

Abstract
Dysfunction of inhibitory neurons in the prefrontal cortex (PFC), represented by decreased expression of GABA-related genes such as the 67 kDa isoform of glutamate decarboxylase (GAD67) and parvalbumin (PV), appears to contribute to cognitive deficits in subjects with schizophrenia. We investigated the involvement of signaling mediated by brain-derived neurotrophic factor (BDNF) and its receptor tyrosine kinase TrkB in producing the altered GABA-related gene expression in schizophrenia. In 15 pairs of subjects with schizophrenia and matched control subjects, both BDNF and TrkB mRNA levels, as assessed by in situ hybridization, were significantly decreased in the PFC of the subjects with schizophrenia, whereas the levels of mRNA encoding the receptor tyrosine kinase for neurotrophin-3, TrkC, were unchanged. In this cohort, within-pair changes in TrkB mRNA levels were significantly correlated with those in both GAD67 and PV mRNA levels. Decreased BDNF, TrkB, and GAD67 mRNA levels were replicated in a second cohort of 12 subject pairs. In the combined cohorts, the correlation between within-pair changes in TrkB and GAD67 mRNA levels was significantly stronger than the correlation between the changes in BDNF and GAD67 mRNA levels. Neither BDNF nor TrkB mRNA levels were changed in the PFC of monkeys after a long-term exposure to haloperidol. Genetically introduced decreases in TrkB expression, but not in BDNF expression, also resulted in decreased GAD67 and PV mRNA levels in the PFC of adult mice; in addition, the cellular pattern of altered GAD67 mRNA expression paralleled that present in schizophrenia. Decreased TrkB signaling appears to underlie the dysfunction of inhibitory neurons in the PFC of subjects with schizophrenia.
Keywords: cortex, GABA, GAD, in situ hybridization, parvalbumin, postmortem, trkB hypomorphic
Introduction
Schizophrenia is characterized by severe and persisting deficits in certain cognitive functions, such as working memory (Weinberger et al., 1986; Goldman-Rakic, 1994). Alterations in the circuitry of the dorsal prefrontal cortex (PFC), which plays a central role in working memory processes (Goldman-Rakic, 1995), appear to contribute to the working memory impairments in schizophrenia (Lewis and Lieberman, 2000). In particular, convergent lines of evidence implicate abnormalities in GABA-containing local circuit neurons in the dysfunction of the PFC in schizophrenia (Blum and Mann, 2002; Volk and Lewis, 2002). For example, postmortem studies from different laboratories have shown decreased expression of mRNAs for GABA-related proteins, including the 67 kDa isoform of glutamate decarboxylase (GAD67) (Akbarian et al., 1995; Guidotti et al., 2000; Volk et al., 2000), a GABA synthesizing enzyme, and the GABA transporter-1 (GAT-1) (Ohnuma et al., 1999; Volk et al., 2001), a presynaptic transporter for the reuptake of GABA, in the PFC of subjects with schizophrenia. Furthermore, these mRNAs appear to be selectively downregulated in the subclass of GABA neurons that contains the calcium-binding protein parvalbumin (PV), and the expression of the mRNA encoding PV is also decreased in these neurons (Hashimoto et al., 2003). In contrast, GABA-related gene expression appears to be unchanged in the calretinin-containing subclass of GABA neurons (Hashimoto et al., 2003).
These alterations may be critical elements in the pathophysiology of cognitive dysfunction in schizophrenia, given the central role that GABA neurotransmission plays in the regulation of PFC information processing during working memory tasks (Sawaguchi et al., 1989; Rao et al., 1999, 2000; Constantinidis et al., 2002). Thus, determining the mechanisms underlying the altered gene expression in PV-containing GABA neurons is essential for understanding the pathogenesis of PFC dysfunction in schizophrenia.
Signaling mediated by brain-derived neurotrophic factor (BDNF) and its receptor tyrosine kinase TrkB regulates the development of GABA neurons. In neuronal cultures, BDNF-TrkB signaling promotes the functional and morphological development of GABA neurons (Ventimiglia et al., 1995; Arenas et al., 1996; Rutherford et al., 1998; Vicario-Abejon et al., 1998; Bolton et al., 2000; Marty et al., 2000; Yamada et al., 2002; Kohara et al., 2003) and induces the expression of GABA-related proteins, including GAD67 and GAT-1 (Mizuno et al., 1994; Bolton et al., 2000; Marty et al., 2000; Yamada et al., 2002). These effects may be selective for PV-containing cortical GABA neurons because TrkB is predominantly expressed by PV-containing neurons and not by calretinin-containing GABA neurons (Cellerino et al., 1996; Gorba and Wahle, 1999). Indeed, in mice genetically engineered to overexpress BDNF, the development of cortical GABA neurons, as assessed by the inhibitory innervation of pyramidal neurons, was accelerated and accompanied by a precocious increase in the number of neurons containing PV (Huang et al., 1999). Furthermore, the number of PV-containing neurons was decreased in the cerebral cortex of developing mice in which bdnf or trkB was deleted (Jones et al., 1994; Alcantara et al., 1997).
This developmental regulation of GABA neurons by BDNF-TrkB signaling suggests that disturbances in BDNF and/or TrkB might be responsible for the altered expression of GABA-related genes in the PFC of subjects with schizophrenia, an idea supported by a report of reduced cortical BDNF expression in schizophrenia (Weickert et al., 2003). To test this hypothesis, we specifically asked (1) whether the mRNA expression for BDNF or its receptor tyrosine kinase TrkB is altered in the PFC of subjects with schizophrenia, (2) whether the changes in BDNF or TrkB mRNA are associated with the changes in GABA-related gene expression, (3) whether the changes in BDNF or TrkB mRNA expression are attributable to the disease process itself or to pharmacological treatments of schizophrenia, and (4) whether GABA-related gene expression is altered in the PFC of adult mice with genetically engineered reductions in the expression of BDNF or TrkB mRNAs.
Materials and Methods
Human subjects. Brain specimens were obtained from two cohorts of human subjects during autopsies performed at the Allegheny County Coroner's Office, Pittsburgh, Pennsylvania, with the approval of the Institutional Review Board for Biomedical Research of the University of Pittsburgh and the consent of the surviving next-of-kin. The two cohorts consisted of 15 (cohort 1) and 12 (cohort 2) subjects with schizophrenia, each of which was paired with one control subject (supplemental Tables 1 and 2, available at www.jneurosci.org as supplemental material). Tissue specimens from subjects in cohort 1 were used in our previous studies (Hashimoto et al., 2003), whereas those in cohort 2 were collected more recently. Because tissue levels of some mRNA species in the PFC may be affected by such factors as sex, age, and postmortem interval (PMI) (the time between death and preservation of brain tissue at -80°C), individual pairs were completely matched for sex and as closely as possible for age and PMI in both cohorts. Paired t tests revealed that the schizophrenia and control groups did not differ in terms of age, PMI, brain pH, or tissue storage time (all t(14) < 1.4, all p > 0.18 for cohort 1; all t(11) < 1.1, all p > 0.29 for cohort 2). All subjects were from a community-based population, and most subjects (12 with schizophrenia and 15 controls in cohort 1; 8 with schizophrenia and 10 controls in cohort 2) died suddenly outside of a hospital setting.
Diagnosis and Statistical Manual of Mental Disorders (1994) diagnoses were made for each subject on the basis of medical records and the results of structured interviews conducted with family members (Glantz and Lewis, 1997). As detailed by Hashimoto et al. (2003) and supplemental Table 3 (available at www.jneurosci.org as supplemental material), five schizophrenia subjects in cohort 1 and six schizophrenia subjects in cohort 2 met criteria for schizoaffective disorder: three subjects with schizophrenia in both cohorts committed suicide; two control subjects in cohort 1 and one control subject in cohort 2 had a history of depressive disorder (not otherwise specified), alcohol abuse (current at the time of death), or posttraumatic stress disorder, respectively; and seven subjects with schizophrenia (four in cohort 1 and three in cohort 2) met criteria for alcohol dependence at the time of death. Toxicology studies for all subjects revealed positive plasma alcohol levels (0.01-0.09%) in three control subjects (pairs 3, 7, and 10) in cohort 1 and in one control (pair 9) and one schizophrenia subject (pair 4) in cohort 2; no other substances of abuse were detected in any subjects. Three schizophrenia subjects (pairs 2, 10, and 14) from cohort 1 (Hashimoto et al., 2003) and one schizophrenia subject (pair 3) from cohort 2 (supplemental Table 3, available at www.jneurosci.org as supplemental material) were free of antipsychotic medications at the time of death. The length of time without these medications before death was 9.6 months, 1.2 months, unknown, and 8.5 years for these subjects, respectively. The mean ± SD age of the schizophrenia subjects at the onset of illness was 25.1 ± 9.0 years (cohort 1) and 25.6 ± 7.1 years (cohort 2), and the average duration of illness was 18.7 ± 9.4 years (cohort 1) and 28.0 ± 16.5 years (cohort 2).
Neuropathological examination revealed an acute infarction in the distribution of the inferior branch of the right middle cerebral artery in the schizophrenia subject in pair 10 from cohort 1 and a previous infarction in the inferior aspect of the frontal lobe in the schizophrenia subject in pair 7 from cohort 2. However, right PFC area 9, the region under study, appeared unaffected in both cases. Other brain disorders were excluded in each subject on the basis of both clinical and neuropathological data.
Haloperidol-treated monkeys. To mimic the clinical treatment of subjects with schizophrenia, four male cynomolgus (Macaca fascicularis) monkeys, matched individually to a control monkey for sex, age, and weight, were exposed chronically to haloperidol (mean ± SD trough serum level, 4.3 ± 1.1 ng/ml) and benztropine mesylate (to relieve extrapyramidal side effects) for 9-12 months, as described previously (Pierri et al., 1999).
Genetically engineered mice. To test the effect of decreased TrkB expression on the expression of GABA-related genes, we used trkB hypomorphic mice in which the trkB gene is mutated into the artificial locus, so-called fBZ, by replacing the first coding exon of trkB with a TrkB cDNA flanked by two loxP sites (Xu et al., 2000a). These mice were generated with the 129 strain-derived embryonic stem cells and C57BL/6 blastocystes (Xu et al., 2000a) and back-crossed into C57BL/6 for at least five generations. Wild-type C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME) were used as control. Homozygous (fBZ/fBZ) and heterozygous (fBZ/+) animals were reported to express ∼25 and 62.5%, respectively, of the TrkB protein levels present in the wild-type animals (Xu et al., 2000a; Rohrer, 2001; Rico et al., 2002). Animals with the fBZ/fBZ genotype do not express the truncated isoforms of TrkB (Xu et al., 2000a). Three groups of animals, including heterozygous and homozygous mice for the fBZ locus and wild-type controls (n = 3 for each group), were killed at 8 weeks of age.
To test the effect of reduced BDNF expression, we used mice with a neuron-specific inducible knock-out of bdnf, in which two transgenes, the tetracycline transactivator (tTA) gene driven by neuron-specific enolase promoter (nse-tTA) (Chen et al., 1998) and the Cre recombinase gene under the control of tTA-responsive tetO promoter (tetO-cre) (Perl et al., 2002; Radomska et al., 2002), regulate the deletion of floxed exon V of bdnf (Rios et al., 2001) in a tetracycline-dependent manner. The two mouse lines with nse-tTA and tetO-cre transgenes were both maintained as homozygotes. These lines were crossed, and the resultant bigenic mice were then crossed with mice with the floxed bdnf allele (Rios et al., 2001). For the deletion of bdnf from embryogenesis, mice were bred in the absence of a tetracycline analog, doxycycline (dox). Previous data using the same mouse line with nse-tTA demonstrated that this transgene is expressed in late embryogenesis (Chen et al., 1998). For deletion of bdnf in adulthood, mice were bred in the presence of dox (1 mg/ml) in drinking water to suppress the recombination. At 3 months of age, dox was removed from the drinking water to induce the recombination. In these mice, recombination was detected 4 weeks after termination of dox treatment (Monteggia et al., 2004); however, to ensure that full recombination had occurred, we allowed for at least a 3 month washout period. Littermates with nse-tTA and tetO-cre transgenes, expressing similar levels of BDNF as wild-type mice (Monteggia et al., 2004), were used as controls. Mice with bdnf deletion induced from embryogenesis or in adulthood, together with the corresponding control animals, were killed at 5 or 6 months of age, respectively (n ≥ 6 for each group).
Tissue preparation and in situ hybridization. As described previously (Hashimoto et al., 2003), the right PFC of each human brain was blocked coronally, frozen immediately, and stored at -80°C. Serial sections (20 μm) containing the superior frontal gyrus were cut at the anteroposterior level corresponding to the middle portion of the superior frontal sulcus, thaw mounted onto glass slides, and stored at -80°C until processed. The location of PFC area 9 was identified by cytoarchitectonic criteria in Nissl-stained sections as described previously (Glantz et al., 2000; Volk et al., 2000). Three sections evenly spaced at ∼400 μm (cohort 1) or 340 μm (cohort 2) were used for analysis of each mRNA of interest. Thirty or twenty-four sections (one section from each subject in cohorts 1 or 2, respectively) were processed in a single run with sections from each pair processed side by side. Three runs were performed for each mRNA in each cohort.
Processing of monkey brains for in situ hybridization was described previously (Hashimoto et al., 2003). Briefly, from each monkey, serial coronal sections (16 μm) were cut from fresh-frozen tissue blocks containing the middle one-third of the principal sulcus. Three sections from each animal, evenly spaced at 400 μm intervals, were selected for analysis of BDNF or TrkB mRNAs.
Mouse brains were frozen immediately after removal from the skull and stored at -80°C. Serial coronal sections (12 μm) were cut from +1.98 to +1.54 bregma (Paxinos and Franklin, 2001), and three sections evenly spaced at ∼180 μm intervals were selected from each mouse and subjected to in situ hybridization for each mRNA of interest.
Templates for synthesis of riboprobes were obtained by PCR. For detection of mRNAs in human and monkey brains, specific primer sets amplified a 363 bp cDNA fragment for BDNF mRNA, a 384 bp fragment for TrkB mRNA, a 408 bp fragment for TrkC mRNA, and a 360 bp fragment for GAD67, corresponding to bases 1043-1405 of the human BDNF mRNA (GenBank accession number M61181), bases 1952-2335 of the human TrkB mRNA (GenBank accession number S76473), bases 1757-2116 of the human GAD67 mRNA (GenBank accession number NM_00817), and bases 1640-2047 of the human TrkC mRNA (GenBank accession number S76473), respectively. The fragments for TrkB and TrkC mRNAs were amplified from the regions encoding the intracellular domains that are specific to the tyrosine kinase-containing isoforms. Antisense riboprobes transcribed from these fragments were shown to detect the specific signal of targeted mRNAs in the primate neocortex (Okuno et al., 1999; Hashimoto et al., 2000). To increase signal intensity for BDNF mRNA, we amplified an additional 358 bp fragment, corresponding to bases 693-1037 of the BDNF mRNA (GenBank accession number M61181), and combined the two antisense riboprobes transcribed from the two fragments. For detection of mRNA in mouse brains, we obtained a 520 bp cDNA fragment for BDNF mRNA, a 345 bp fragment for TrkB mRNA, a 311 bp fragment for GAD67 mRNA, a 339 bp fragment for PV mRNA, and a 488 bp fragment for calretinin mRNA, corresponding to bases 843-1362 of the mouse BDNF mRNA (GenBank accession number AY057908), bases 2567-2911 of the mouse TrkB mRNA (GenBank accession number X17647), bases 151-461 of the mouse GAD67 mRNA (GenBank accession number Y12257), bases 256-594 of the mouse PV mRNA (GenBank accession number X59382), and bases 225-712 of the mouse calretinin mRNA (GenBank accession number NM_007586). The fragment for TrkB mRNA was amplified within the tyrosine kinase-containing isoform-specific region. Nucleotide sequencing revealed 100% homologies between the amplified fragments and the previously reported sequences.
Amplified fragments were subcloned into the plasmid pSTBlue-1 (Novagen, Madison, WI), and antisense and sense probes were transcribed in vitro in the presence of 35S-CTP (Amersham Biosciences, Piscataway, NJ), using T7 or SP6 RNA polymerase.
Hybridization was performed as described previously (Mirnics et al., 2000; Middleton et al., 2002; Hashimoto et al., 2003). After fixation with 4% paraformaldehyde in PBS, the sections were acetylated, dehydrated through a graded ethanol series, and de-fatted in chloroform for 10 min. The sections were then hybridized with 35S-labeled riboprobes (2 × 107 dpm/ml) in a hybridization buffer at 56°C for 16 hr. The sections were washed in a solution containing 0.3 m NaCl, 20 mm Tris-HCl, pH 8.0, 1 mm EDTA, pH 8.0, and 50% formamide at 63°C, treated with RNase A (20 μg/ml) at 37°C, and washed in 0.1× SSC (150 mm NaCl and 15 mm sodium citrate) at 67°C. Sections were then dehydrated through a graded ethanol series, air dried, and exposed to BioMax MR film (Eastman Kodak, Rochester, NY). After the exposure to film, sections were coated with NTB2 emulsion (Eastman Kodak) diluted 1:1 with water, exposed at 4°C, developed with D-19 (Eastman Kodak), and counterstained with cresyl violet.
Quantification of mRNA expression levels. Quantification was performed without knowledge of subject diagnosis by random coding of the sections. Trans-illuminated autoradiographic film images were captured by a video camera under precisely controlled conditions, digitized, and analyzed using a Microcomputer Imaging Device system (Imaging Research, London, Ontario, Canada). Images of adjacent sections stained with cresyl violet were also captured and superimposed onto the autoradiographic images to draw contours of the full thickness of the cortex. Optical density was measured within the contours and expressed as nanoCuries per gram tissue by reference to radioactive standards (Carbon-14 standards; American Radiolabeled Chemicals, St. Louis, MO) exposed on the same film. In the human study, we sampled the zone of PFC area 9 cut perpendicular to the pial surface. The mean ± SD total of sampled areas per subject was 321 ± 130, 310 ± 121, 356 ± 104, and 247 ± 68.1 mm2 for BDNF, TrkB, GAD67, and TrkC mRNAs, respectively. For laminar analyses of mRNA expression in human PFC area 9, three cortical traverses, 1-2 mm in width, were sampled from each section (nine traverses per subject). Within these traverses, the ratios of the depth of each laminar border to the total cortical thickness, determined from adjacent Nissl-stained sections, were applied to the autoradiograms to obtain optical densities for each layer. In the haloperidol-exposed monkeys and their controls, the optical density of each mRNA signal was measured in PFC areas 9 and 46. In the mouse study, quantification was done in the PFC, including the cingulate and prelimbic cortices. All cortical optical density measures were corrected by subtracting background measures in the white matter.
To investigate the anatomical substrate for the observed GABA-related mRNA expression changes in trkB hypomorphic mice, we determined neuron density, GAD67 mRNA-positive neuron density, and the level of GAD67 mRNA expression per neuron in the PFC of wild-type, fBZ/+, and fBZ/fBZ mice using a previously published approach (Hashimoto et al., 2003). Three sections hybridized with antisense GAD67 mRNA probe and processed for emulsion autoradiography were analyzed for each animal. Using a microscope equipped with a motor-driven stage, sampling frames with a size of 120 × 120 μm were placed systematically and randomly within the cingulate and prelimbic cortices. The mean ± SD number of sampling frames per animal was 55.3 ± 1.5, 54.0 ± 1.0, and 54.7 ± 1.2 for wild-type, fBZ/+, and fBZ/fBZ animals, respectively. Within each sampling frame, Nissl-stained neuronal nuclei were counted for the measurement of neuron density. A circle with a fixed diameter of 16 μm was centered over every neuronal nucleus, and the number of silver grains was determined as a measure of the cellular expression level of GAD67 mRNA. The size of the circle was determined to cover the largest GAD67-positive neurons in the mouse cortex (Solberg et al., 1988). Background grain number was determined for each section by counting grain number in the same size circle centered over glial nuclei. Small glial nuclei, stained darkly with cresyl violet, were easily discriminated from large and faintly stained neuronal nuclei. The mean ± SD number of PFC neurons sampled per animal was 1256 ± 48, 1235 ± 48, and 1215 ± 33 for wild-type, fBZ/+, and fBZ/fBZ mice, respectively. To identify GAD67 mRNA-positive neurons, we set a threshold of grain density at five times the background, which corresponded to the point of rarity in the bimodal distribution histograms of grain number per neuron (Volk et al., 2000). The mean ± SD number of GAD67 mRNA-positive neurons sampled per animal was 201.0 ± 7.0, 160.3 ± 5.5, and 143.7 ± 11.9 for wild-type, fBZ/+, and fBZ/fBZ mice, respectively.
Statistical analyses. Analysis of covariance (ANCOVA) models (Neter et al., 1996) were performed to examine the differences in BDNF, TrkB, TrkC, and GAD67 mRNA expression levels between the schizophrenia and control groups for the full thickness and for each layer of the cortex. Because three sections were measured for every subject, the dependent variable (e.g., film optical density) was represented by three observations for each subject. Thus, the three observations for each subject can be considered to be exchangeably correlated and treated as repeated measures with compound symmetric covariance structure (Neter et al., 1996). Because no data were missing and the numbers of observations for each subject were identical, the ANCOVA models produce the same results as the repeated-measures analysis of covariance in terms of testing the diagnosis effect. The first ANCOVA model, used for each dependent variable, had diagnostic group as a main effect, subject pair as a blocking effect, and brain pH as a covariate. The inclusion of pair effect reflects the matching of individual subject pairs for sex, age, and PMI. Postmortem brain pH was included as a covariate because it might reflect the integrity of some mRNA species (Harrison et al., 1995). Subject pairing may be considered an attempt to balance the two diagnostic groups with regard to the experimental factors instead of a true statistical paired design. Thus, to validate the first model, a second ANCOVA model was performed with a main effect of diagnostic group and covariates of all pairing factors (sex, age, and PMI), as well as brain pH. Because these two ANCOVAs produced similar results for all of the variables analyzed, only the results from the first analysis are reported. These analyses were implemented in SAS PROC GLM (SAS Institute, Cary, NC).
To test the association between the change in BDNF or TrkB mRNA expression and the GABA-related gene expression changes in schizophrenia, the percentage of differences within subject pairs was calculated for BDNF and TrkB mRNAs and for GAD67, PV, and calretinin mRNAs across subject pairs (Hashimoto et al., 2003). Correlations between within-pair percentage of differences were assessed by Pearson's correlation analysis. For comparison of two correlation coefficients, we performed an asymptotic test using Fisher's Z transformation (Meng et al., 1992).
The influence of sex, history of substance abuse/dependence, history of antidepressant medications, diagnosis of schizoaffective disorder, and cause of death on the within-pair differences in gene expression levels was assessed by two-sample t test.
For the haloperidol-treated monkeys, paired t tests were used to determine the effect of treatment group on BDNF and TrkB mRNA expression in the PFC.
For the genetically engineered mice, the effects of genotype on mRNA expression levels and neuronal densities were evaluated by single-factor ANOVAs. When significance (p < 0.05) was indicated by ANOVA, these measures were compared across different genotypes by using Tukey's multiple comparison test. Pearson's correlation analysis was used to examine the relationship among gene expression levels.
Results
Expression of BDNF, TrkB, and TrkC mRNAs in the PFC of subjects with schizophrenia
To examine the involvement of BDNF-TrkB signaling in the GABA-related gene expression deficits in schizophrenia, we first tested whether BDNF and TrkB mRNA expression levels were altered in PFC area 9 of subjects with schizophrenia using 15 subject pairs in cohort 1 (supplemental Table 1, available at www.jneurosci.org as supplemental material) (for details, see Hashimoto et al., 2003).
In the PFC of control subjects, BDNF mRNA was expressed in a layer-specific pattern; signal intensity was high in layers V/VI, moderate in layers II/III, low in layer IV, and absent in layer I (Fig. 1A). In contrast, TrkB mRNA was homogeneously distributed across layers II-VI (Fig. 1C). BDNF mRNA-positive neurons, represented by accumulations of silver grains over neuronal nuclei in emulsion-coated slides, were scattered in layers II/III and V/VI, rarely observed in layer IV, and not found in layer I (data not shown). The TrkB mRNA was expressed by the majority of neurons across layers II-VI and by a few in layer I (data not shown). These expression patterns in control subjects were consistent with previously published observations in monkey and human association cortices (Okuno et al., 1999; Hashimoto et al., 2000; Romanczyk et al., 2002; Weickert et al., 2003).
Figure 1.
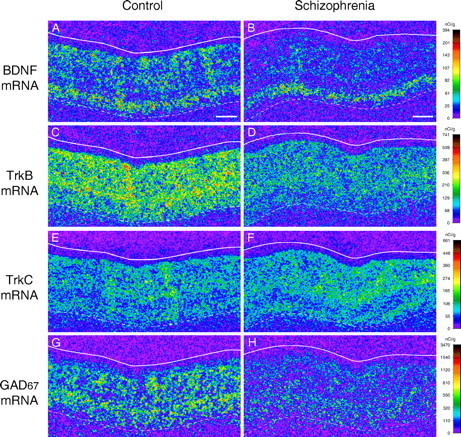
Gene expression in the PFC of control and schizophrenia subjects. Representative autoradiograms illustrating the expression of BDNF (A, B), TrkB (C, D), TrkC (E, F), and GAD67 (G, H) mRNAs in PFC area 9 of a control subject (A, C, E, G) and an age-, sex-, and PMI-matched subject with schizophrenia (B, D, F, H) from the subject pair indicated by the triangle in Figures 2 and 3 are shown. The densities of hybridization signals are presented in a pseudocolor manner according to the calibration scales (right) for each mRNA. Expression levels of BDNF and TrkB mRNAs are reduced in the schizophrenia subject (B, D) compared with the control subject (A, C), whereas TrkC mRNA expression levels appear similar between the control (E) and schizophrenia (F) subjects. GAD67 mRNA expression is also reduced in the schizophrenia subject (H) compared with the control subject (G). Solid and broken lines indicate the pial surface and the border between gray matter and white matter, respectively. Scale bars (in A, B), 1 mm.
In PFC area 9 of subjects with schizophrenia (Fig. 1B,D), mean BDNF and TrkB mRNA expression levels were significantly decreased by 28% (F(1,13) = 15.0; p = 0.002) and 27% (F(1,13) = 23.3; p < 0.001), respectively, compared with the matched control subjects (Fig. 2A,B). Analyses of the mRNA expression levels by cortical layer revealed significant effects of diagnostic group on the expression levels of BDNF mRNA in layers II, III, and V/VI (all F(1,13) > 12.7; all p < 0.004) and on the expression levels of TrkB mRNA in layers II-V/VI (all F(1,13) > 17.2; all p < 0.002) (Fig. 2C,D). BDNF mRNA expression levels in layer IV were not different between the subject groups (F(1,13) = 4.05; p = 0.07) (Fig. 2C), perhaps reflecting its low expression level in this layer in control subjects. Consistent with previous reports of age-dependent declines in BDNF and TrkB mRNA expression levels (Hayashi et al., 1997; Romanczyk et al., 2002), we detected significant negative correlations between BDNF mRNA expression and age for both the control (r = -0.62; p = 0.015) and schizophrenia (r = -0.52; p = 0.049) groups and trends for negative correlations between TrkB mRNA and age (r = -0.49, p = 0.065 for the control group; r = -0.48, p = 0.070 for the schizophrenia group). The regression lines for subjects with schizophrenia were parallel to and shifted downward from those for control subjects (Fig. 2E,F), suggesting that the decreased expression of these mRNAs in schizophrenia is similar in magnitude across adult life.
Figure 2.
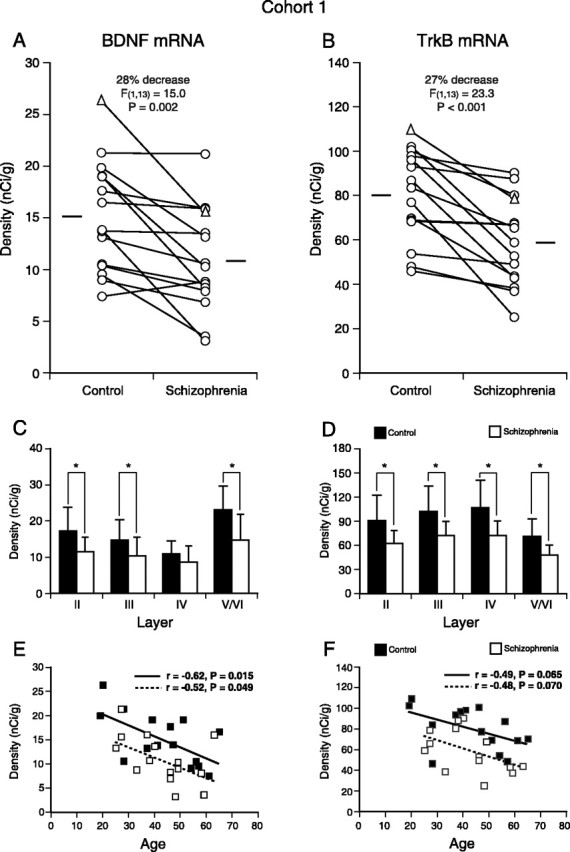
Reduced BDNF and TrkB mRNA expression in the PFC of subjects with schizophrenia in cohort 1. A, B, Comparisons of the mRNA expression levels for BDNF (A) and TrkB (B) by film optical density in PFC area 9 of subjects with schizophrenia and age-, sex-, and PMI-matched control subjects. The triangles represent the data for the pair presented in Figure 1. The expression levels of BDNF and TrkB mRNAs are decreased in the subjects with schizophrenia for 14 and 15 of the pairs, respectively. The mean values for each subject group are indicated by horizontal bars. C, D, Mean ± SD expression levels of BDNF (C) and TrkB (D) mRNAs for each cortical layer. BDNF mRNA expression levels were significantly decreased by 33, 29, and 37% in layers II, III, and V/VI, respectively. The expression levels of TrkB mRNA were significantly decreased by 31, 29, 33, and 32% in layers II, III, IV, and V/VI, respectively. *p < 0.005. E, F, The expression levels of BDNF (E) and TrkB (F) mRNAs are plotted against age for each subject. For both mRNAs, expression levels are negatively correlated with age, and the regression lines for schizophrenia subjects are parallel to and shifted downward from those for control subjects, suggesting that the decreased expression of these mRNAs is similar in magnitude across adult life. These age-dependent changes, similarly affecting both control and schizophrenia subjects, may contribute to the relatively large overlaps in the distributions of the mRNA expression levels between the control and schizophrenia groups in A and B.
To determine whether the decreased mRNA levels were selective for BDNF-TrkB signaling, we examined mRNA expression for TrkC, a receptor tyrosine kinase for neurotrophin-3 (NT-3), in the same 15 subject pairs. Consistent with previous observations in primate association cortices (Okuno et al., 1999; Hashimoto et al., 2000), the hybridization signal for TrkC mRNA was detected in layers II, III, V, and VI, mainly in neurons, in area 9 of control and schizophrenia subjects (Fig. 1E,F). No significant differences were detected in the pattern or level (F(1,13) = 0.004; p = 0.950) of TrkC mRNA expression between the control and schizophrenia groups (Fig. 3A).
Figure 3.

Comparisons of TrkC and GAD67 mRNA expression in the PFC in cohort 1. The expression level of TrkC mRNA did not differ between the control and schizophrenia groups (A), whereas the expression of GAD67 mRNA (B) was significantly decreased in the schizophrenia group. In all 15 subject pairs, GAD67 mRNA expression levels were decreased in the subjects with schizophrenia relative to the matched control subjects. The triangles represent the data for the pair presented in Figure 1. The mean values for each subject group are indicated by horizontal bars.
Association of altered expression of BDNF and TrkB mRNAs with changes in GABA-related mRNA levels
If altered GABA-related gene expression is driven by reduced BDNF-TrkB signaling in schizophrenia subjects, then the changes in BDNF or TrkB mRNA expression should be positively correlated with the changes in GABA-related gene expression across the same subject pairs. To test this hypothesis, we first examined GAD67 mRNA expression in PFC area 9 using the same 15 subject pairs (Fig. 1G,H). Consistent with previous studies (Akbarian et al., 1995; Guidotti et al., 2000; Volk et al., 2000), the expression of GAD67 mRNA was significantly (F(1,13) = 18.6; p = 0.001) decreased by 28% in the schizophrenia group (Fig. 3B). Correlation analyses revealed that the within-pair percentage of changes in GAD67 mRNA expression was significantly correlated with the changes in TrkB mRNA expression (r = 0.67; p = 0.006) and marginally correlated with the changes in BDNF mRNA expression (r = 0.51; p = 0.055). Interestingly, similar correlations were present for the changes in PV (Hashimoto et al., 2003) and TrkB (r = 0.58; p = 0.024) mRNA expression and for PV and BDNF mRNA expression (r = 0.51; p = 0.051). In contrast, calretinin mRNA expression (Hashimoto et al., 2003) was not significantly correlated with BDNF (r = 0.26; p = 0.341) or TrkB (r = 0.38; p = 0.163) mRNAs.
Replication of the gene expression changes in a second cohort of subjects
To confirm these observations, we analyzed the expression of BDNF, TrkB, and GAD67 mRNAs in PFC area 9 using the subjects in cohort 2 (supplemental Table 2, available at www.jneurosci.org as supplemental material). Among these 12 pairs, one subject with schizophrenia (pair 5) showed a strikingly high BDNF mRNA expression level (>7 SDs above the mean of control subjects) (Fig. 4A). This particular subject committed suicide by an overdose of a tricyclic antidepressant, doxepin (supplemental Table 3, available at www.jneurosci.org as supplemental material), and had a serum level >100 times the normal therapeutic range. Because systemic administration of tricyclic antidepressants was reported to induce BDNF expression in the rodent cerebral cortex (Nibuya et al., 1995), we excluded this subject from the analysis of BDNF mRNA expression between the control and schizophrenia groups. In cohort 2, BDNF, TrkB, and GAD67 mRNA expression levels in the schizophrenia group were significantly decreased by 44% (F(1,9) = 8.66; p = 0.016), 18% (F(1,10) = 7.13; p = 0.024), and 24% (F(1,10) = 5.06; p = 0.048), respectively, compared with the control group (Fig. 4).
Figure 4.

Replication of the gene expression differences in cohort 2. A, Decreased BDNF mRNA expression in PFC area 9 of schizophrenia subjects in cohort 2. Pair 5 was excluded from the analysis because the schizophrenia subject showed an abnormally high level of BDNF mRNA expression in association with an antidepressant overdose. B, C, Decreased expression levels of TrkB (B) and GAD67 (C) mRNAs in schizophrenia subjects of cohort 2. The mean mRNA values for each subject group are indicated by solid horizontal bars. The broken lines in A indicate mean values across subjects, including pair 5.
Correlation analyses across 26 subject pairs, including the 15 pairs from cohort 1 and 11 pairs (without pair 5) in cohort 2, revealed a remarkably strong association between the within-pair percentage of changes in TrkB and GAD67 mRNA expression levels (r = 0.74; p < 0.001), as well as a significant correlation between the changes in BDNF and GAD67 mRNA expression levels (r = 0.52; p = 0.007) (Fig. 5). The correlation between the changes in TrkB and GAD67 mRNA expression levels was significantly (p = 0.043) stronger than the correlation between the changes in BDNF and GAD67 mRNA expression levels.
Figure 5.
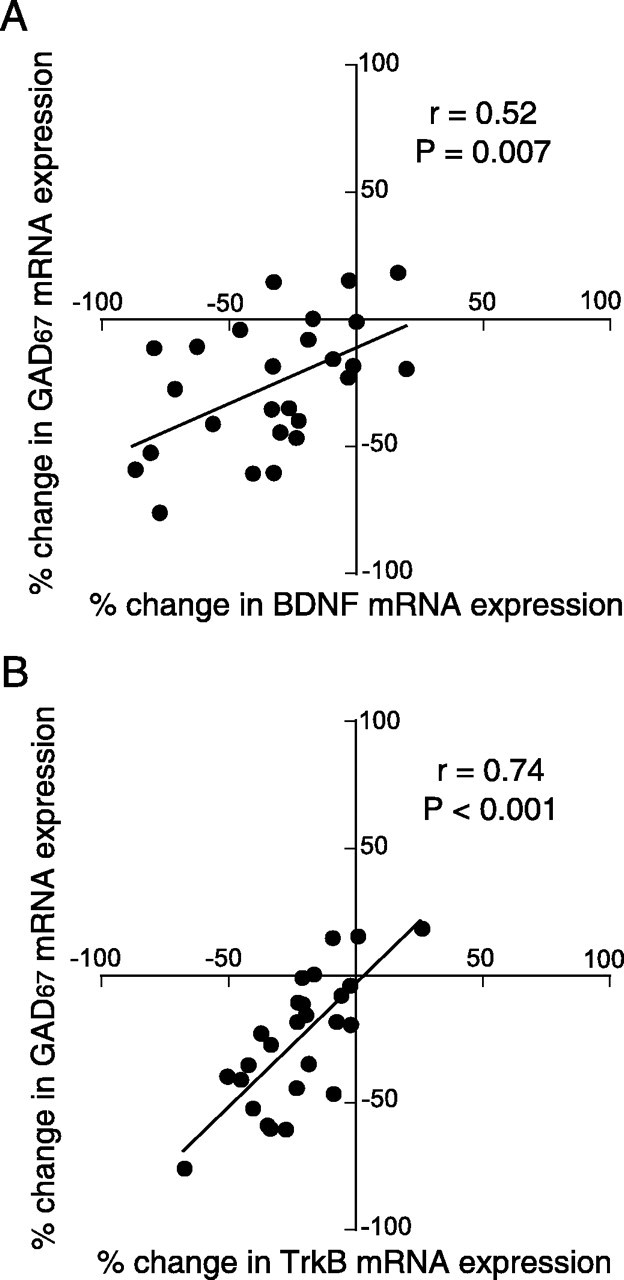
Associations between the differences in BDNF or TrkB mRNA expression and in GAD67 mRNA expression across subject pairs. The within-pair percentage of changes in GAD67 mRNA expression is plotted against those for BDNF (A) or TrkB (B) mRNA expression for 26 subject pairs (excluding pair 5 in cohort 2). The changes in both BDNF and TrkB mRNA expression are significantly correlated with the change in GAD67 mRNA expression across the 26 subject pairs. The expression changes in TrkB mRNA were more highly correlated with the expression changes in GAD67 mRNA than were the expression changes in BDNF mRNA.
Potential influence of other factors on the expression of BDNF and TrkB mRNAs
In the combined data set from both subject cohorts (Fig. 6), the within-pair percentage of decreases in BDNF and TrkB mRNA expression did not differ as a function of sex, history of substance abuse and/or dependence, a history of antidepressant treatments, a diagnosis of schizoaffective disorder, or suicide (all t(24) < 1.56, all p > 0.132 for BDNF mRNA; all t(25) < 1.04, all p > 0.310 for TrkB mRNA). Thus, the observed mRNA expression changes in subjects with schizophrenia were not likely to be influenced by any of these factors.
Figure 6.
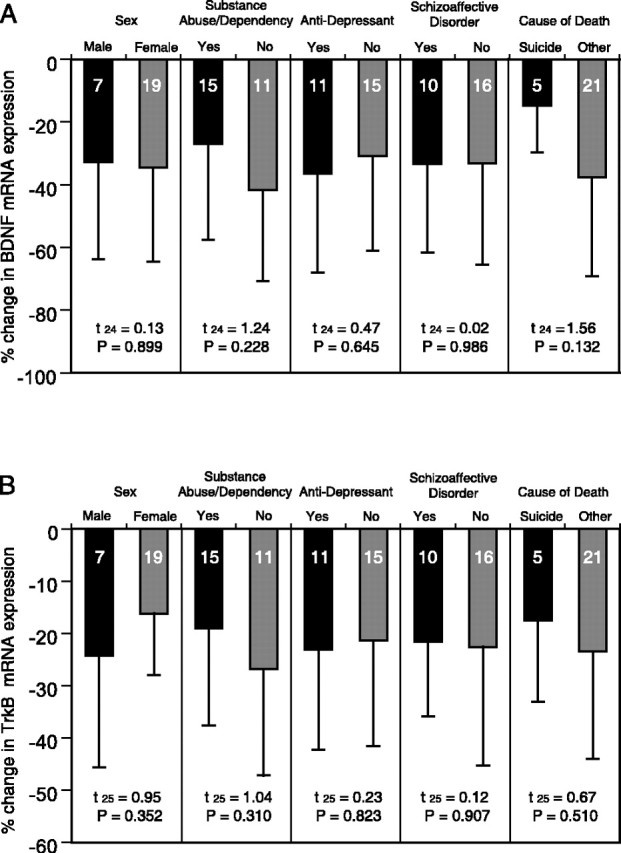
The effects of confounding factors on the expression changes in BDNF and TrkB mRNAs in schizophrenia. Mean ± SD percentage of differences from control subjects for BDNF (A) and TrkB (B) mRNAs within subject pairs grouped by potential confounding factors in subjects with schizophrenia. Neither sex, substance abuse/dependence history, history of antidepressant medications, diagnosis of schizoaffective disorder, nor cause of death significantly affected the expression changes in BDNF or TrkB mRNAs. For the comparison of BDNF mRNA expression change (A), pair 5 in cohort 2 was excluded. The numbers within each bar indicate the number of subject pairs.
To test the potential effect of antipsychotic medication, we evaluated the expression of BDNF and TrkB mRNAs in PFC areas 9 and 46 of monkeys exposed to haloperidol and benztropine mesylate (administered to relieve extra-pyramidal side effects) for 9-12 months (Pierri et al., 1999). The expression of BDNF and TrkB mRNAs (Fig. 7) did not differ between haloperidol-exposed monkeys and their sex-, age-, and weight-matched control monkeys in either areas 9 or 46 (all t(3) < 1.06; all p > 0.369). These findings suggest that the alterations in BDNF and TrkB mRNA expression in the PFC of subjects with schizophrenia are not likely to be attributable to antipsychotic medications. Consistent with this interpretation, the mean percentage of decreases in these mRNAs across the four schizophrenia subjects who were not receiving antipsychotic medications at the time of death (25% for BDNF and 30% for TrkB) did not differ (t(24) = 0.59, p = 0.559 for BDNF mRNA; t(25) = 0.96, p = 0.347 for TrkB mRNA) from those in the schizophrenia subjects who were receiving antipsychotics at the time of death (35% for BDNF mRNA and 21% for TrkB mRNA).
Figure 7.
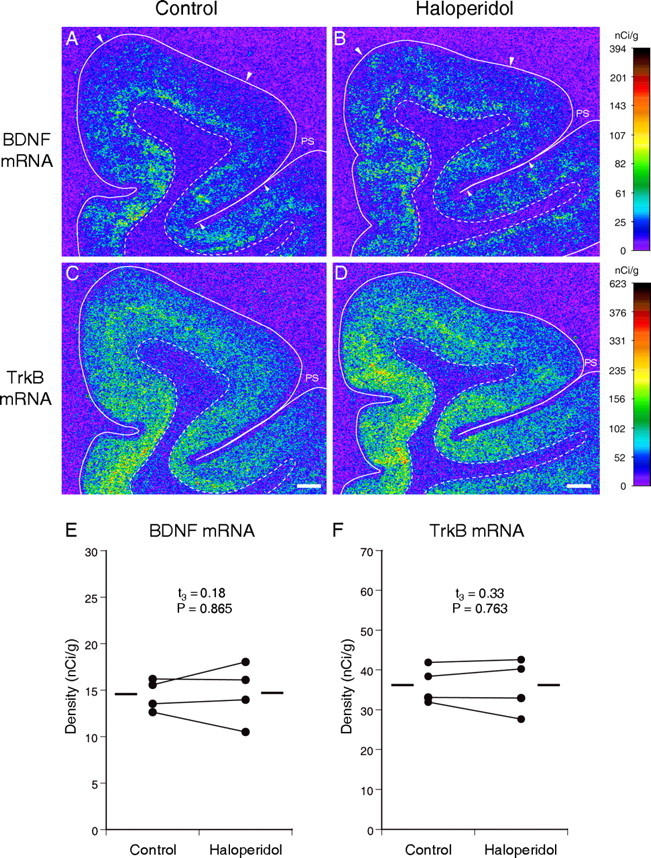
The effects of long-term exposure to haloperidol on expression of BDNF and TrkB mRNAs in monkey PFC. Representative autoradiograms illustrating the expression of BDNF (A, B) and TrkB (C, D) mRNAs in a control monkey (A, C) and an age-, sex-, and body weight-matched monkey exposed chronically to haloperidol (B, D). The densities of hybridization signals are presented in a pseudocolor manner according to the calibration scales (right) for each mRNA. For both mRNAs, the signal densities and distribution patterns appear to be unchanged in the PFC of the haloperidol-exposed monkey (B, D) compared with the control monkey (A, C). Solid and broken lines indicate the pial surface and the border between gray and white matter, respectively. PS, Principal sulcus. Pairs of large and small arrowheads (A, B) indicate the quantified regions in areas 9 and 46, respectively. Scale bars (in C, D), 1 mm. E, F, Pairwise comparisons of the mRNA expression levels for BDNF (E) and TrkB (F) in PFC area 9 between monkeys exposed to haloperidol and their sex-, age-, and weight-matched controls. The mean values for each group are indicated by horizontal bars.
GABA-related gene expression in genetically engineered mice
The strikingly strong correlations between the changes in TrkB mRNA expression and GABA-related gene expression suggested the importance of TrkB in the regulation of GABA-related gene expression changes in the PFC of subjects with schizophrenia. To test this hypothesis, we used trkB hypomorphic mice (Xu et al., 2000a) in which the disruption of trkB by floxed TrkB cDNA (fBZ locus) resulted in decreased TrkB expression in a gene dose-dependent manner (see Materials and Methods). We compared TrkB, GAD67, and PV mRNA expression levels in the PFC across three genotypes [wild-type, heterozygous (fBZ/+), and homozygous (fBZ/fBZ) mice for fBZ locus] at 8 weeks of age (Fig. 8). A single-factor ANOVA revealed a significant effect of genotype on the expression levels of TrkB (F(2,6) = 51.1; p < 0.001), GAD67 (F(2,6) = 11.1; p = 0.010), and PV (F(2,6) = 9.92; p = 0.013) mRNAs (Fig. 8J-L). Consistent with the previous reports (Xu et al., 2000a; Rohrer, 2001; Rico et al., 2002), TrkB mRNA expression levels were significantly (p < 0.05) decreased by 42 and 75% in mice with fBZ/+ and fBZ/fBZ genotypes, respectively, compared with wild-type mice (Fig. 8J). In fBZ/fBZ mice, PFC expression levels of GAD67 and PV mRNAs were significantly decreased (p < 0.05) by 23 and 40%, respectively, compared with wild-type mice (Fig. 8K,L). Additionally, in the fBZ/+ mice, the expression levels of GAD67 and PV mRNAs were intermediate between the wild-type control and fBZ/fBZ mice (Fig. 8K,L). In contrast, genotype had no significant effect on the expression of calretinin mRNA in the PFC (F(2,6) = 1.07; p = 0.401) (data not shown). In addition, BDNF mRNA expression levels did not differ between wild-type control and fBZ/fBZ mice (t(4) = 0.38; p = 0.722).
Figure 8.
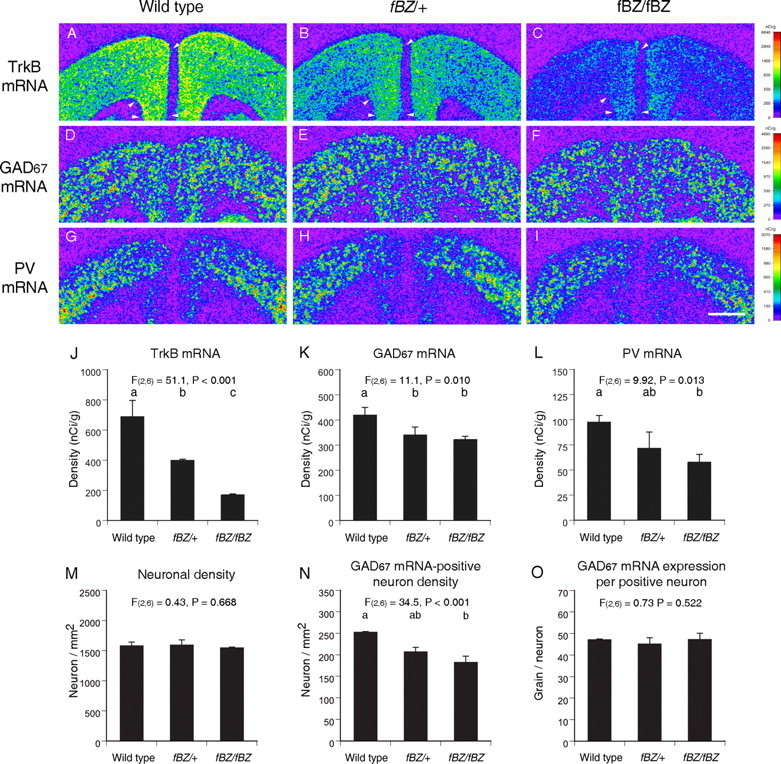
Gene expression changes in the PFC of mice with decreased TrkB expression. A-I, Representative autoradiograms illustrating TrkB (A-C), GAD67 (D-F), and PV (G-I) mRNA expression in a wild-type mouse (left), a mouse heterozygous for the fBZ locus (fBZ/+) (middle), and a mouse homozygous for the fBZ locus (fBZ/fBZ) (right) in adulthood. The densities of hybridization signals are presented in a pseudocolor manner according to the calibration scales (right) for each mRNA. Note that TrkB mRNA is expressed in proportion to the gene dose (A-C). GAD67 and PV mRNA expression levels appear to be decreased in the PFC of the fBZ/fBZ mouse (F, I), in which the expression of TrkB mRNA is remarkably decreased (C). The arrowheads in A-C indicate the quantified regions in the PFC (applies to all sections from each mouse). Scale bar (in I), 1 mm. J-L, Mean ± SD expression levels of TrkB (J), GAD67 (K), and PV (L) mRNAs in the PFC of adult mice with wild-type, fBZ/+, or fBZ/fBZ genotypes. M-O, Mean ± SD neuronal density (M), GAD67 mRNA-positive neuron density (N), and GAD67 mRNA expression per positive neuron (O). Bars not sharing the same alphabetical letter are statistically different (p < 0.05; post hoc Tukey's multiple comparison).
At the cellular level, decreased TrkB expression did not affect total neuron density (F(2,6) = 0.43; p = 0.668) (Fig. 8M). The density of neurons with detectable levels of GAD67 mRNA, however, was significantly decreased in a manner directly related to the trkB gene dose (F(2,6) = 34.5; p < 0.001) (Fig. 8N). In contrast, grain density per GAD67 mRNA-positive neuron did not differ across the three groups of mice (F(2,6) = 0.73; p = 0.522) (Fig. 8O), indicating that the deficit in GAD67 mRNA expression was restricted to a subpopulation of GABA neurons.
We next assessed the effect of decreased BDNF expression on GABA-related gene expression using mice with a neuron-specific inducible knock-out of bdnf (Fig. 9). As expected, BDNF mRNA expression was dramatically decreased (F(1,10) = 34.7; p < 0.001) by 80% in the PFC of mice with the bdnf knock-out induced from embryogenesis at 5 months of age (Fig. 9A,B,I). However, in contrast to the mice with decreased TrkB expression, no difference in the expression levels of GAD67 (F(1,10) = 0.26; p = 0.620) or PV (F(1,10) = 0.52; p = 0.489) mRNAs were present between the knock-out and wild-type control animals (Fig. 9K,L). Interestingly, the average TrkB mRNA expression level was increased by 20% in the knock-out mice compared with the wild-type controls (Fig. 9J), although this increase did not achieve statistical significance (F(1,10) = 4.29; p = 0.065). We obtained similar results in mice with the bdnf knock-out induced in adulthood. Mean BDNF mRNA expression levels in the PFC of these mice were decreased by 80% (F(1,14) = 40.4; p < 0.001) compared with the wild-type controls. However, no change was detected in expression levels of TrkB (F(1,14) = 0.82; p = 0.382), GAD67 (F(1,14) = 0.02; p = 0.888), or PV (F(1,14) = 0.18; p = 0.675) mRNAs.
Figure 9.
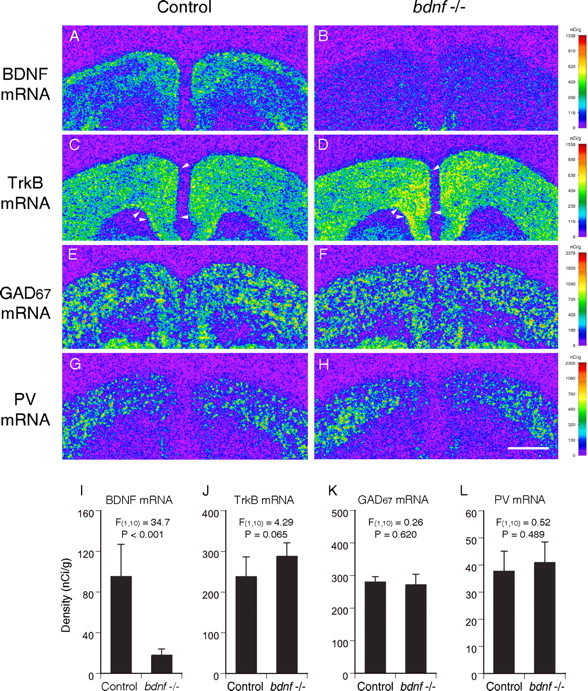
Gene expression in the PFC of mice with an inducible neuron-specific bdnf knock-out in embryogenesis. Representative autoradiograms (A-H) illustrating the expression of BDNF (A, B), and TrkB (C, D), GAD67 (E, F), and PV (G, H) mRNAs in a control mouse (A, C, E, G) and a mouse with embryonic bdnf knock-out (B, D, F, H) at 5 month of age are shown. The densities of hybridization signals are presented in a pseudocolor manner according to the calibration scales (right) for each mRNA. Note that BDNF mRNA expression was dramatically decreased in the knock-out mouse (B) compared with the control (A). GAD67 (E, F) and PV (G, H) mRNA expression levels appear to be similar between knock-out and control mice, whereas the expression level of TrkB mRNA is higher in the knock-out mouse (D) than the control mouse (C). The arrowheads in C and D indicate the quantified regions in the PFC in all sections from each mouse. Scale bar (in H), 1 mm. I-L, Mean ± SD expression levels of BDNF (I), TrkB (J), GAD67 (K), and PV (L) mRNAs in control and embryonic bdnf knock-out mice. Although BDNF mRNA expression levels are significantly decreased by 80% in embryonic knock-out mice (I), the expression levels of both GAD67 and PV mRNAs are unchanged (K, L). The expression level of TrkB mRNA is increased by 20% in embryonic knock-out mice with marginal significance.
Together, these findings suggested that changes in TrkB expression, and not in BDNF expression, play a primary role in the regulation of GABA-related gene expression in the adult PFC. Indeed, across the animals with three trkB genotypes (wild type, fBZ/+, and fBZ/fBZ), the expression levels of GAD67 and PV mRNAs were highly correlated with the level of TrkB mRNA (r = 0.91, p = 0.001 and r = 0.89, p = 0.001, respectively).
Discussion
In this study, expression levels of both BDNF and TrkB mRNAs were found to be significantly decreased in the PFC of two cohorts of subjects with schizophrenia. The following lines of evidence suggest that alterations in BDNF and TrkB mRNA expression are specific to the disease process of schizophrenia, or at least not a consequence of other factors commonly associated with the illness. First, the reduced levels of BDNF and TrkB mRNAs do not appear to be attributable to the pharmacological treatment of schizophrenia, because the expression of these mRNAs was unchanged in the PFC of monkeys after long-term exposure to haloperidol (Fig. 7). These findings are consistent with previous studies reporting no effects of chronic exposure to antipsychotic drugs on the levels of BDNF mRNA and protein or the expression of TrkB mRNA in the rodent frontal cortex (Nibuya et al., 1995; Linden et al., 2000; Takahashi et al., 2000; Lipska et al., 2001) (but see Angelucci et al., 2000; Dawson et al., 2001). The results from animal experiments are also complemented by the findings of reduced levels of both BDNF and TrkB mRNAs in the subjects with schizophrenia who were not receiving antipsychotic medications at the time of death.
Second, the available data indicate that comorbid substance abuse and/or dependence did not contribute to the decreases in BDNF or TrkB mRNA expression. For example, relative to their matched controls, the change in BDNF and TrkB mRNA levels did not differ between schizophrenia subjects with or without history of substance abuse/dependence (Fig. 6). In addition, a control subject with a history of alcohol abuse had greater expression levels for both BDNF and TrkB mRNAs than the matched schizophrenia subject. Finally, the four control subjects with positive plasma alcohol levels at the time of death still had higher expression levels of both mRNAs than their matched schizophrenia subjects.
Third, although reductions in BDNF and TrkB mRNA levels have been reported in the PFC and hippocampus of suicide victims (Dwivedi et al., 2003), reduced levels of these mRNAs were still present in our study in those subjects with schizophrenia who did not die by suicide (Fig. 6). Some subjects with schizophrenia had been treated with antidepressants. However, because these agents have been reported to increase BDNF expression (Nibuya et al., 1995), they are more likely to have obscured rather than to have caused the observed decreases in gene expression. In fact, a history of antidepressant treatment did not have a significant effect on the expression changes in BDNF or TrkB mRNAs in subjects with schizophrenia (Fig. 6).
Finally, premortem agonal state events associated with hypoxia and acidosis may affect postmortem levels of some RNA species. Brain pH, an inverse correlate of the length/severity of the agonal state (Harrison et al., 1995), was included as a covariate in testing the effect of the diagnosis on gene expression and did not differ between the control and schizophrenia groups. In addition, the observed mRNA expression changes were not attributable to a general deterioration of mRNA integrity in subjects with schizophrenia, because the expression of both TrkC and calretinin (Hashimoto et al., 2003) mRNAs and multiple other transcripts (Glantz et al., 2000; Mirnics et al., 2000; Middleton et al., 2002) did not differ between the subject groups in the first cohort.
Because BDNF-TrkB signaling has been reported to influence the development of cortical GABA neurons, including the expression of both GAD67 and PV (Jones et al., 1994; Alcantara et al., 1997; Huang et al., 1999; Yamada et al., 2002; Cotrufo et al., 2003), we hypothesized that altered BDNF-TrkB signaling might be an “upstream” event that contributed to the altered expression of GABA-related genes in the PFC of subjects with schizophrenia. Consistent with this hypothesis, the changes in GAD67 mRNA expression were significantly correlated with those in both BDNF and TrkB mRNA expression across subject pairs. Interestingly, the correlation was significantly higher between TrkB and GAD67 mRNA than between BDNF and GAD67 mRNA, suggesting the importance of TrkB in the regulation of GABA-related gene expression. However, these correlational data from human studies cannot reveal the mechanistic relationships between the decreased TrkB expression and the deficits in GABA-related gene expression in the PFC of subjects with schizophrenia.
To directly test the effect of reduced TrkB expression on GABA-related gene expression, we used mice heterozygous and homozygous for the fBZ locus, in which PFC TrkB mRNA expression levels were 58 and 25% of wild-type animals, respectively. As predicted, both GAD67 and PV mRNA expression levels were decreased in a manner directly related to trkB gene dose, confirming the importance of TrkB signaling in the regulation of gene expression in PV-containing GABA neurons. In addition, consistent with the selective expression of TrkB in PV-containing GABA neurons, mRNA levels for calretinin were not altered in either the mice with reduced TrkB expression or the human subjects with schizophrenia. Thus, reduced expression of TrkB mRNA appears to be sufficient to produce the altered expression of GAD67 and PV mRNAs in the PFC of subjects with schizophrenia.
In contrast, despite an 80% reduction in BDNF mRNA levels in the PFC of mice with an inducible knock-out of bdnf, the expression levels of GAD67 and PV were not altered. Recently, He et al. (2004) reported that a conditional knock-out of bdnf (which induced an 80% decrease in hippocampal BDNF levels) caused only a slight change in epileptogenesis in the kindling model, whereas a conditional knock-out of trkB suppressed it. Importantly, in the hippocampus of the bdnf knock-out mice, neither the expression level of TrkB nor its activation was changed compared with wild-type animals. Together, these findings suggest that the decreased availability of BDNF alone has a limited effect on downstream outcomes compared with the decreased expression of TrkB.
He et al. (2004) also detected an increased expression of NT-3, but not of NT-4, in the hippocampus of their bdnf knock-out mice. Although NT-3 expression may be increased in the PFC of subjects with schizophrenia, any activation of TrkB by NT-3 may be expected to be limited when the expression of TrkB itself is decreased (Table 1).
Table 1.
Summary of mRNA expression changes
|
|
Schizophrenia |
fBZ/fBZ |
bdnf-/-Em |
bdnf-/-Ad |
|---|---|---|---|---|
| BDNF | ↓ 35%a | ↔ | ↓ 80% | ↓ 80% |
| TrkB | ↓ 23%b | ↓ 75% | ↑ 20% (p = 0.07) | ↔ |
| GAD67 | ↓ 26%b | ↓ 23% | ↔ | ↔ |
| PV |
↓ 30%c
|
↓ 40%
|
↔ |
↔ |
Percentages of changes from control subjects or from wild-type animals are presented with arrows indicating the direction of changes. Statistically significant changes (p < 0.05) are expressed in bold.
Average across 26 pairs in cohorts 1 and 2 (without pair 5 in cohort 2).
Average across 27 pairs in cohorts 1 and 2.
Average across 15 pairs in cohort 1 [bdnf-/-Em knock-out induced from late embryogenesis and bdnf-/-Ad knock-out induced in adulthood (12 weeks of age)].
Previous postmortem studies, including our analyses of the same subjects used in this study, provide insight into the neuronal substrate of the observed mRNA changes. For example, total neuron number in the PFC of subjects with schizophrenia has been reported to be unchanged (Thune et al., 2001). Furthermore, in PFC area 9, the density of pyramidal neurons has been reported to be slightly increased across cortical layers (Selemon et al., 1995) or to be unchanged in layer III (Pierri et al., 2003). Because BDNF mRNA is expressed by cortical pyramidal neurons, the decrease in BDNF mRNA appears to be attributable to a downregulation of BDNF mRNA in existing pyramidal neurons, and not to a reduction in the number of these neurons. Likewise, the density of all nonpyramidal neurons has been reported to be slightly increased (Selemon et al., 1995) or unchanged (Akbarian et al., 1995) in the PFC of subjects with schizophrenia, and, in particular, the densities of PV-immunoreactive (Woo et al., 1997; Beasley et al., 2002) and PV mRNA-positive (Hashimoto et al., 2003) neurons were not altered, indicating that the decreases in GAD67 and PV mRNA expression levels are attributable to decreased gene expression in existing GABA neurons. The evidence that neither pyramidal nor nonpyramidal neurons in the PFC are lost in schizophrenia indicates that the decreased TrkB mRNA expression is attributable to downregulation in existing pyramidal and nonpyramidal neurons. Similar to these observations in the PFC of subjects with schizophrenia, neuronal density was not reduced in the PFC of trkB hypomorphic mice, indicating that the decreases in mRNA expression were not attributable to the loss of neurons. Furthermore, the significant decrease in GAD67 mRNA-positive neuron density without a change in the mRNA expression levels per positive neuron indicates that GAD67 mRNA expression was reduced in a subset of GABA neurons in the trkB hypomorphic mice. These findings directly parallel the pattern of altered GAD67 mRNA expression in schizophrenia (Volk et al., 2000). Thus, the alterations in GABA neurons in trkB hypomorphic mice replicate those found in subjects with schizophrenia at both the tissue and cellular levels.
In both subjects with schizophrenia and trkB hypomorphic mice, the observed deficits in expression of GAD67 or PV mRNAs may be the direct result of reduced TrkB expression in GABA neurons and/or an indirect consequence of alterations in pyramidal neurons. BDNF-TrkB signaling has been reported to enhance the somatodendritic development (McAllister et al., 1995; Xu et al., 2000b; Horch and Katz, 2002; Gorski et al., 2003) and spine formation (Horch et al., 1999) of pyramidal neurons. Interestingly, pyramidal neurons have been reported to have decreased somal size, dendritic length, and spine density in the PFC of subjects with schizophrenia (Garey et al., 1998; Rajkowska et al., 1998; Glantz and Lewis, 2000; Pierri et al., 2001), consistent with an effect of reduced BDNF-TrkB signaling on pyramidal neurons in the illness. These findings, in concert with evidence that BDNF-TrkB signaling directly affects the efficacy of excitatory neurotransmission among pyramidal neurons (Kang and Schuman, 1995; Xu et al., 2000a), suggest that the reduced expression of TrkB in pyramidal neurons may cause a decrease in their activity. Such a reduction in the activity of pyramidal neurons may, in turn, alter gene expression in GABA neurons, especially those containing PV, which receive direct excitatory inputs from neighboring pyramidal neurons (Melchitzky and Lewis, 2003).
The findings of this study provide both convergent evidence in the disease state and a proof of concept test in genetically modified animals that a reduction in signaling via the TrkB receptor contributes to altered gene expression in the PV-containing subpopulation of PFC GABA neurons in schizophrenia. The importance of this pathogenetic mechanism is demonstrated by the evidence suggesting that these gene expression alterations in PV-containing GABA neurons have functional consequences that likely contribute to the cognitive deficits of schizophrenia (Lewis et al., 2004).
Footnotes
This work was supported by National Institutes of Health Grants MH43784 (D.A.L.), MH45156 (D.A.L.), and NS16033, by an investigator-initiated grant from Pfizer (D.A.L.), and by a National Alliance for Research on Schizophrenia and Depression Young Investigator Award (T.H.). We thank Sarah Ng, Aniko Evans, Leah M. Konopka, Erastus Allen, and Mary L. Brady for technical assistance.
Correspondence should be addressed to Dr. David A. Lewis, Department of Psychiatry, University of Pittsburgh, W1650 Biomedical Science Tower, 3811 O'Hara Street, Pittsburgh, PA 15213. E-mail: lewisda@upmc.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250372-12$15.00/0
References
- Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney Jr WE, Jones EG (1995) Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry 52: 258-266. [DOI] [PubMed] [Google Scholar]
- Alcantara S, Frisen J, del Rio JA, Soriano E, Barbacid M, Silos-Santiago I (1997) TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell death. J Neurosci 17: 3623-3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelucci F, Mathe AA, Aloe L (2000) Brain-derived neurotrophic factor and tyrosine kinase receptor trkB in rat brain are significantly altered after haloperidol and risperidone administration. J Neurosci Res 60: 783-794. [DOI] [PubMed] [Google Scholar]
- Arenas E, Akerud P, Wong V, Boylan C, Persson H, Lindsay RM, Altar CA (1996) Effects of BDNF and NT-4/5 on striatonigral neuropeptides or nigral GABA neurons in vivo. Eur J Neurosci 8: 1707-1717. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Zhang ZJ, Patten I, Reynolds GP (2002) Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry 52: 708-715. [DOI] [PubMed] [Google Scholar]
- Blum BP, Mann JJ (2002) The GABAergic system in schizophrenia. Int J Neuropsychopharmacol 5: 159-179. [DOI] [PubMed] [Google Scholar]
- Bolton MM, Pittman AJ, Lo D (2000) Brain-derived neurotrophic factor differentially regulates excitatory and inhibitory synaptic transmission in hippocampal cultures. J Neurosci 20: 3221-3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellerino A, Maffei L, Domenici L (1996) The distribution of brain-derived neurotrophic factor and its receptor trkB in parvalbumin-containing neurons of the rat visual cortex. Eur J Neurosci 8: 1190-1197. [DOI] [PubMed] [Google Scholar]
- Chen J, Kelz MB, Zeng G, Sakai N, Steffen C, Shockett PE, Picciotto MR, Duman RS, Nestler EJ (1998) Transgenic animals with inducible, targeted gene expression in brain. Mol Pharmacol 54: 495-503. [DOI] [PubMed] [Google Scholar]
- Constantinidis C, Williams GV, Goldman-Rakic PS (2002) A role for inhibition in shaping the temporal flow of information in prefrontal cortex. Nat Neurosci 5: 175-180. [DOI] [PubMed] [Google Scholar]
- Cotrufo T, Viegi A, Berardi N, Bozzi Y, Mascia L, Maffei L (2003) Effects of neurotrophins on synaptic protein expression in the visual cortex of darkreared rats. J Neurosci 23: 3566-3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson NM, Hamid EH, Egan MF, Meredith GE (2001) Changes in the pattern of brain-derived neurotrophic factor immunoreactivity in the rat brain after acute and subchronic haloperidol treatment. Synapse 39: 70-81. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN (2003) Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry 60: 804-815. [DOI] [PubMed] [Google Scholar]
- Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, Barnes TRE, Hirsch SR (1998) Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry 65: 446-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA (1997) Reduction of synaptophysin immunoreactivity in the prefrontal cortex of subjects with schizophrenia: regional and diagnostic specificity. Arch Gen Psychiatry 54: 943-952. [DOI] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA (2000) Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57: 65-73. [DOI] [PubMed] [Google Scholar]
- Glantz LA, Austin MC, Lewis DA (2000) Normal cellular levels of synaptophysin mRNA expression in the prefrontal cortex of subjects with schizophrenia. Biol Psychiatry 48: 389-397. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS (1994) Working memory dysfunction in schizophrenia. J Neuropsychiatry Clin Neurosci 6: 348-357. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS (1995) Cellular basis of working memory. Neuron 14: 477-485. [DOI] [PubMed] [Google Scholar]
- Gorba T, Wahle P (1999) Expression of trkB and trkC but not BDNF mRNA in neurochemically identified interneurons in rat visual cortex in vivo and in organotypic cultures. Eur J Neurosci 11: 1179-1190. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Zeiler SR, Tamowski S, Jones KR (2003) Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci 23: 6856-6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A, Auta J, Davis JM, Gerevini VD, Dwivedi Y, Grayson DR, Impagnatiello F, Pandey G, Pesold C, Sharma R, Uzunov D, Costa E (2000) Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder. Arch Gen Psychiatry 57: 1061-1069. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Heath PR, Eastwood SL, Burnet PWJ, McDonald B, Pearson RCA (1995) The relative importance of premortem acidosis and postmortem interval for human brain gene expression studies: selective mRNA vulnerability and comparison with their encoded proteins. Neurosci Lett 200: 151-154. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Okuno H, Tokuyama W, Li YX, Miyashita Y (2000) Expression of brain-derived neurotrophic factor, neurotrophin-3 and their receptor messenger RNAs in monkey rhinal cortex. Neuroscience 95: 1003-1010. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA (2003) Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci 23: 6315-6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Yamashita A, Shimizu K (1997) Somatostatin and brainderived neurotrophic factor mRNA expression in the primate brain: decreased levels of mRNAs during aging. Brain Res 749: 283-289. [DOI] [PubMed] [Google Scholar]
- He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO (2004) Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron 43: 31-42. [DOI] [PubMed] [Google Scholar]
- Horch HW, Katz LC (2002) BDNF release from single cells elicits local dendritic growth in nearby neurons. Nat Neurosci 5: 1177-1184. [DOI] [PubMed] [Google Scholar]
- Horch HW, Krüttgen A, Portbury SD, Katz LC (1999) Destabilization of cortical dendrites and spines by BDNF. Neuron 23: 353-364. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S (1999) BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98: 739-755. [DOI] [PubMed] [Google Scholar]
- Jones KR, Farinas I, Backus C, Reichardt LF (1994) Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 76: 899-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Schuman EM (1995) Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 267: 1658-1662. [DOI] [PubMed] [Google Scholar]
- Kohara K, Kitamura A, Adachi N, Nishida M, Itami C, Nakamura S, Tsumoto T (2003) Inhibitory but not excitatory cortical neurons require presynaptic brain-derived neurotrophic factor for dendritic development, as revealed by chimera cell culture. J Neurosci 23: 6123-6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Lieberman JA (2000) Catching up on schizophrenia: natural history and neurobiology. Neuron 28: 325-334. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Volk DW, Hashimoto T (2004) Selective alterations in prefrontal cortical GABA neurotransmission in schizophrenia: a novel target for the treatment of working memory dysfunction. Psychopharmacology 174: 143-150. [DOI] [PubMed] [Google Scholar]
- Linden AM, Vaisanen J, Lakso M, Nawa H, Wong G, Castren E (2000) Expression of neurotrophins BDNF and NT-3, and their receptors in rat brain after administration of antipsychotic and psychotrophic agents. J Mol Neurosci 14: 27-37. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Khaing ZZ, Weickert CS, Weinberger DR (2001) BDNF mRNA expression in rat hippocampus and prefrontal cortex: effects of neonatal ventral hippocampal damage and antipsychotic drugs. Eur J Neurosci 14: 135-144. [DOI] [PubMed] [Google Scholar]
- Marty S, Wehrle R, Sotelo C (2000) Neuronal activity and brain-derived neurotrophic factor regulate the density of inhibitory synapses in organotypic slice cultures of postnatal hippocampus. J Neurosci 20: 8087-8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Lo DC, Katz LC (1995) Neurotrophins regulate dendritic growth and developing visual cortex. Neuron 15: 791-803. [DOI] [PubMed] [Google Scholar]
- Melchitzky DS, Lewis DA (2003) Pyramidal neuron local axon terminals in monkey prefrontal cortex: differential targeting of subclasses of GABA neurons. Cereb Cortex 13: 452-460. [DOI] [PubMed] [Google Scholar]
- Meng X-L, Rosenthal R, Rubin DB (1992) Comparing correlated correlation coefficients. Psychol Bull 111: 172-175. [Google Scholar]
- Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P (2002) Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci 22: 2718-2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P (2000) Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron 28: 53-67. [DOI] [PubMed] [Google Scholar]
- Mizuno K, Carnahan J, Nawa H (1994) Brain-derived neurotrophic factor promotes differentiation of striatal GABAergic neurons. Dev Biol 165: 243-256. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, Meuth S, Nagy A, Greene RW, Nestler EJ (2004) Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci USA 101: 10827-10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neter J, Kutner MH, Nachtsheim CJ, Wasserman W (1996) Applied linear statistical models, Ed 4. Boston: McGraw-Hill.
- Nibuya M, Morinobu S, Duman RS (1995) Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci 15: 7539-7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnuma T, Augood SJ, Arai H, McKenna PJ, Emson PC (1999) Measurement of GABAergic parameters in the prefrontal cortex in schizophrenia: focus on GABA content, GABAA receptor α-1 subunit messenger RNA and human GABA transporter-1 (HGAT-1) messenger RNA expression. Neuroscience 93: 441-448. [DOI] [PubMed] [Google Scholar]
- Okuno H, Tokuyama W, Li YX, Hashimoto T, Miyashita Y (1999) Quantitative evaluation of neurotrophin and trk mRNA expression in visual and limbic areas along the occipito-temporo-hippocampal pathway in adult macaque monkeys. J Comp Neurol 408: 378-398. [PubMed] [Google Scholar]
- Paxinos G, Franklin K (2001) The mouse brain in stereotaxic coordinates, Ed 2. San Diego: Academic.
- Perl AK, Wert SE, Nagy A, Lobe CG, Whitsett JA (2002) Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci USA 99: 10482-10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierri JN, Chaudry AS, Woo T-UW, Lewis DA (1999) Alterations in chandelier neuron axon terminals in the prefrontal cortex of schizophrenic subjects. Am J Psychiatry 156: 1709-1719. [DOI] [PubMed] [Google Scholar]
- Pierri JN, Volk CLE, Auh S, Sampson A, Lewis DA (2001) Decreased somal size of deep layer 3 pyramidal neurons in the prefrontal cortex of subjects with schizophrenia. Arch Gen Psychiatry 58: 466-473. [DOI] [PubMed] [Google Scholar]
- Pierri JN, Volk CLE, Auh S, Sampson A, Lewis DA (2003) Somal size of prefrontal cortical pyramidal neurons in schizophrenia: differential effects across neuronal subpopulations. Biol Psychiatry 54: 111-120. [DOI] [PubMed] [Google Scholar]
- Radomska HS, Gonzalez DA, Okuno Y, Iwasaki H, Nagy A, Akashi K, Tenen DG, Huettner CS (2002) Transgenic targeting with regulatory elements of the human CD34 gene. Blood 100: 4410-4419. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Selemon LD, Goldman-Rakic PS (1998) Neuronal and glial somal size in the prefrontal cortex: a postmortem morphometric study of schizophrenia and Huntington disease. Arch Gen Psychiatry 55: 215-224. [DOI] [PubMed] [Google Scholar]
- Rao SG, Williams GV, Goldman-Rakic PS (1999) Isodirectional tuning of adjacent interneurons and pyramidal cells during working memory: evidence for microcolumnar organization in PFC. J Neurophysiol 81: 1903-1916. [DOI] [PubMed] [Google Scholar]
- Rao SG, Williams GV, Goldman-Rakic PS (2000) Destruction and creation of spatial tuning by disinhibition: GABAA blockade of prefrontal cortical neurons engaged by working memory. J Neurosci 20: 485-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico B, Xu B, Reichardt LF (2002) TrkB receptor signaling is required for establishment of GABAergic synapses in the cerebellum. Nat Neurosci 5: 225-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R (2001) Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol 15: 1748-1757. [DOI] [PubMed] [Google Scholar]
- Rohrer B (2001) Gene dosage effect of the TrkB receptor on rod physiology and biochemistry in juvenile mouse retina. Mol Vis 7: 288-296. [PubMed] [Google Scholar]
- Romanczyk TB, Weickert CS, Webster MJ, Herman MM, Akil M, Kleinman JE (2002) Alterations in trkB mRNA in the human prefrontal cortex throughout the lifespan. Eur J Neurosci 15: 269-280. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG (1998) BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron 21: 521-530. [DOI] [PubMed] [Google Scholar]
- Sawaguchi T, Matsumura M, Kubota K (1989) Delayed response deficits produced by local injection of bicuculline into the dorsolateral prefrontal cortex in Japanese macaque monkeys. Exp Brain Res 75: 457-469. [DOI] [PubMed] [Google Scholar]
- Selemon LD, Rajkowska G, Goldman-Rakic PS (1995) Abnormally high neuronal density in the schizophrenic cortex: a morphometric analysis of prefrontal area 9 and occipital area 17. Arch Gen Psychiatry 52: 805-818. [DOI] [PubMed] [Google Scholar]
- Solberg Y, White EL, Keller A (1988) Types and distribution of glutamic acid decarboxylase (GAD)-immunoreactive neurons in mouse motor cortex. Brain Res 459: 168-172. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Shirakawa O, Toyooka K, Kitamura N, Hashimoto T, Maeda K, Koizumi S, Wakabayashi K, Takahashi H, Someya T, Nawa H (2000) Abnormal expression of brain-derived neurotrophic factor and its receptor in the corticolimbic system of schizophrenic patients. Mol Psychiatry 5: 293-300. [DOI] [PubMed] [Google Scholar]
- Thune JJ, Uylings HBM, Pakkenberg B (2001) No deficit in total number of neurons in the prefrontal cortex in schizophrenics. J Psychiatry Res 35: 15-21. [DOI] [PubMed] [Google Scholar]
- Ventimiglia R, Mather PE, Jones BE, Lindsay RM (1995) The neurotrophins BDNF, NT-3 and NT-4/5 promote survival and morphological and biochemical differentiation of striatal neurons in vitro. Eur J Neurosci 7: 213-222. [DOI] [PubMed] [Google Scholar]
- Vicario-Abejon C, Collin C, McKay RDG, Segal M (1998) Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J Neurosci 18: 7256-7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk DW, Lewis DA (2002) Impaired prefrontal inhibition in schizophrenia: relevance for cognitive dysfunction. Physiol Behav 77: 501-505. [DOI] [PubMed] [Google Scholar]
- Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA (2000) Decreased GAD67 mRNA expression in a subset of prefrontal cortical GABA neurons in subjects with schizophrenia. Arch Gen Psychiatry 57: 237-245. [DOI] [PubMed] [Google Scholar]
- Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA (2001) GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. Am J Psychiatry 158: 256-265. [DOI] [PubMed] [Google Scholar]
- Weickert CS, Hyde TM, Lipska BK, Herman MM, Weinberger DR, Kleinman JE (2003) Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol Psychiatry 8: 592-610. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, Berman KF, Zec RF (1986) Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Arch Gen Psychiatry 43: 114-124. [DOI] [PubMed] [Google Scholar]
- Woo T-U, Miller JL, Lewis DA (1997) Shizophrenia and the parvalbumin-containing class of cortical local circuit neurons. Am J Psychiatry 154: 1013-1015. [DOI] [PubMed] [Google Scholar]
- Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF (2000a) The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci 20: 6888-6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Ruff NL, Zang YA, McConnell SK, Strycker MP, Reichardt LF (2000b) Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron 26: 233-245. [DOI] [PubMed] [Google Scholar]
- Yamada MK, Nakanishi K, Ohba S, Nakamura T, Ikegaya Y, Nishiyama N, Matsuki N (2002) Brain-derived neurotrophic factor promotes the maturation of GABAergic mechanisms in cultured hippocampal neurons. J Neurosci 22: 7580-7585. [DOI] [PMC free article] [PubMed] [Google Scholar]