

Abstract
Noncompetitive NMDA receptor antagonists, such as ketamine, induce a transient schizophrenia-like state in healthy individuals and exacerbate psychosis in schizophrenic patients. In rodents, noncompetitive NMDA receptor antagonists induce a behavioral syndrome that represents an experimentally valid model of schizophrenia. Current experimental evidence has implicated the nucleus accumbens in the pathophysiology of schizophrenia and the psychomimetic actions of ketamine. In this study, we have demonstrated that acute systemic administration of ketamine, at a dose known to produce hyperlocomotion and stereotypy, depressed the amplitude of the monosynaptic component of fimbria-evoked field potentials recorded in the nucleus accumbens. A similar effect was observed using the more selective antagonist dizocilpine maleate, indicating the depression was NMDA receptor dependent. Paired-pulse facilitation was enhanced concomitantly with, and in proportion to, ketamine-induced depressed synaptic efficacy, indicative of a presynaptic mechanism of action. Notably, the depression of field potentials recorded in the nucleus accumbens was markedly reduced after a focal 6-hydroxydopamine lesioning procedure in the nucleus accumbens. More specifically, pretreatment with the D2/D4 antagonist haloperidol, but not the D1 antagonist SCH23390, blocked ketamine-induced depression of nucleus accumbens responses. Our findings provide supporting evidence for the contemporary theory of schizophrenia as aberrant excitatory neurotransmission at the level of the nucleus accumbens.
Keywords: ketamine, NMDA, nucleus accumbens, evoked field potentials, dopamine, schizophrenia
Introduction
Subanaesthetic doses of noncompetitive NMDA receptor antagonists, such as ketamine, in healthy individuals can lead to the development of behavior similar to the positive and negative symptoms of schizophrenia (Krystal et al., 1994; Adler et al., 1999; Newcomer et al., 1999; Morgan et al., 2004) and exacerbate psychosis in stabilized schizophrenic patients (Lahti et al., 1995; Malhotra et al., 1997). In rodents, noncompetitive NMDA receptor antagonists induce a behavioral syndrome characterized by hyperlocomotion, stereotypy, cognitive impairments, and abnormal social interaction (Mansbach, 1991; Sams-Dodd, 1995; Abi-Saab et al., 1998; Becker et al., 2003). Additionally, noncompetitive NMDA receptor antagonists have also been shown to disrupt latent inhibition (Turgeon et al., 1998; Traverso et al., 2003) and prepulse inhibition (Bakshi et al., 1999), both of which are disrupted in the acute phase of schizophrenia (Rascle et al., 2001; Ludewig et al., 2003).
Substantial evidence has accumulated to implicate the nucleus accumbens (NAc) in the pathophysiology of schizophrenia (for review, see Gray, 1998; Grace, 2000; Chambers et al., 2001) and the psychomimetic activity of NMDA receptor antagonists (French et al., 1985; Carboni et al., 1989; McCullough and Salamone, 1992; Steinpreis and Salamone, 1993). The NAc receives excitatory glutamatergic projections from functionally and anatomically distinct brain regions that can converge on the same NAc neuron: the hippocampus, prefrontal cortex (PFC), basolateral amygdala, and thalamus (O'Donnell and Grace, 1995; Finch, 1996; Mulder et al., 1998; French and Totterdell, 2002). The NAc also receives a dense dopamine (DA) projection arising from the ventral tegmental area (VTA) (Oades and Halliday, 1987). By a complex mechanism, almost certainly involving DA, an important modulator of glutamatergic synapses, information from excitatory pathways is believed to be integrated at the level of the NAc (DeFrance et al., 1980; Yim and Mogenson, 1982; Mulder et al., 1996; Floresco et al., 2001; Brady and O'Donnell, 2004). It has been suggested that reduced excitatory neurotransmission of NAc afferents may modify the tonic level of DA release in the NAc producing a DA-hypersensitive state (Grace, 1991, 2000). In this primed system, dysregulated bursting of DA neurons may lead to the assignment of salience to behaviorally irrelevant events, which has been hypothesized as a mechanism of psychosis (Grace, 2000; Kapur, 2003).
In this study, we tested the hypothesis that excitatory neurotransmission to the NAc would be depressed using the ketamine rodent model of schizophrenia. Stimulation of the fimbria, which is know to primarily contain efferents from the subiculum of the hippocampus, produces a characteristic field potential in the NAc, which has been electrophysiologically characterized (Boeijinga et al., 1990, 1993; Pennartz and Kitai, 1991; Mulder et al., 1996; Hugues et al., 2003). The early (p10) component of the field potential represents monosynaptic activation (Boeijinga et al., 1993). Throughout this study, the amplitude of the p10 component was used as a measure of synaptic efficacy in the hippocampal-NAc pathway.
Materials and Methods
Surgery and placement of electrodes. Male Wistar rats (250-350 gm) were anesthetized with sodium pentobarbital (60 mg/kg, i.p.). Rats were mounted in a stereotaxic frame with blunt ear bars. The skull was exposed, and five burr holes were drilled, three for skull screws and two for electrodes made of twisted platinum-iridium (90-10%) wires insulated except at the tip. Electrodes were implanted ipsilaterally according to the coordinates of the stereotaxic atlas (Paxinos and Watson, 1986): stimulating electrodes were placed, with respect to the bregma, in the fimbria (posterior, 1.5 mm; lateral, 1.3 mm; ventral, 4 mm) and recording electrodes in the NAc shell (anterior, 1.6 mm; lateral, 0.8 mm; ventral, 5-7 mm). The electrodes were moved along a dorsal-ventral axis until the characteristic biphasic response was recorded (Boeijinga et al., 1993). Electrodes and screws were secured firmly in place with dental cement. Animals were allowed to recover for at least 5 d in the holding room. All experiments were conducted in accordance with the European Community guidelines on the care and use of laboratory animals (86/609/EEC).
Stimulation and data acquisition. Rats were placed in a recording chamber (35 cm wide, 35 cm long, 42 cm high), and electrophysiological activity was recorded through a junction field effect transistor (JFET) operational amplifier connected to the headstage. Cables from the JFET were relayed at the top of the box by a multichannel rotating connector, allowing the animal free movement inside the recording chamber. Responses were amplified (gain, 1000; bandpass, 0.001-1 kHz) and digitalized using a Power 1401 interface (CED, Cambridge, UK) connected to a personal computer for off-line analysis with Spike 2 (CED). NAc field potentials were evoked by fimbria single-pulse stimulation (0.1 msec rectangular monophasic pulses). Rats were habituated for 2 d to the process of headstage connection and transport to the recording chamber. Input-output curves were obtained at seven increasing intensities (100-800 μA; three field potentials/intensity recorded at 0.2 Hz). In this way, the stimulation intensity corresponding to 60-70% maximum amplitude was calculated and used as the test stimulus. In all studies, input-output curves were recorded for at least two more days to ensure that baseline responses were stable.
On the test day, rats were placed in the recording chamber, and a stimulus intensity corresponding to 60-70% maximum amplitude was delivered at 30 sec intervals. After a baseline period of at least 40 min, 25 mg/kg (±)-ketamine (Sigma, Lyon, France) or 0.9% saline was injected intraperitoneally. Responses were recorded for an additional 90 min after injection. In a separate study, full input-output curves were recorded before and 30, 60, and 90 min after injection. The effect of systemic injection of dizocilpine maleate (MK-801) (0.1 mg/kg; Sigma) was evaluated after 40 min baseline (corresponding to 60-70% maximum amplitude) and continued to be recorded up to 90 min after injection. In the same rats, complete input-output curves were obtained at baseline and 90 min after injection, at which point the experiment was terminated. Paired-pulse facilitation (PPF) of NAc field potentials was examined at an interstimulus interval of 50 msec before and 30, 60, and 90 min after injection of 25 mg/kg ketamine.
Dopaminergic lesion. Stimulating and recording electrodes were implanted using the same procedure as described above. A 25 gauge guide was placed adjacent to the recording electrodes that was 1.5 mm shorter than the tip of the recording electrodes. This method provided a way to deliver 6-hydroxydopamine (6-OHDA; Sigma) adjacent to the recording electrode. Lesioning was performed unilaterally based on the protocol by Mulder et al. (1996), which has been shown to lesion DA projection fibers in the NAc. Briefly, rats received an intra-NAc injection of 2 μl of 4 μg/μl 6-OHDA, prepared in 0.9% saline solution containing 0.1% ascorbic acid (Sigma). Sham animals were injected with an equal volume of vehicle. To prevent destruction of noradrenergic fibers, 25 mg/kg desipramine (Sigma) was injected intraperitoneally 30 min before the intra-NAc injection. Two weeks after the lesioning procedure, complete input-output curves were recorded over at least 3 d. When stable baseline values were obtained, we tested the effect of 25 mg/kg ketamine on 6-OHDA-lesioned and sham rats. A stimulus intensity corresponding to 60-70% maximum amplitude was delivered at intervals of 30 sec. After a baseline period of 30 min, 25 mg/kg ketamine or 0.9% saline was injected (intraperitoneally). Responses continued to be recorded for an additional 90 min after injection.
Dopaminergic antagonists. Rats implanted with stimulating and recorded electrodes were pretreated for 30 min with either the selective D1 receptor antagonist SCH23390 (0.5 mg/kg, i.p., prepared in 0.9% saline; Sigma) or the D2/D4 receptor antagonist haloperidol (1.5 mg/kg, i.p., prepared in 0.9% saline and 25 μl of glacial acetic acid; Sigma). Complete input-output curves were obtained immediately before pretreatment and 60 min after injection of 25 mg/kg ketamine.
Statistical analysis. Unless stated otherwise, all data were subjected to repeated-measures ANOVA. When the ANOVA revealed a significant difference, Bonferroni post hoc test was used to find individual points of significance between input-output curves. For experiments in which responses were evoked every 30 sec, data points were pooled over consecutive 2 min periods. Dunnett's post hoc test was used to find within-group changes with respect to baseline values. Differences were considered statistically significant if p ≤ 0.05.
Results
Ketamine induces depression of excitatory synaptic transmission in the nucleus accumbens
Stimulation of the fimbria produced a characteristic biphasic response in the NAc consisting of an early monosynaptic peak (8-10 msec) and later polysynaptically driven peak (∼20 msec). These latencies are similar to our previous findings (Hugues et al., 2003) and those reported by others (Boeijinga et al., 1993; Mulder et al., 1996). Histological verification of electrode placement showed that recording electrodes were located in the medial shell or shell/core border, and the stimulating electrodes were located in the fimbria, the majority of which were located in the dorsal aspect (Fig. 1A,B). An example of a fimbria-evoked field potential recorded in the NAc is shown in Figure 1C.
Figure 1.

Placement of electrodes. A, Stimulating electrodes were placed in the fimbria. B, Recording electrodes were located in the medial shell (NAcSh) or shell/core (NAcC) border of the NAc. C, Example of a fimbria-evoked field potential recorded in the NAc with positive peak latencies at 10 and 21 msec.
In the first series of experiments, we examined the effect of ketamine (25 mg/kg, i.p.) on fimbria-evoked field responses in the NAc. Pilot studies involving a small number of animals showed that this dose reliably induced a behavioral syndrome characterized by hyperlocomotion and stereotypy. Additionally, this dose has been shown to produce robust changes in brain metabolism (Duncan et al., 1999), disruption of attentional performance (Nelson et al., 2002), and is in the dose range that produces neurochemical changes (Nelson et al., 2002). Continuous responses recorded in the NAc, corresponding to 60-70% saturation level, were obtained during a baseline period of at least 40 min and for an additional 90 min after injection of 25 mg/kg ketamine or saline (0.9%). ANOVA of these data revealed that ketamine produced a depression of the monosynaptic component of field potentials recorded in the NAc with a main effect of group (F(1,11) = 12.45; p < 0.05), time (F(66,726) = 7.21; p < 0.01), and group × time interaction (F(66,726) = 10.02; p < 0.0001). Independent post hoc pairwise comparisons between saline- and ketamine-injected rats did not reveal any individual point of significance. However, within-group changes in amplitude revealed a significant depression between 14 and 64 min after injection of ketamine, with respect to baseline (Fig. 2A). Injection of saline did not significantly alter the amplitude of field potentials recorded in the NAc. Maximal depression (∼18%) developed ∼20 min after injection of ketamine, and by 90 min, the amplitudes had returned to their original baseline, indicating that depression was fully reversible. In a separate study, the effect of ketamine was evaluated by complete input-output curves collected before and 30, 60, and 90 min after injection. ANOVA of these data revealed an effect of time (F(3,63) = 2.07; p < 0.0001) and stimulus intensity (F(6,63) = 59.66; p < 0.0001) with no interaction (F(18,189) = 0.89; NS). Post hoc analysis revealed significant depression at 30 and 60 min after injection of ketamine (Fig. 2 B). No significant difference was found at 90 min with respect to baseline. Examples of fimbria-evoked NAc field potentials are shown in Figure 2C.
Figure 2.

Effect of ketamine on fimbria-evoked field potentials recorded in the NAc. A, Ketamine (25 mg/kg; n = 7) or saline (n = 6) was injected 40 min after baseline responses (indicated by the arrow). Depression of the p10 amplitude of field potentials recorded in the NAc was detected after injection of ketamine but not saline. Injection of saline did not modify the p10 amplitude offield potentials. B, Comparison of complete input-output curves of the p10 amplitude obtained at baseline (BL) and 30, 60, and 90 min after injection of ketamine (n = 10). Representative examples of field potentials recorded in the NAc are shown in C. Values are expressed as mean ± SEM. *p < 0.05 and **p < 0.01 indicate a significant difference with respect to baseline, and ##p < 0.01 indicates a significant difference 60 min after injection of ketamine with respect to baseline.
Depression of field potentials in the nucleus accumbens is NMDA receptor mediated
In addition to activity at NMDA receptors, ketamine is known to have a number of other sites of action (Kapur and Seeman, 2002). To determine whether depression of excitatory field potentials was mediated by noncompetitive antagonism at NMDA receptors, we used the more selective antagonist MK-801 (0.1 mg/kg, i.p.). MK-801 induced a gradual depression of fimbria-evoked field potentials recorded in the NAc (one-way ANOVA; F(63,315) = 26.47; p < 0.0001) (Fig. 3A). Comparison of input-output curves obtained before and 90 min after injection of MK-801, at which point the experiment was terminated, revealed a significant effect of MK-801 (F(1,28) = 3.66; p < 0.0001), stimulus intensity (F(6,28) = 79.45; p < 0.0001), and interaction (F(6,28) = 1.46; p < 0.05) (Fig. 3B).
Figure 3.

A, Time course of MK-801-induced depression of the p10 component of fimbria-evoked field potentials recorded in the NAc (n = 6). B, Complete input-output curves were obtained for five of these rats at baseline (BL) and 90 min after injection of MK-801. **p < 0.01 indicates a significant difference from baseline. Values are expressed as mean ± SEM.
Ketamine induces depression by reducing the probability of glutamate release
We next determined whether ketamine-induced depression of excitatory neurotransmission to the NAc was mediated by a presynaptic and/or a postsynaptic mechanism. PPF is a form of activity-dependent synaptic plasticity that is elicited by delivering a pair of synaptic responses with a short interstimulus interval. The probability of neurotransmitter release is thought to be inversely proportional to PPF (Anwyl et al., 1989; Schulz et al., 1994). For this study, an interstimulus interval of 50 msec was chosen; pilot studies showed this interval to give the most reproducible potentiation in freely moving animals. The NAc exhibited robust PPF in response to fimbria stimulation at all the intensities tested (F(6,28) = 11.00; p < 0.0001). Our findings are in accordance with previous reports using anesthetized rats and slice preparations that the NAc exhibits PPF (Boeijinga et al., 1990; Pennartz et al., 1990, 1991; Mulder et al., 1997). An enhancement of PPF was found 30 and 60 min after injection of ketamine, which coincided with the period of depressed field potentials recorded in the NAc (Fig. 4A). Furthermore, the percentage of PPF positively correlated with depressed neurotransmission (Fig. 4B). Notably, the maximal postsynaptic response determined by the amplitude of the second pulse was not significantly different before or 30, 60, and 90 min after injection of ketamine (1.033 ± 0.039, 1.064 ± 0.081, 0.955 ± 0.056, 0.938 ± 0.032 mV, respectively). This suggests there was no decrease in the postsynaptic sensitivity to glutamate. Figure 4C shows typical field potentials elicited by paired-pulse stimulation in the NAc.
Figure 4.

Effect of ketamine on PPF. A, Ketamine significantly increased PPF of the p10 component at 30 and 60 min after injection (n = 9). B, Enhancement of PPF correlated with ketamine-induced depression at the highest stimulation intensity (800 μA). Representative PPF at baseline (BL) and 60 min after injection of ketamine are shown in C. *p < 0.05 and **p < 0.01 indicate a significant difference with respect to baseline. Values are expressed as the mean ± SEM percentage change with respect to baseline.
Ketamine-induced depression of synaptic transmission is mediated by dopamine
Noncompetitive NMDA receptor antagonists are known to elevate the extracellular levels of DA in the NAc (Imperato et al., 1990; Bristow et al., 1993). It has been shown previously, using anesthetized rats, that a train of pulses delivered to the VTA attenuates hippocampal-evoked firing of NAc units (Yang and Mogenson, 1984); therefore, we hypothesized that DA may contribute to the ketamine-induced depression of fimbria-evoked field potentials recorded in the NAc.
The effect of DA depletion on the ketamine-induced depression of NAc field potentials was examined 2 weeks after focal intra-NAc injection of 6-OHDA. This procedure has been shown previously to modify fimbria-evoked field potentials in the NAc in anesthetized rats (Mulder et al., 1996). After a 30 min baseline period, 6-OHDA- and sham-treated rats were injected with ketamine and recorded for an additional 90 min. ANOVA revealed a main effect of group (F(1,11) = 14.41; p < 0.01) and time (F(55,605) = 11.55; p < 0.0001) with a significant interaction (F(55,605) = 9.61; p < 0.0001). Post hoc analysis between the groups did not yield individual points of significance; however, within-group analysis revealed points of significance in the sham- but not 6-OHDA-treated rats between 8 and 64 min with respect to baseline after injection of ketamine (Fig. 5A). Notably, two of seven 6-OHDA-treated rats displayed a short-term (∼30 min) depression after injection of ketamine. To determine which DA receptor subtype(s) mediated ketamine-induced depression of NAc field potentials, we pretreated rats (30 min) with the selective D1 receptor antagonist SCH23390 (0.5 mg/kg) or the D2/D4 receptor antagonist haloperidol (1.5 mg/kg). In the presence of haloperidol, complete input-output curves obtained at baseline and 60 min after injection of ketamine revealed a significant effect of stimulus intensity (F(6,21) = 9.85; p < 0.0001) but no effect of time (F(1,21) = 4.3; NS) or interaction (F(6,21) = 4.3; NS). In contrast, in the presence of SCH23390, a significant effect of stimulus intensity (F(6,28) = 59.66; p < 0.0001) and time (F(1,28) = 2.04; p < 0.0001) with no significant interaction (F(6,28) = 0.12; NS) was observed (Fig. 5B). In our study, we found that SCH23390 induced a small depression of fimbria-evoked NAc field potentials in two of five rats during pretreatment. One-way ANOVA of these data revealed a significant effect of time (F(53,212) = 2.76; p < 0.0001), and post hoc analysis revealed a significant depression occurred after injection of ketamine. The change in amplitude of NAc field potentials in the presence of SCH23390 over time is shown in Figure 5C.
Figure 5.
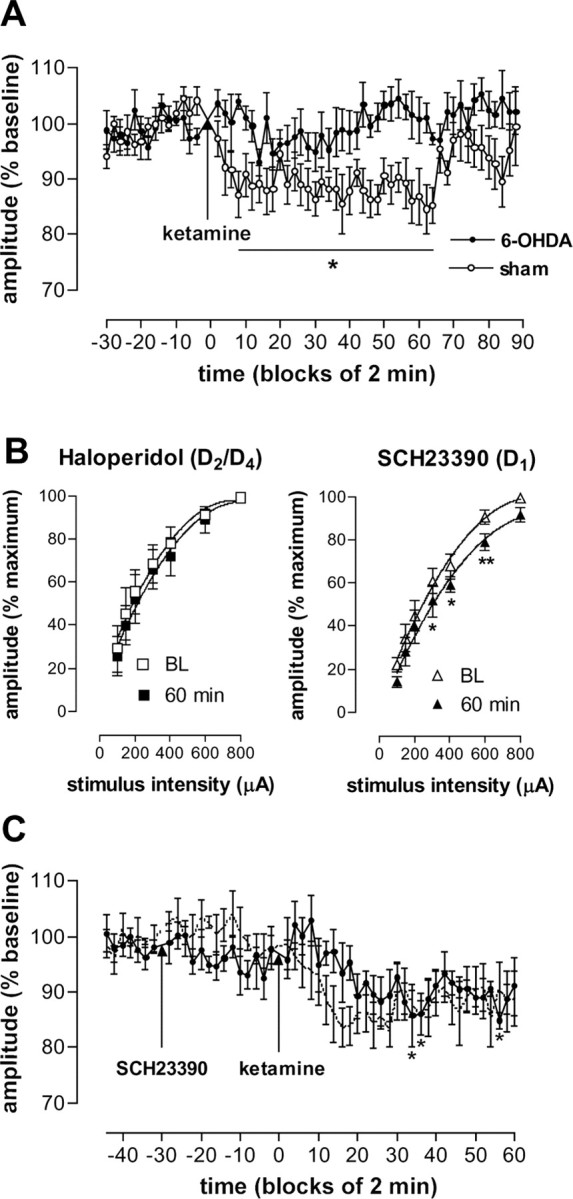
DA mediates ketamine-induced depression of the p10 component of fimbria-evoked field potentials recorded in the NAc. A, Field potentials from 6-OHDA-treated rats (n = 7) were not significantly modified after injection of ketamine. A reduction in the amplitude of field potentials from sham-treated rats (n = 6) occurred after injection of ketamine. B, Pretreatment with haloperidol (n = 4) but not SCH23390 (n = 5) blocked ketamine-induced depression of field potentials. C, A time course of the effect of ketamine on the p10 component in rats pretreated with SCH23390. For comparison, the time course of ketamine alone is shown (adapted from Fig. 2 A). *p < 0.05 and **p < 0.01 indicate a significant difference from baseline (BL). Values represent mean ± SEM activity.
Discussion
We have shown, in freely moving rats, that a subanaesthetic dose of the noncompetitive NMDA receptor antagonist ketamine depressed the p10 component of fimbria-evoked field potentials recorded in the NAc. We have presented evidence that ketamine-induced depression of field potentials is mediated presynaptically and is DA dependent. The duration of depressed synaptic efficacy ≥60 min well exceeded the much shorter behavioral syndrome, characterized by hyperlocomotion and stereotypy that had normally subsided 20 min after injection of ketamine. The effect of ketamine was reversible, and amplitudes had returned to values approaching their original baseline ∼90 min after injection.
Intracellular recordings in vivo have shown that NAc neurons are bistable existing in a depolarized “active” state or hyperpolarized “inactive” state (O'Donnell and Grace, 1995). Depolarization of NAc neurons has been shown to be dependent on excitation from the hippocampus, and transection of the fimbria blocks the bistable state of NAc neurons (O'Donnell and Grace, 1995). In accordance with our finding that ketamine depressed the activity of neurotransmission in the fimbria-NAc pathway, systemic administration of the related noncompetitive NMDA receptor antagonist phencyclidine (PCP) reduced the frequency and duration of the depolarized state of NAc neurones compared with control animals, indicative of reduced excitatory drive by the hippocampus (O'Donnell and Grace, 1998). The influence of the hippocampus on the bistable state of NAc neurons is reported to function as a gating mechanism for PFC input to the NAc (O'Donnell and Grace, 1995). Accordingly, reduced hippocampal drive is predicted to reduce PFC throughput, thereby producing a state of hypofrontalilty (O'Donnell and Grace, 1995) that has been associated with schizophrenia (for review, see Weinberger and Berman, 1996). In rats, a neonatal excitotoxic lesion of the ventral hippocampus can produce a complex array of postpubertal molecular and behavioral abnormalities including impairments in working memory, social interaction, and increased sensitivity to noncompetitive NMDA receptor antagonists (Lipska et al., 1993; Sams-Dodd et al., 1997; Al-Amin et al., 2001; Lipska et al., 2002). Of note, at an age when behavioral abnormalities are detectable, a significant reduction in the release of amino acids in hippocampal slices was found with respect to control rats (Schroeder et al., 1999). Together, these findings suggest that dysfunction of hippocampal neurotransmission may participate in the pathophysiology of schizophrenia.
We determined whether ketamine-induced depression of synaptic transmission in the NAc occurred by a presynaptic and/or postsynaptic mechanism using the PPF paradigm. PPF is a form of short-term synaptic plasticity (Anwyl et al., 1989; Schulz et al., 1994) evoked by delivering two stimuli of short interstimulus intervals. Residual presynaptic calcium, after the first stimulation, is thought to enhance neurotransmitter release in response to the second stimulation and the magnitude of the facilitation thought to be inversely proportional to the probability of neurotransmitter release. Our findings suggest that ketamine-induced depression of excitatory synapses in the NAc was mediated primarily by a presynaptic mechanism for several reasons. First, an enhancement of PPF occurred concomitantly with depression of evoked neurotransmission in the NAc. PPF normalized as the amplitude of the NAc response returned to baseline. Second, a positive correlation was found between ketamine-induced depression of synaptic transmission and enhancement of PPF. Third, reduced synaptic efficacy occurred in the absence of a modified maximal evoked response to the test pulse, which remained within the normal range throughout the study. This suggests that the sensitivity to glutamate was not markedly changed throughout the course of the experiment.
In addition to noncompetitive antagonism of NMDA receptors, ketamine has many other sites of action including D2, 5-HT2 (Kapur and Seeman, 2002), opiate receptors (Smith et al., 1980), and acetylcholinesterase (Cohen et al., 1974). MK-801, the more selective NMDA receptor antagonist, progressively reduced the amplitude of NAc field potentials, suggesting that the depressed activity was NMDA receptor mediated. Consistent with reports that MK-801 has greater affinity for NMDA receptors than ketamine (Wong et al., 1988; Sun and Wessinger, 2004), we found that field potentials remained depressed for a longer duration. Indeed, at the end of the experiment (90 min), marked depression was observed in the presence of MK-801, whereas the responses of ketamine-treated rats had frequently returned to their original baseline values.
It appears unlikely that local antagonism of NMDA receptors within the NAc accounted for the depression of NAc field potentials. Studies using NAc slice preparations demonstrated that evoked activity of NAc neurons is principally mediated by AMPA/kainate receptors (Horne et al., 1990; Pennartz et al., 1990, 1991; Pennartz and Kitai, 1991). Furthermore, an in vivo microiontophoresis study has also shown that the excitatory effects of glutamate on single units in the NAc involve non-NMDA receptors (Hu and White, 1996). Indeed, intra-NAc injection of ketamine may in fact increase the amplitude of fimbria-evoked field potentials (our unpublished observation). However, in both NAc slice and anesthetized rat preparations, potentiation of neuronal excitability in the hippocampal-NAc pathway has been shown to be NMDA receptor dependent (Pennartz et al., 1993; Feasey-Truger and ten Bruggencate, 1994; Floresco et al., 2001).
The NAc receives dense DA projections from the VTA (Oades and Halliday, 1987), and in anesthetized rats, noncompetitive NMDA receptor antagonists induce synchronous bursting of VTA neurons (French and Ceci, 1990). Accordingly, noncompetitive NMDA receptor antagonists are known to elevate the concentrations of extracellular DA in the NAc (Carboni et al., 1989; Bristow et al., 1993; Adams and Moghaddam, 1998). In our study, a 6-OHDA lesioning procedure, which has been shown previously to modify fimbria-evoked field potentials in the NAc (Mulder et al., 1996), substantially reduced ketamine-induced depression of synaptic transmission in the NAc. It is unlikely that nonspecific damage contributed to the reduced depression, because sham-lesioned rats were injected with the same volume and at the same speed. More specifically, we found that pretreatment with the D2/D4 antagonist haloperidol abolished ketamine-induced depression of NAc field potentials. These findings are consistent with reports that activation of D2 receptors can depress neurotransmission at excitatory synapses in the NAc, thought to be mediated by a presynaptic mechanism of action (Yang and Mogenson, 1986; O'Donnell and Grace, 1994; Brady and O'Donnell, 2004). In support of this, a recent electron microscopy study has confirmed the existence of D2 receptors at excitatory terminals in the NAc (Delle Donne et al., 2004). However, the role of DA in the NAc is complex and DA-mediated depression of excitatory neurotransmission can also occur by a mechanism involving D1 receptors (Pennartz et al., 1992; Harvey and Lacey, 1996; Nicola et al., 1996, 2000). It is noteworthy that in anesthetized rats, the competitive NMDA receptor antagonist (±)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid did not modify baseline activity of fimbria-evoked field potentials in the NAc (Feasey-Truger and ten Bruggencate, 1994; Floresco et al., 2001). This is not necessarily surprising, because noncompetitive NMDA receptor antagonists, such as ketamine, and competitive antagonists induce different behavioral syndromes in rodents (Tricklebank et al., 1989). Indeed, activation of mesolimbic DA neurons, which appears to be critical for the depression we observed, is not associated with competitive NMDA receptor antagonists (French et al., 1991).
Our findings contrast with the effect of NMDA receptor antagonists in the PFC. In freely moving rats, systemic administration of PCP is reported to increase the rate of firing of neurons in the PFC (Suzuki et al., 2002; Jodo et al., 2003, 2004). The increased firing rate may contribute to the elevated levels of glutamate, which have been reported to occur in the NAc after injection of PCP (Adams and Moghaddam, 1998). Local injection of PCP into the ventral hippocampus also markedly increased the firing rate of PFC neurons, indicating a hippocampal origin for this activity (Jodo et al., 2004). However, PCP injected in the ventral subiculum did not modify the overall bistable state of NAc neurons, although a reduction was reported in one of four NAc neurons (O'Donnell and Grace, 1998). This indicates that the responsiveness of hippocampal-NAc and hippocampal-PFC pathways after administration of noncompetitive NMDA receptor antagonists may be substantially different.
In addition to the putative role of the NAc in schizophrenia, this brain region is an important site for the psychomotor and rewarding properties of drugs of abuse (Carboni et al., 1989). Our finding that ketamine depressed excitatory neurotransmission in the NAc appears to be a common neurophysiological mechanism of diverse classes of drugs of abuse, including amphetamines, cocaine, and cannabinoids (Nicola et al., 1996; Pistis et al., 2002). Repeated exposure to psychostimulants and psychomimetics can produce a syndrome characterized by paranoid schizophrenia-like symptoms and persistent structural modification in the NAc and PFC, which may last several months (Robinson and Kolb, 1997). The high rates of comorbidity of schizophrenia and substance abuse suggests there may be an associated physiological mechanism (Chambers et al., 2001). Because the rewarding properties of substances of abuse act on a common brain region implicated in the pathogenesis of schizophrenia, it is conceivable that exposure may prime the mesolimbic system and consequently produce a vulnerable population that may precipitate the first episode of psychotic symptoms.
Footnotes
This work was supported by the Institut National de la Santé et de la Recherche Médicale.
Correspondence should be addressed to Mark J. Hunt, Neurobiologie Comportementale, Equipe Avenir, Institut National de la Santé et de la Recherche Médicale, Université de Nice-Sophia Antipolis, 06108 Nice, France. E-mail: hunt@unice.fr.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250524-08$15.00/0
References
- Abi-Saab WM, D'Souza DC, Moghaddam B, Krystal JH (1998) The NMDA antagonist model for schizophrenia: promise and pitfalls. Pharmacopsychiatry 31 [Suppl 2]: 104-109. [DOI] [PubMed] [Google Scholar]
- Adams B, Moghaddam B (1998) Corticolimbic dopamine neurotransmission is temporally dissociated from the cognitive and locomotor effects of phencyclidine. J Neurosci 18: 5545-5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler CM, Malhotra AK, Elman I, Goldberg T, Egan M, Pickar D, Breier A (1999) Comparison of ketamine-induced thought disorder in healthy volunteers and thought disorder in schizophrenia. Am J Psychiatry 156: 1646-1649. [DOI] [PubMed] [Google Scholar]
- Al-Amin HA, Shannon Weickert C, Weinberger DR, Lipska BK (2001) Delayed onset of enhanced MK-801-induced motor hyperactivity after neonatal lesions of the rat ventral hippocampus. Biol Psychiatry 49: 528-539. [DOI] [PubMed] [Google Scholar]
- Anwyl R, Mulkeen D, Rowan MJ (1989) The role of N-methyl-d-aspartate receptors in the generation of short-term potentiation in the rat hippocampus. Brain Res 503: 148-151. [DOI] [PubMed] [Google Scholar]
- Bakshi VP, Tricklebank M, Neijt HC, Lehmann-Masten V, Geyer MA (1999) Disruption of prepulse inhibition and increases in locomotor activity by competitive N-methyl-d-aspartate receptor antagonists in rats. J Pharmacol Exp Ther 288: 643-652. [PubMed] [Google Scholar]
- Becker A, Peters B, Schroeder H, Mann T, Huether G, Grecksch G (2003) Ketamine-induced changes in rat behaviour: a possible animal model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 27: 687-700. [DOI] [PubMed] [Google Scholar]
- Boeijinga PH, Pennartz CM, Lopes da Silva FH (1990) Paired-pulse facilitation in the nucleus accumbens following stimulation of subicular inputs in the rat. Neuroscience 35: 301-311. [DOI] [PubMed] [Google Scholar]
- Boeijinga PH, Mulder AB, Pennartz CM, Manshanden I, Lopes da Silva FH (1993) Responses of the nucleus accumbens following fornix/fimbria stimulation in the rat. Identification and long-term potentiation of mono- and polysynaptic pathways. Neuroscience 53: 1049-1058. [DOI] [PubMed] [Google Scholar]
- Brady AM, O'Donnell P (2004) Dopaminergic modulation of prefrontal cortical input to nucleus accumbens neurons in vivo. J Neurosci 24: 1040-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristow LJ, Hutson PH, Thorn L, Tricklebank MD (1993) The glycine/NMDA receptor antagonist, R-(+)-HA-966, blocks activation of the mesolimbic dopaminergic system induced by phencyclidine and dizocilpine (MK-801) in rodents. Br J Pharmacol 108: 1156-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carboni E, Imperato A, Perezzani L, Di Chiara G (1989) Amphetamine, cocaine, phencyclidine and nomifensine increase extracellular dopamine concentrations preferentially in the nucleus accumbens of freely moving rats. Neuroscience 28: 653-661. [DOI] [PubMed] [Google Scholar]
- Chambers RA, Krystal JH, Self DW (2001) A neurobiological basis for substance abuse comorbidity in schizophrenia. Biol Psychiatry 50: 71-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MG, Chan SL, Bhargava HN, Trevor AJ (1974) Inhibition of mammalian brain acetylcholinesterase by ketamine. Biochem Pharmacol 23: 1647-1652. [DOI] [PubMed] [Google Scholar]
- DeFrance JF, Marchand JE, Stanley JC, Sikes RW, Chronister RB (1980) Convergence of excitatory amygdaloid and hippocampal input in the nucleus accumbens septi. Brain Res 185: 183-186. [DOI] [PubMed] [Google Scholar]
- Delle Donne KT, Chan J, Boudin H, Pelaprat D, Rostene W, Pickel VM (2004) Electron microscopic dual labeling of high-affinity neurotensin and dopamine D2 receptors in the rat nucleus accumbens shell. Synapse 52: 176-187. [DOI] [PubMed] [Google Scholar]
- Duncan GE, Miyamoto S, Leipzig JN, Lieberman JA (1999) Comparison of brain metabolic activity patterns induced by ketamine, MK-801 and amphetamine in rats: support for NMDA receptor involvement in responses to subanesthetic dose of ketamine. Brain Res 843: 171-183. [DOI] [PubMed] [Google Scholar]
- Feasey-Truger KJ, ten Bruggencate G (1994) The NMDA receptor antagonist CPP suppresses long-term potentiation in the rat hippocampalaccumbens pathway in vivo. Eur J Neurosci 6: 1247-1254. [DOI] [PubMed] [Google Scholar]
- Finch DM (1996) Neurophysiology of converging synaptic inputs from the rat prefrontal cortex, amygdala, midline thalamus, and hippocampal formation onto single neurons of the caudate/putamen and nucleus accumbens. Hippocampus 6: 495-512. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Blaha CD, Yang CR, Phillips AG (2001) Modulation of hippocampal and amygdalar-evoked activity of nucleus accumbens neurons by dopamine: cellular mechanisms of input selection. J Neurosci 21: 2851-2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French ED, Ceci A (1990) Non-competitive N-methyl-d-aspartate antagonists are potent activators of ventral tegmental A10 dopamine neurons. Neurosci Lett 119: 159-162. [DOI] [PubMed] [Google Scholar]
- French ED, Pilapil C, Quirion R (1985) Phencyclidine binding sites in the nucleus accumbens and phencyclidine-induced hyperactivity are decreased following lesions of the mesolimbic dopamine system. Eur J Pharmacol 116: 1-9. [DOI] [PubMed] [Google Scholar]
- French ED, Ferkany J, Abreu M, Levenson S (1991) Effects of competitive N-methyl-d-aspartate antagonists on midbrain dopamine neurons: an electrophysiological and behavioral comparison to phencyclidine. Neuropharmacology 30: 1039-1046. [DOI] [PubMed] [Google Scholar]
- French SJ, Totterdell S (2002) Hippocampal and prefrontal cortical inputs monosynaptically converge with individual projection neurons of the nucleus accumbens. J Comp Neurol 446: 151-165. [DOI] [PubMed] [Google Scholar]
- Grace AA (1991) Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience 41: 1-24. [DOI] [PubMed] [Google Scholar]
- Grace AA (2000) Gating of information flow within the limbic system and the pathophysiology of schizophrenia. Brain Res Brain Res Rev 31: 330-341. [DOI] [PubMed] [Google Scholar]
- Gray JA (1998) Integrating schizophrenia. Schizophr Bull 24: 249-266. [DOI] [PubMed] [Google Scholar]
- Harvey J, Lacey MG (1996) Endogenous and exogenous dopamine depress EPSCs in rat nucleus accumbens in vitro via D1 receptors activation. J Physiol (Lond) 492: 143-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne AL, Woodruff GN, Kemp JA (1990) Synaptic potentials mediated by excitatory amino acid receptors in the nucleus accumbens of the rat, in vitro. Neuropharmacology 29: 917-921. [DOI] [PubMed] [Google Scholar]
- Hu XT, White FJ (1996) Glutamate receptor regulation of rat nucleus accumbens neurons in vivo. Synapse 23: 208-218. [DOI] [PubMed] [Google Scholar]
- Hugues S, Kessal K, Hunt MJ, Garcia R (2003) A conditioned stressful environment causes short-term metaplastic-like changes in the rat nucleus accumbens. J Neurophysiol 90: 3224-3231. [DOI] [PubMed] [Google Scholar]
- Imperato A, Scrocco MG, Bacchi S, Angelucci L (1990) NMDA receptors and in vivo dopamine release in the nucleus accumbens and caudatus. Eur J Pharmacol 187: 555-556. [DOI] [PubMed] [Google Scholar]
- Jodo E, Suzuki Y, Takeuchi S, Niwa S, Kayama Y (2003) Different effects of phencyclidine and methamphetamine on firing activity of medial prefrontal cortex neurons in freely moving rats. Brain Res 962: 226-231. [DOI] [PubMed] [Google Scholar]
- Jodo E, Suzuki Y, Katayama T, Hoshino KY, Takeuchi S, Niwa SI, Kayama Y (2004) Activation of medial prefrontal cortex by phencyclidine is mediated via a hippocampo-prefrontal pathway. Cereb Cortex, in press. [DOI] [PubMed]
- Kapur S (2003) Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry 160: 13-23. [DOI] [PubMed] [Google Scholar]
- Kapur S, Seeman P (2002) NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D(2) and serotonin 5-HT(2)receptors-implications for models of schizophrenia. Mol Psychiatry 7: 837-844. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers Jr MB, Charney DS (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51: 199-214. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Koffel B, LaPorte D, Tamminga CA (1995) Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 13: 9-19. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Jaskiw GE, Weinberger DR (1993) Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: a potential animal model of schizophrenia. Neuropsychopharmacology 9: 67-75. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Aultman JM, Verma A, Weinberger DR, Moghaddam B (2002) Neonatal damage of the ventral hippocampus impairs working memory in the rat. Neuropsychopharmacology 27: 47-54. [DOI] [PubMed] [Google Scholar]
- Ludewig K, Geyer MA, Vollenweider FX (2003) Deficits in prepulse inhibition and habituation in never-medicated, first-episode schizophrenia. Biol Psychiatry 54: 121-128. [DOI] [PubMed] [Google Scholar]
- Malhotra AK, Pinals DA, Adler CM, Elman I, Clifton A, Pickar D, Breier A (1997) Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology 17: 141-150. [DOI] [PubMed] [Google Scholar]
- Mansbach RS (1991) Effects of NMDA receptor ligands on sensorimotor gating in the rat. Eur J Pharmacol 202: 61-66. [DOI] [PubMed] [Google Scholar]
- McCullough LD, Salamone JD (1992) Increases in extracellular dopamine levels and locomotor activity after direct infusion of phencyclidine into the nucleus accumbens. Brain Res 577: 1-9. [DOI] [PubMed] [Google Scholar]
- Morgan CJ, Mofeez A, Brandner B, Bromley L, Curran HV (2004) Acute effects of ketamine on memory systems and psychotic symptoms in healthy volunteers. Neuropsychopharmacology 29: 208-218. [DOI] [PubMed] [Google Scholar]
- Mulder AB, Manshanden I, Vos PE, Wolterink G, van Ree JM, Lopes da Silva FH (1996) Modifications in glutamatergic transmission after dopamine depletion of the nucleus accumbens. A combined in vivo/in vitro electrophysiological study in the rat. Neuroscience 72: 1009-1021. [DOI] [PubMed] [Google Scholar]
- Mulder AB, Arts MP, Lopes da Silva FH (1997) Short- and long-term plasticity of the hippocampus to nucleus accumbens and prefrontal cortex pathways in the rat, in vivo. Eur J Neurosci 9: 1603-1611. [DOI] [PubMed] [Google Scholar]
- Mulder AB, Hodenpijl MG, Lopes da Silva FH (1998) Electrophysiology of the hippocampal and amygdaloid projections to the nucleus accumbens of the rat: convergence, segregation, and interaction of inputs. J Neurosci 18: 5095-5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CL, Burk JA, Bruno JP, Sarter M (2002) Effects of acute and repeated systemic administration of ketamine on prefrontal acetylcholine release and sustained attention performance in rats. Psychopharmacology (Berl) 161: 168-179. [DOI] [PubMed] [Google Scholar]
- Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T, Craft S, Olney JW (1999) Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology 20: 106-118. [DOI] [PubMed] [Google Scholar]
- Nicola SM, Kombian SB, Malenka RC (1996) Psychostimulants depress excitatory synaptic transmission in the nucleus accumbens via presynaptic D1-like dopamine receptors. J Neurosci 16: 1591-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola SM, Surmeier J, Malenka RC (2000) Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci 23: 185-215. [DOI] [PubMed] [Google Scholar]
- Oades RD, Halliday GM (1987) Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res 434: 117-165. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA (1994) Tonic D2-mediated attenuation of cortical excitation in nucleus accumbens neurons recorded in vitro. Brain Res 634: 105-112. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA (1995) Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci 15: 3622-3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA (1998) Phencyclidine interferes with the hippocampal gating of nucleus accumbens neuronal activity in vivo. Neuroscience 87: 823-830. [DOI] [PubMed] [Google Scholar]
- Paxinos W, Watson C (1986) The rat brain in stereotaxic coordinates, Ed 2. San Diego: Academic.
- Pennartz CM, Kitai ST (1991) Hippocampal inputs to identified neurons in an in vitro slice preparation of the rat nucleus accumbens: evidence for feed-forward inhibition. J Neurosci 11: 2838-2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennartz CM, Boeijinga PH, Lopes da Silva FH (1990) Locally evoked potentials in slices of the rat nucleus accumbens: NMDA and non-NMDA receptor mediated components and modulation by GABA. Brain Res 529: 30-41. [DOI] [PubMed] [Google Scholar]
- Pennartz CM, Boeijinga PH, Kitai ST, Lopes da Silva FH (1991) Contribution of NMDA receptors to postsynaptic potentials and paired-pulse facilitation in identified neurons of the rat nucleus accumbens in vitro. Exp Brain Res 86: 190-198. [DOI] [PubMed] [Google Scholar]
- Pennartz CM, Dolleman-Van der Weel MJ, Kitai ST, Lopes da Silva FH (1992) Presynaptic dopamine D1 receptors attenuate excitatory and inhibitory limbic inputs to the shell region of the rat nucleus accumbens studied in vitro. J Neurophysiol 67: 1325-1334. [DOI] [PubMed] [Google Scholar]
- Pennartz CM, Ameerun RF, Groenewegen HJ, Lopes da Silva FH (1993) Synaptic plasticity in an in vitro slice preparation of the rat nucleus accumbens. Eur J Neurosci 5: 107-117. [DOI] [PubMed] [Google Scholar]
- Pistis M, Muntoni AL, Pillolla G, Gessa GL (2002) Cannabinoids inhibit excitatory inputs to neurons in the shell of the nucleus accumbens: an in vivo electrophysiological study. Eur J Neurosci 15: 1795-1802. [DOI] [PubMed] [Google Scholar]
- Rascle C, Mazas O, Vaiva G, Tournant M, Raybois O, Goudemand M, Thomas P (2001) Clinical features of latent inhibition in schizophrenia. Schizophr Res 51: 149-161. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Kolb B (1997) Persistent structural modifications in nucleus accumbens and prefrontal cortex neurons produced by previous experience with amphetamine. J Neurosci 17: 8491-8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sams-Dodd F (1995) Distinct effects of d-amphetamine and phencyclidine on the social behaviour of rats. Behav Pharmacol 6: 55-65. [PubMed] [Google Scholar]
- Sams-Dodd F, Lipska BK, Weinberger DR (1997) Neonatal lesions of the rat ventral hippocampus result in hyperlocomotion and deficits in social behaviour in adulthood. Psychopharmacology (Berl) 132: 303-310. [DOI] [PubMed] [Google Scholar]
- Schroeder H, Grecksch G, Becker A, Bogerts B, Hoellt V (1999) Alterations of the dopaminergic and glutamatergic neurotransmission in adult rats with postnatal ibotenic acid hippocampal lesion. Psychopharmacology (Berl) 145: 61-66. [DOI] [PubMed] [Google Scholar]
- Schulz PE, Cook EP, Johnston D (1994) Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J Neurosci 14: 5325-5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Pekoe GM, Martin LL, Coalgate B (1980) The interaction of ketamine with the opiate receptor. Life Sci 26: 789-795. [DOI] [PubMed] [Google Scholar]
- Steinpreis RE, Salamone JD (1993) The role of nucleus accumbens dopamine in the neurochemical and behavioral effects of phencyclidine: a microdialysis and behavioral study. Brain Res 612: 263-270. [DOI] [PubMed] [Google Scholar]
- Sun W, Wessinger WD (2004) Characterization of the non-competitive antagonist binding site of the NMDA receptor in dark Agouti rats. Life Sci 75: 1405-1415. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Jodo E, Takeuchi S, Niwa S, Kayama Y (2002) Acute administration of phencyclidine induces tonic activation of medial prefrontal cortex neurons in freely moving rats. Neuroscience 114: 769-779. [DOI] [PubMed] [Google Scholar]
- Traverso LM, Ruiz G, De la Casa LG (2003) Latent inhibition disruption by MK-801 in a conditioned taste-aversion paradigm. Neurobiol Learn Mem 80: 140-146. [DOI] [PubMed] [Google Scholar]
- Tricklebank MD, Singh L, Oles RJ, Preston C, Iversen SD (1989) The behavioural effects of MK-801: a comparison with antagonists acting noncompetitively and competitively at the NMDA receptor. Eur J Pharmacol 167: 127-135. [DOI] [PubMed] [Google Scholar]
- Turgeon SM, Auerbach EA, Heller MA (1998) The delayed effects of phencyclidine (PCP) disrupt latent inhibition in a conditioned taste aversion paradigm. Pharmacol Biochem Behav 60: 553-558. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, Berman KF (1996) Prefrontal function in schizophrenia: confounds and controversies. Philos Trans R Soc Lond B Biol Sci 351: 1495-1503. [DOI] [PubMed] [Google Scholar]
- Wong EH, Knight AR, Woodruff GN (1988) [3H]MK-801 labels a site on the N-methyl-d-aspartate receptor channel complex in rat brain membranes. J Neurochem 50: 274-281. [DOI] [PubMed] [Google Scholar]
- Yang CR, Mogenson GJ (1984) Electrophysiological responses of neurones in the nucleus accumbens to hippocampal stimulation and the attenuation of the excitatory responses by the mesolimbic dopaminergic system. Brain Res 324: 69-84. [DOI] [PubMed] [Google Scholar]
- Yang CR, Mogenson GJ (1986) Dopamine enhances terminal excitability of hippocampal-accumbens neurons via D2 receptor: role of dopamine in presynaptic inhibition. J Neurosci 6: 2470-2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim CY, Mogenson GJ (1982) Response of nucleus accumbens neurons to amygdala stimulation and its modification by dopamine. Brain Res 239: 401-415. [DOI] [PubMed] [Google Scholar]