

Abstract
Central glucocorticoid receptors (GRs) and NMDA receptors (NMDARs) have been shown to play a significant role in the mechanisms of neuropathic pain after peripheral nerve injury; however, how central GRs and NMDARs interact in this process remains unknown. Here we show that the expression and function of spinal NMDARs after peripheral nerve injury were modulated by central GRs. Chronic constriction nerve injury (CCI) in rats induced a time-dependent upregulation of NR1 and NR2 subunits of the NMDAR within the spinal cord dorsal horn ipsilateral to CCI. The upregulation of NMDARs was significantly diminished by intrathecal administration (twice daily for postoperative days 1-6) of either the GR antagonist RU38486 or an antisense oligonucleotide against GRs. Moreover, this CCI-induced expression of NMDARs was significantly attenuated in rats receiving intrathecal treatment with an interleukin-6 (IL-6) antiserum and in mice with protein kinase Cγ (PKCγ) knock-out. Because IL-6 and PKCγ mediated the upregulation of central GRs after CCI as demonstrated previously, the results suggest that IL-6 and PKCγ served as cellular mediators contributing to the GR-mediated expression of NMDARs after CCI. Functionally, nociceptive behaviors induced by NMDAR activation and CCI were reversed by a single intrathecal administration of the GR antagonist RU38486. Conversely, a single intrathecal injection with the noncompetitive NMDAR antagonist MK-801 reversed neuropathic pain behaviors exacerbated by the GR agonist dexamethasone in CCI rats. These data suggest that interactions between central GRs and NMDARs through genomic and nongenomic regulation may be an important mechanism critical to neuropathic pain behaviors in rats.
Keywords: glucocorticoid receptor, NMDA receptor, neuropathic pain, nerve injury, RU38486, interleukin, protein kinase C
Introduction
A large number of studies have shown that antagonizing the NMDA receptor (NMDAR) reduces neuropathic pain behaviors induced by peripheral nerve injury (Dubner, 1991; Yamamoto and Yaksh, 1992; Mao et al., 1995; Woolf and Mannion, 1999). Mechanistically, activation of NMDARs is considered to initiate intracellular cascades through calcium influx and activation of protein kinases, which in turn modulates cell membrane excitability and enhances nociceptive transmission (Mao et al., 1995; Woolf and Mannion, 1999). More recently, central glucocorticoid receptors (GRs) have been shown to be upregulated within the spinal cord dorsal horn after peripheral nerve injury, which also contributed to the mechanisms of neuropathic pain behaviors in rats (Wang et al., 2004). Although both GRs and NMDARs play a crucial role in the neural and molecular mechanisms of neuropathic pain, how central GRs and NMDARs interact in this process remains unknown.
An increasing body of evidence indicates that the function of NMDARs can be regulated by activation of central GRs. For example, activation of central GRs has been shown to potentiate NMDA-induced responses in dopamine-sensitive neurons within the ventral tegmental area (Cho and Little, 1999) and to regulate NMDAR-mediated long-term depression (Coussens et al., 1997). Moreover, neuronal degeneration in the rat magnocellular nucleus basalis induced by exogenous NMDA was enhanced by a GR agonist (Abraham et al., 2000), and conversely, neuronal apoptosis in the hippocampus resulting from a glucocorticoid challenge was mediated at least in part through NMDARs (Lu et al., 2003), supporting a functional interaction between GRs and NMDARs. Thus, it is possible that central GRs may modulate NMDARs after peripheral nerve injury, contributing to the mechanisms of neuropathic pain behaviors.
Cytokines play a significant role in inflammatory responses through intracellular mediators such as protein kinase C (PKC) and cAMP (Barkhudaryan and Dunn, 1999). Specifically, interleukins (ILs), such as IL-6, have been implicated in both peripheral and central responses to peripheral nerve injury (Deleo et al., 1996; Arruda et al., 1998; Murphy et al., 1999; Okamoto et al., 2001; Raghavendra et al., 2002; Schafers et al., 2003, Wang et al., 2004). Indeed, ILs have been shown to enhance conditioned fear memory mediated in part through the effects of glucocorticoids (Song et al., 2003) and contribute to the development of neuropathic pain behaviors in several animal models (Opree and Kress, 2000; Sweitzer et al., 2001; Milligan et al., 2003; Schafers et al., 2003; Watkins and Maier, 2003). Of interest is that the upregulation of GRs within the spinal cord dorsal horn after peripheral nerve injury was mediated at least in part through IL-6 and PKCγ (Wang et al., 2004), suggesting that cellular mediators such as IL-6 and PKCγ may contribute to the proposed interaction between GRs and NMDARs after peripheral nerve injury.
In a rat model of neuropathic pain behaviors induced by chronic constriction nerve injury (CCI) (Bennett and Xie, 1988), we examined the hypotheses that (1) peripheral nerve injury would alter the expression of spinal NMDARs, (2) altered expression of NMDARs would be modulated through activation of spinal GRs and cellular mediators such as IL-6 and PKCγ, and (3) a functional interaction between spinal GRs and NMDARs would contribute to nociceptive behaviors in rats.
Materials and Methods
Experimental animals and the CCI model. Adult male Sprague Dawley rats weighing 275-325 gm (Charles River Laboratories, Wilmington, MA) were used. The animal room was artificially lighted from 7 A.M. until 7 P.M.; water and food pellets were available ad libitum. The experimental protocol was approved by our Institutional Animal Care and Use Committee. CCI rats were produced by loosely ligating one common sciatic nerve under pentobarbital (50 mg/kg, i.p.) anesthesia according to the method of Bennett and Xie (1988). Sham rats were subjected to the same surgical procedure except for nerve ligation. In one experiment, PKCγ knock-out (KO) mice were used to examine the contribution of PKCγ to NMDAR expression after CCI. Both PKCγ knock-out mice and the matched wild-type (WT) mice were purchased from The Jackson Laboratory (Bar Harbor, ME).
Intrathecal catheterization and drug delivery. An intrathecal catheter was implanted in each rat under the same surgical conditions, and a PE10 catheter was inserted in the lumbar enlargement area (∼8.5 cm from the incision site for this rat age group) according to the method described previously (Mao et al., 2002). Those rats exhibiting postoperative neurological deficits (e.g., paralysis) or poor grooming were excluded from the experiments as previously described (Mao et al., 2002). The following drugs were purchased from Sigma (St. Louis, MO): RU38486, NMDA, MK-801, dexamethasone, and IL-6. RU38486 or dexamethasone was dissolved in 10% ethanol solution. NMDA, MK-801, or IL-6 was dissolved in normal saline. IL-6 antiserum was purchased from R & D Systems (Minneapolis, MN). The antibody was raised in goat (goat against rat), affinity purified, and diluted in PBS according to the manufacturer's information sheet. This antibody, showing <1% cross-reactivity with recombinant human IL-6, was selected for its ability to neutralize the biological activity of recombinant rat IL-6, and the neutralization dose50 is 0.03-0.09 ng/μl (0.03-0.09 μg/ml) (R & D Systems). There was 0.5 μg of IL-6 IgG in 10 μl, and a goat IgG, diluted in the same solution for IL-6 IgG with the same concentration, was used as control serum. All drugs were injected intrathecally in a 10 μl volume followed by a 10 μl saline flush. A vehicle or saline was used as control according to each agent.
For those experiments using GR oligonucleotides (ONDs), antisense sequences overlapping the respective initiation codon were designed for targeting GR mRNA: GR antisense (TGG AGT CCA TTG GCA AAT), GR sense (ATT TGC CAA TGG ACT CCA), and a mixed base (TGA AGT TCA GTG TCA ACT). Sequencing was performed by MWG-Biotech (High Point, NC). Oligonucleotides were dissolved in 0.9% saline and injected (5 μmol, i.t.) twice a day through an intrathecal catheter.
Behavioral tests and statistical analysis. Animals were habituated to the test environment daily (a 60 min session) for 2 d before the baseline testing. Testing for thermal hyperalgesia was performed according to a previously published method (Hargreaves et al., 1988). Temperature was set and maintained across testing day to result in a baseline latency of 10-12 sec and a cutoff latency of 20 sec throughout the experiment to avoid tissue damage. Mechanical allodynia was examined by using von Frey filaments applied to the plantar surface of each hindpaw (Tal and Bennett, 1994). The cutoff force was 20 gm. All tests were conducted between 9 A.M. and noon. Data from both thermal hyperalgesia (with-drawal latency in seconds) and mechanical allodynia (threshold bending force in grams) tests were analyzed by using repeated measure two-way ANOVA across testing time points to detect overall differences among treatment groups. Whenever applicable, the data were also examined by using repeated measure two-way ANOVA across treatment groups to examine overall differences among testing time points. In both cases, when significant main effects were observed, the post hoc Newman-Keuls tests were performed to determine sources of differences. Differences were considered to be statistically significant at the level of α = 0.05. The experiments were performed with the investigators being blind to treatment conditions.
Immunocytochemical staining. Immunocytochemical staining was used to detect GR (1:1000, rabbit polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA) and NR1 (1:500, mouse monoclonal; United States Biological) within the spinal cord. Lumbar spinal cord segments were mounted on a block and cut into 20-μm-thick sections using a cryostat. These sections were blocked with 1% goat serum in 0.3% Triton X-100 for 1 hr at room temperature and incubated overnight at 4°C with a primary antibody. For controls, a primary antibody was omitted and antigen absorption (for GR) was used in control sections. The sections were then incubated for 1 hr at room temperature with a corresponding FITC- or Cy3-conjugated secondary antibody (1:400; Chemicon, Temecula, CA). For double staining, a second primary antibody was added after incubation with the first primary antibody following the same procedure described above. Four to six nonadjacent spinal sections were randomly selected, analyzed using a fluorescence microscope (Olympus, Tokyo, Japan), recorded with a digital camera, and processed using Adobe Photoshop.
Western blot. Rats were killed rapidly (<1 min) by decapitation after anesthesia, and the lumbar spinal cord segments were removed. The lumbar segments were harvested because CCI has a main impact at these spinal segments and the intrathecal drug delivery also was aimed at this site. Lumbar spinal segments were divided into the ipsilateral and contralateral side as well as the dorsal and ventral part and homogenized in an SDS sample buffer containing a mixture of proteinase inhibitors (Sigma). Protein samples were separated on SDS-PAGE gel (4-15% gradient gel; Bio-Rad, Richmond, CA) and transferred to polyvinylidene difluoride filters (Millipore, Bedford, MA). The filters were blocked with 3% milk and incubated overnight at 4°C with a primary antibody (NR1 or NR2, 1:2000; mouse monoclonal from Novus Biological) and 1 hr at room temperature with HRP-conjugated secondary antibody (1:7000; Amersham Biosciences, Arlington Heights, IL).
The blots were visualized in ECL solution (DuPont NEN, Boston, MA) for 1 min and exposed to hyperfilms (Amersham Biosciences) for 1-10 min. The blots were then incubated in a stripping buffer (67.5 mm Tris, pH 6.8, 2% SDS, 0.7% β-mercaptoethanol) for 30 min at 50°C and reprobed with a polyclonal rabbit anti-β-actin antibody (1:20,000; Alpha Diagnostic International, San Antonio, TX) as loading controls. The Western analysis was performed in triplicate. The density of specific bands was measured with a computer-assisted imaging analysis system and normalized against corresponding loading control bands. Differences were compared using repeated measure one-way ANOVA followed by post hoc Newman-Keuls tests.
RT-PCR. Total RNA was extracted from the lumbar spinal cord segments and divided into the dorsal and ventral horn and ipsilateral and contralateral side, by using Trizol reagent (Tel-Test, Friendswood, TX). After incubation for 15 min at 4°C, chloroform was added for the phase separation. The upper aqueous phase was collected, and RNA was precipitated through mixing the sample with isopropyl alcohol. The RNA pellet was washed once with 75% ethanol, air dried, and finally redissolved in RNase-free water. The A260/A280 ratio was between 1.6 and 1.8.
RT-PCR was performed using the Titan One Tube RT-PCR System (Roche, Indianapolis, IN). Each PCR amplification contained a volume of 25 μl with 100 ng of total RNA, 0.4 μm each primer, 0.2 mm each deoxynucleotide (dNTP), 5 mm dithiothreitol, 5 U RNase inhibitor, 1.5 mm MgCl2, and 0.5 μl of enzyme mix. The reactions were performed in a thermocycler (MJ Research) using the following programs: NR1: forward primer (CTC ATC TCT AGC CAG GTC TA), reverse primer (TCG CAT CAT CTC AAA CCA GAC), program (94°C, 1 min; 55°C, 1 min; 72°C, 1 min); NR2: forward primer (TCC ATT CTT CTG TCA TCC TGC), reverse primer (AAG ACC GTC TCT CAC TCT TGC), program (95°C, 50 sec; 55°C, 1 min; 72°C, 1 min). There were 32-35 cycles for each program, and each program ended with 7 min at 68°C and was stored at 4°C. Every PCR was accompanied by one negative control reaction without template RNA. PCR products were analyzed by gel electrophoresis on an ethidium bromide-stained 1% agarose gel (Sigma) in a Trisborate-EDTA buffer. The amount of RNA per RT-PCR sample was normalized using PCR with primers specific for β-actin: forward primer (TAC AAC CTC CTT GCA CC), reverse primer (ACA ATG CCG TGT TCA ATG G), program (95°C, 1 min; 55°C, 1 min; 72°C, 1 min). Each band was then measured with a computer-assisted imaging analysis system and normalized against the loading control. Differences were compared using a one-way ANOVA followed by post hoc Newman-Keuls tests.
Results
Increased NMDAR expression after CCI
CCI but not sham operation increased the expression of both NR1 and NR2 subunits of the NMDAR within the spinal cord dorsal horn ipsilateral to the site of nerve injury as revealed by RT-PCR (Fig. 1a,b) (n = 4-6). The mRNA level for both NR1 and NR2 began to increase on postoperative day 3 in CCI rats and remained elevated when examined on postoperative days 7 and 14 (Fig. 1a,b) (p < 0.05). Because there were no significant differences between the expression of NR1 and NR2 subunits by RT-PCR, we focused on the expression of the NR1 subunit in the following experiment. Consistently, a similar pattern of NR1 expression was detected by Western blot in CCI rats compared with sham control (Fig. 1c,d) (n = 4-6; p < 0.05). There were no differences in the NR1 expression on the contralateral spinal cord dorsal horn between CCI and sham rats (Fig. 1a) (NR1 only). Thus, CCI induced a time-dependent and region-specific expression of NMDARs at both the mRNA level and the protein level.
Figure 1.
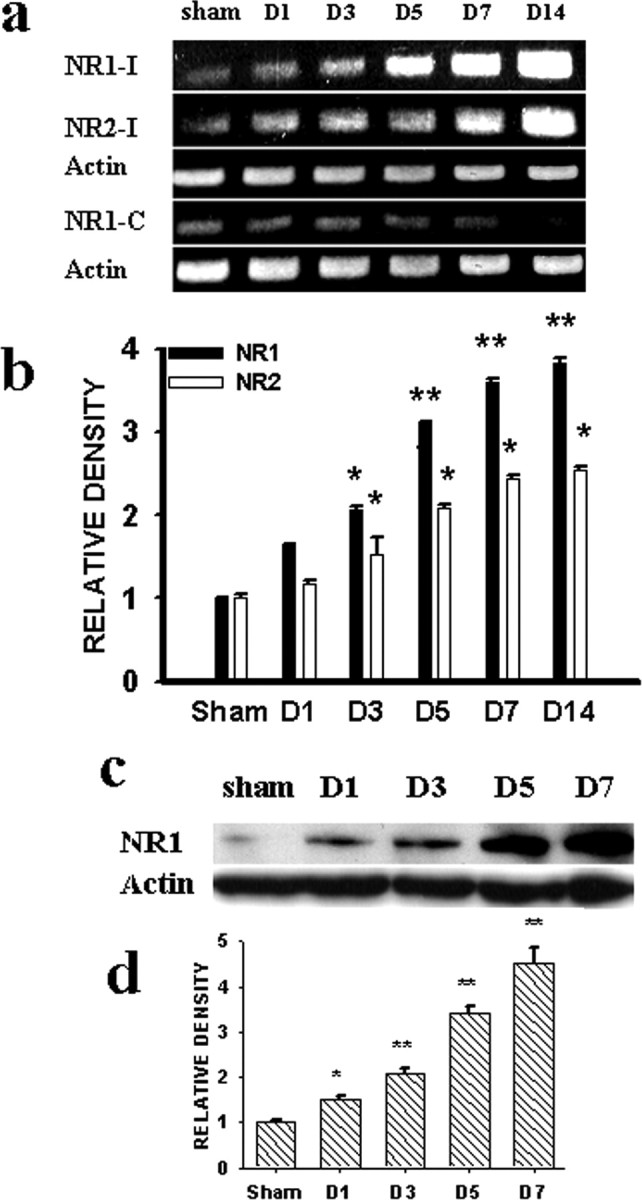
a, b, RT-PCR (a) and statistical analysis (b) showed the time course of NR1 and NR2 expression after CCI within the ipsilateral (I) and contralateral (C) (NR1 only) spinal cord dorsal horn. c, d, Western blot (c) (100 kDa), as well as statistical analysis (d), also showed an increased NR1 expression within the ipsilateral spinal cord dorsal horn in the CCI rats. In a and b, *p < 0.05 and **p < 0.01 compared with sham rats. In this figure, the data from day 7 after sham operation were presented as sham control because there were no differences in the expression of NR1 or NR2 in sham rats across postoperative days 1, 3, 5, and 7 in our pilot experiments. For all figures, the number of animals per group is given in Results. Actin, β-Actin for loading control in all panels.
Attenuated NMDAR expression by the GR antagonist RU38486
To examine the role of central GRs in the expression of NMDARs in CCI rats, we first asked whether there was a colocalization between central GRs and NMDARs by using an immunocytochemical technique. In this regard, a large number of cell profiles expressing NR1 immunoreactivity (NR1-ir) also expressed GR-ir within the spinal cord dorsal horn (Fig. 2a-c), indicating a close cellular association between GRs and NMDARs.
Figure 2.

a-c, Colocalization of GRs (a) and NR1 (b), as shown in the emerged image (c), was observed in the ipsilateral spinal cord dorsal horn of a representative CCI rat. The sample was taken at day 7 after CCI. DL, Dorsal lateral part of the dorsal horn. Scale bar, 60 μm. d, e, The GR antagonist RU38486 (2 μg, i.t.) given twice daily on postoperative days 1-6 attenuated the expression of NR1 (Western blot) within the ipsilateral spinal cord dorsal horn of CCI rats, when examined on postoperative day 7. *p < 0.05, compared with sham rats. S/V, Sham plus vehicle; C/V, CCI plus vehicle; C/RU, CCI plus RU38486. RU38486 alone did not change the baseline NR1 expression (data not shown).
Furthermore, the effect of the GR antagonist RU38486 on the expression of NMDARs after CCI was examined in four groups of rats (n = 4-5), including CCI plus vehicle, CCI plus RU38486 (2 μg), sham plus vehicle, and sham plus RU38486 (2 μg). Each agent was given intrathecally twice daily on postoperative days 1-6 beginning immediately after operation. The RU38486 dose (2 μg) was selected based on our pilot experiment using 0.5, 1, 2, or 4 μg of RU38486 (n = 2-3; data not shown) that showed a stable effect of 2 μg RU38486 on the NR1 expression. The NR1 expression was significantly attenuated in the CCI plus RU38486 group when examined on postoperative day 7 (Fig. 2d,c) (p < 0.05; n = 4-5), whereas RU38486 (2 μg) alone did not significantly change the NR1 expression in sham rats (p > 0.05; n = 4).
Attenuated NMDAR expression by a GR antisense OND
To further examine whether the interruption of GR expression would influence NMDAR expression in CCI rats, we used an antisense OND against GRs. The OND technique was used because GR knock-out mice are mostly lethal and unavailable commercially (Tronche et al., 1999; Gartner et al., 2002; St. Hilaire et al., 2003). In this experiment, CCI rats each received 5 μmol each of a GR antisense OND, sense OND, mixed-base OND, or vehicle (n = 8-10), given twice daily via an intrathecal catheter on postoperative day 1-6. The spinal cord samples were harvested on postoperative day 1 or 7 (n = 4-5 per group per day). The expression of both NR1 and NR2 subunits within the ipsilateral spinal cord dorsal horn was significantly reduced on postoperative day 7 in CCI rats receiving the GR antisense OND, compared with the group receiving either a sense or mixed-base OND (Fig. 3a,b) (p < 0.05; n = 5). When examined on day 7, the same antisense OND treatment resulted in a substantial reduction of GR expression within the spinal cord dorsal horn compared with the vehicle group (Fig. 3c,d) (p < 0.05; n = 4-5), a finding similar to that reported in a previous study (Wang et al., 2004). In addition, the GR antisense OND treatment did not change the NR1 and NR2 expression on the contralateral spinal cord dorsal horn in CCI rats (Fig. 3a), indicating a selective modulatory effect of central GRs on NMDAR expression after CCI.
Figure 3.
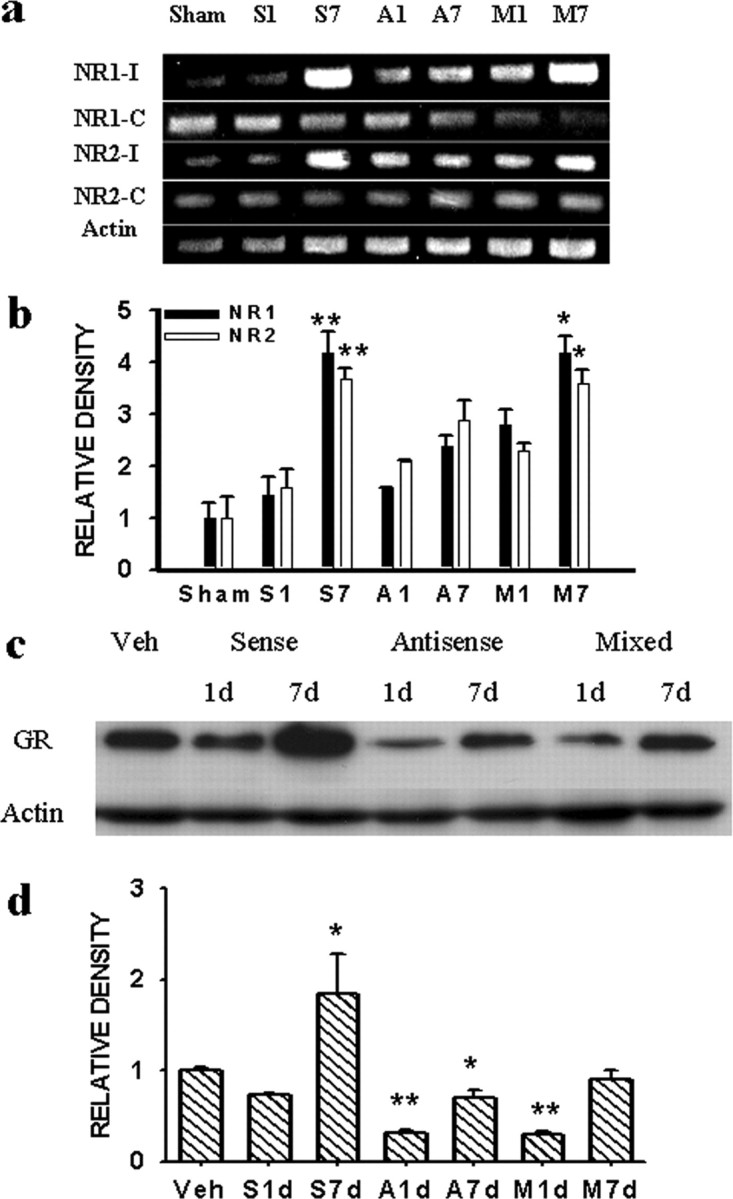
a, b, The expression of NR1 and NR2 (RT-PCR) was attenuated substantially in CCI rats treated with a GR antisense OND but not a sense or mixed-base OND. *p < 0.05 and **p < 0.01, compared with day 1 of each corresponding group. S1, S7; A1, A7; M1, M7: day 1 and day 7 from CCI rats treated with a GR sense (S), antisense (A), and mixed-base (M) OND, respectively. Sham, Sham control. c, d, GR expression (Western blot) (95 kDa) within the ipsilateral spinal cord dorsal horn was substantially attenuated in CCI rats treated with a GR antisense OND. *p < 0.05 and **p < 0.01, compared with CCI rats treated with vehicle (Veh). Actin, β-Actin (42kDa) for loading control. S1d-M7d: see legend for a and b.
Effect of IL-6 and PKCγ on NMDAR expression
Because both IL-6 and PKCγ mediated the expression of central GRs after CCI (Wang et al., 2004), we examined whether IL-6 or PKCγ would also modulate the expression of NMDARs in CCI rats. In this set of experiments, IL-6 antiserum (0.5 μg, i.t.), a dose referenced and modified from previous studies including ours (Milligan et al., 2003; Wang et al., 2004), or control serum was given once daily on postoperative days 1-6 beginning immediately after operation. IL-6 antiserum, but not control serum, significantly attenuated the NR1 expression in CCI rats when examined on postoperative day 7 (Fig. 4a) (p < 0.05, n = 4-5).
Figure 4.

a, IL-6 antiserum (0.5 μg, i.t.) given once daily on postoperative days 1-6 attenuated the NR1 expression within the ipsilateral spinal cord dorsal horn in CCI rats, when examined on day 7. *p < 0.05 compared with both sham rats and CCI rats treated with the IL-6 antiserum. C/V, CCI plus vehicle; S/V, sham plus vehicle; C/IS, CCI plus IL-6 antiserum. b, Time course of NR1 and NR2 expression (RT-PCR) within the spinal cord dorsal horn of naive rats induced by IL-6 (33 ng) or a vehicle given intrathecally once daily for 6 consecutive days. c, NR1 expression was also observed using Western blot, and NR1 upregulation returned to baseline at 7 d after the discontinuation of IL-6 treatment (D14). *p < 0.05 and **p < 0.01 compared with the corresponding vehicle (Veh) treatment group. D1-D14 refers to the samples taken on days 1-14 of the experimental period.
The role of IL-6 in NMDAR expression was further examined in naive rats receiving either exogenous IL-6 [33 ng, i.t.; a dose modified from the reports of Deleo et al. (1996) and Wang et al. (2004)] or vehicle once daily for 6 d. In those rats treated with exogenous IL-6, the expression of NR1 and NR2 (RT-PCR) was increased substantially within the spinal cord dorsal horn compared with the vehicle group (Fig. 4b) (p < 0.05; n = 4-5), which mimicked the time course of NMDAR expression after CCI (Fig. 1). The IL-6-induced NR1 expression returned to the baseline level at 7 d after the discontinuation of the IL-6 treatment (Fig. 4c) (p > 0.05; n = 5), indicating a specific effect of exogenous IL-6 on NMDAR expression.
Moreover, the NR1 expression within the ipsilateral spinal cord dorsal horn was nearly abolished in PKCγ KO mice compared with that in the matched PKCγ WT mice, when both groups were examined on postoperative day 7 (Fig. 5a,b) (p < 0.05; n = 5). Because there were no differences in the NR1 expression after CCI within the contralateral spinal cord dorsal horn between PKCγ KO and the matched PKCγ WT mice, the results indicated a critical role of intracellular PKCγ in the expression of NMDARs after CCI.
Figure 5.

a,b, NR1 expression (RT-PCR) within the ipsilateral spinal cord dorsal horn after CCI was nearly prevented in PKCγ KO mice compared with the matched PKCγ WT mice. Con., Contralateral; Ipsi., ipsilateral.
Reversal by RU38486 of NMDAR-mediated nociceptive behaviors
The role of central GRs in NMDAR-mediated nociceptive behaviors was examined in naive rats receiving either acute (a single injection) or chronic (twice daily injections for 3 d) treatment with the NMDAR agonist NMDA. Thermal hyperalgesia and mechanical allodynia induced by a single injection with NMDA (5 μg, i.t.) in naive rats, observed at 30 min after the NMDA injection, was prevented when RU38486 (2 μg, i.t., but not vehicle) was given 30 min before the NMDA injection (Fig. 6a,b) (p < 0.05; n = 5). The RU38486 dose was chosen based on our previous study showing that the same RU38486 dose attenuated the development of neuropathic pain behaviors in CCI rats (Wang et al., 2004). Similarly, persistent thermal hyperalgesia and mechanical allodynia induced by the 3 d NMDA (5 μg, i.t.) treatment in naive rats was reversed at 30 min after a single injection of RU38486 (2 μg, i.t.) on day 4 (Fig. 6c,d) (p < 0.05; n = 5). RU38486 alone did not change the baseline nociceptive threshold (p > 0.05; n = 4). The results indicated that RU38486 reversed NMDA-induced nociceptive behaviors in naive rats.
Figure 6.

a, b, A single injection of the NMDAR agonist NMDA (5 μg, i.t.) induced both thermal hyperalgesia (a) and mechanical allodynia (b) in naive rats when examined at 30 min after the NMDA injection, which was prevented by pretreatment with 2 μg of RU38486 (RU) at 30 min before the NMDA injection. c, d, A single injection of RU38486 (2 μg, i.t.) on day 4 reversed both thermal hyperalgesia (c) and mechanical allodynia (d) induced by repeated treatment with 5 μg of NMDA (twice daily for 3 d), when examined at 30 min after the RU38486 injection. *p < 0.05 compared with the baseline in each corresponding group. FWL, Foot-with-drawal latency. Threshold force, Threshold bending force from von Frey filament stimulation.
Reversal of neuropathic pain behaviors by RU38486
To examine whether RU38486 would reverse neuropathic pain behaviors in CCI rats, RU38486 (2 μg) or a vehicle was given intrathecally on postoperative day 7 when both thermal hyperalgesia and mechanical allodynia were clearly established in CCI rats. RU38486 reversed thermal hyperalgesia and mechanical allodynia when examined 30 min after the injection (Fig. 7a,b) (p < 0.05; n = 5), whereas RU38486 alone did not change the baseline nociceptive threshold in sham rats. Given that RU38486 (4 = 2 > 1 = 0.5 μg) also attenuated the development of neuropathic pain behaviors when given repeatedly in CCI rats (Wang et al., 2004), the results indicated that GRs contributed to both induction and maintenance of neuropathic pain behaviors after CCI.
Figure 7.

a, b, A single injection of RU38486 (2 μg, i.t.), but not vehicle, on postoperative day 7 reversed thermal hyperalgesia (a) and mechanical allodynia (b) in CCI rats when examined at 30 min after the RU38486 injection. *p < 0.05 compared with sham rats. C/RU, CCI plus RU38486; C/V, CCI plus vehicle. c, d, A single injection of the noncompetitive NMDAR antagonist MK-801 (10 nmol, i.t.) on day 7 reversed thermal hyperalgesia (c) and mechanical allodynia (d) exacerbated by the dexamethasone treatment (DEX) (4 μg, i.t.; twice daily for 6 d) in CCI rats, when examined at 30 min after the MK-801 injection. *p < 0.05 compared with sham rats. CCI, CCI rats; C/D/V, CCI plus DEX plus vehicle; C/D/MK, CCI plus DEX plus MK-801. FWL/Threshold force: see Figure 6 legend.
Reversal of neuropathic pain behaviors by MK-801
Thermal hyperalgesia and mechanical allodynia after CCI were exacerbated by the synthetic GR agonist dexamethasone (4 μg, i.t.) given twice daily for postoperative days 1-6 (Fig. 7c,d) (p < 0.05; n = 5). Dexamethasone was chosen as the GR agonist because this glucocorticoid has an almost exclusive effect on GRs (>30×) compared with its negligible effect on mineralocorticoid receptors, and our previous study (Wang et al., 2004) has shown that mineralocorticoid receptors within the spinal cord were not changed after peripheral nerve injury in the same CCI model. A single injection with the noncompetitive NMDAR antagonist MK-801 (10 nmol, i.t.) (Mao et al., 1992) reversed both thermal hyperalgesia and mechanical allodynia that were exacerbated by the GR agonist dexamethasone in CCI rats (Fig. 7c,d) (p < 0.05; n = 5). Neither dexamethasone nor MK-801 alone changed the baseline thermal or mechanical nociceptive threshold (p > 0.05; n = 4). The behavioral tests were made in each group before and at 30 min after the MK-801 treatment on day 7 after CCI.
Discussion
The present study demonstrates that (1) peripheral nerve injury induced a time-dependent and region-specific upregulation of the NR1 and NR2 subunits of the NMDAR within the spinal cord dorsal horn ipsilateral to CCI; (2) the upregulation of NMDARs was attenuated by either pharmacological inhibition of central GRs with RU38486 or interrupted GR expression using an antisense OND; (3) cellular mediators such as IL-6 and PKCγ contributed to the GR-mediated regulation of NMDAR expression; and (4) an interaction between central GRs and NMDARs contributed to the manifestation of nociceptive behaviors after CCI. These results indicate that the interaction between central GRs and NMDARs through genomic and nongenomic regulation may be an important mechanism critical to neuropathic pain behaviors in rats.
The rationale for examining the relationship between central GRs and NMDARs in CCI rats is twofold. First, a considerable number of previous studies support the notion that NMDARs play a pivotal role in the mechanisms of neuropathic pain (Yamamoto and Yaksh, 1992; Mao et al., 1992, 1995; Woolf and Mannion, 1999). Second, studies have shown that central GRs also contribute to the mechanisms of neuropathic pain (Wang et al., 2004) and activation of central GRs regulates the function of NMDARs. For example, GR activation has been shown to (1) potentiate NMDA-induced responses in dopamine-sensitive neurons within the ventral tegmental area (Cho and Little, 1999), (2) regulate NMDAR-mediated long-term depression (Coussens et al., 1997), (3) modulate Ca2+ channel conductance (Nair et al., 1998), (4) elevate intracellular Ca2+ concentrations in hippocampal neurons (Takahashi et al., 2002), and (5) mediate NMDA-induced neuronal degeneration in the rat magnocellular nucleus basalis (Abraham et al., 2000). At the cellular level, the modulatory effects of central GRs on NMDARs could take place through genomic and/or nongenomic mechanisms. The genomic mechanism of GR actions could modulate NMDAR expression through transcriptional and/or translational regulation, whereas the nongenomic mechanism would involve interactions at the receptor level between central GRs and NMDARs. Our data indicate that both genomic and nongenomic mechanisms are likely to be involved in the GR-mediated regulation of NMDARs after peripheral nerve injury.
Action of GRs through a genomic mechanism begins with activation of cytosolic GRs (Neeck et al., 2002). Activation of GRs requires the formation of a GR homodimer after dissociation from its cytosolic complex consisting of heat shock proteins (Drouin et al., 1992). A GR homodimer binds to nuclear-specific DNA responsive elements and regulates the gene transcription and translation of many cellular elements (Drouin et al., 1992). For instance, GR activation has been shown to induce the expression of a Ca2+ channel subunit associated with neurons in the basolateral amygdale through genomic regulation (Karst et al. 2002). In the present study, several lines of evidence support a genomic regulation of NMDARs by central GRs: (1) the expression of NMDARs after CCI, as revealed by both RT-PCR and Western blot, was attenuated by the GR antagonist RU38486; (2) interruption of GR expression using an antisense OND also prevented the upregulation of NMDARs after CCI; and (3) there was a colocalization of central GRs and NMDARs within the spinal cord dorsal horn, indicating a close cellular association between central GRs and NMDARs. Of interest is that our pervious study (Wang et al., 2004) has shown abundant colocalization between GRs and a neuronal marker (neuronal-specific nuclear protein), indicating the association of GRs with neuronal cells, although this finding does not exclude the possible association of GRs with spinal glial cells. It should be pointed out that although the current data examined a regulatory role of central GRs in the expression of NMDARs, these results do not exclude the possibilities of additional regulation on NMDARs. For example, phosphorylation of the NR2 subunit of the NMDAR has been observed in association with the development and maintenance of inflammatory hyperalgesia (Guo et al., 2002). Whether central GRs also participate in a similar regulation after CCI remains to be investigated.
Our data suggest that cellular mediators such as IL-6 and PKCγ are contributory to the GR-mediated regulation of NMDAR expression after CCI. First, the expression of NMDARs was attenuated by an IL-6 antiserum. Second, exogenous IL-6 mimicked the upregulation of NMDARs in naive rats with a time course similar to that after CCI. Third, the IL-6-induced NMDAR expression returned to the baseline after discontinuation of exogenous IL-6. Fourth, the NMDAR expression was substantially diminished in PKCγ KO mice, and PKCγ has been known to contribute to the mechanisms of neuropathic pain behaviors at least in part through the regulation of NMDARs (Mao et al., 1995; Malmberg et al., 1997). Importantly, our recent study has shown that central GRs are upregulated after CCI, and both IL-6 and PKCγ are critical cellular mediators leading to the GR upregulation in CCI rats (Wang et al., 2004). Although additional cytokines and intracellular signaling pathways (Opree and Kress, 2000; Sweitzer et al., 2001; Milligan et al., 2003) might be involved in the GR-mediated regulation of NMDARs, our results indicate that IL-6 and PKCγ may be such cellular mediators that are critical to the mechanisms underlying the genomic regulation of NMDARs by central GRs after CCI. Thus, both previous and present results suggest a cellular mechanism of GR and NMDAR interactions after peripheral nerve injury that involves a sequence of events including IL-6 and PKCγ, upregulation of central GRs, and the downstream regulation of NMDAR expression. It is of interest to note that the exact relationship between IL-6 and PKCγ in this process remains to be elucidated. It is possible that the effect of IL-6 on GR-mediated NMDAR expression is mediated through PKCγ, because our previous study using the same CCI model has shown that the IL-6 antiserum prevented the upregulation of PKCγ after CCI, whereas exogenous IL-6 increased the expression of PKCγ within the spinal cord dorsal horn (Wang et al., 2004). In addition, although a previous study has shown the expression of IL-6 receptors in neurons in human fetus brain and spinal cord (Dame and Juul, 2000), future studies may confirm the presence of IL-6 receptors within spinal cord neurons.
A number of previous studies have provided compelling evidence regarding GR actions through nongenomic mechanisms. For example, (1) interactions between GRs and activating protein-1 or nuclear factor κB have been shown to be involved in the process of immunosuppression and anti-inflammation (Barrett and Vedeckis, 1996; Refojo et al., 2001); (2) GRs mediate the inhibition of bradykinin-induced Ca2+ influx in PC12 cells (Qiu et al., 2003); (3) the regulatory role of cortisol in prolactin release is mediated through GRs (Borski et al., 2002); (4) GRs regulate spinal glutamate binding to glutamate receptors after spinal cord injury (Gonzalez et al., 1995); (5) GRs mediate acute elevation of the intracellular Ca2+ concentration induced by corticosterone in hippocampal neurons (Takahashi et al., 2002); and (6) activation of GRs potentiates NMDAR-mediated responses in dopamine-sensitive neurons within the ventral tegmental area (Cho and Little, 1999) as well as long-term depression (Coussens et al., 1997). Collectively, these previous studies strongly indicate that activation of GRs could directly interact with cellular components through nongenomic regulation, thereby modulating cellular events.
A similar nongenomic regulatory effect of central GRs on NMDAR function is indicated from our behavioral data examining nociceptive behaviors in both naive and CCI rats. First, hyperalgesia and allodynia induced by NMDA were prevented by a pretreatment (30 min before) with the GR antagonist RU38486, suggesting a tonic functional interaction between central GRs and NMDARs. Second, a single injection with the GR antagonist RU38486 on postoperative day 7 reversed neuropathic pain behaviors in CCI rats. Third, the noncompetitive NMDAR antagonist MK-801 also reversed hyperalgesia and allodynia exacerbated by treatment with the GR agonist dexamethasone in CCI rats. These results are consistent with our previous observation that RU38486 prevented the development of neuropathic pain behaviors after CCI (Wang et al., 2004). Although the exact nature of interactions between central GRs and NMDARs through a nongenomic mechanism remains to be elucidated, these results indicate that nongenomic interactions between central GRs and NMDARs played a significant role in the maintenance of neuropathic pain behaviors in CCI rats.
Neuropathic pain is a major chronic pain condition that remains difficult to treat. Investigation of the mechanisms of neuropathic pain has been a major focus of interest for decades (Wall et al., 1974; Devor, 1983, Dubner, 1991; Dougherty and Willis, 1991; Yamamoto and Yaksh, 1992; Mao et al., 1995; Woolf and Mannion, 1999). On the other hand, high doses of systemic corticosteroids are often used to reduce signs of regional tissue inflammation, including edema and neurogenic extravasation after a traumatic injury; this provides beneficial effects during the acute phase of such injuries. Our data suggest that the effects of steroids through central GRs may activate the same mechanisms known to be involved in the development and maintenance of neuropathic pain behaviors. This consequence could be of particular significance when no overt tissue inflammation and edema occur after injury, such as shown in this CCI model in which changes occurred within the spinal cord dorsal horn after peripheral nerve injury.
Thus, the present findings may have significant clinical implications for two reasons. First, the results indicate a link between central GRs and the expression of NMDARs, which is consistent with recent studies indicating a detrimental effect of GR agonists on NMDA-induced neuronal degeneration (Abraham et al., 2000) as well as on neuronal apoptosis during autoimmunerelated inflammation (Diem et al., 2003). Thus, prevention of the NMDAR upregulation by blocking GRs may regulate the cellular mechanisms of neuropathic pain and prevent the development of neuropathic pain mediated by NMDARs (Mao et al., 1995; Woolf and Mannion, 1999). Second, a nongenomic interaction between GRs and NMDARs at the receptor level may directly disrupt the function of NMDARs and reverse neuropathic pain behaviors as demonstrated in the present study, making it possible that a GR antagonist, when used alone or in combination with an NMDAR antagonist, may have an improved therapeutic role in the clinical management of neuropathic pain.
Footnotes
This work was supported by United States Public Health Service RO1 Grants NS 42661, NS 45681, and DA08835 to J.M.
Correspondence should be addressed to Dr. Jianren Mao, Pain Research Group, Massachusetts General Hospital Pain Center, WACC 324, Massachusetts General Hospital, Harvard Medical School, 15 Parkman Street, Boston, MA 02114. E-mail: jmao@partners.org.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250488-08$15.00/0
References
- Abraham I, Harkany T, Horvath K, Veenema AH, Penke B, Nyakas C, Luiten PG (2000) Chronic corticosterone administration dose-dependently modulates abeta(1-42)- and NMDA-induced neurodegeneration in rat magnocellular nucleus basalis. J Neuroendocrinol 12: 486-494. [DOI] [PubMed] [Google Scholar]
- Arruda JL, Colburn RW, Rickman AJ, Rutkoski MD, Deleo JA (1998) Increase of interleukin-6 mRNA in the spinal cord following peripheral injury in the rat: potential role of IL-6 in neuropathic pain. Brain Res Mol Brain Res 62: 228-235. [DOI] [PubMed] [Google Scholar]
- Barkhudaryan N, Dunn AJ (1999) Molecular mechanisms of actions of interleukin-6 on the brain, with special reference to serotonin and the hypothalamo-pituitary-adrenocortical axis. Neurochem Res 24: 1169-1180. [DOI] [PubMed] [Google Scholar]
- Barrett TJ, Vedeckis WV (1996) Occupancy and composition of proteins bound to the AP-1 sites in the glucocorticoid receptor and c-jun promoters after glucocorticoid treatment and in different cell types. Recept Signal Transduct 6: 179-193. [PubMed] [Google Scholar]
- Bennett GJ, Xie YK (1988) A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33: 87-107. [DOI] [PubMed] [Google Scholar]
- Borski RJ, Hyde GN, Fruchtman S (2002) Signal transduction mechanisms mediating rapid, nongenomic effects of cortisol on prolactin release. Steroids 67: 539-548. [DOI] [PubMed] [Google Scholar]
- Cho K, Little HJ (1999) Effects of corticosterone on excitatory amino acid responses in dopamine-sensitive neurons in the ventral tegmental area. Neuroscience 88: 837-845. [DOI] [PubMed] [Google Scholar]
- Coussens CM, Kerr DS, Abraham WC (1997) Glucocorticoid receptor activation lowers the threshold for NMDA-receptor-dependent homosynaptic long-term depression in the hippocampus through activation of voltage-dependent calcium channels. J Neurophysiol 78: 1-9. [DOI] [PubMed] [Google Scholar]
- Dame JB, Juul SE (2000) The distribution of receptors for the proinflammatory cytokine interleukin (IL)-6 and IL-8 in the developing human fetus. Early Hum Dev 58: 25-39. [DOI] [PubMed] [Google Scholar]
- Deleo JA, Colburn RW, Nichols M, Malhotra A (1996) Interleukin-6-mediated hyperalgesia/allodynia and increased spinal IL-6 expression in a rat mononeuropathy model. J Interferon Cytokine Res 16: 695-700. [DOI] [PubMed] [Google Scholar]
- Devor M (1983) Nerve pathophysiology and mechanisms of pain in causalgia. J Auton Nerve Syst 7: 371-384. [DOI] [PubMed] [Google Scholar]
- Diem R, Hobom M, Maier K, Weissert R, Storch MK, Meyer R, Bahr M (2003) Methylprednisolone increases neuronal apoptosis during autoimmune CNS inflammation by inhibition of an endogenous neuroprotective pathway. J Neurosci 23: 6993-7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty PM, Willis WD (1991) Modification of the responses of primate spinothalamic neurons to mechanical stimulation by excitatory amino acids and an N-methyl-d-aspartate antagonist. Brain Res 542: 15-22. [DOI] [PubMed] [Google Scholar]
- Drouin J, Sun YL, Tremblay S, Lavender P, Schmidt TJ, de Lean A, Nemer M (1992) Homodimer formation is rate-limiting for high affinity DNA binding by glucocorticoid receptor. Mol Endocrinol 6: 1299-1309. [DOI] [PubMed] [Google Scholar]
- Dubner R (1991) Neuronal plasticity and pain following peripheral tissue inflammation or nerve injury. In: Proceedings of 10th World Congress on pain. Pain research and clinical management, Vol 5 (Bond M, Charlton E, Woolf CJ, eds), pp 263-276. Amsterdam: Elsevier. [Google Scholar]
- Gartner H, Graul MC, Oesterreicher TJ, Finegold MJ, Henning SJ (2002) Development of the fetal intestine in mice lacking the glucocorticoid receptor (GR). J Cell Physiol 194: 80-87. [DOI] [PubMed] [Google Scholar]
- Gonzalez S, Coirini H, Gonzalez Deniselle MC, Gonzalez S, Calandra R, De Nicola AF (1995) Time-dependent effects of dexamethasone on glutamate binding, ornithine decarboxylase activity and polyamine levels in the transected spinal cord. J Steroid Biochem Mol Biol 55: 85-92. [DOI] [PubMed] [Google Scholar]
- Guo W, Zou S, Guan Y, Ikeda T, Tal M, Dubner R, Ren K (2002) Tyrosine phosphorylation of the NR2B subunit of the NMDA receptor in the spinal cord during the development and maintenance of inflammatory hyperalgesia. J Neurosci 22: 6208-6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32: 77-88. [DOI] [PubMed] [Google Scholar]
- Karst H, Nair S, Velzing E, Rumpff-van Essen L, Slagter E, Shinnick-Gallagher P, Joels M (2002) Glucocorticoids alter calcium conductances and calcium channel subunit expression in basolateral amygdale neurons. Eur J Neurosci 16: 1083-1089. [DOI] [PubMed] [Google Scholar]
- Lu J, Goula D, Sousa N, Almeida OF (2003) Ionotropic and metabotropic glutamate receptor mediation of glucocorticoid-induced apoptosis in hippocampal cells and the neuroprotective role of synaptic N-methyl-d-aspartate receptors. Neuroscience 121: 123-131. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Chen C, Tonegawa S, Basbaum AI (1997) Preserved acute pain and reduced neuropathic pain in mice lacking PKC gamma. Science 278: 279-283. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ, Lu J, Hayes RL (1992) Intrathecal MK-801 and local nerve anesthesia synergistically reduce nociceptive behaviors in rats with experimental peripheral mononeuropathy. Brain Res 576: 254-262. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ (1995) Mechanisms of hyperalgesia and opiate tolerance: a current view of their possible interactions. Pain 62: 259-274. [DOI] [PubMed] [Google Scholar]
- Mao J, Sung B, Ji RR, Lim G (2002) Chronic morphine induces down-regulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 22: 8312-8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Twining C, Chacur M, Biedenkapp J, O'Connor K, Poole S, Tracey K, Martin D, Maier S, Watkins LR (2003) Spinal glia and proinflammatory cytokine mediate mirror-image neuropathic pain in rats. J Neurosci 23: 1026-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PG, Ramer MS, Borthwick L, Gauldie J, Richardson PM, Bisby MA (1999) Endogenous interleukin-6 contributes to hypersensitivity to cutaneous stimuli and changes in neuropeptides associated with chronic nerve constriction in mice. Eur J Neurosci 11: 2243-2253. [DOI] [PubMed] [Google Scholar]
- Nair SM, Werkman TR, Craig J, Finnell R, Joels M, Eberwine JH (1998) Corticosterone regulation of ion channel conductances and mRNA levels in individual hippocampal CA1 neurons. J Neurosci 18: 2685-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeck G, Renkawitz R, Eggert M (2002) Molecular aspects of glucocorticoid hormone action in rheumatoid arthritis. Cytokines Cell Mol Ther 7: 61-69. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Martin DP, Schmelzer JD, Mitsui Y, Low PA (2001) Pro- and anti-inflammatory cytokines gene expression in rat sciatic nerve chronic constriction injury model of neuropathic pain. Exp Neurol 169: 386-391. [DOI] [PubMed] [Google Scholar]
- Opree A, Kress M (2000) Involvement of the proinflammatory cytokines tumor necrosis factor-α, IL-1β, and IL-6 but not IL-8 in the development of heat hyperalgesia: effects on heat-evoked calcitonin gene-related peptide release from rat skin. J Neurosci 20: 6289-6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Wang CG, Haung XY, Chen YZ (2003) Nongenomic mechanism of glucocorticoid inhibition of bradykinin-induced calcium influx in PC12 cells: possible involvement of protein kinase C. Life Sci 72: 2533-2542. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Rutkowski MD, Deleo JA (2002) The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci 22: 9980-9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refojo D, Liberman AC, Holsboer F, Arzt E (2001) Transcription factor-mediated molecular mechanisms involved in the functional cross-talk between cytokines and glucocorticoids. Immunol Cell Biol 79: 385-394. [DOI] [PubMed] [Google Scholar]
- Schafers M, Svensson CI, Sommer C, Sorkin LS (2003) Tumor necrosis factor-α induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci 23: 2517-2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Phillips AG, Leonard B (2003) Interleukin 1 beta enhances conditioned fear memory in rats: possible involvement of glucocorticoids. Eur J Neurosci 18: 1739-1743. [DOI] [PubMed] [Google Scholar]
- St. Hilaire M, Tremblay PO, Levesque D, Barden N, Rouillard C (2003) Effects of cocaine on c-Fos and NGFI-B mRNA expression in transgenic mice underexpressing glucocorticoid receptors. Neuropsychopharmacology 28: 478-489. [DOI] [PubMed] [Google Scholar]
- Sweitzer S, Martin D, DeLeo JA (2001) Intrathecal interleukin-1 receptor antagonist in combination of soluble tumor necrosis factor receptor exhibits an anti-allodynia action in a rat model of neuropathic pain. Neuroscience 103: 529-539. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kimoto T, Tanabe N, Hattori TA, Yasumatsu N, Kawata T (2002) Corticosterone acutely prolonged N-methyl-d-aspartate receptor mediated Ca2+ elevation in cultured rat hippocampal neurons. J Neurochem 83: 1441-1451. [DOI] [PubMed] [Google Scholar]
- Tal M, Bennett GJ (1994) Extra-territorial pain in rats with a peripheral mononeuropathy: mechano-hyperalgesia and mechano-allodynia in the territory of an uninjured nerve. Pain 57: 275-382. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Shutz G (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23: 99-103. [DOI] [PubMed] [Google Scholar]
- Wall PD, Waxman SG, Basbaum AI (1974) Ongoing activity in peripheral nerve: injury discharge. Exp Neurol 45: 576-589. [DOI] [PubMed] [Google Scholar]
- Wang S, Lim G, Zeng Q, Sung B, Ai Y, Guo G, Yang L, Mao J (2004) Expression of central glucocorticoid receptors after peripheral nerve injury contributes to neuropathic pain behaviors in rats. J Neurosci 24: 8595-8605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Maier SF (2003) Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov 2: 973-985. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ (1999) Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 353: 1959-1964. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Yaksh TL (1992) Spinal pharmacology of thermal hyperalgesia induced by constriction injury of sciatic nerve: excitatory amino acid antagonists. Pain 49: 121-128. [DOI] [PubMed] [Google Scholar]