

Abstract
The breathing motor pattern in mammals originates in brainstem networks. Whether pacemaker neurons play an obligatory role remains a key unanswered question. We performed whole-cell recordings in the preBötzinger Complex in slice preparations from neonatal rodents and tested for pacemaker activity. We observed persistent Na+ current (INaP)-mediated bursting in ∼5% of inspiratory neurons in postnatal day 0 (P0)-P5 and in P8-P10 slices. INaP-mediated bursting was voltage dependent and blocked by 20 μm riluzole (RIL). We found Ca2+ current (ICa)-dependent bursting in 7.5% of inspiratory neurons in P8-P10 slices, but in P0-P5 slices these cells were exceedingly rare (0.6%). This bursting was voltage independent and blocked by 100 μm Cd2+ or flufenamic acid (FFA) (10-200 μm), which suggests that a Ca2+-activated inward cationic current (ICAN) underlies burst generation. These data substantiate our observation that P0-P5 slices exposed to RIL contain few (if any) pacemaker neurons, yet maintain respiratory rhythm. We also show that 20 nm TTX or coapplication of 20 μm RIL + FFA (100-200 μm) stops the respiratory rhythm, but that adding 2 μm substance P restarts it. We conclude that INaP and ICAN enhance neuronal excitability and promote rhythmogenesis, even if their magnitude is insufficient to support bursting-pacemaker activity in individual neurons. When INaP and ICAN are removed pharmacologically, the rhythm can be maintained by boosting neural excitability, which is inconsistent with a pacemaker-essential mechanism of respiratory rhythmogenesis by the preBötzinger complex.
Keywords: breathing, preBötzinger Complex, brainstem, bursting, central pattern generator, calcium-activated nonspecific cation current (ICAN)
Introduction
Behaviors like breathing, walking, and eating emanate from central pattern generator networks of the brainstem and spinal cord. Pacemaker neurons may play a central role in generating these rhythms (Marder, 2001), which we evaluate here in the mammalian respiratory network.
The cellular mechanisms that underlie respiratory rhythm can be examined in neonatal rodent slice preparations that generate respiratory-related motor output in vitro, where the critical neurons are localized within the preBötzinger Complex (preBötC) (Smith et al., 1991). This rhythm continues after blocking postsynaptic inhibition, which rules out mechanisms based on reciprocal inhibition and favors pacemaker-driven models (Feldman and Smith, 1989). The pacemaker hypothesis posits that a kernel of neurons with intrinsic bursting properties are essential to generate synchronized rhythmic activity that is distributed to follower respiratory neurons (Feldman and Smith, 1989; Smith et al., 1991, 2000; Rekling and Feldman, 1998; Butera et al., 1999; Koshiya and Smith, 1999; Del Negro et al., 2001).
If preBötC pacemaker neurons are essential for rhythmogenesis in slices, then removing them from the network should disrupt or abolish respiratory activity. Two mechanisms for bursting have been described: one based on a voltage-dependent persistent Na+ current (INaP) (Feldman and Smith, 1989; Smith et al., 1991; Johnson et al., 1994; Del Negro et al., 2001, 2002b; Rybak et al., 2003) and another that depends on Ca2+ currents (ICa) and Ca2+-activated nonspecific cationic current (ICAN) (Thoby-Brisson and Ramirez, 2001; Pena et al., 2004). Here, we pharmacologically abolish INaP-mediated pacemaker activity, and characterize a developmental period during which ICa-dependent pacemaker neurons are extremely sparse, and we show that respiratory rhythm persists (with unchanged frequency) in their apparent absence. We also show that pharmacological attenuation of both INaP and ICAN stops the respiratory rhythm at any age, but that enhancing neural excitability restarts it. We conclude that respiratory rhythm in slices is not pacemaker driven but rather emerges when neurons are sufficiently excitable and there is recurrent excitation in the network. Given the critical role preBötC neurons play in normal rhythmogenesis in intact rodents (Gray et al., 2001), we suggest that this is also the case in intact mammals.
Materials and Methods
We used neonatal C57BL/6 mice [postnatal day 0 (P0)-P10] and Sprague Dawley rats (P0-P10) for experiments in vitro. The Office for the Protection of Research Subjects (University of California Animal Research Committee) and the Institutional Animal Care and Use Committee (The College of William and Mary) approved all protocols. Transverse slices (550 μm thick) containing the preBötC (Smith et al., 1991) and hypoglossal (XII) motoneurons were dissected in normal artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 3 KCl, 1.5 CaCl2, 1 MgSO4, 25 NaHCO3, 0.5 NaH2PO4, and 30 d-glucose, equilibrated with 95% O2 and 5% CO2 (27°C), pH 7.4. Slices were perfused with ACSF at 4 ml/min and maintained at 27°C. ACSF K+ concentration was raised to 9 mm, and respiratory motor output was recorded from XII nerve roots using suction electrodes and a differential amplifier (bandpass filtered at 0.3-1 kHz, full wave-rectified, and smoothed for display).
Whole-cell patch electrodes (3-6 MΩ) contained the following (in mm): 140 CsMeSO4, 5 NaCl, 0.1 EGTA, 10 HEPES, 2 Mg ATP, and 0.3 Na3 GTP, pH 7.25, for voltage-clamp experiments or 140 K gluconate, 1 CaCl2, 1 MgCl2, either 0.1 or 10 EGTA, 10 HEPES, 2 Mg ATP, and 0.3 Na3 GTP, pH 7.25, for current clamp. Pipettes were visually guided to the preBötC using video microscopy. The z-axis of a Sutter Instruments (Novato, CA) MP-285 robotic micromanipulator was set to 0 μmatthe slice surface to monitor the depth of subsequent whole-cell recordings. A modified ACSF was used in voltage-clamp experiments to isolate Na+ currents containing the following (in mm): 114.5 NaCl, 10 TEA Cl, 3 KCl, 0.5 CaCl2, 2 MgCl2, 25 NaHCO3, and 30 d-glucose. Descending voltage ramps from +10 to -90 mV (20 mV/s) were used to measure noninactivating INaP. We calculated input resistance from the slope of the current-voltage (I-V) relationship generated by the voltage ramp in the linear region negative to -60 mV. Cell capacitance (CM) was computed from the integral of the transient capacity current (IC, leak-subtracted), which was evoked by 15 ms hyperpolarizing voltage step commands (ΔVC), using the formula CM = ∫IC/ΔVC. Series (access) resistance (RS) was calculated from the decay-time constant (τ) of transient IC because τ =RS * CM in voltage clamp, where τ is the fitted exponential IC decay time. An acceptable voltage clamp requires RN ≥ 10 * RS. Cells that failed to meet this criterion were discarded. We applied analog RS compensation without whole-cell capacity compensation to continuously monitor and ensure stationary voltage-clamp conditions.
We bath-applied the following drugs obtained from Sigma (St. Louis, MO) (in μm): 1-200 riluzole (RIL) (2-amino-6-trifluoromethoxy benzothiazole), 10-200 flufenamic acid (FFA), 10 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 20 dl-2-amino-5-phosphonopentanoic acid (APV), 2-5 bicuculline (BIC) or picrotoxin (PTX), 2-5 strychnine (STR), 0.5-2 substance P (SP), and 0.5 AMPA. In some experiments, we applied 100 μm CdCl2 to block ICa; NaH2PO4 was omitted from the ACSF to avoid precipitation.
We monitored the effects of RIL on respiratory motor output by plotting the period and amplitude of XII discharge on a cycle-to-cycle basis (see Fig. 3). We then divided each experiment into contiguous 2 min segments and computed the mean period during each segment. The mean period and amplitude during each segment for three experiments is reported in Figure 5.
Figure 3.

Effects of RIL on respiratory rhythm in rat slice preparations. Increasing concentrations of RIL plotted from top to bottom. Respiratory period (gray circles) and XII amplitude (black triangles) are plotted versus time. RIL was applied for 30 min in each bout. Period is scaled from 0-24 s (left ordinate), and XII amplitude is scaled uniformly (right ordinate) for each experiment in arbitrary normalized units.
Figure 5.

Effects of RIL on INaP and XII respiratory period. INaP was measured every 20 s and plotted with open circles (left). The scale bar shows 20 μm RIL application. The current-voltage curve (right) from a mouse experiment is shown at three time points during the protocol. The mean respiratory period at RIL concentrations from 10-200 μm is plotted as a function of time from n = 3 rat slices tested at each concentration (no error bars).
In control experiments, we found that RIL had very similar effects on neonatal rats and mice during P0-P5. RIL at 1-200 μm caused the same dose-dependent and time-dependent effects on XII amplitude, area, and period, regardless of species (n = 10 mice; n = 8 rats). RIL at 10-20 μm attenuated INaP to the same extent in voltage clamp (n = 6 mice; n = 5 rats). Therefore, we pooled systems-level data and INaP voltage-clamp experiments (see Figs. 3, 4, 5). RIL-sensitive pacemaker neurons were found in both rats (n = 46) and mice (n = 14); this database contains neurons from previous studies (Del Negro et al., 2002a). Pacemaker neurons that depend on ICa and ICAN were found only in mice, consistent with the first published report of Cd2+-sensitive pacemaker neurons in juvenile mice (Thoby-Brisson and Ramirez, 2001).
Figure 4.

Dose-response curves for RIL. Closed circles show mean time to abolish XII activity obtained from rat experiments (n = 3 at each dose, no error bars), EC50 = 32 μm. Open circles show attenuation of INaP inrats and mice. Some data points were originally reported in the study by Del Negro et al. (2002a).
We used the Wilcoxon signed-rank test to assess the effects of RIL on respiratory frequency (see Fig. 5), and we used the Fisher exact test to assess the relative prevalence of pacemaker neurons in rodent slices of different age ranges (see Results for details). We applied the Wilcoxon signed rank test and the paired t test to evaluate the cellular and network effects of 20 nm TTX (see Fig. 6), which yielded the same results. We tested whether RIL caused significant respiratory period fluctuations (before cessation) using the Kolmogorov-Smirnov test (K-S test). Significance was set at p ≤ 0.05.
Figure 6.

The effects of 20 nm TTX on respiratory rhythm in vitro. A, XII output is shown in control, 20 nm TTX (15 and 20 min elapsed time), and recovery conditions. B, Evoked action potentials in control and 20 nm TTX conditions. Baseline VM was -60 mV. The calibration bar applies to both VM and stimulus current (Ia). C, Bar charts showing the mean effects of TTX on cellular and systems level respiratory activity (n = 9 mouse slices tested). Frequency in TTX was taken as the average of three to five bursts just before TTX-induced cessation of the rhythm. The amplitude of XII motor output is scaled in arbitrary units (a.u.). Rheobase and spike “active fraction” are defined in Results. Error bars indicate SEM for n = 9 mouse slice preparations. Single asterisk indicates statistical significance at p < 0.01, and double asterisk indicates p < 0.005.
Results
INaP-mediated pacemaker neurons
Figure 1 depicts typical INaP-mediated bursting in a mouse pre-BötC inspiratory neuron (Smith et al., 1991; Johnson et al., 1994; Koshiya and Smith, 1999; Thoby-Brisson and Ramirez, 2001; Del Negro et al., 2002a,b). Here, we used patch solution containing 0.1 mm EGTA. Respiratory drive potentials occurred at membrane potentials below the INaP activation threshold (Fig. 1A, left) (Rybak et al., 2003). Depolarization evoked INaP and caused ectopic bursts in the intervals between XII discharges (Fig. 1A, middle). Intrinsic bursting was confirmed by its persistence after blocking synaptic transmission with CNQX, APV, BIC, and STR in either 9 mm (Fig. 1A, right) or 3 mm (Fig. 1B) K+ concentration in the ACSF. Bursting was voltage dependent. Depolarizing the baseline membrane potential using bias current (Ia) caused the cell to move from quiescence to bursting, and further depolarization increased burst frequency (Fig. 1B). However, 20 μm RIL blocked INaP-mediated bursting within 90 s, and no amount of depolarizing Ia could restore it (Fig. 1C). All rhythmically active preBötC neurons with stable baseline membrane potentials and overshooting spikes were tested for pacemaker activity and are included in our database. INaP-mediated bursting was detected in eight of 178 P0-P5 preBötC neurons (4.5%) and in three of 54 P8-P10 preBötC neurons (5.6%), which was not significantly different (Fisher exact test, p = 0.3).
Figure 1.

INaP-mediated bursting in a mouse preBötC neuron. A, Respiratory activity at -60 mV baseline membrane potential (VM). ACSF contained 9 mm [K+]. Ectopic bursts emerged after adding 50 pA of bias current (Ia). Intrinsic bursting after CNQX, APV, BIC, and STR eliminated respiratory network activity (XII). B, Intrinsic bursting in the same cell as A after reducing ACSF [K+] to 3 mm. Baseline VM is -55 mV. C, Adding 20 μm RIL blocked bursting, which could not be restored after 60-90 s by depolarization via Ia. Calibration applies to all traces.
Calcium current-dependent pacemaker neurons
Bursting in preBötC neurons in mice >P5 that depends on ICa is a recent discovery (Thoby-Brisson and Ramirez, 2001). In the past, we never observed this phenotype in P0-P5 rats or mice (Del Negro et al., 2002a), so here we recorded in juvenile mice ages P8-P10 to see whether developmental effects could explain this disparity. After identifying a pacemaker neuron in CNQX, APV, PTX, and STR (Fig. 2A), we applied Cd2+ to test whether ICa was required. We found four ICa-dependent pacemaker neurons in 54 neurons sampled (7.5%) during P8-P10, whereas we found only one ICa-dependent pacemaker neuron in 178 neurons sampled during P0-P5 (0.6%), which indicates significantly higher prevalence of bursting cells in the older neonates (Fisher exact test, p = 0.01). Of the 54 preBötC neurons recorded at P8-P10, 40 were recorded with 10 mm EGTA patch solution (Thoby-Brisson and Ramirez, 2001) and 14 with 0.1 mm EGTA patch solution. ICa-dependent bursting neurons in these older neonates made up 7.5% of the sample regardless of EGTA concentration (n = 3 of 40 and 1 of 14, respectively), which suggests that slow calcium buffering by 10 mm EGTA does not influence pacemaker activity (Fisher exact test, p = 0.43). The ICa-dependent pacemaker neurons often showed ramp-like trajectories during the interburst interval, i.e., expiration, and a strong inspiratory discharge that was enhanced by depolarization (Fig. 2A). Bursting continued in CNQX and RIL but ceased in Cd2+ (Fig. 2A,D), which demonstrates dependence on ICa. Bursting was also blocked by 10 μm FFA (Fig. 2B), which suggests that a Ca2+-activated nonspecific cationic current (ICAN) (Gogelein et al., 1990; Guinamard et al., 2004) carries significant burstgenerating inward current in addition to ICa.
Figure 2.
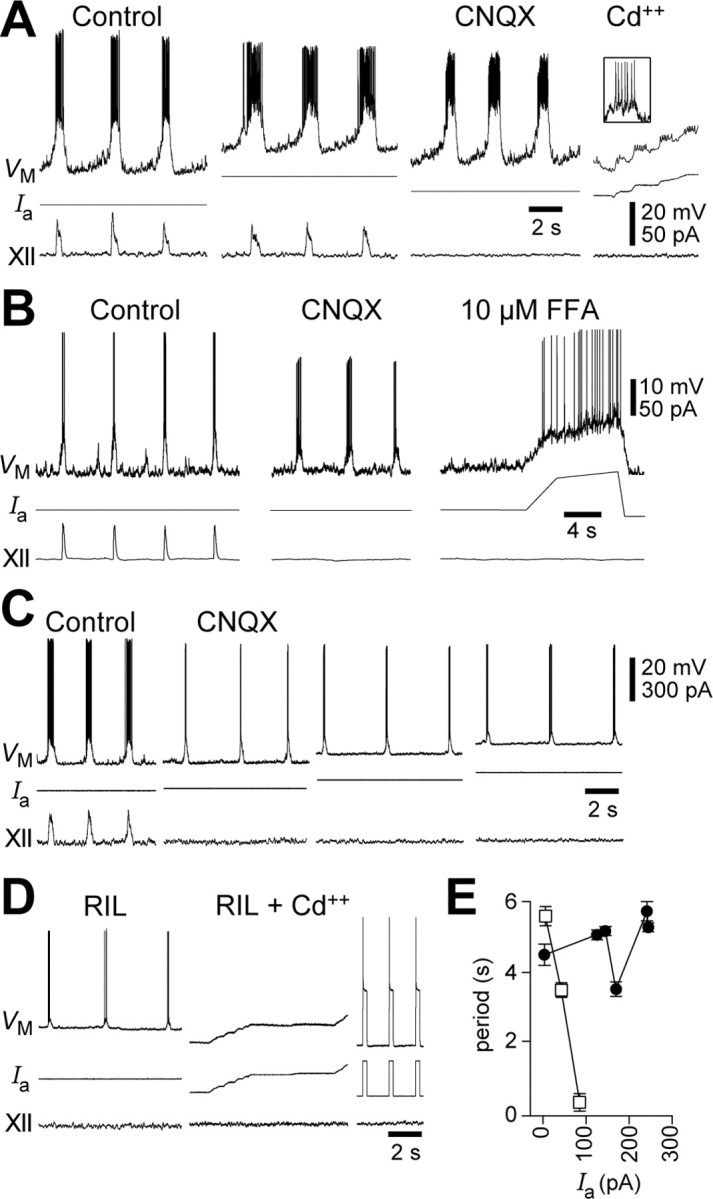
ICa-dependent bursting in mouse preBötC neurons (P8). A, Respiratory discharge from a baseline VM of -60 mV; adding 30 pA depolarized the cell and strengthened ramp-like VM trajectory and respiratory drive potentials and spike discharge. Intrinsic bursting continued in CNQX, APV, PTX, and STR. Cd2+ blocked bursting but not action potentials (inset). B, A similar experiment, but here the bursting was blocked by 10 μm FFA. C, Bursting behavior in CNQX, APV, PTX, and STR was subjected to depolarizing bias current (Ia). D, RIL at 20 μm did not block bursting but Cd2+ did (same cell as C). Ia pulses were used to evoke action potentials in RIL plus Cd2+ conditions to ensure that the cell could still generate spikes. E, Burst period from the cell in C (circles) and the voltage-dependent INaP pacemaker neuron from Figure 1 B (squares), plotted versus Ia.
ICa-dependent bursting was extremely rare in P0-P5 mice (1 of 178; 0.6%), in agreement with Pena et al. (2004). In a P2 mouse we blocked bursting in this one neuron using 200 μm FFA and then added 20 μm RIL, which additionally hyperpolarized the cell by 2-3 mV and thus suggests that this neuron expressed INaP in addition to ICAN.
Unlike INaP-mediated bursting, ICa-dependent bursting was not voltage dependent. For example, the pacemaker neuron in Figure 2C was subjected to increasing levels of Ia but maintained a ∼4 s burst period (Fig. 2E, filled circles). Figure 2E further compares this cell with INaP-mediated voltage-dependent bursting from Figure 1B (open squares). Nevertheless, Ia increased the number of action potentials per burst. For example, depolarization increased the mean number of spikes per burst from 21 to 37 in Figure 2A (in the context of network activity) and from 4 to 7 in Figure 2C (in the isolated pacemaker neuron after CNQX, APV, PTX, and STR).
RIL applications in vitro
We applied RIL for 30 min and monitored respiratory motor output in P0-P5 slices from rats and mice (Figs. 3, 4, 5). RIL at 10 μm reduced XII amplitude (Fig. 3, black triangles) but did not change the mean period (Figs. 3, 5, gray circles) (for n = 3 experiments, Wilcoxon test, p = 0.6). RIL at 10 μm also caused cycle-to-cycle variability in the period that was noticeable but not statistically significant (K-S test, p = 0.08).
RIL at 20 μm decreased XII amplitude monotonically and caused periodic fluctuation but did not change the mean period until after the XII amplitude decreased to baseline, which occurred at 40 min. Afterward, the period rapidly increased (Fig. 3, note the upward thrust of gray circles in the row labeled 20 μm) and within a dozen cycles rhythmic output ceased altogether. Similar results were obtained at RIL concentrations from 50 to 200 μm: motor output disappeared after a precipitous increase in period, which always coincided with XII amplitude reaching barely detectable levels (Fig. 3). Before the abrupt cessation of rhythm, the mean period for n = 3 experiments at each RIL concentration was not significantly different from control (Wilcoxon tests, all p > 0.05). RIL consistently caused periodic fluctuations, which were statistically significant at 50 and 200 μm (K-S test, p < 0.01).
Higher RIL doses accelerated the rate of decline in motor output. The mean time required to abolish XII output decreased sigmoidally with an EC50 value of 32 μm, whereas INaP in rats and mice was blocked by much lower doses (EC50 = 3 μm) and within 9 min (Fig. 4) (Del Negro et al., 2002a). This suggests that either respiratory rhythm did not require INaP and RIL abolished motor output via some other effect or that INaP was required but that RIL affected rhythmogenic neurons only at high doses (or in prolonged exposures).
The second scenario might arise if RIL has difficulty penetrating the slice preparation. To evaluate this, we measured the effects of 20 μm RIL on INaP recorded in preBötC neurons from rats and mice at various depths. We previously reported rapid blockade (<9 min) of INaP by RIL in neurons within 125 μm of the surface (Del Negro et al., 2002a). Here, we recorded neurons 250-360 μm deep and applied descending voltage ramps that inactivate the fast Na+ current responsible for action potentials (but do not completely inactivate INaP) every 20 s during RIL application (Del Negro et al., 2002a). Using a cesium-based patch solution in neurons that satisfied our criteria for adequate voltage clamp, a region of negative slope was always present in the steady-state I-V curve because of INaP (Del Negro et al., 2002b). RIL always blocked this negative slope region in <9 min (n = 7) such that the I-V curve was monotonic with positive slope between -60 and -40 mV. Under these conditions, voltage-dependent INaP bursting is impossible (Smith et al., 1975). In three cells, the time course of INaP blockade could be fitted by an exponential decay function with a time constant of 2 min (Fig. 5). Figure 5 also plots the mean respiratory period computed from three slice experiments like Figure 3, which shows that the mean respiratory period was undiminished within the time frame needed for 20 μm RIL to block INaP at depths ≥250 μm.
Low doses of TTX
Low doses of TTX preferentially antagonize INaP in respiratory neurons (Koizumi and Smith, 2003). We found that 20 nm TTX reversibly abolished respiratory rhythm in 20-40 min in 15 mouse slices (P0-P5) tested (Fig. 6A). An average of the last three to five respiratory bursts showed that XII amplitude decreased 62%, and frequency declined 94% before the rhythm ceased (Fig. 6C). TTX at 20 nm hyperpolarized respiratory neurons by 5-10 mV, which is consistent with blockade of a Na+ current active at rest, such as INaP.To determine whether 20 nm TTX also affected spike generation, we applied 20 ms step currents from a baseline membrane potential of -60 mV to evoke action potentials (during the expiratory phase). Rheobase increased from 277 ± 177 pA in control to 399 ± 187 pA in TTX (≥30 min exposure to 20 nm TTX) (p < 0.001; n = 9) (Fig. 6B,C). Spike amplitude declined in TTX. Changes in rheobase required more passive charging of the membrane potential to reach spike threshold, and this passive charging progressively encompassed a greater percentage of the total spike amplitude in TTX. Therefore, we defined the active fraction of the evoked spike as the difference between the maximum spike amplitude and the peak of the passive membrane response. Active fraction decreased from 67 ± 10 mV in control to 29 ± 13 mV in 20 nm TTX (p < 0.0005; n = 9) (Fig. 6B,C). All of the changes induced by 20 nm TTX were statistically significant by Wilcoxon signed rank or paired t tests and were reversible in washout (Fig. 6C). In one preparation, 20 nm TTX completely abolished spikes (data not shown).
Because 20 nm TTX hyperpolarized preBötC neurons, increased rheobase and decreased spike amplitude, we posited that TTX-induced decreases in excitability might be responsible for respiratory rhythm cessation, rather than (highly) selective attenuation of INaP. After at least 2 min of quiescence, we attempted to recover network function in the presence of 20 nm TTX by adding the excitatory neuropeptide SP (0.5-2 μm). SP revived the rhythm in five of nine preparations tested (Fig. 7). The recovered rhythm in SP and TTX conditions was stable for 5-10 min and then gradually became unstable and ceased after 40-60 min exposure to TTX. Respiratory rhythm recovered fully in washout (Fig. 6A).
Figure 7.

SP at 2 μm restores respiratory rhythm in a mouse slice exposed to 20 nm TTX. TTX hyperpolarized this inspiratory neuron by 7 mV; thus, 220 pA of depolarizing bias current (Ia) was applied to restore baseline VM to -60 mV (20 nm TTX, 5 min). Additional depolarizing Ia was required to maintain VM of -60 mV by the time TTX abolished rhythmic activity (20 nm TTX, 15 min). After the rhythm ceased, adding 2 μm SP to the bath revived it. The amplitude of the cellular drive potentials and XII amplitude recovered in SP, but spike discharge did not occur with baseline VM at -60 mV (attributable to increases in rheobase, see Results and Fig. 6).
FFA and RIL coapplication experiments
Although ICa-dependent pacemaker neurons are extremely sparse in P0-P5 mice, one could argue that during bouts of RIL (e.g., Figure 3), a small number of RIL-insensitive pacemaker neurons sustains the rhythm (Pena et al., 2004). We tested this possibility in FFA and RIL coapplication experiments. In P0-P5 mouse slices, first we applied 100 μm FFA, which did not affect XII frequency or amplitude (Fig. 8A,B) but at the cellular level reversibly reduced (but did not abolish) respiratory drive potentials and spike amplitude (Fig. 8B). After washout, we coapplied 100 μm FFA and 20 μm RIL, which blocked respiratory rhythm within 5 min; the last cycle of cellular and XII activity is shown in Figure 8B. After more than 2 min of quiescence in the presence of FFA and RIL, adding 0.5-2 μm SP to the bath restored rhythmic activity: XII output resembled the first FFA washout, but respiratory drive potentials were significantly attenuated (n = 5 slices). We were also able to rescue the rhythm after it ceased in 100 μm FFA and 20 μm RIL using low concentrations of bathapplied AMPA (500 nm; n = 2).
Figure 8.

Rhythm generation in the presence of 100 μm FFA and 20 μm RIL. A, Continuous segments of the experiment showing control, FFA, recovery, and then FFA and RIL coapplication, as well as FFA plus RIL plus SP conditions in a mouse slice. Rhythm cessation in FFA plus RIL was rescued by SP. B, Examples of cellular respiratory drive and XII output from A are plotted with greater time resolution. Broken line facilitates XII amplitude comparisons.
Discussion
Conditions that eliminated bursting in INaP- and ICa-dependent pacemaker neurons did not prevent respiratory rhythm generation in medullary slices in vitro. When INaP and ICAN are pharmacologically blocked the rhythm ceases but a boost in neural excitability restores it. We propose that INaP and ICAN normally enhance excitability and promote inspiratory bursts in all pre-BötC respiratory neurons, regardless of whether or not a small subset of these neurons (which express INaP and ICAN abundantly) support intrinsic bursting.
INaP pacemaker neurons
RIL blocks bursting and INaP in preBötC neurons within minutes at low doses (EC50 = 3 μm), whereas it abolishes respiratory rhythm in slices only after long exposures or when concentration is >10 μm (Del Negro et al., 2002a). RIL caused periodic fluctuation at all concentrations, thus INaP and pacemaker neurons probably help stabilize network rhythmicity. The time to block XII output was dose dependent (EC50 = 32 μm, corresponding to 35 min for XII discharge to disappear), which is an order of magnitude higher than the EC50 value of 3 μm to block INaP within 9 min in neurons recorded at depths up to 350 μm in slices. The disparity in dose and time dependence suggests that the effects of RIL that abolish respiratory rhythm include more than just blocking INaP.
INaP-mediated bursting in preBötC neurons does not appear to be developmentally regulated because these cells were found at ∼5% in P0-P5 and P8-P10 cohorts. Is 5% a reasonable estimate of the fractional content of INaP bursting neurons in the preBötC? Pena et al. (2004) reported 16-29% INaP pacemaker neurons in P1-P15 mice. However, these authors preferentially selected neurons that exhibited pacemaker-like characteristics in the cell-attached mode (Pena et al., 2004). Their sampling favors neurons that discharge many spikes per XII cycle and more frequently exhibit pacemaker properties after application of CNQX and other blockers, which significantly overestimates the prevalence of INaP pacemaker neurons. Moreover, because they reported that the likelihood of finding pacemaker neurons of any type in a given slice was <10%, then the 16-29% figure cannot be representative of their actual relative prevalence. In contrast, we sampled preBötC neurons without regard to activity pattern in cell-attached mode. All rhythmic neurons were tested for pacemaker properties in the presence of CNQX, APV, BIC/PTX, and STR. We avoided using low Ca2+ solution to block synaptic transmission because it also blocks ICAN and Ca2+-dependent K+ current and can induce silent cells to burst (Pena et al., 2004). Therefore, we estimate that the actual fraction of INaP pacemaker neurons in the preBötC is closer to 5%.
Calcium-dependent pacemaker neurons
We detected only one ICa-dependent pacemaker neuron of 178 neurons (0.6%) during P0-P5, whereas we found them 7.5% of the time at P8-P10, which is significantly different. This suggests that either ICa (Onimaru et al., 1996, 2003; Elsen and Ramirez, 1998) or ICAN are developmentally regulated and that Cd2+-sensitive bursting behavior emerges predominantly after P5. Independent reports from our group and Pena et al. (2004) show that these cells increase in numbers by an order of magnitude after P6. Despite differences in sampling protocols, these two reports estimate the fraction of ICAN pacemaker neurons to be between 7.5 and ∼9% in juvenile mice.
ICa is necessary for bursting in these neurons because Cd2+ blocks pacemaker activity (Thoby-Brisson and Ramirez, 2001). However, the voltage-independent nature of the bursting suggests that Ca2+ activates ICAN, which dominates the burst-generating mechanism. TRPM4- or TRPM5-like ion channels engender ICAN and are blocked by 30-100 μm FFA (Teulon, 2000; Launay et al., 2002; Montell et al., 2002; Guinamard et al., 2004; Moran et al., 2004). We abolished bursting with 10-200 μm FFA, which is consistent with a role for TRPM4/5-mediated ICAN (Schiller, 2004). FFA also blocks gap junctions (Srinivas and Spray, 2003; Ye et al., 2003), enhances K+ currents (Stumpff et al., 2001), with EC50 < 50 μm so that doses >100 μm could have side effects that impact respiratory network activity (Pena et al., 2004).
Regardless of age, ICa-dependent pacemaker neurons showed very strong inspiratory bursts in control and FFA always attenuated drive potentials (Figs. 2A, 8 B). These data suggest that ICAN contributes significantly to inspiratory bursts even at young ages when the actual fraction of cells with ICAN pacemaker properties is extremely low.
Low doses of TTX depress excitability in preBötC neurons
Low doses of TTX simultaneously diminished the frequency and amplitude of XII motor output, whereas RIL primarily decreased XII amplitude. TTX (20 nm) hyperpolarized preBötC neurons, increased spike threshold and rheobase, and significantly diminished (or abolished) action potentials. These effects decrease neuronal excitability and could ultimately cause the cessation of respiratory rhythm independent of any direct effects on bursting properties. Unlike TTX, RIL does not significantly alter spike threshold or rheobase at concentrations that block INaP (Del Negro et al., 2002a).
Despite the general depression of excitability caused by 20 nm TTX, SP added to the bath restarted respiratory rhythm in most slices. SP depolarizes synaptically isolated rhythmogenic preBötC neurons by closing K+ channels and activating a TTX-insensitive linear Na+ current, but SP does not induce a region of negative slope conductance nor induce intrinsic bursting behavior in pre-BötC neurons (Gray et al., 1999; Pena and Ramirez, 2004). Here, SP was applied in the presence of TTX. Therefore, spiking properties and INaP would remain attenuated, and we conclude that depolarization of preBötC neurons by SP was directly responsible for restarting rhythmic activity, and not the restoration of pacemaker properties.
Rhythm generation in the presence of FFA and RIL
Similar to 20 nm TTX, coapplication of RIL and FFA suppressed respiratory rhythm. Again, the rhythm could be rescued with SP or low concentrations of AMPA, which depolarize cells and boost excitability but do not directly cause pacemaker properties (Gray et al., 1999; Pena and Ramirez, 2004). SP presumably compensates for the hyperpolarization caused by the loss of INaP and ICAN, which suggests that the ability of INaP and ICAN to give rise to bursting-pacemaker activity in a subset of isolated neurons is not essential for rhythmogenesis. Rather, we conclude that normally INaP and ICAN contribute to rhythmogenesis by enhancing the general excitability of the network and promoting inspiratory burst generation in all respiratory preBötC neurons without requiring bursting pacemaker activity.
Emergent network properties
Our data suggest that pacemaker neurons per se are not essential to generate the respiratory rhythm, but that INaP and ICAN contribute to rhythmogenesis by promoting excitability. We favor an emergent network mechanism of respiratory rhythmogenesis such as the group pacemaker (Rekling et al., 1996a,b, 2000; Rekling and Feldman, 1998) in which pacemaker neurons can be embedded but are not obligatory for rhythm generation. We posit that recurrent synaptic excitation in preBötC neurons triggers inward currents carried by INaP and ICAN, thereby amplifying depolarization through a positive feedback cycle (a little synaptic excitation leads to a lot of neuronal output). INaP and ICAN are present in cells without pacemaker activity and serve to boost membrane potential from baseline to suprathreshold levels in response to synaptic activity. Recurrent excitation causes cascades of synaptic activity in cultured spinal cord networks, which are enhanced by intrinsic cellular currents and excitability (Streit, 1993; Streit et al., 2001; Darbon et al., 2002). Respiratory neurons use burst-promoting currents like INaP and ICAN, but the majority of cells are not pacemakers because they do not burst unless synaptically activated. In the absence of INaP and ICAN, we conclude that a compensatory boost in excitability depolarizes constituent neurons closer to spike threshold so that excitatory synaptic drive can still evoke the inspiratory burst without the amplification normally provided by INaP and ICAN.
We propose that inspiratory burst termination results from a collection of intrinsic cellular properties such as INaP inactivation (Del Negro et al., 2002b; Rybak et al., 2003), ICAN deactivation, recruitment of calcium-dependent potassium channels, or electrogenic ion pumps (Ballerini et al., 1997; Darbon et al., 2003). These cellular processes can activate based on Na+ and Ca2+ accumulation during inspiration. Burst termination does not require coupling, whereas network burst initiation is impossible without the excitatory coupling (Rekling et al., 1996b; Rekling and Feldman, 1998).
Finally, neurons rostral to the preBötC may contribute to rhythm generation (Mellen et al., 2003; Onimaru and Homma, 2003) and some have pacemaker properties (Ballanyi et al., 1999). Because these neurons are not contained in slices, we cannot predict how such cells might contribute to the final pattern of respiratory activity when the preBötC is embedded in the more extensive brainstem respiratory network.
Footnotes
This work was supported by National Institutes of Health Grant HL-40959, the Jeffress Memorial Trust, and the Parker B. Francis Fellowship in Pulmonary Research.
Correspondence should be addressed to Christopher A. Del Negro, Department of Applied Science, McGlothlin-Street Hall, Room 303, The College of William and Mary, Williamsburg, VA 23187-8795. E-mail: cadeln@wm.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250446-08$15.00/0
C.A.D. and C.M.-V. contributed equally as first author.
J.A.H., D.D.M., and R.W.P. made equal contributions and are listed alphabetically.
References
- Ballanyi K, Onimaru H, Homma I (1999) Respiratory network function in the isolated brainstem-spinal cord of newborn rats. Prog Neurobiol 59: 583-634. [DOI] [PubMed] [Google Scholar]
- Ballerini L, Bracci E, Nistri A (1997) Pharmacological block of the electrogenic sodium pump disrupts rhythmic bursting induced by strychnine and bicuculline in the neonatal rat spinal cord. J Neurophysiol 77: 17-23. [DOI] [PubMed] [Google Scholar]
- Butera Jr RJ, Rinzel J, Smith JC (1999) Models of respiratory rhythm generation in the pre-Bötzinger complex. II. Populations of coupled pacemaker neurons. J Neurophysiol 82: 398-415. [DOI] [PubMed] [Google Scholar]
- Darbon P, Scicluna L, Tscherter A, Streit J (2002) Mechanisms controlling bursting activity induced by disinhibition in spinal cord networks. Eur J Neurosci 15: 671-683. [DOI] [PubMed] [Google Scholar]
- Darbon P, Tscherter A, Yvon C, Streit J (2003) Role of the electrogenic Na/K pump in disinhibition-induced bursting in cultured spinal networks. J Neurophysiol 90: 3119-3129. [DOI] [PubMed] [Google Scholar]
- Del Negro CA, Johnson SM, Butera RJ, Smith JC (2001) Models of respiratory rhythm generation in the pre-Bötzinger complex. III. Experimental tests of model predictions. J Neurophysiol 86: 59-74. [DOI] [PubMed] [Google Scholar]
- Del Negro CA, Morgado-Valle C, Feldman JL (2002a) Respiratory rhythm: an emergent network property? Neuron 34: 821-830. [DOI] [PubMed] [Google Scholar]
- Del Negro CA, Koshiya N, Butera Jr RJ, Smith JC (2002b) Persistent sodium current, membrane properties and bursting behavior of pre-botzinger complex inspiratory neurons in vitro J Neurophysiol 88: 2242-2250. [DOI] [PubMed] [Google Scholar]
- Elsen FP, Ramirez JM (1998) Calcium currents of rhythmic neurons recorded in the isolated respiratory network of neonatal mice. J Neurosci 18: 10652-10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Smith JC (1989) Cellular mechanisms underlying modulation of breathing pattern in mammals. Ann NY Acad Sci 563: 114-130. [DOI] [PubMed] [Google Scholar]
- Gogelein H, Dahlem D, Englert HC, Lang HJ (1990) Flufenamic acid, mefenamic acid and niflumic acid inhibit single nonselective cation channels in the rat exocrine pancreas. FEBS Lett 268: 79-82. [DOI] [PubMed] [Google Scholar]
- Gray PA, Rekling JC, Bocchiaro CM, Feldman JL (1999) Modulation of respiratory frequency by peptidergic input to rhythmogenic neurons in the preBötzinger complex. Science 286: 1566-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray PA, Janczewski WA, Mellen N, McCrimmon DR, Feldman JL (2001) Normal breathing requires preBötzinger complex neurokinin-1 receptor-expressing neurons. Nat Neurosci 4: 927-930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinamard R, Chatelier A, Demion M, Potreau D, Patri S, Rahmati M, Bois P (2004) Functional characterization of a Ca2+-activated non-selective cation channel in human atrial cardiomyocytes. J Physiol (Lond) 558: 75-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SM, Smith JC, Funk GD, Feldman JL (1994) Pacemaker behavior of respiratory neurons in medullary slices from neonatal rat. J Neurophysiol 72: 2598-2608. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Smith JC (2003) Respiratory rhythm and pattern generation in vitro involves persistent sodium current (INaP). Soc Neurosci Abstr 29: 503.19. [Google Scholar]
- Koshiya N, Smith JC (1999) Neuronal pacemaker for breathing visualized in vitro Nature 400: 360-363. [DOI] [PubMed] [Google Scholar]
- Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R, Kinet JP (2002) TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 109: 397-407. [DOI] [PubMed] [Google Scholar]
- Marder E (2001) Moving rhythms. Nature 410: 755. [DOI] [PubMed] [Google Scholar]
- Mellen NM, Janczewski WA, Bocchiaro CM, Feldman JL (2003) Opioid-induced quantal slowing reveals dual networks for respiratory rhythm generation. Neuron 37: 821-826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell C, Birnbaumer L, Flockerzi V (2002) The TRP channels, a remarkably functional family. Cell 108: 595-598. [DOI] [PubMed] [Google Scholar]
- Moran MM, Xu H, Clapham DE (2004) TRP ion channels in the nervous system. Curr Opin Neurobiol 14: 362-369. [DOI] [PubMed] [Google Scholar]
- Onimaru H, Homma I (2003) A novel functional neuron group for respiratory rhythm generation in the ventral medulla. J Neurosci 23: 1478-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onimaru H, Ballanyi K, Richter DW (1996) Calcium-dependent responses in neurons of the isolated respiratory network of newborn rats. J Physiol (Lond) 491: 677-695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onimaru H, Ballanyi K, Homma I (2003) Contribution of Ca2+-dependent conductances to membrane potential fluctuations of medullary respiratory neurons of newborn rats in vitro J Physiol (Lond) 552: 727-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena F, Ramirez JM (2004) Substance P-mediated modulation of pacemaker properties in the mammalian respiratory network. J Neurosci 24: 7549-7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena F, Parkis MA, Tryba AK, Ramirez JM (2004) Differential contribution of pacemaker properties to the generation of respiratory rhythms during normoxia and hypoxia. Neuron 43: 105-117. [DOI] [PubMed] [Google Scholar]
- Rekling JC, Feldman JL (1998) PreBötzinger complex and pacemaker neurons: hypothesized site and kernel for respiratory rhythm generation. Annu Rev Physiol 60: 385-405. [DOI] [PubMed] [Google Scholar]
- Rekling JC, Champagnat J, Denavit-Saubie M (1996a) Thyrotropin-releasing hormone (TRH) depolarizes a subset of inspiratory neurons in the newborn mouse brain stem in vitro J Neurophysiol 75: 811-819. [DOI] [PubMed] [Google Scholar]
- Rekling JC, Champagnat J, Denavit-Saubie M (1996b) Electroresponsive properties and membrane potential trajectories of three types of inspiratory neurons in the newborn mouse brain stem in vitro J Neurophysiol 75: 795-810. [DOI] [PubMed] [Google Scholar]
- Rekling JC, Shao XM, Feldman JL (2000) Electrical coupling and excitatorysynaptic transmission between rhythmogenic respiratory neurons in the preBötzinger complex. J Neurosci 20: RC113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak IA, Ptak K, Shevtsova NA, McCrimmon DR (2003) Sodium currents in neurons from the rostroventrolateral medulla of the rat. J Neurophysiol 90: 1635-1642. [DOI] [PubMed] [Google Scholar]
- Schiller Y (2004) Activation of a calcium-activated cation current during epileptiform discharges and its possible role in sustaining seizure-like events in neocortical slices. J Neurophysiol 92: 862-872. [DOI] [PubMed] [Google Scholar]
- Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL (1991) Pre-Bötzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science 254: 726-729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Butera RJ, Koshiya N, Del Negro C, Wilson CG, Johnson SM (2000) Respiratory rhythm generation in neonatal and adult mammals: the hybrid pacemaker-network model. Respir Physiol 122: 131-147. [DOI] [PubMed] [Google Scholar]
- Smith Jr TG, Barker JL, Gainer H (1975) Requirements for bursting pacemaker potential activity in molluscan neurones. Nature 253: 450-452. [DOI] [PubMed] [Google Scholar]
- Srinivas M, Spray DC (2003) Closure of gap junction channels by arylaminobenzoates. Mol Pharmacol 63: 1389-1397. [DOI] [PubMed] [Google Scholar]
- Streit J (1993) Regular oscillations of synaptic activity in spinal networks in vitro J Neurophysiol 70: 871-878. [DOI] [PubMed] [Google Scholar]
- Streit J, Tscherter A, Heuschkel MO, Renaud P (2001) The generation of rhythmic activity in dissociated cultures of rat spinal cord. Eur J Neurosci 14: 191-202. [DOI] [PubMed] [Google Scholar]
- Stumpff F, Boxberger M, Thieme H, Strauss O, Wiederholt M (2001) Flufenamic acid enhances current through maxi-K channels in the trabecular meshwork of the eye. Curr Eye Res 22: 427-437. [DOI] [PubMed] [Google Scholar]
- Teulon J (2000) Ca2+-activated nonselective cation channels. In: Pharmacology of ionic channel function: activators and inhibitors (Endo M, Kurachi Y, Mishina M, eds), pp 625-649. Berlin: Springer.
- Thoby-Brisson M, Ramirez JM (2001) Identification of two types of inspiratory pacemaker neurons in the isolated respiratory neural network of mice. J Neurophysiol 86: 104-112. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR (2003) Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci 23: 3588-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]