et al., 2002), studies with recombinant receptors showed that GluR4-containing AMPAR are insensitive to subanesthetic concentrations of ethanol (Lovinger, 1993). Thus, the preferential expression of this subunit could not fully explain the high sensitivity of neonatal AMPARs to ethanol. Differences in the phosphorylation state of neonatal AMPARs or their association with other proteins could further contribute to their higher sensitivity to ethanol.
In agreement with a previous report (Weiner et al., 1999), we found that NMDARs are acutely inhibited by 10–50 mm ethanol in CA3 pyramidal neurons from juvenile rats but, surprisingly, are insensitive to these concentrations of ethanol in neonatal neurons. The subunit composition of NMDARs changes significantly during hippocampal development, and this could be responsible for the age-dependent changes in the sensitivity of these receptors to ethanol. The results of our immunoblotting and pharmacological studies suggest that the presence of both NR2A and NR2B subunits confers sensitivity to ethanol in CA3 pyramidal neurons from juvenile animals, whereas expression of receptors containing NR2B and NR2D subunits confers insensitivity in neurons from neonatal rats. Interestingly, it was discovered recently that acute ethanol exposure can inhibit NMDAR function by inducing internalization of NR2A (Suvarna et al., 2005). Moreover, studies with recombinant receptors expressed in Xenopus oocytes showed that NR1b/NR2D heteromeric channels are significantly less sensitive to ethanol inhibition than NR1b/NR2A, NR1b/NR2B, or NR1b/NR2C heteromers (Chu et al., 1995).
It should be noted that, given that intracellular signaling pathways are likely to be affected during whole-cell patch-clamp recordings and these modulate sensitivity of NMDARs to ethanol (Ron, 2004), it is possible that the sensitivity of these receptors is altered under whole-cell patch clamping. This uncertainty must be kept in mind when interpreting our results.
Ethanol affects glutamatergic synaptic currents in neonatal neurons via both presynaptic and postsynaptic mechanisms
We show here that ethanol acutely reduces the amplitude of both AMPAR- and NMDAR-mediated EPSCs in neonatal CA3 pyramidal neurons but not in neurons from more mature rats. We also found that ethanol increases the paired-pulse ratio for both AMPAR- and NMDAR-mediated currents, suggesting that it decreases glutamate release probability (Zucker and Regehr, 2002). This effect was confirmed in our mEPSC recordings in which we detected a reversible decrease in event frequency. Importantly, the decrease in frequency could only be detected under conditions of KCl-induced axonal depolarization and not in the presence of sucrose, which enhances transmitter release via a Ca2+-independent mechanism (Stevens and Tsujimoto, 1995; Stevens and Sullivan, 1998). Together, these findings indicated that ethanol acts via an interaction with presynaptic VGCCs.
To the best of our knowledge, this is the first demonstration of a presynaptic effect of ethanol on glutamate release in the CA3 region. However, there is evidence indicating that ethanol also affects glutamate release in other hippocampal regions. Maldve et al. (2004) found that ethanol inhibits KCl-induced vesicular FM1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino) styryl) pyridinium dibromide] destaining in the CA1 stratum radiatum of P21–P28 rats, and this effect was occluded by blockers of N-type and P/Q-type VGCCs. They also found that ethanol inhibits AMPA mEPSCs in the presence, but not in the absence, of KCl, in general agreement with the results of our neonatal slice experiments. Importantly, the same group of investigators found that ethanol decreases the frequency of asynchronous NMDA mEPSCs and increases the paired-pulse ratio of NMDA EPSCs in CA1 pyramidal neurons (Hendricson et al., 2004). Collectively, the results of these studies support the notion that ethanol inhibits glutamate release from Schaffer collaterals/commissural axons to CA1 pyramidal neurons. However, their findings are inconsistent with those of other studies indicating that ethanol does not affect AMPAR-mediated EPSCs evoked by Schaffer collateral/commissural pathway stimulation in CA1 pyramidal neurons and interneurons in slices from rats older than P12 (Lovinger et al., 1990; Morrisett et al., 1991; Morrisett and Swartzwelder, 1993; Nelson et al., 1999; Carta et al., 2003). Future work will be required to establish the reasons for the discrepancies between these studies.
Given that ethanol inhibits AMPA receptor function via both presynaptic and postsynaptic mechanisms in neonatal neurons, it is surprising that it did not exert a more robust effect on AMPA EPSCs than NMDA EPSCs (compare Figs. 5C, 6C). A possible explanation is that ethanol postsynaptically affects a select population of AMPARs that is stimulated only in the experiments with exogenous agonist and the mEPSC studies.
Ethanol reduces glutamate release by depressing N-type Ca2+ channels
Our data suggest that the reason for the preferential ethanol sensitivity of glutamate release in neonatal slices is that N-type VGCC contribution to glutamate release is larger at early developmental stages (Wheeler et al., 1994; Scholz and Miller, 1995; Reid et al., 2003). However, N-type VGCCs also had a detectable contribution in slices from more mature rats (Kamiya et al., 1988; Castillo et al., 1994; Tokunaga et al., 2004) so it is surprising that ethanol did not produce some inhibition of glutamate release in these animals. Thus, there may be other factors, such as differences in the phosphorylation state of N-type VGCCs between neonatal and juvenile rats that modulate sensitivity to ethanol. Indeed, activation of PKA was shown to antagonize ethanol modulation of N- and P/Q-type VGCCs in PC12 cells (Solem et al., 1997). N-type channel subunit composition modifications during development could be an additional explanation for the different sensitivity to ethanol throughout maturation (Jones et al., 1997; Vance et al., 1998; Lin et al., 1999; Pan and Lipscombe, 2000)
Our findings are consistent with several reports indicating that these channels are important targets of ethanol. Through their modulation, ethanol inhibits dopamine release from rat striatal synaptosomes (Woodward et al., 1990), vasopressin release from rat neurohypophysial nerve terminals (Wang et al., 1991), and depolarization-induced rises in [Ca2+]i in PC12 cells (Solem et al., 1997). Importantly, mice lacking N-type channels display reduced voluntary ethanol consumption, decreased sensitivity to its hypnotic effects, and a mild increase in ethanol-induced ataxia (Newton et al., 2004).
Implications for FASD
It has been shown that exposure to ethanol during the third-trimester equivalent in the rat produces profound functional and structural alterations in the CA3 region (Livy et al., 2003; Galindo et al., 2005), and it has been proposed that this may contribute to the behavioral deficits that are associated with FASD. Blockade of NMDARs during development was shown to produce apoptotic neurodegeneration in several regions of the CNS, including the hippocampus (Ikonomidou et al., 1999, 2001). It has been therefore assumed that the neuroteratogentic effects of alcohol are mediated, in part, via this NMDAR-dependent mechanism. However, our data demonstrate that, in the CA3 region of the hippocampus of neonatal rats, postsynaptic NMDARs are insensitive to subanesthetic concentrations of ethanol. Our findings suggest that ethanol may actually damage CA3 pyramidal neurons via inhibition of postsynaptic AMPARs and a decrease in glutamate release, and that this is secondary to inhibition of presynaptic N-type VGCCs. Thus, we have identified a novel mechanism that may be involved in the pathophysiology of FASD.
Footnotes
This work was supported by National Institutes of Health Grant AA15614. Additional support was provided by United States Army Grant DAMD17-00-1-0579. We thank Drs. L. D. Partridge, A. S. Maita, and R. Glew for their assistance and comments, and Dr. D. T. Monaghan (Department of Pharmacology, University of Nebraska, Lincoln, NE) for generously providing PPDA. We are also grateful to Dr. D. Benke (Institute of Pharmacology and Toxicology, University of Zurich, Zurich, Switzerland) for kindly providing anti-NR2D antibodies and Dr. M. D. Browning (Department of Pharmacology, University of Colorado Health Sciences Center, Denver, CO) for providing anti-NR2A and NR2B antibodies.
Correspondence should be addressed to Dr. C. Fernando Valenzuela, Department of Neurosciences, MSC08 4740, 1 University of New Mexico, Albuquerque, NM 87131-0001. E-mail: fvalenzuela@salud.unm.edu.
M. Carta's present address: Department of Experimental Biology, Section of Neuroscience, University of Cagliari, 09123 Cagliari, Italy.
DOI:10.1523/JNEUROSCI.2434-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258027-10$15.00/0
References
- Berzhanskaya J, Urban NN, Barrionuevo G (1998) Electrophysiological and pharmacological characterization of the direct perforant path input to hippocampal area CA3. J Neurophysiol 79: 2111–2118. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA (1995) Regulation of hippocampal transmitter release during development and long-term potentiation. Science 269: 1730–1734. [DOI] [PubMed] [Google Scholar]
- Buchhalter JR, Fieles A, Dichter MA (1990) Hippocampal commissural connections in the neonatal rat. Brain Res Dev Brain Res 56: 211–216. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Rozov A (2000) Genomic control of receptor function. Cell Mol Life Sci 57: 1499–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Ariwodola OJ, Weiner JL, Valenzuela CF (2003) Alcohol potently inhibits the kainate receptor-dependent excitatory drive of hippocampal interneurons. Proc Natl Acad Sci USA 100: 6813–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Weisskopf MG, Nicoll RA (1994) The role of Ca2+ channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron 12: 261–269. [DOI] [PubMed] [Google Scholar]
- Chu B, Anantharam V, Treistman SN (1995) Ethanol inhibition of recombinant heteromeric NMDA channels in the presence and absence of modulators. J Neurochem 65: 140–148. [DOI] [PubMed] [Google Scholar]
- Costa ET, Soto EE, Cardoso RA, Olivera DS, Valenzuela CF (2000a) Acute effects of ethanol on kainate receptors in cultured hippocampal neurons. Alcohol Clin Exp Res 24: 220–225. [PubMed] [Google Scholar]
- Costa ET, Savage DD, Valenzuela CF (2000b) A review of the effects of prenatal or early postnatal ethanol exposure on brain ligand-gated ion channels. Alcohol Clin Exp Res 24: 706–715. [PubMed] [Google Scholar]
- Crowder TL, Ariwodola OJ, Weiner JL (2002) Ethanol antagonizes kainate receptor-mediated inhibition of evoked GABA(A) inhibitory postsynaptic currents in the rat hippocampal CA1 region. J Pharmacol Exp Ther 303: 937–944. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7–61. [PubMed] [Google Scholar]
- Durand GM, Kovalchuk Y, Konnerth A (1996) Long-term potentiation and functional synapse induction in developing hippocampus. Nature 381: 71–75. [DOI] [PubMed] [Google Scholar]
- Feng B, Tse HW, Skifter DA, Morley R, Jane DE, Monaghan DT (2004) Structure-activity analysis of a novel NR2C/NR2D-preferring NMDA receptor antagonist: 1-(phenanthrene-2-carbonyl) piperazine-2,3-dicarboxylic acid. Br J Pharmacol 141: 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo R, Zamudio PA, Valenzuela CF (2005) Alcohol is a potent stimulant of immature neuronal networks: implications for fetal alcohol spectrum disorder. J Neurochem, in press. [DOI] [PubMed]
- Hendricson AW, Thomas MP, Lippmann MJ, Morrisett RA (2003) Suppression of L-type voltage-gated calcium channel-dependent synaptic plasticity by ethanol: analysis of miniature synaptic currents and dendritic calcium transients. J Pharmacol Exp Ther 307: 550–558. [DOI] [PubMed] [Google Scholar]
- Hendricson AW, Sibbald JR, Morrisett RA (2004) Ethanol alters the frequency, amplitude, and decay kinetics of Sr2+-supported, asynchronous NMDAR mEPSCs in rat hippocampal slices. J Neurophysiol 91: 2568–2577. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Malenka RC, Nicoll RA (1998) Development of excitatory circuitry in the hippocampus. J Neurophysiol 79: 2013–2024. [DOI] [PubMed] [Google Scholar]
- Hua JY, Smith SJ (2004) Neural activity and the dynamics of central nervous system development. Nat Neurosci 7: 327–332. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283: 70–74. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Koch C, Genz K, Hoerster F, Felderhoff-Mueser U, Tenkova T, Dikranian K, Olney JW (2001) Neurotransmitters and apoptosis in the developing brain. Biochem Pharmacol 62: 401–405. [DOI] [PubMed] [Google Scholar]
- Jonas P, Major G, Sakmann B (1993) Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus. J Physiol (Lond) 472: 615–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones OT, Bernstein GM, Jones EJ, Jugloff DG, Law M, Wong W, Mills LR (1997) N-Type calcium channels in the developing rat hippocampus: subunit, complex, and regional expression. J Neurosci 17: 6152–6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Sawada S, Yamamoto C (1988) Synthetic omega-conotoxin blocks synaptic transmission in the hippocampus in vitro. Neurosci Lett 91: 84–88. [DOI] [PubMed] [Google Scholar]
- Lei S, McBain CJ (2003) GABA B receptor modulation of excitatory and inhibitory synaptic transmission onto rat CA3 hippocampal interneurons. J Physiol (Lond) 546: 439–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Wilson WA, Swartzwelder HS (2002) Differential effect of ethanol on NMDA EPSCs in pyramidal cells in the posterior cingulate cortex of juvenile and adult rats. J Neurophysiol 87: 705–711. [DOI] [PubMed] [Google Scholar]
- Lin Z, Lin Y, Schorge S, Pan JQ, Beierlein M, Lipscombe D (1999) Alternative splicing of a short cassette exon in α1B generates functionally distinct N-type calcium channels in central and peripheral neurons. J Neurosci 19: 5322–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livy DJ, Miller EK, Maier SE, West JR (2003) Fetal alcohol exposure and temporal vulnerability: effects of binge-like alcohol exposure on the developing rat hippocampus. Neurotoxicol Teratol 25: 447–458. [DOI] [PubMed] [Google Scholar]
- Lovinger DM (1993) High ethanol sensitivity of recombinant AMPA-type glutamate receptors expressed in mammalian cells. Neurosci Lett 159: 83–87. [DOI] [PubMed] [Google Scholar]
- Lovinger DM (1997) Alcohols and neurotransmitter gated ion channels: past, present and future. Naunyn Schmiedebergs Arch Pharmacol 356: 267–282. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF (1989) Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 243: 1721–1724. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF (1990) NMDA receptor-mediated synaptic excitation selectively inhibited by ethanol in hippocampal slice from adult rat. J Neurosci 10: 1372–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozovaya NA, Grebenyuk SE, Tsintsadze T, Feng B, Monaghan DT, Krishtal OA (2004) Extrasynaptic NR2B and NR2D subunits of NMDA receptors shape “superslow” afterburst EPSC in rat hippocampus. J Physiol (Lond) 558: 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SM, Yeh HH (1999) Ethanol modulates AMPA-induced current responses of primary somatosensory cortical neurons. Neurochem Int 35: 175–183. [DOI] [PubMed] [Google Scholar]
- Maldve RE, Chen X, Zhang TA, Morrisett RA (2004) Ethanol selectively inhibits enhanced vesicular release at excitatory synapses: real-time visualization in intact hippocampal slices. Alcohol Clin Exp Res 28: 143–152. [DOI] [PubMed] [Google Scholar]
- Mameli M, Carta M, Partridge LD, Valenzuela CF (2005) Neurosteroid-induced plasticity of immature synapses via retrograde modulation of presynaptic NMDA receptors. J Neurosci 25: 2285–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal C, Mulle C (2004) Postnatal maturation of mossy fibre excitatory transmission in mouse CA3 pyramidal cells: a potential role for kainate receptors. J Physiol (Lond) 561: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa T, Yagi T, Kitazawa H, Yasuda M, Kawai N, Tsuboi K, Niki H (1997) Fyn-kinase as a determinant of ethanol sensitivity: relation to NMDA-receptor function. Science 278: 698–701. [DOI] [PubMed] [Google Scholar]
- Molnar E, Pickard L, Duckworth JK (2002) Developmental changes in ionotropic glutamate receptors: lessons from hippocampal synapses. The Neuroscientist 8: 143–153. [DOI] [PubMed] [Google Scholar]
- Morrisett RA, Swartzwelder HS (1993) Attenuation of hippocampal long-term potentiation by ethanol: a patch-clamp analysis of glutamatergic and GABAergic mechanisms. J Neurosci 13: 2264–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrisett RA, Martin D, Oetting TA, Lewis DV, Wilson WA, Swartzwelder HS (1991) Ethanol and magnesium ions inhibit N-methyl-d-aspartate-mediated synaptic potentials in an interactive manner. Neuropharmacology 30: 1173–1178. [DOI] [PubMed] [Google Scholar]
- Moykkynen T, Korpi ER, Lovinger DM (2003) Ethanol inhibits alpha-amino-3-hydyroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor function in central nervous system neurons by stabilizing desensitization. J Pharmacol Exp Ther 306: 546–555. [DOI] [PubMed] [Google Scholar]
- Nelson TE, Ur CL, Gruol DL (1999) Chronic intermittent ethanol exposure alters CA1 synaptic transmission in rat hippocampal slices. Neuroscience 94: 431–442. [DOI] [PubMed] [Google Scholar]
- Newton PM, Orr CJ, Wallace MJ, Kim C, Shin HS, Messing RO (2004) Deletion of N-type calcium channels alters ethanol reward and reduces ethanol consumption in mice. J Neurosci 24: 9862–9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Madamba SG, Siggins GR (1994) Ethanol inhibits glutamatergic neurotransmission in nucleus accumbens neurons by multiple mechanisms. J Pharmacol Exp Ther 271: 1566–1573. [PubMed] [Google Scholar]
- Okabe S, Collin C, Auerbach JM, Meiri N, Bengzon J, Kennedy MB, Segal M, McKay RD (1998) Hippocampal synaptic plasticity in mice overexpressing an embryonic subunit of the NMDA receptor. J Neurosci 18: 4177–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW (2004) Fetal alcohol syndrome at the cellular level. Addict Biol 9: 137–149, discussion 151. [DOI] [PubMed] [Google Scholar]
- Pan JQ, Lipscombe D (2000) Alternative splicing in the cytoplasmic II–III loop of the N-type Ca channel α1B subunit: functional differences are β subunit-specific. J Neurosci 20: 4769–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard L, Noel J, Henley JM, Collingridge GL, Molnar E (2000) Developmental changes in synaptic AMPA and NMDA receptor distribution and AMPA receptor subunit composition in living hippocampal neurons. J Neurosci 20: 7922–7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard L, Noel J, Duckworth JK, Fitzjohn SM, Henley JM, Collingridge GL, Molnar E (2001) Transient synaptic activation of NMDA receptors leads to the insertion of native AMPA receptors at hippocampal neuronal plasma membranes. Neuropharmacology 41: 700–713. [DOI] [PubMed] [Google Scholar]
- Reid CA, Bekkers JM, Clements JD (2003) Presynaptic Ca2+ channels: a functional patchwork. Trends Neurosci 26: 683–687. [DOI] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR (2004) Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci 24: 1594–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D (2004) Signaling cascades regulating NMDA receptor sensitivity to ethanol. The Neuroscientist 10: 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz D, Mellor J, Nicoll RA (2001) Presynaptic kainate receptor mediation of frequency facilitation at hippocampal mossy fiber synapses. Science 291: 1972–1976. [DOI] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ (1995) Developmental changes in presynaptic calcium channels coupled to glutamate release in cultured rat hippocampal neurons. J Neurosci 15: 4612–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuttleworth CW, Connor JA (2001) Strain-dependent differences in calcium signaling predict excitotoxicity in murine hippocampal neurons. J Neurosci 21: 4225–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel SJ, Brose N, Janssen WG, Gasic GP, Jahn R, Heinemann SF, Morrison JH (1994) Regional, cellular, and ultrastructural distribution of N-methyl-d-aspartate receptor subunit 1 in monkey hippocampus. Proc Natl Acad Sci USA 91: 564–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol RJ, Delaney-Black V, Nordstrom B (2003) Fetal alcohol spectrum disorder. JAMA 290: 2996–2999. [DOI] [PubMed] [Google Scholar]
- Solem M, McMahon T, Messing RO (1997) Protein kinase A regulates inhibition of N- and P/Q-type calcium channels by ethanol in PC12 cells. J Pharmacol Exp Ther 282: 1487–1495. [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM (1998) Regulation of the readily releasable vesicle pool by protein kinase C. Neuron 21: 885–893. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Tsujimoto T (1995) Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc Natl Acad Sci USA 92: 846–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strawn JR, Cooper RL (2002) The effects of ethanol on pre-synaptic components of synaptic transmission in a model glutamatergic synapse: the crayfish neuromuscular junction. Comp Biochem Physiol C Toxicol Pharmacol 131: 395–404. [DOI] [PubMed] [Google Scholar]
- Suvarna N, Borgland SL, Wang J, Phamluong K, Auberson YP, Bonci A, Ron D (2005) Ethanol alters trafficking and functional NMDA receptor NR2 subunit ratio via H-RAS. J Biol Chem, in press. [DOI] [PubMed]
- Swartzwelder HS, Wilson WA, Tayyeb MI (1995) Differential sensitivity of NMDA receptor-mediated synaptic potentials to ethanol in immature versus mature hippocampus. Alcohol Clin Exp Res 19: 320–323. [DOI] [PubMed] [Google Scholar]
- Tokunaga T, Miyazaki K, Koseki M, Mobarakeh JI, Ishizuka T, Yawo H (2004) Pharmacological dissection of calcium channel subtype-related components of strontium inflow in large mossy fiber boutons of mouse hippocampus. Hippocampus 14: 570–585. [DOI] [PubMed] [Google Scholar]
- van Zundert B, Yoshii A, Constantine-Paton M (2004) Receptor compartmentalization and trafficking at glutamate synapses: a developmental proposal. Trends Neurosci 27: 428–437. [DOI] [PubMed] [Google Scholar]
- Vance CL, Begg CM, Lee WL, Haase H, Copeland TD, McEnery MW (1998) Differential expression and association of calcium channel alpha1B and beta subunits during rat brain ontogeny. J Biol Chem 273: 14495–14502. [DOI] [PubMed] [Google Scholar]
- Walter HJ, Messing RO (1999) Regulation of neuronal voltage-gated calcium channels by ethanol. Neurochem Int 35: 95–101. [DOI] [PubMed] [Google Scholar]
- Wang XM, Lemos JR, Dayanithi G, Nordmann JJ, Treistman SN (1991) Ethanol reduces vasopressin release by inhibiting calcium currents in nerve terminals. Brain Res 551: 338–341. [DOI] [PubMed] [Google Scholar]
- Warren KR, Foudin LL (2001) Alcohol-related birth defects—the past, present, and future. Alcohol Res Health 25: 153–158. [PMC free article] [PubMed] [Google Scholar]
- Wasling P, Hanse E, Gustafsson B (2004) Developmental changes in release properties of the CA3-CA1 glutamate synapse in rat hippocampus. J Neurophysiol 92: 2714–2724. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Fukaya M, Sakimura K, Manabe T, Mishina M, Inoue Y (1998) Selective scarcity of NMDA receptor channel subunits in the stratum lucidum (mossy fibre-recipient layer) of the mouse hippocampal CA3 subfield. Eur J Neurosci 10: 478–487. [DOI] [PubMed] [Google Scholar]
- Weiner JL, Dunwiddie TV, Valenzuela CF (1999) Ethanol inhibition of synaptically evoked kainate responses in rat hippocampal CA3 pyramidal neurons. Mol Pharmacol 56: 85–90. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Nicoll RA (1995) Presynaptic changes during mossy fibre LTP revealed by NMDA receptor-mediated synaptic responses. Nature 376: 256–259. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Scheurer L, Kunzi R, Fritschy JM, Mohler H, Benke D (1995) Distribution of NMDA receptor subunit proteins NR2A, 2B, 2C and 2D in rat brain. NeuroReport 7: 45–48. [PubMed] [Google Scholar]
- Wenzel A, Villa M, Mohler H, Benke D (1996) Developmental and regional expression of NMDA receptor subtypes containing the NR2D subunit in rat brain. J Neurochem 66: 1240–1248. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Fritschy JM, Mohler H, Benke D (1997) NMDA receptor heterogeneity during postnatal development of the rat brain: differential expression of the NR2A, NR2B, and NR2C subunit proteins. J Neurochem 68: 469–478. [DOI] [PubMed] [Google Scholar]
- Wheeler DB, Randall A, Tsien RW (1994) Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science 264: 107–111. [DOI] [PubMed] [Google Scholar]
- Woodward JJ, Machu T, Leslie SW (1990) Chronic ethanol treatment alters omega-conotoxin and Bay K 8644 sensitive calcium channels in rat striatal synaptosomes. Alcohol 7: 279–284. [DOI] [PubMed] [Google Scholar]
- Zhang LI, Poo MM (2001) Electrical activity and development of neural circuits. Nat Neurosci [Suppl] 4: 1207–1214. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R (2000) Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci 3: 1098–1106. [DOI] [PubMed] [Google Scholar]
- Ziskind-Conhaim L, Gao BX, Hinckley C (2003) Ethanol dual modulatory actions on spontaneous postsynaptic currents in spinal motoneurons. J Neurophysiol 89: 806–813. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355–405. [DOI] [PubMed] [Google Scholar]