

Abstract
Both the acquisition and extinction of conditioned fear appear to require the basolateral amygdala (BLA). Because these two forms of learning have opposing effects on the expression of conditioned fear, we hypothesized that they may modulate GABAergic tone differentially within the BLA. Previously, we reported that gene expression for the GABAA receptor clustering protein gephyrin was significantly downregulated in the BLA after fear acquisition (Ressler et al., 2002). Here we demonstrate an analogous decrease in BLA gephyrin protein levels, together with a decrease in the surface expression of GABAA receptors in the BLA after fear acquisition, as evidenced by decreased binding of H3-flunitrazepam. In marked contrast, gephyrin mRNA and protein levels in the BLA significantly increased after extinction training, as did H3-flunitrazepam binding. These results implicate the protein gephyrin in both fear acquisition and extinction and suggest that the modulation of gephyrin and GABAA receptor expression in the BLA may play a role in the experience-dependent plasticity underlying both of these types of learning. Furthermore, these results demonstrate that physiologically relevant, dynamic alterations of GABAergic synapses occur during the consolidation phase of BLA-dependent learning and may interact with previously described alterations in glutamatergic transmission to initiate and stabilize memory formation in vivo.
Keywords: amygdala, autoradiography, extinction, fear, GABA (γ-aminobutyric acid), learning
Introduction
The basolateral complex of the amygdala (BLA) is a critical player in the acquisition of conditioned fear (Davis, 2000; LeDoux, 2000) and also has been implicated in the learned suppression of conditioned fear responses (CRs) in extinction (Falls et al., 1992). Extinction refers both to a training procedure involving exposure of a previously fear-conditioned animal to repeated presentations of the conditioned stimulus (CS) in the absence of the unconditioned stimulus (US) (hereafter referred to as “extinction training”) and to the outcome of that procedure, a gradual disappearance of the CR (hereafter referred to as “extinction”). Several lines of evidence suggest that extinction is a form of learning that is distinct from and operates in parallel to fear acquisition (Myers and Davis, 2002).
Acquisition and extinction of conditioned fear are disrupted by a similar array of pharmacological manipulations, including NMDA receptor antagonism (Miserendino et al., 1990; Falls et al., 1992), inhibition of MAP kinase activation (Schafe et al., 1999; Lu et al., 2001; Lin et al., 2003b), and protein synthesis inhibition in the amygdala (Nader et al., 2000; Lin et al., 2003b). Additionally, both fear acquisition and extinction appear to promote the activation of the transcription factor cAMP response element-binding protein (Josselyn et al., 2001; Lin et al., 2003a). Nevertheless, because these two forms of learning have opposite behavioral consequences, it seems reasonable to expect divergences in their neural underpinnings as well, perhaps in the form of differential plastic alterations of excitatory and inhibitory neurotransmission within the amygdala.
In fact, there is some evidence to suggest that GABAergic neurotransmission is modulated differentially after acquisition and extinction of conditioned fear. For example, it has been shown that presentation of a feared CS to previously fear-conditioned animals leads to a decrease in extracellular GABA levels, indicating that GABAergic tone is reduced by fear acquisition (Stork et al., 2002). Perhaps consistent with this, patients suffering from combat-related posttraumatic stress disorder exhibit persistent reductions in GABAA receptor binding (Bremner et al., 2000), and GABAA receptor expression within the amygdala is reduced in animals by manipulations that increase measures of anxiety and fear, including early life stressors (Caldji et al., 2003). Conditioned fear extinction also appears to involve GABA transmission via GABAA receptors, because systemic administration of N-methyl-β-carboline-3-carboxamide [which acts at the benzodiazepine (BZD)-binding site to decrease GABA transmission] blocks the development and expression of extinction (Harris and Westbrook, 1998).
The present study seeks to address more directly the modulation of GABAergic transmission after conditioned fear acquisition and extinction and follows from our previous finding that mRNA for the GABAA receptor clustering protein gephyrin is significantly downregulated in the BLA after fear acquisition in rats (Ressler et al., 2002). Gephyrin promotes the stabilization of GABAA receptors at synapses through interactions with the γ2 subunit of the GABAA receptor and other cytoskeletal elements and, in so doing, serves as an important regulator of GABAergic neurotransmission (Betz, 1998; Sassoe-Pognetto and Fritschy, 2000; Kneussel, 2002). Indeed, deletion of the gene encoding gephyrin leads to a dramatic reduction in GABAA receptors at synaptic sites and at the cell surface (Kneussel et al., 1999).
In the present study, we address the hypothesis that opposing alterations in GABAergic tone may take place after fear acquisition and extinction, as suggested by their opposing effects on behavior. We have focused our examination on the plasticity-dependent recruitment of GABAA receptors to postsynaptic sites by studying alterations in the levels of protein and mRNA for gephyrin, as well as measuring changes in GABAA receptor binding in the BLA after fear conditioning and extinction.
Materials and Methods
Fear conditioning. Fear conditioning was performed as described previously (Falls et al., 1992). Briefly, animals were preexposed to the training chambers for 10 min on each of 3 d. They were returned to the chambers on each of the following 2 d and presented with 30 startle stimuli (50 msec, 95 dB white noise bursts) at a 30 sec interstimulus interval (ISI). The animals were matched into experimental groups exhibiting equivalent mean startle amplitudes in the second of these sessions. Twenty-four hours later, rats received 15 pairings of a light (3.7 sec) coterminating with a 0.4 mA, 0.5 sec shock (2 min intertrial interval). Animals to be used for in situ, Western blot, or autoradiographic analyses were killed 2 or 6 hr after training. A separate set of animals was tested for potentiated startle 24 hr after training (see Fig. 1A). Behavioral testing involved the presentation of 30 startle stimuli (“leaders”) at a 30 sec ISI to habituate the startle response to a stable baseline. The data from these trials were not examined further. Thirty additional startle stimuli followed immediately, 15 of which were presented 3.2 sec after the onset of the 3.7 sec light. Fear-potentiated startle (FPS) was defined as a significant increase in the amplitude of the startle response when startle was elicited in the presence versus in the absence of the light (see Fig. 1).
Figure 1.
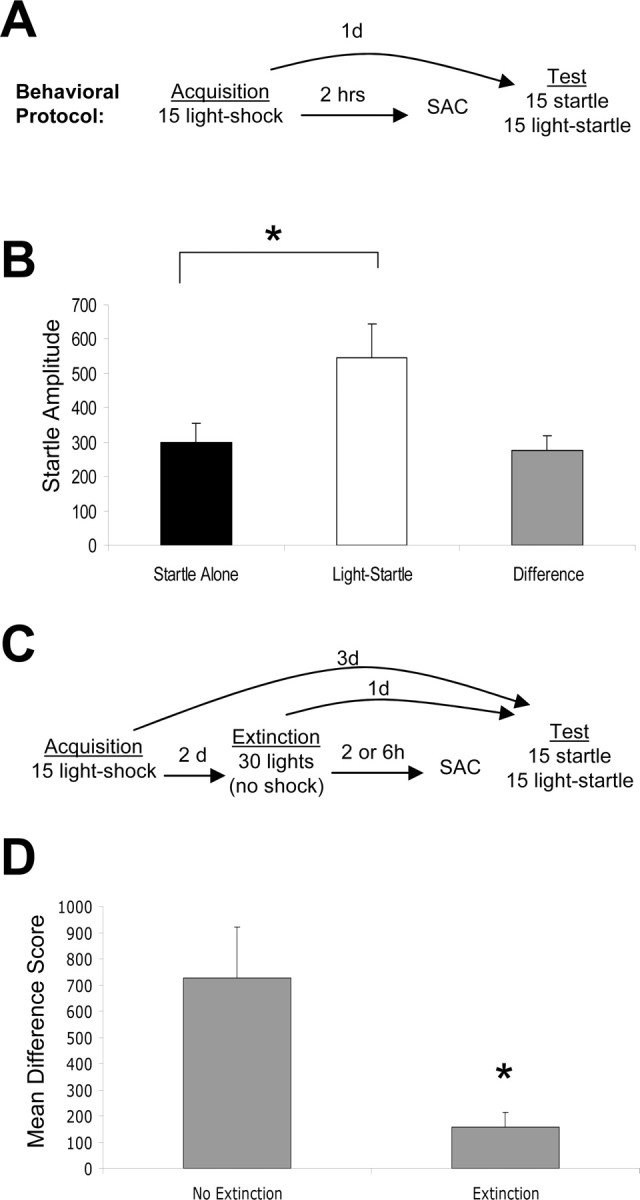
Behavioral paradigms. A, Schematic of fear-conditioning protocol. B, Control animals tested for FPS 24 hr after training demonstrate robust FPS (defined as a significant increase in the light-startle condition compared with startle alone). C, Schematic of extinction protocol. D, Control animals tested 24 hr after extinction training show a significant reduction in FPS. *p < 0.05. Error bars indicate ±SEM. See Results for statistics.
Extinction training. Fear conditioning proceeded as described above. Extinction training began 48 hr later and consisted of the presentation of 30 lights in the absence of shock (30 sec ISI). Animals for use in gephyrin assays were killed 2 or 6 hr later. Animals slated for behavioral testing were returned to the chambers 24 hr later and presented with the same test sequence used after fear conditioning. Control animals received fear conditioning but no extinction training or context exposure (see Fig. 1 D, nonextinguished control) or were behaviorally naive (see Fig. 3A, home cage control).
Figure 3.

Gephyrin mRNA is increased after extinction training. A, Gephyrin mRNA (2 hr) is increased in the BLA of extinction-trained animals compared with animals who were fear conditioned but not extinction trained (no extinction group) and animals who were neither fear-conditioned nor extinction trained (home cage group; n = 6 per group). B, Increasing the strength of extinction training leads to greater increases in gephyrin mRNA 2 hr after extinction (n = 6 per group). C, Gephyrin protein is increased 6 hr after extinction compared with controls (extinction 2 hr, n = 6; extinction 6 hr, n = 11; no extinction, n = 6). D, Qualitative comparison of the in situ hybridization study quantified in B. Arrows highlight the location of the BLA complex. *p < 0.05. Error bars indicate ±SEM. See Results for statistics.
Western blotting. Brains were blocked rapidly over ice into 2-mm-thick coronal sections. The BLA was removed bilaterally using a brain punch tool, and punches from each side were pooled and homogenized in buffer (5 mm HEPES, 1 μm EDTA, and protease inhibitors) and kept frozen at -80°C until Western blot assay. Whole-cell lysed samples were tested for protein concentration using a BCA assay (Pierce, Rockford, IL). Fifteen micrograms of protein per animal were loaded onto polyacrylamide-SDS mini-gels, separated electrophoretically, blotted onto polyvinylidene difluoride membranes (Osmonics, Minnetonka, MN), and blocked for 1 hr in 5% nonfat dry milk and 0.5% Tween 20 in PBS (PBS-T). Membranes were incubated overnight at 4°C in a 1:500 dilution of mouse anti-gephyrin antibody (clone 45; BD Biosciences, San Diego, CA) in 5% BSA-PBS-T buffer and then incubated in a 1:4000 dilution of an HRP-labeled secondary antibody for 90 min. Bound antibody was detected using ECL+, and luminescence was recorded onto Kodak BioMax MR film. Antibody detection of eukaryotic translation initiation factor 4E or glyceraldehyde-3-phosphate dehydrogenase was used to control for variations in protein loading.
In situ hybridization. In situ hybridization was performed as described previously (Ressler et al., 2002). The gephyrin cDNA clone (Integrated Molecular Analysis of Genome Expression Consortium expressed sequence tag clone; GenBank identifier accession number 880230) was linearized after sequence verification, and the antisense riboprobe was generated with T3 polymerase. Slides were postfixed and proteinase was digested and blocked, followed by overnight hybridization of the tissue at 52°C with 35S-UTP-labeled riboprobes. After a stringent wash protocol, slides were apposed to autoradiography film, and hybridization density was quantified. The level of hybridization in each section was compared with a linear 14C radiation standard using Adobe Photoshop. Relative levels of mRNA were determined as follows: (1) regions of interest (ROI) were determined based on qualitative analysis; (2) density was determined for the ROI and for an adjacent background area of the same size; (3) normalized density was calculated as the difference between the ROI density and background density; (4) two different sections per brain were averaged to give the density for each section; and (5) hybridization density was reported as the average density of all individual animals for that condition ±SEM.
Autoradiography. Fresh-frozen tissue was sectioned at 30 μm and thaw-mounted onto slides (SuperFrost Plus; Fisher Scientific, Houston, TX). Slides were incubated for 30 min at 4°C in a Tris-HCl wash buffer (in mm: 50 Tris-HCl, 120 NaCl, and 5 KCl, pH 7.4) before binding to remove endogenous ligands. Sections were then incubated in 15 nm H3-flunitrazepam (H3-Flu) (TRK-590; Amersham, Piscataway, NJ) diluted in binding buffer (50 mm Tris, 120 mm NaCl, 5 mm KCl, 10 mm MgCl, and 0.1% BSA) for 60 min at 4°C. After binding, sections were washed in wash buffer (in mm: 50 Tris, 120 NaCl, 5 KCl, and 10 MgCl), dried at 37°C, and apposed to Kodak BioMax MR film. To assay for the specificity of H3-Flu for the BZD site, several parallel sections were incubated in a similar manner, but with the inclusion of 30 μm clonazepam in the binding buffer. Residual binding of H3-Flu in the presence of 30 μm clonazepam was observed to be minimal (see Fig. 2D), demonstrating the specificity of H3-Flu for the BZD-binding site. Films were exposed for 4 weeks and then developed. Quantification was performed as for in situ hybridization.
Figure 2.

Gephyrin and GABAA binding are decreased after fear acquisition. A, Gephyrin mRNA in the BLA is significantly decreased 2 hr after fear conditioning in trained animals (light-shock) compared with animals presented with lights alone or unpaired lights and shocks during training (n = 5 per group). B, Gephyrin protein in the BLA is significantly decreased 2 hr after fear acquisition in trained (light-shock) animals compared with animals who received only lights or shocks during training (n = 6 per group) C, F, Binding of H3-Flu is significantly decreased 6 hr after fear conditioning in trained animals compared with naive controls (nonconditioned controls). A similar reduction in H3-Flu binding was not observed in the ventral striatum (F; n = 6 per group). Da, Db, Low-power image of 15 nm H3-Flu binding in a fear-conditioned animal in the absence (Da) and presence (Db) of 30 μm clonazepam. Ea, Eb, Example autoradiographs of home cage control (Ea) or fear-conditioned (Eb) animals. Ec, Ed, Pseudo-color images of Ea and Eb, respectively, demonstrating qualitative decreases in GABAA binding within the BLA after fear conditioning. Arrows highlight the location of the BLA complex. *p < 0.05. Error bars indicate ±SEM. See Results for statistics.
Results
Fear conditioning decreases gephyrin mRNA and protein levels in the BLA
To examine whether fear conditioning leads to alterations of GABAergic tone in the BLA, rats were exposed to 15 light-shock pairings (paired group), 15 lights without shocks [light alone (LA) group], or 15 shocks without lights [shock alone (SA) group]. We did not include a group in which lights and shocks were unpaired because animals under these conditions can learn that the light is a safety signal (Rescorla, 1967), comparable in some respects to extinction. After training, the paired group exhibited significant potentiated startle in the presence of the light (Fig. 1B) (p < 0.001), whereas the LA and SA groups did not (data not shown).
BLA gephyrin mRNA was decreased in the paired group 2 hr after training (Fig. 2A)(F(2,13) = 7.20; p < 0.01) but not in the LA or SA groups. No group differences in hippocampal gephyrin mRNA were observed, indicating that paired training did not decrease gephyrin mRNA in all brain regions involved in fear conditioning (Ressler et al., 2002). Western blot analysis of gephyrin protein was performed to determine whether a similar decrease in gephyrin protein was observed after fear acquisition. Western blot analysis of tissue homogenates taken 2 hr after training indicated that BLA gephyrin protein decreased in the paired group but not in the LA or SA groups. Because there were no significant differences observed between the LA or SA control groups, these control groups were pooled for comparison to the LS group (Fig. 2B)(F(1,17) = 5.41; p < 0.05).
Fear conditioning reduces the availability of BZD-sensitive GABAA receptors in the BLA
Together, these results suggest that there may be decreased trafficking of GABAA receptors to synaptic sites after associative fear conditioning. We tested this hypothesis by examining the binding of tritiated H3-Flu, an agonist of the GABAA receptor at the BZD-binding site, to GABAA receptors in animals killed 2 or 6 hr after fear acquisition. There was a significant reduction in H3-Flu binding in all fear-conditioned animals compared with nonconditioned controls (F(1,17) = 5.05; p < 0.05). Specificity of flunitrazepam for the BZD site was assayed by incubating slides in the presence (Fig. 2Db) or absence (Fig. 2Da) of 30 μm clonazepam, another agonist at the BZD site. As expected, H3-Flu binding was almost completely abolished in the presence of 30 μm clonazepam, indicating that H3-Flu binding is quite specific for the BZD site. Comparison of H3-flu binding between nonconditioned animals and animals killed 2 or 6 hr after training indicated a significant linear decrease in H3-Flu binding after training (linear contrast term; F(1,17) = 4.77; p < 0.05) as well as a significant reduction in binding at 6 hr (post hoc LSD; p < 0.05; 2 hr vs control; p = 0.13) compared with controls. Qualitative comparisons of GABAA binding further suggest decreased BLA GABAA binding in paired animals (Fig. 2Eb,d) compared with controls (Fig. 2Ea,c). No significant group differences in GABAA binding were observed in the ventral striatum (Fig. 2F).
Extinction training increases gephyrin mRNA and protein levels in the BLA
We next examined whether gephyrin mRNA and protein are modulated differentially after extinction training versus fear conditioning. Animals were exposed to 15 light-shock pairings and were returned to the conditioning chambers 48 hr later, where they were presented with 30 lights in the absence of footshock (extinguished group) or no explicit CS presentations (Fig. 1C, context control group). When tested 24 hr later, the extinguished group exhibited a significant reduction in FPS relative to controls (p < 0.01) (Fig. 1D).
In contrast to the decrease seen after fear acquisition, BLA gephyrin mRNA increased significantly 2 hr after extinction training (Fig. 3A) (main effect of group; F(2,17) = 7.88; p < 0.05). Pairwise comparisons indicated that gephyrin mRNA expression was significantly greater in extinguished animals than in home cage (i.e., untrained; p < 0.05) and nonextinguished (p < 0.05) controls. Additionally, as shown in the previous experiments, exposure to lights in the absence of shock did not alter BLA gephyrin levels in animals not previously fear conditioned (LA group).
We examined whether altering the strength of extinction training by varying the number of nonreinforced light presentations would affect the magnitude of the observed increase in gephyrin. The gephyrin mRNA increase was greater in animals receiving 120 lights than in animals receiving 10 lights or context exposure alone (Fig. 3B) (linear contrast term; F(1,17) = 4.83; p < 0.05). Qualitative comparisons between the 120 light extinction group and the context exposure group illustrate this effect (Fig. 3D, pseudocolor image). Previous reports have demonstrated that extended nonreinforced CS exposure produces greater reduction in FPS (Falls et al., 1992), which, together with the present findings, suggests that the magnitude of the upregulation of BLA gephyrin mRNA correlates with the extent of extinction.
BLA gephyrin protein expression increased significantly 6 hr after extinction training in extinguished but not in nonextinguished animals, suggesting that the change in gephyrin mRNA correlated with a change in protein levels (Fig. 3C) (ANOVA linear contrast term; F(1,20) = 5.408; p < 0.05).
Extinction increases the availability of BZD-sensitive GABAA receptors in the BLA
We next determined whether extinction alters the availability of BZD-sensitive GABAA receptors. H3-Flu binding in the BLA increased after extinction training (Fig. 4A) (main effect of group; F(2,13) = 10.40; p < 0.05). Post hoc comparisons indicated that this increase was significant at 2 hr (p < 0.05) and 6 hr (p < 0.05) after extinction training relative to nonextinguished controls. Qualitative comparison of GABAA binding further suggests that binding is increased after extinction (Figs. 4Ba-d). There was no significant change in GABAA receptor binding in the ventral striatum (Fig. 4C) after extinction training.
Figure 4.

GABAA binding is increased after extinction training. A, B, Binding of H3-Flu in the BLA is significantly increased 2 and 6 hr after extinction in trained animals (2 and 6 hr, n = 4 per group) compared with animals who were fear conditioned but not extinction trained (no extinction group, n = 6). A similar reduction in H3-Flu binding was not observed in the ventral striatum (C). Ba, Bb, Example autoradiographs from no extinction (Ba) and extinction-trained (Bb) animals. Bc, Bd, Pseudocolor images of Ba and Bb, respectively, demonstrating qualitative increases in GABAA binding within the BLA after extinction. Arrows highlight the location of the BLA complex. *p < 0.05. Error bars indicate ±SEM. See Results for statistics.
Discussion
We report that the mRNA and protein levels for the GABAA receptor clustering protein gephyrin are differentially regulated after acquisition and extinction of conditioned fear in rats, being downregulated after light-shock pairings (fear conditioning) and upregulated after nonreinforced, post-acquisition light presentations (extinction training). Given the role of gephyrin in the promotion and stabilization of postsynaptic GABAA clusters, this suggests that GABAergic synaptic transmission may be decreased after fear acquisition and increased after extinction. Consistent with this, autoradiographic analyses of BZD-sensitive GABAA receptor binding demonstrated decreases in the number of GABAA receptors available at the cell surface after fear acquisition and increases after extinction training. The similar patterns of changes in gephyrin and GABAA binding suggest that learning-related plastic changes in the GABA system occur at synaptic sites receiving direct GABAergic inhibition.
These results implicate the protein gephyrin in both fear conditioning and extinction and suggest a role for this protein in the alterations in synaptic weight that are thought to underlie some forms of associative learning. The observed concordance of gephyrin and GABAA binding at several, but not all, of the time points examined suggests that regulation of gephyrin protein may be an important component of a sophisticated cellular process underlying dynamic alterations in GABAergic tone in the BLA. However, it is very likely that numerous other proteins are critically involved in the plasticity-induced changes in GABAA receptor trafficking and stabilization. More work is needed to more directly address the issue of whether alterations in gephyrin levels are necessary and/or sufficient to support similar changes in GABAA receptor surface expression at mature inhibitory synapses and to determine the interplay of gephyrin with other proteins regulating the trafficking and expression of GABAA receptors, such as collybistin and GABARAP (Betz, 1998; Luscher and Keller, 2004).
The downregulation of gephyrin mRNA and protein expression and the decrease in the number of BZD-sensitive GABAA receptors that we have observed in the BLA after light-shock pairings seems consistent with currently favored mechanistic accounts of fear acquisition. For example, a number of studies have demonstrated a potentiation of excitatory inputs to the amygdala after CS-US pairings in rodents (Rogan and LeDoux, 1995; McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997), and it has been reported that exposure to CS-US pairings occludes long-term potentiation (LTP) of cortical inputs into the amygdala (Tsvetkov et al., 2002). Given that post-fear acquisition decreases in gephyrin mRNA do not seem to persist 48 hr after acquisition (Fig. 3A, compare home cage and no extinction groups), additional studies are needed to examine the time course of GABAergic changes and whether these changes may be compensatory responses to neural activity during behavioral training, or whether these changes may allow for the appropriate types of BLA plasticity to take place during fear acquisition.
Notably, many in vitro studies of LTP in the amygdala make use of the GABAA antagonist picrotoxin to achieve potentiation (Tsvetkov et al., 2002), suggesting that a window of reduced GABAA-mediated inhibition is required to achieve LTP. Because cortical and thalamic inputs to the BLA have been shown to synapse on both glutamatergic projection neurons and onto GABAergic interneurons (Szinyei et al., 2000), alterations in inhibition might alter the balance of these convergent inputs in favor of excitation or inhibition (for review, see Pape and Stork, 2003). Indeed, the necessity of reducing inhibition of glutamatergic neurons to achieve LTP has been demonstrated elegantly by Luthi and colleagues (Bissiere et al., 2003), who observed that the induction of amygdalar LTP required the presence of either picrotoxin or dopamine-mediated suppression of feedforward inhibition. In this context, the observed reduction in GABAA receptor expression after fear acquisition may indicate that an experience-dependent disinhibition of glutamatergic BLA neurons normally occurs after fear acquisition in vivo, perhaps creating a window in which LTP can occur. This, in turn, suggests that alterations in GABAergic tone may be permissive in allowing the plastic changes underlying BLA-dependent learning to occur. Such a role seems consistent with the time course of the changes observed here, because dynamic changes in GABAergic tone appear to be taking place within the consolidation phase of learning.
From a behavioral perspective, the present findings support the notion that fear acquisition and extinction are distinct forms of learning that exert opposing effects on the expression of the CR. Indeed, several theoretical accounts of extinction have suggested that CS- and situation-specific control of GABAergic transmission within the amygdala during and after extinction training is a central mechanism of extinction (cf. Harris and Westbrook, 1998; Royer and Pare, 2002; Quirk et al., 2003).
Despite the evidence against an “unlearning” account of extinction (cf. Myers and Davis, 2002), it has been suggested that extinction involves a “depotentiation” of the same synapses that are potentiated after acquisition and a reversal of the original molecular signaling (Lin et al., 2003a,b). The extinction-induced increases in GABAergic tone observed here are partially consistent with a “depotentiation” hypothesis, because both would suggest a decrease in BLA pyramidal cell excitability after extinction. However, the depotentiation effect also may be mediated in vivo by a strengthening of inhibitory neurotransmission onto pyramidal cells (i.e., a heterosynaptic long-term depression-like phenomenon), rather than by a decrease in the efficacy of excitatory inputs into the BLA. In either case, it may be that both fear acquisition and extinction lead to plasticity-dependent alterations within the same circuit but at different synapses and that plastic changes in the GABA system may play a more central role in amygdala-dependent learning than previously thought.
Footnotes
This work was supported by National Institutes of Health Grants MH070218 (J.P.C.), MH69884 (K.J.R.), and MH47840 (M.D.) and by the Science and Technology Center Program (Center for Behavioral Neuroscience) of the National Science Foundation under Agreement IBN-987675. We thank Dr. Darlene Francis and Kate Sharer for their expertise and assistance with GABAA autoradiography.
Correspondence should be addressed to Dr. Michael Davis, Department of Psychiatry and Behavioral Sciences, Yerkes Neuroscience Building, Room 5200, 954 Gatewood Road Northeast, Atlanta, GA 30322. E-mail: mdavis4@emory.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250502-05$15.00/0
References
- Betz H (1998) Gephyrin, a major player in GABAergic postsynaptic membrane assembly? Nat Neurosci 1: 541-543. [DOI] [PubMed] [Google Scholar]
- Bissiere S, Humeau Y, Luthi A (2003) Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat Neurosci 6: 587-592. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Innis RB, Southwick SM, Staib L, Zoghbi S, Charney DS (2000) Decreased benzodiazepine receptor binding in prefrontal cortex in combat-related posttraumatic stress disorder. Am J Psychiatry 157: 1120-1126. [DOI] [PubMed] [Google Scholar]
- Caldji C, Diorio J, Meaney MJ (2003) Variations in maternal care alter GABAA receptor subunit expression in brain regions associated with fear. Neuropsychopharmacology 28: 1950-1959. [DOI] [PubMed] [Google Scholar]
- Davis M (2000) The role of the amygdala in conditioned and unconditioned fear and anxiety. In: The amygdala, Vol 2 (Aggleton JP, ed), pp 213-287. Oxford: Oxford UP. [Google Scholar]
- Falls WA, Miserendino MJD, Davis M (1992) Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci 12: 854-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JA, Westbrook RF (1998) Evidence that GABA transmission mediates context-specific extinction of learned fear. Psychopharmacology 140: 105-115. [DOI] [PubMed] [Google Scholar]
- Josselyn SA, Shi C, Carlezon WA, Neve RL, Nestler EJ, Davis M (2001) Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci 21: 2404-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneussel M (2002) Dynamic regulation of GABAA receptors at synaptic sites. Brain Res Rev 39: 74-83. [DOI] [PubMed] [Google Scholar]
- Kneussel M, Brandstatter JH, Laube B, Stahl S, Muller U, Betz H (1999) Loss of postsynaptic GABAA receptor clustering in gephyrin-deficient mice. J Neurosci 19: 9289-9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE (2000) The amygdala and emotion: a view through fear. In: The amygdala (Aggleton JP, ed), pp 289-310. New York: Oxford UP.
- Lin C-H, Yeh S-H, Leu T-H, Chang W-C, Wang S-T, Gean P-W (2003a) Identification of calcineurin as a key signal in the extinction of fear memory. J Neurosci 23: 1574-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C-H, Yeh S-H, Lu H-Y, Gean P-W (2003b) The similarities and diversities of signal pathways leading to consolidation of acquisition and consolidation of extinction of fear memory. J Neurosci 23: 8310-8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K-T, Walker DL, Davis M (2001) Mitogen-activated protein kinase cascade in the basolateral nucleus of the amygdala is involved in extinction of fear-potentiated startle. J Neurosci 21: RC162(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher B, Keller CA (2004) Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther 102: 195-221. [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P (1997) Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390: 607-611. [DOI] [PubMed] [Google Scholar]
- Miserendino MJD, Sananes CB, Melia KR, Davis M (1990) Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature 345: 716-718. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M (2002) Behavioral and neural analysis of extinction. Neuron 36: 567-584. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE (2000) Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406: 722-726. [DOI] [PubMed] [Google Scholar]
- Pape HC, Stork O (2003) Genes and mechanisms in the amygdala involved in the formation of fear memory. Ann NY Acad Sci 985: 92-105. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Likhtik E, Pelletier JG, Pare D (2003) Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J Neurosci 23: 8800-8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescorla RA (1967) Pavlovian conditioning and its proper control procedures. Psychol Rev 74: 71-80. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Paschall GY, Zhou XL, Davis M (2002) Regulation of synaptic plasticity genes during consolidation of fear learning. J Neurosci 22: 7892-7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, LeDoux JE (1995) LTP is accompanied by commensurate enhancement of auditory-evoked responses in a fear conditioning circuit. Neuron 15: 127-136. [DOI] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE (1997) Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390: 604-607. [DOI] [PubMed] [Google Scholar]
- Royer S, Pare D (2002) Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience 115: 455-462. [DOI] [PubMed] [Google Scholar]
- Sassoe-Pognetto M, Fritschy JM (2000) Mini-review: gephyrin, a major postsynaptic protein of GABAergic synapses. Eur J Neurosci 12: 2205-2210. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Nadel NV, Sullivan GM, Harris A, LeDoux JE (1999) Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn Mem 6: 97-110. [PMC free article] [PubMed] [Google Scholar]
- Stork O, Ji FY, Obata K (2002) Reduction of extracellular GABA in the mouse amygdala during and following confrontation with a conditioned fear stimulus. Neurosci Lett 327: 138-142. [DOI] [PubMed] [Google Scholar]
- Szinyei C, Heinbockel J, Montagne J, Pape HC (2000) Activation of putative cortical and thalamic inputs elicits convergent excitation in a population of GABAergic interneurons of the lateral amygdala. J Neurosci 20: 8909-8915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov E, Carlezon WA, Benes FM, Kandel ER, Bolshakov VY (2002) Fear conditioning occludes LTP-induced presynaptic enhancement of synaptic transmission in the cortical pathway to the lateral amygdala. Neuron 34: 289-300. [DOI] [PubMed] [Google Scholar]