

Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disorder pathologically characterized by deposition of β-amyloid (Aβ) peptides as senile plaques in the brain. Recent studies suggest that green tea flavonoids may be used for the prevention and treatment of a variety of neurodegenerative diseases. Here, we report that (-)-epigallocatechin-3-gallate (EGCG), the main polyphenolic constituent of green tea, reduces Aβ generation in both murine neuron-like cells (N2a) transfected with the human “Swedish” mutant amyloid precursor protein (APP) and in primary neurons derived from Swedish mutant APP-overexpressing mice (Tg APPsw line 2576). In concert with these observations, we find that EGCG markedly promotes cleavage of the α-C-terminal fragment of APP and elevates the N-terminal APP cleavage product, soluble APP-α. These cleavage events are associated with elevated α-secretase activity and enhanced hydrolysis of tumor necrosis factor α-converting enzyme, a primary candidate α-secretase. As a validation of these findings in vivo, we treated Tg APPsw transgenic mice overproducing Aβ with EGCG and found decreased Aβ levels and plaques associated with promotion of the nonamyloidogenic α-secretase proteolytic pathway. These data raise the possibility that EGCG dietary supplementation may provide effective prophylaxis for AD.
Keywords: aging, Alzheimer's disease, β-amyloid, green tea, PKC, protease
Introduction
Amyloid precursor protein (APP) proteolysis is the fundamental process for the production of β-amyloid (Aβ) peptides implicated in Alzheimer's disease (AD) pathology (Golde et al., 2000; Huse and Doms, 2000; Sambamurti et al., 2002; Funamoto et al., 2004). APP proteolytic products arise from the coordinated action of α-, β-, and γ-secretases. In the amyloidogenic pathway, Aβ peptides are produced by the initial action of β-secretase (BACE) cleaveage, which creates an Aβ-containing C-terminal fragment (CTF) known as β-CTF or C99 (Sinha and Lieberburg, 1999; Yan et al., 1999). This proteolysis also generates an N-terminal, soluble APP-β (sAPP-β) fragment, which is released extracellularly. Intracellularly, β-CTF is then cleaved by a multiprotein γ-secretase complex that results in generation of the Aβ peptide and a smaller γ-CTF, also known as C57 (De Strooper et al., 1998; Steiner et al., 1999). Conversely, in the nonamyloidogenic pathway, APP is first cleaved at the α-secretase site, which results in the release of N-terminal sAPP-α and the generation of α-CTF or C83 (Hooper and Turner, 2002), events that are indicative of α-secretase activity (Hooper and Turner, 2002). Because of the limiting amount of APP in the cell and the failure to saturate the BACE pathway during APP overexpression, it is believed that the above-mentioned amyloidogenic and nonamyloidogenic pathways compete for substrate in the process of APP proteolysis (Gandhi et al., 2004). Therefore, it is often inferred that extracellular elevation of sAPP-α (resulting from nonamyloidogenic pathway activation) can be taken as indirect evidence of inhibition of BACE and the associated amyloidogenic pathway. However, because the extracellular secretion of these various fragments can be regulated independently of APP cleavage, it is important to fully characterize the effects of treatment on both pathways concurrently before making inferences about underlying mechanisms (Rossner et al., 2000).
Over the past decade, intense focus has been given to investigating the processes of APP proteolysis and Aβ metabolism as possible targets for AD therapy (Hardy and Selkoe, 2002). Various synthetic and naturally occurring compounds have been analyzed for their efficacy in the modulation of these pathological events. One such naturally occurring compound achieving worldwide popularity for its therapeutic application is green tea. Green tea contains polyphenolic structures categorized as flavonoids, which are believed to be the active components accounting for the therapeutic properties of green tea. Arguably, one of the most promising green tea compounds being analyzed is (-)-epigallocatechin-3-gallate (EGCG), which has been extensively studied primarily because of its reported anticarcinogenic effects (Lin and Liang, 2000; Moyers and Kumar, 2004). Recently, EGCG has been found to modulate protein kinase C (PKC) activity and to consequently increase secreted levels of sAPP-α (Levites et al., 2002; Levites et al., 2003). Additionally, EGCG has been shown to inhibit various activities of proinflammatory cytokines (Ahmed et al., 2002; Han, 2003; Li et al., 2004). Accordingly, signal transducer and activator of transcription 1 and nuclear factor κB responses are inhibited by EGCG (Han, 2003; Aktas et al., 2004). Elucidation of these molecular actions of EGCG substantiates the compound as a versatile modulator of cellular responses that may contribute to disease pathogenesis.
In the present study, we demonstrate that EGCG treatment of both murine N2a cells transfected with the human “Swedish” mutant form of APP (SweAPP N2a cells) and primary neuronal cells derived from Alzheimer Swedish mutant APP overexpressing mice (Tg APPsw line 2576) (Hsiao et al., 1996) leads to a significant reduction in Aβ production. Furthermore, Tg APPsw mice treated with EGCG show decreased Aβ levels and β-amyloid plaques in the brain. These effects are associated with increased generation of α-CTF and sAPP-α and elevated α-secretase cleavage activity, showing that EGCG promotes the nonamyloidogenic α-secretase proteolytic pathway both in vitro and in vivo.
Materials and Methods
Reagents. Green tea-derived flavonoids (>95% purity by HPLC), including EGCG, (-) EC, (+) EC, GC, and C, were purchased from Sigma (St. Louis, MO). Tumor necrosis factor-α (TNF-α) protease inhibitor-1 (TAPI-1) and β-secretase inhibitor were obtained from Calbiochem (San Diego, CA). Green tea extract (75% polyphenols) was obtained from the Vitamin Shoppe (North Bergen, NJ). Antibody against the C terminus of APP was obtained from Chemicon (Temecula, CA), and those against the N terminus of APP (22C11) and against actin were purchased from Roche (Basel, Switzerland).
ELISA. Cultured cells were lysed in ice-cold lysis buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% v/v Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerolphosphate, 1 mm Na3VO4, 1 μg/ml leupeptin, 1 mm PMSF) as described previously (Tan et al., 2002). Mouse brains were isolated under sterile conditions on ice and placed in ice-cold lysis buffer. Brains were then sonicated on ice for ∼3 min, allowed to stand for 15 min at 4°C, and centrifuged at 15,000 rpm for 15 min. Aβ1–40,42 species were detected by acid extraction of brain homogenates in 5 m guanidine buffer (Johnson-Wood et al., 1997), followed by a 1:10 dilution in lysis buffer. Soluble Aβ1–40,42 was directly detected in cultured cell lysates or brain homogenates prepared with lysis buffer described above by a 1:4 or 1:10 dilution, respectively. Aβ1–40,42 was quantified in these samples using the Aβ1–40,42 ELISA kits (IBL-American, Minneapolis, MN) in accordance with the instructions of the manufacturer, except that standards included 0.5 m guanidine buffer in some cases.
Western blot and immunoprecipitation. Cultured cells or mouse brains were lysed in ice-cold lysis buffer described above, and an aliquot corresponding to 50 μg of total protein was electrophoretically separated using 16.5% Tris-tricine gels. Electrophoresed proteins were then transferred to polyvinylidene difluoride membranes (Bio-Rad, Richmond, CA), washed in dH2O, and blocked for 2 h at ambient temperature in Tris-buffered saline (TBS; Bio-Rad) containing 5% (w/v) nonfat dry milk. After blocking, membranes were hybridized for 2 h at ambient temperature with various primary antibodies. Membranes were then washed three times for 5 min each in dH2O and incubated for 1 h at ambient temperature with the appropriate HRP-conjugated secondary antibody (1:1000; Pierce Biotechnology, Rockford, IL). All antibodies were diluted in TBS containing 5% (w/v) nonfat dry milk. Blots were developed using the luminol reagent (Pierce Biotechnology). Densitometric analysis was done as described previously using a FluorS Multiimager with Quantity One software (Bio-Rad) (Tan et al., 2002). Immunoprecipitation was performed for detection of sAPP-α, sAPP-β, and Aβ by incubating 200 μg of total protein of each sample with various sequential combinations of 6E10 (1:100; Signet Laboratories, Dedham, MA) and/or 22C11 (1:100; Roche) antibodies overnight with gentle rocking at 4°C and 10 μl of 50% protein A-Sepharose beads were then added to the sample (1:10; Sigma) before gentle rocking for an additional 4 h at 4°C. After washes with 1 × cell lysis buffer, samples were subjected to Western blot analysis as described above. Antibodies used for Western blot analysis included the APP-C-terminal antibody 369 (1:1000), C-terminal APP antibody (1:500; Calbiochem), N-terminal APP antibody (clone 22C11), N-terminal Aβ antibodies BAM-10 (β-amyloid 10) (1:1000; Sigma) or 6E10 (1:1000; Signet Laboratories), ADAM-10 (a disintegrin and metalloprotease 10) antibody (Calbiochem), TNF-α converting enzyme (TACE) antibody (Calbiochem), or actin antibody (1:1500; as an internal reference control; Roche). α-, β-, and γ-secretase activities were quantified in cell lysates and mouse brain homogenates using available kits based on secretase-specific peptides conjugated to fluorogenic reporter molecules (R&D Systems, Minneapolis, MN).
Mice. Tg APPsw mice (line 2576) were purchased from Taconic (Germantown, NY). For intraperitoneal administration of EGCG, a total of 10 female Tg APPsw mice were used; five mice received EGCG, and the other five received PBS. Beginning at 12 months of age, Tg APPsw mice were intraperitoneally injected with EGCG (20 mg/kg) or PBS daily for 60 d based on methods described previously (Chyu et al., 2004). These mice were then killed at 14 months of age for analyses of Aβ levels and Aβ load in the brain according to methods described previously (Tan et al., 2002). In an additional experiment, 10 female Tg APPsw mice at 7 months of age were subjected to a similar treatment regimen as described above, in which six mice received EGCG and the other four received PBS. These mice were killed at 9 months of age for analysis of amyloidosis (Tan et al., 2002). For intracerebroventricular injection (n = 3 females for each group), mice were injected in the lateral ventricle with EGCG [(5 μl (10 μg)/mouse)] or PBS once (Tan et al., 2002; Chauhan and Siegel, 2003). Twenty-four hours after injection, these mice were killed for analysis of cerebral Aβ levels as described previously (Tan et al., 2002). Animals were housed and maintained at the College of Medicine Animal Facility of the University of South Florida, and all experiments were in compliance with protocols approved by the University of South Florida Institutional Animal Care and Use Committee.
Immunohistochemistry. Mice were anesthetized with isofluorane and transcardially perfused with ice-cold physiological saline containing heparin (10 U/ml). Brains were rapidly isolated and quartered using a mouse brain slicer (Muromachi Kikai, Tokyo, Japan). The first and second anterior quarters were homogenized for Western blot analysis, and the third and fourth posterior quarters were used for microtome or cryostat sectioning. Brains were then fixed in 4% paraformaldehyde in PBS at 4°C overnight and routinely processed in paraffin in a core facility at the Department of Pathology (University of South Florida College of Medicine). Five coronal sections from each brain (5 μm thickness) were cut with a 150 μm interval. Sections were routinely deparaffinized and hydrated in a graded series of ethanol before preblocking for 30 min at ambient temperature with serum-free protein block (Dako Cytomation, Carpinteria, CA). Aβ immunohistochemical staining was performed using anti-human amyloid-β antibody (clone 4G8; 1:100; Signet Laboratories) in conjunction with the VectaStain Elite ABC kit (Vector Laboratories, Burlingame, CA) coupled with diaminobenzidine substrate. 4G8-positive Aβ deposits were examined under bright field using an Olympus (Tokyo, Japan) BX-51 microscope. For thioflavin S histochemistry, sections were routinely deparaffinized and rinsed in 70% (v/v) ethanol before staining with fresh-filtered 1% (w/v) thioflavin S diluted in 70% ethanol for 5 min. These sections were rinsed three times for 5 min each in 70% ethanol, hydrated for 5 min in PBS, and mounted in Vectashield fluorescence mounting media (Vector Laboratories). Thioflavin S-positive β-amyloid plaques were visualized under dark field using an Olympus BX-51 microscope.
Image analysis. Quantitative image analysis (conventional “Aβ burden” analysis) was performed for 4G8 immunohistochemistry and thioflavin S histochemistry in Tg APPsw mice injected with EGCG or PBS. Images were obtained using an Olympus BX-51 microscope and digitized using an attached MagnaFire imaging system (Olympus). Briefly, images of five 5 μm sections (150 μm apart) through each anatomic region of interest (hippocampus or cortical areas) were captured, and a threshold optical density was obtained that discriminated staining from background. Manual editing of each field was used to eliminate artifacts. Data are reported as a percentage of immunolabeled area captured (positive pixels) divided by the full area captured (total pixels). Quantitative image analysis was performed by a single examiner (T.M.) blinded to sample identities.
Statistical analysis. All data were normally distributed; therefore, in instances of single mean comparisons, Levene's test for equality of variances followed by a t test for independent samples was used to assess significance. In instances of multiple mean comparisons, ANOVA was used, followed by post hoc comparison using Bonferonni's method. α Levels were set at 0.05 for all analyses. The statistical package for the social sciences release 10.0.5 (SPSS, Chicago, IL) was used for all data analyses.
Results
EGCG inhibits Aβ1–40,42 generation from SweAPP N2a cells and Tg APPsw mouse-derived primary neuronal cells
HPLC analysis of green tea shows that EGCG is the main polyphenolic constituent, although other compounds, including (-)-epicatechin [(-) EC], (+)-epicatechin [(+) EC], (-)-gallocatechin (GC), and (-)-catechin (C), are present in relatively lesser quantities (Moyers and Kumar, 2004). To examine the effects of the polyphenolic constituents of green tea on APP cleavage, we first treated SweAPP N2a cells and primary neuronal cells derived from Tg APPsw mice with a wide dose range of each of these compounds. We found that EGCG reduces Aβ generation (both Aβ1–40 and Aβ1–42 peptides) in either SweAPP N2a cells or primary Tg APPsw-derived neuronal cells in a dose-dependent manner (Fig. 1a,b). Most importantly, EGCG (20 μm) reduces Aβ generation from SweAPP N2a cells by 61% (Fig. 1a) and from primary Tg APPsw-derived neuronal cells by 38% (Fig. 1b) compared with untreated cells. It should be noted that both enantiomeric species of another green tea component, EC, inhibit Aβ generation by nearly 20–30% in both cell types, albeit at relatively high doses (Fig. 1a,b). However, two other components of green tea, GC and C, modestly promote Aβ production by ∼20–30% (for SweAPP N2a cells) or 10–15% (for primary Tg APPsw-derived neuronal cells) at a relatively high (80 μm) concentration. To test whether GC and/or C could oppose the inhibition of Aβ generation by EGCG, SweAPP N2a cells were cotreated with EGCG (20 μm) plus GC (80 μm), C (80 μm), or both for 12 h. As expected, data show that the presence of GC or C, and particularly the combination of both, markedly inhibits the ability of EGCG to reduce Aβ generation from SweAPP N2a cells (Fig. 1c). Thus, these data suggest that a purified preparation of EGCG is more capable of reducing Aβ generation in vitro than when it is present in a mixture in whole green tea extract (GT). To further address this hypothesis, we incubated SweAPP N2a cells with GT at various doses and with a similar dose of EGCG as present in GT. As shown in Figure 1d, data indicate that the various levels of EGCG alone elicit more profound effects on reduction of Aβ generation versus whole GT. Thus, the ability of the purified EGCG alone to inhibit Aβ generation is much greater than that of GT.
Figure 1.

EGCG treatment inhibits Aβ generation in cultured neuronal cells. Aβ1–40,42 peptides were analyzed in conditioned media from SweAPP N2a cells (a, c, d) or TgAPPsw mouse-derived primary neurons (b) by ELISA (n = 3 for each condition). Data are represented as a percentage of Aβ1–40,42 peptides secreted 12 h after EGCG treatment relative to control (untreated). a, b, One-way ANOVA followed by post hoc comparison revealed significant differences between EGCG and the other compounds at 40, 20, 10, and 5 μm treatment concentrations (p < 0.001). c, When comparing EGCG (20 μm) treatment with cotreatment of SweAPP N2a cells with EGCG (20 μm) plus GC (80 μm), C (80 μm), or GC/C, a significant difference was noted for each comparison (p < 0.001). d, SweAPP N2a cells were treated with EGCG at a comparable concentration with that found in GT (GT contains 30% EGCG), and a significant difference was noted between GT and EGCG treatments (40 μg/ml vs 20 μm; 20 μg/ml vs 10 μm; 10 μg/ml vs 5 μm) on inhibition of Aβ generation (p < 0.001 for each comparison). Reduction for each treatment condition is indicated for c and d.
EGCG activates nonamyloidogenic processing of APP in SweAPP N2a cells
To elucidate the mode of action of green tea components on APP cleavage, we examined APP cleavage profiles after treatment of SweAPP N2a cells with EGCG, EC, GC, and C using Western blot and immunoprecipitation analyses. In concert with reduced Aβ generation (Fig. 1a,b), EGCG treatment results in greatly increased nonamyloidogenic α-CTF generation and augmented α-CTF to β-CTF band density ratio in cell lysates (Fig. 2a). Accordingly, sAPP-α (but not sAPP-β) is elevated in SweAPP N2a cell culture media (Fig. 2b), an effect that is clearly associated with increased α-CTF in corresponding cell lysates after the treatment (Fig. 2c). Most notably, these effects are both time and dose dependent (Fig. 2c,d). Additionally, as shown in Figure 2e (lower left) (-) EC treatment only increases α-CTF generation at high doses (similar results were obtained with (+) EC; data not shown). In contrast, both GC and C treatment result in decreased α-CTF relative to β-CTF at 80 μm as indicated (Fig. 2e, right). More importantly, at this dose, GC, C, or GC/C significantly oppose the effect of EGCG on α-CTF cleavage (Fig. 2f). In agreement with Aβ ELISA data as shown in Figure 1d, purified EGCG demonstrates a marked effect on α-CTF generation versus an equivalent amount of GT (Fig. 2g). It should be noted that we did not observe changes in full-length holo APP expression after any of these treatments (see Western blot analysis as shown in Fig. 2), suggesting that these treatments promote nonamyloidogenic APP cleavage as opposed to altered APP expression in SweAPP N2a cells.
Figure 2.
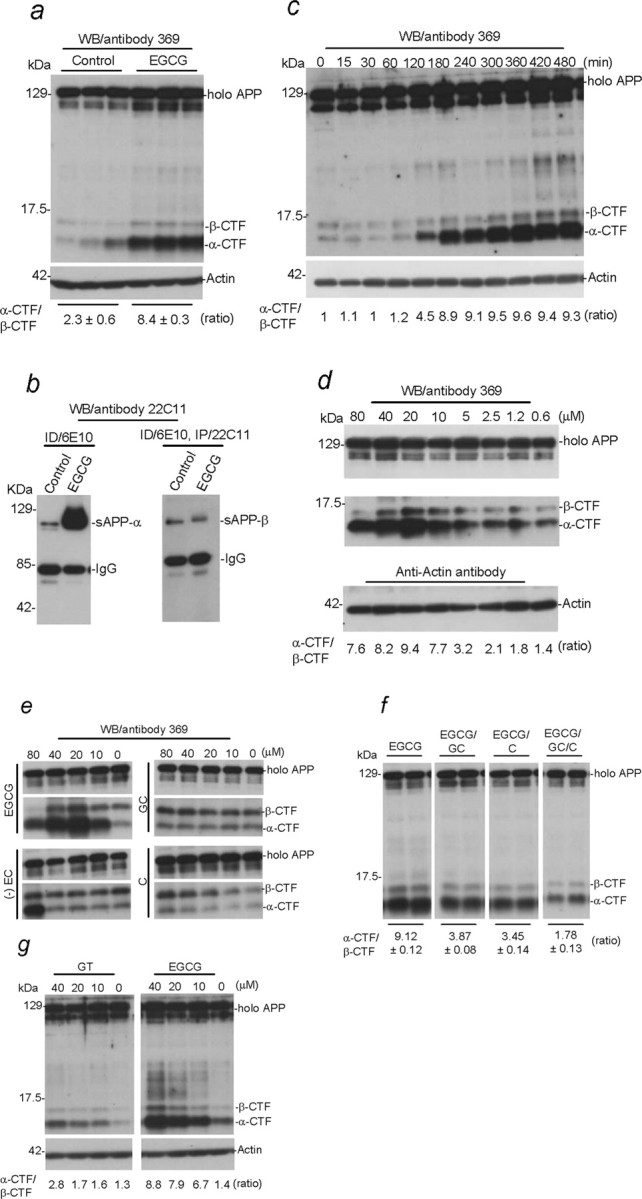
EGCG treatment alters APP cleavage in vitro. a, b, SweAPP N2 a cells were treated with EGCG at 20 μm or PBS (control) for 12 h. Cell lysates were prepared and subjected to Western blot (WB) analysis of APP CTFs (a), and conditioned media were collected for immunoprecipitation (IP)/WB (b). c, d, Cell lysates were prepared from SweAPP N2a cells treated with EGCG at 20μm for a range of time points (c) or EGCG at various doses for 12 h (d) and subjected to WB for APP CTFs. e, g, Cell lysates were prepared from SweAPP N2a cells treated with EGCG (-) EC, (+) EC, GC, C, or GT at the doses indicated for 12 h. For f, SweAPP N2a cells were cotreated with EGCG (20 μm) and GC, C, or GC/C at 80 μm for 12 h. For a and c–g, WB analysis using antibody 369 against the cytoplasmic tail of APP shows full-length holo APP and two bands corresponding to β-CTF (C99) and α-CTF (C83). For b, WB analysis using antibody 22C11 against the N terminus of APP shows sAPP-α (IP with antibody 6E10 directed against Aβ1–17) and sAPP-β [following immunodepletion (ID) with 6E10 and subsequent IP with 22C11]. a, c, d, g, Western blot analysis with anti-actin antibody shows actin protein (as an internal reference control). Densitometry analysis shows the ratio of α-CTF to β-CTF as indicated below the figures. a, A t test revealed a significant difference between EGCG treatment and control (n = 3 for each condition; p < 0.001). f, One-way ANOVA showed significant between-groups differences (p < 0.01) with n = 4 for each condition, and post hoc comparison revealed a significant difference between EGCG and EGCG/GC/C treatments (p < 0.001). Similar results were observed in N2a cells transfected with human wild-type APP695 using 369 antibody and in SweAPP N2a cells using Calbiochem polyclonal APP C-terminal APP antibody (data not shown).
EGCG promotes α-secretase cleavage of APP in SweAPP N2a cells
As illustrated in Figure 2c, Western blot analysis clearly shows a time-dependent increase in α-CTF generation in EGCG-treated SweAPP N2a cells. Most notably, α-CTF generation dramatically increases at 3–4 h up to 8 h after EGCG treatment. To test whether the alteration of APP α-CTF was attributable to enhanced α-secretase cleavage activity, we first examined expression of TACE, an APP α-secretase candidate (Skovronsky et al., 2001; Allinson et al., 2003), in cell lysates by Western blot analysis. Results show that TACE expression is significantly increased at 2–4 h after EGCG treatment and then rapidly degraded through to 8 h (Fig. 3a). To test whether this EGCG-induced alteration in TACE expression correlated with modulation of α-secretase cleavage activity, we evaluated the latter in cell lysates prepared from EGCG-treated or vehicle (PBS)-treated SweAPP N2a cells. Results reveal that α-secretase cleavage activity is markedly elevated at the first 1–3 h after EGCG treatment of SweAPP N2a cells (Fig. 3b). Finally, we cotreated SweAPP N2a cells with EGCG and TAPI-1, a selective TACE inhibitor (Slack et al., 2001; Robert et al., 2005), for 4 h. Data show that the presence of TAPI-1 significantly diminishes the ability of EGCG to increase α-CTF generation (Fig. 3c), attenuates EGCG-induced elevation of α-secretase cleavage activity (Fig. 3d), and blunts EGCG-mediated reduction of detergent-soluble Aβ (Fig. 3e). Together, these data suggest that elevation of TACE expression in response to EGCG treatment is likely responsible for increased α-secretase activity in SweAPP N2a cells.
Figure 3.

EGCG treatment promotes α-secretase cleavage of APP in vitro. a, b, Cell lysates were prepared from SweAPP N2a cells treated with EGCG (20μm) for different time points as indicated. a, Western blot analysis by anti-TACE antibody shows TACE and cleaved fragments. b, α-, β-, and γ-secretase cleavage activities were analyzed in cell lysates using secretase cleavage activity assays. Data are presented as a percentage of fluorescence units/milligrams protein activated 1, 2, or 3 h after EGCG treatment relative to control (PBS). A t test revealed a significant difference betweenα-secretase and either β- or γ-secretase cleavage activity at 1, 2, and 3 h after EGCG treatment (p < 0.001). c–e, SweAPP N2a cells were treated with EGCG (20 μm) or PBS (control) in the presence or absence of TAPI-1 at various doses (c) or at 25 μm (d, e) for 4 h. Cell-cultured supernatants were collected, and cell lysates were prepared from cultured cells. c, Western blot analysis by antibody 369 shows holo APP and two bands corresponding to β-CTF and α-CTF. d, Data are represented as percentage of α-secretase cleavage activity calculated in terms of fluorescence units/milligrams protein after EGCG treatment relative to control (PBS) 4 h after EGCG treatment in the presence or absence of TAPI-1. A t test revealed a significant difference between EGCG treatment and cotreatment with EGCG and TAPI-1 (p < 0.001); increased levels of activity are indicated. e, Data are presented as percentage of Aβ secretion relative to PBS control 4 h after EGCG treatment in the presence or absence of TAPI-1. A t test revealed a significant difference between EGCG and EGCG plus TAPI-1 treatment (p < 0.001); reduction for each treatment condition is indicated.
Treatment of Tg APPsw mice with EGCG results in nonamyloidogenic APP processing and reduction of cerebral amyloidosis
We validated in vivo whether EGCG treatment could promote nonamyloidogenic APP processing/α-secretase proteolysis and impact cerebral Aβ levels/β-amyloid plaques in Alzheimer transgenic mice. We intraperitoneally administered EGCG to Tg APPsw mice, a well-established transgenic mouse model of AD, at an age when Aβ deposits are accumulating (12 months). EGCG was administered based on a treatment schedule that produces benefit in an atherosclerosis mouse model (Chyu et al., 2004). Tg APPsw mice at 12 months of age were intraperitoneally injected with EGCG or PBS daily for 60 d. Nonamyloidogenic APP fragments, including α-CTF and sAPP-α, are markedly increased in brains of Tg APPsw mice treated with EGCG versus PBS (Fig. 4a). Accordingly, both detergent soluble Aβ1–40,42 levels are reduced by ∼54 and 44%, respectively; and insoluble Aβ1–40,42 (prepared by acid extraction in 5 m guanidine) levels are reduced by 47 and 38%, respectively, in EGCG-treated Tg APPsw mice, as determined by Aβ ELISAs (Fig. 4b,c). A similar magnitude of reduction was noted when considering each Aβ species alone (data not shown). We also found an ∼40% increase in α-secretase cleavage activity (Fig. 4d) that was inversely associated with reduced total Aβ levels.
Figure 4.
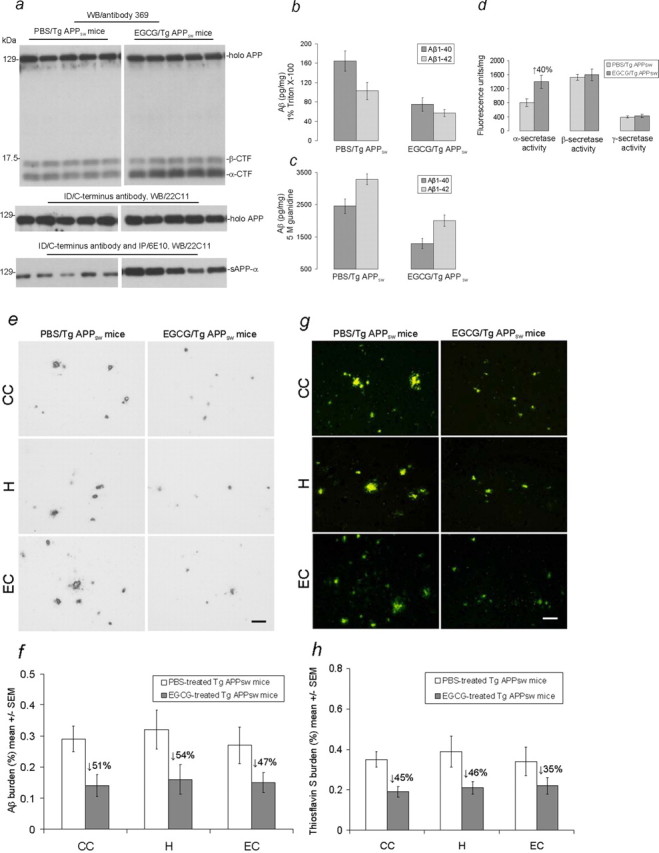
EGCG promotes nonamyloidogenic APP processing and reduces cerebral amyloidosis in TgAPPsw mice. Brain homogenates were prepared from female TgAPPsw mice treated with EGCG (n = 11) or PBS (n = 9). a, Top, Western blot analysis by antibody 369 shows holo APP and two bands corresponding to β-CTF and α-CTF. a, Middle and bottom, Western blot analysis by antibody 22C11 shows holo APP (middle; following ID/C-terminal APP antibody) and sAPP-α (bottom; following ID/C-terminal APP antibody and IP/6E10). Detergent-soluble Aβ1–40,42 (b) and insoluble Aβ1–40,42 prepared with 5 m guanidine (c) were analyzed by ELISA. Data are presented as mean ± 1 SEM of Aβ1–40 or Aβ1–42 (pg/mg protein) separately. For b and c, a t test revealed a significant between-groups difference for either soluble or insoluble Aβ1–40,42 (p < 0.001 for each comparison). d, α-, β-, and γ-secretase cleavage activities were analyzed by secretase cleavage activity assays. Data are presented as mean ± 1 SEM of fluorescence units/mg protein. A t test revealed a significant difference between EGCG- and PBS-treated Tg APPsw mice for α-secretase activity (p < 0.001). e, Mouse brain coronal paraffin sections were stained with anti-human Aβ antibody (4G8). Left, Control PBS-treated mice. Right, EGCG-treated mice. The top panels are from the cingulate cortex (CC), the middle panels are from the hippocampus (H), and the bottom panels are from the entorhinal cortex (EC). f, Percentages of 4G8-immunoreactive Aβ plaques (mean ± 1 SEM) were calculated by quantitative image analysis, and reduction for each brain region is indicated. g, Mouse brain sections from the indicated brain regions were stained with thioflavin S. Left, Control PBS-treated mice. Right, EGCG-treated mice. h, Percentage of thioflavin S plaques (mean ± 1 SEM) were quantified by image analysis, and reduction for each brain region is indicated. A t test for independent samples revealed significant differences (p < 0.005) between groups for each brain region examined in f and h. Scale bar, 50 μm.
The above data suggest that EGCG may act as an α-secretase agonist in a transgenic mouse model of AD. Yet, because EGCG was administered intraperitoneally, we tested whether these EGCG effects are attributable to action of the compound in the periphery and/or the CNS. We administered EGCG intracerebroventricularly to Tg APPsw mice. With this treatment regimen, EGCG-treated Tg APPsw mice show significant reduction in cerebral detergent soluble Aβ1–40,42 levels by 39% [384.65 ± 22.49 vs 235.60 ± 13.04 (mean pg/mg total protein ± SEM)], an effect that is associated with increased production of α-CTF/sAPP-α (data not shown), and elevated α-secretase cleavage activity by 32% [601.8 ± 38.13 vs 890.29 ± 104.41 (mean fluorescence units/mg total protein ± SEM)]. Importantly, cerebral soluble Aβ1–40,42 levels are reduced by a comparable magnitude in Tg APPsw mice treated with EGCG via intracerebroventricular and intraperitoneal routes, suggesting that the in vivo effect of EGCG on nonamyloidogenic processing of APP is mainly owed to CNS action of the compound.
Finally, we examined β-amyloid plaques by 4G8 immunohistochemistry and thioflavin S histochemistry in brains of mice that received peripheral injection of EGCG or vehicle (PBS) (Fig. 4e,g). At 14 months of age, 4G8 immunoreactive and thioflavin S-positive Aβ deposits were significantly reduced by 47–54% (Fig. 4f) and 35–46% (Fig. 4h) across the hippocampal and cortical brain regions examined. These data are supported by Aβ ELISA results examining insoluble 5 m guanidine-extracted brain homogenates, in which a 41% reduction was noted (Fig. 4c). Together, the above lines of evidence demonstrate that EGCG promotes nonamyloidogenic processing of APP and attenuates cerebral amyloidosis in a transgenic mouse model of AD.
Discussion
Previous reports have shown that green tea components such as EGCG have neuroprotective properties; however, a clear cellular/ molecular mechanism underlying these effects has hitherto not been established (Mandel et al., 2004). Our study shows that EGCG promotes nonamyloidogenic APP processing in vitro and in vivo, and that it accomplishes this by promoting α-secretase cleavage of APP. The mechanism for increased α-secretase cleavage activity by an induction of TACE appears transient as shown in Figure 3. In addition, we measured TACE protein levels in EGCG-versus PBS-treated Tg APPsw mice using Western blot analysis and observed that the expression of TACE was not significantly altered in brain homogenates derived from EGCG-treated Tg APPsw mice when compared with control PBS-treated mice (data not shown). These data suggest that, on a chronic EGCG treatment basis in vivo, elevated α-secretase cleavage activity may not require an increase in TACE protein expression. We intend to fully explore this mechanism of elevated α-secretase activity in vivo in a future study.
It is well known that TACE is a multifunctional molecule that has been shown to be critically involved in TNF-α maturation associated with proinflammatory responses (Moro et al., 2003). Additionally, TACE has been identified as a key α-secretase candidate molecule, and our data suggest that TACE is responsible for the increased α-secretase cleavage of APP observed after EGCG treatment of SweAPP N2a cells.
Because TACE has not been shown to exhibit substrate preference for TNF-α versus APP, we wondered whether EGCG, by increasing TACE levels, could promote TNF-α maturation and release in cultured primary murine microglial cells. Interestingly, despite the observation that TACE transient expression increases with EGCG treatment, we saw no associated elevation of TNF-α levels in cultured media of primary microglial cells (data not shown). This observation is consistent with other reports, in which EGCG treatment actually inhibits TNF-α expression and subsequently neuronal damage (Suganuma et al., 2000; Li et al., 2004). Because microglia must first undergo coordinated activation in response to innate immune stimuli to cleave pro-TNF-α and subsequently secrete TNF-α, a likely explanation for why EGCG does not elicit TNF-α release from murine microglia is that it does not trigger microglial activation and may in fact be immunosuppressive. Based on the data presented in our study of cultured neurons and neuron-like cells and in vivo in Tg APPsw mice, it is now possible to identify and dissect the mechanism through which EGCG exerts its anti-amyloidogenic effects.
A previous study showed that EGCG might act as a β-secretase inhibitor in a cell-free system (Jeon et al., 2003), raising the possibility that EGCG-mediated inhibition of Aβ generation may be accomplished via blockade of BACE activity. To address this possibility, we cotreated SweAPP N2a cells with the BACE inhibitor, β-secretase inhibitor II. Results show that treatment withβ-secretase inhibitor II at a dose range reported to block β-secretase activity (0.5–1.5 μm) (Abbenante et al., 2000) failed to mimic (data not shown) increased α-CTF and sAPP-α levels after EGCG treatment as shown in Figure 2, a and b. However, TAPI-1, a TACE inhibitor (Slack et al., 2001), significantly attenuated the effect of EGCG on promoting α-secretase cleavage of APP (Fig. 3d,e). In addition, as shown in Figures 3b and 4d, EGCG treatment markedly increased α-secretase cleavage activity both in vitro and in vivo but did not decrease β-secretase cleavage activity. Rather, it appears that there may actually be a slight increase in β-secretase cleavage activity, as depicted in Figure 2, a and c. This increase may be an indication of the extent of competition for APP between the two pathways or an attempt toward homeostasis. However, because of the sheer amount of α-secretase cleavage as illustrated by α-CTF/β-CTF ratios in Figure 2, a and c, and relatively unchanged γ-secretase cleavage (Fig. 3b), the nonamyloidogenic pathway seems to be the default cascade and, certainly after EGCG treatment, dominates APP proteolysis. Moreover, in two other studies (Levites et al., 2002, 2003), EGCG was found to modulate PKC activity and to consequently increase secreted levels of sAPP-α, further supporting this conclusion. However, these studies failed to measure the effects of EGCG on Aβ generation, which is important given that secretion of sAPP-α can be regulated independently of Aβ production (Rossner et al., 2000). Our data show that EGCG both promotes α-secretase activity and attenuates Aβ production, thereby augmenting nonamyloidogenic APP processing.
Green tea contains numerous polyphenolic flavonoids, and studies that have shown therapeutic benefits of green tea generally do not identify the active component(s) responsible. We found that EGCG confers nonamyloidogenic processing of APP; yet, the observation that other flavonoids, including GC and C, can actually oppose this effect of EGCG (Figs. 1c, 2f) suggests that the mixture of these polyphenolic flavonoids as found in whole green tea may actually oppose or mask the beneficial properties of EGCG. This may explain why research involving green tea extracts or combinations of flavonoids results with such variable findings (Chung et al., 2003). These observations showing that EGCG promotes α-secretase activity may lead to the creation of a new generation of the green tea extract yielding the greatest therapeutic benefit for AD. Although we found that pure EGCG reduces Aβ generation in vivo, it is still unclear whether equivalent doses in humans would be safe and effective. Based on a previous safety and pharmacokinetic study, it is likely that a daily 1500–1600 mg bolus of EGCG in humans will approximately achieve levels similar to those in sera of EGCG (20 mg/kg) intraperitoneally injected mice (Chow et al., 2003). Although not administered on a chronic basis, oral doses of similar magnitudes have been used in a clinical trial (Ullmann et al., 2003). Thus, our data prompt the need for future clinical trials with pure EGCG or its analogs, which demonstrate more effective α-secretase activity-enhancing properties and/or have better bioavailability. Structure-activity relationship studies are currently underway in our laboratory to determine the feasibility of these potentially promising therapeutic approaches.
When taken together, our data have shown that EGCG treatment leads to a reduction of Aβ levels in both SweAPP N2a cells and Tg APPsw mouse-derived primary neuronal cells. Furthermore, we found that α-CTF generation and sAPP-α secretion are markedly increased in these cells after EGCG treatment, effects that are correlated with elevated α-secretase cleavage activity. To demonstrate the in vivo therapeutic relevance of these observations, we intraperitoneally administered EGCG to Tg APPsw mice and found a significant reduction in cerebral Aβ levels concomitant with reduced β-amyloid plaques. In addition to intraperitoneal administration, intracerebroventricular injection of EGCG into Tg APPsw mice shows comparable reduction of cerebral Aβ levels associated with increased α-secretase cleavage activity, suggesting that the therapeutic effects of peripherally administered EGCG are mainly derived from direct CNS action of the compound. If Aβ pathology in this transgenic model is representative of disease pathology in the clinical syndrome, then EGCG administration to AD patients might be an effective prophylactic strategy for reduction of cerebral amyloidosis.
Footnotes
This work was supported by the Johnnie B. Byrd Senior Alzheimer's Center and Research Institute (J.T.) and University of South Florida College of Medicine Faculty Start-Up funds (J.T.). T.T. was supported by a Ruth L. Kirschstein National Institutes of Health–National Research Service Award–National Institute on Aging postdoctoral fellowship and an Alzheimer Association grant. We thank S. Gandy (Nathan S. Kline Institute for Psychiatry Research, New York University) and H. Steiner (Adolf Butenandt-Institute, Ludwig-Maximilians-University) for providing antibodies against the C terminus of APP (369 antibody) and S. Gandy for providing the N2a cell lines that stably overexpress human “Swedish”-mutated APP-695 or wild-type APP-695.
Correspondence should be addressed to Dr. Jun Tan, Neuroimmunology Laboratory, Department of Psychiatry, University of South Florida, 3515 East Fletcher Avenue, Tampa, FL 33613. E-mail: jtan@hsc.usf.edu.
DOI:10.1523/JNEUROSCI.1521-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258807-08$15.00/0
K.R.-Z., R.D.S., and N.S. contributed equally to this work.
References
- Abbenante G, Kovacs DM, Leung DL, Craik DJ, Tanzi RE, Fairlie DP (2000) Inhibitors of beta-amyloid formation based on the beta-secretase cleavage site. Biochem Biophys Res Commun 268: 133–135. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Rahman A, Hasnain A, Lalonde M, Goldberg VM, Haqqi TM (2002) Green tea polyphenol epigallocatechin-3-gallate inhibits the IL-1 beta-induced activity and expression of cyclooxygenase-2 and nitric oxide synthase-2 in human chondrocytes. Free Radic Bio Med 33: 1097–1105. [DOI] [PubMed] [Google Scholar]
- Aktas O, Prozorovski T, Smorodchenko A, Savaskan NE, Lauster R, Kloetzel PM, Infante-Duarte C, Brocke S, Zipp F (2004) Green tea epigallocatechin-3-gallate mediates T cellular NF-kappaB inhibition and exerts neuroprotection in autoimmune encephalomyelitis. J Immunol 173: 5794–5800. [DOI] [PubMed] [Google Scholar]
- Allinson TM, Parkin ET, Turner AJ, Hooper NM (2003) ADAMs family members as amyloid precursor protein alpha-secretases. J Neurosci Res 74: 342–352. [DOI] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ (2003) Intracerebroventricular passive immunization with anti-Abeta antibody in Tg2576. J Neurosci Res 74: 142–147. [DOI] [PubMed] [Google Scholar]
- Chow HH, Cai Y, Hakim IA, Crowell JA, Shahi F, Brooks CA, Dorr RT, Hara Y, Alberts DS (2003) Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin Cancer Res 9: 3312–3319. [PubMed] [Google Scholar]
- Chung FL, Schwartz J, Herzog CR, Yang YM (2003) Tea and cancer prevention: studies in animals and humans. J Nutr 133: 3268S–3274S. [DOI] [PubMed] [Google Scholar]
- Chyu KY, Babbidge SM, Zhao X, Dandillaya R, Rietveld AG, Yano J, Dimayuga P, Cercek B, Shah PK (2004) Differential effects of green tea-derived catechin on developing versus established atherosclerosis in apolipoprotein E-null mice. Circulation 109: 2448–2453. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: 387–390. [DOI] [PubMed] [Google Scholar]
- Funamoto S, Morishima-Kawashima M, Tanimura Y, Hirotani N, Saido TC, Ihara Y (2004) Truncated carboxyl-terminal fragments of beta-amyloid precursor protein are processed to amyloid beta-proteins 40 and 42. Biochemistry 43: 13532–13540. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Refolo LM, Sambamurti K (2004) Amyloid precursor protein compartmentalization restricts beta-amyloid production: therapeutic targets based on BACE compartmentalization. J Mol Neurosci 24: 137–143. [DOI] [PubMed] [Google Scholar]
- Golde TE, Eckman CB, Younkin SG (2000) Biochemical detection of Abeta isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer's disease. Biochim Biophys Acta 1502: 172–187. [DOI] [PubMed] [Google Scholar]
- Han MK (2003) Epigallocatechin gallate, a constituent of green tea, suppresses cytokine-induced pancreatic beta-cell damage. Exp Mol Med 35: 136–139. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353–356. [DOI] [PubMed] [Google Scholar]
- Hooper NM, Turner AJ (2002) The search for alpha-secretase and its potential as a therapeutic approach to Alzheimer's disease. Curr Med Chem 9: 1107–1119. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99–102. [DOI] [PubMed] [Google Scholar]
- Huse JT, Doms RW (2000) Closing in on the amyloid cascade: recent insights into the cell biology of Alzheimer's disease. Mol Neurobiol 22: 81–98. [DOI] [PubMed] [Google Scholar]
- Jeon SY, Bae K, Seong YH, Song KS (2003) Green tea catechins as a BACE1 (beta-secretase) inhibitor. Bioorg Med Chem Lett 13: 3905–3908. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L (1997) Amyloid precursor protein processing and Abeta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA 94: 1550–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y, Amit T, Youdim MB, Mandel S (2002) Involvement of protein kinase C activation and cell survival/cell cycle genes in green tea polyphenol (-)-epigallocatechin 3-gallate neuroprotective action. J Biol Chem 277: 30574–30580. [DOI] [PubMed] [Google Scholar]
- Levites Y, Amit T, Mandel S, Youdim MB (2003) Neuroprotection and neurorescue against Abeta toxicity and PKC-dependent release of nonamyloidogenic soluble precursor protein by green tea polyphenol (-)-epigallocatechin-3-gallate. FASEB J 17: 952–954. [DOI] [PubMed] [Google Scholar]
- Li R, Huang YG, Fang D, Le WD (2004) (-)-Epigallocatechin gallate inhibits lipopolysaccharide-induced microglial activation and protects against inflammation-mediated dopaminergic neuronal injury. J Neurosci Res 78: 723–731. [DOI] [PubMed] [Google Scholar]
- Lin JK, Liang YC (2000) Cancer chemoprevention by tea polyphenols. Proc Natl Sci Counc Repub China 1: 1–13. [PubMed] [Google Scholar]
- Mandel S, Weinreb O, Amit T, Youdim MB (2004) Cell signaling pathways in the neuroprotective actions of the green tea polyphenol (-)-epigallocatechin-3-gallate: implications for neurodegenerative diseases. J Neurochem 88: 1555–1569. [DOI] [PubMed] [Google Scholar]
- Moro MA, Hurtado O, Cardenas A, Romera C, Madrigal JL, Fernandez-Tome P, Leza JC, Lorenzo P, Lizasoain I (2003) Expression and function of tumour necrosis factor-alpha-converting enzyme in the central nervous system. Neurosignals 12: 53–58. [DOI] [PubMed] [Google Scholar]
- Moyers SB, Kumar NB (2004) Green tea polyphenols and cancer chemoprevention: multiple mechanisms and endpoints for phase II trials. Nutr Rev 62: 204–211. [DOI] [PubMed] [Google Scholar]
- Robert S, Maillet M, Morel E, Launay JM, Fischmeister R, Mercken L, Lezoualc'h F (2005) Regulation of the amyloid precursor protein ectodomain shedding by the 5-HT4 receptor and Epac. FEBS Lett 579: 1136–1142. [DOI] [PubMed] [Google Scholar]
- Rossner S, Beck M, Stahl T, Mendla K, Schliebs R, Bigl V (2000) Constitutive overactivation of protein kinase C in guinea pig brain increases alpha-secretory APP processing without decreasing beta-amyloid generation. Eur J Neurosci 12: 3191–3200. [DOI] [PubMed] [Google Scholar]
- Sambamurti K, Greig NH, Lahiri DK (2002) Advances in the cellular and molecular biology of the beta-amyloid protein in Alzheimer's disease. Neuromolecular Med 1: 1–31. [DOI] [PubMed] [Google Scholar]
- Sinha S, Lieberburg I (1999) Cellular mechanisms of beta-amyloid production and secretion. Proc Natl Acad Sci USA 96: 11049–11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovronsky DM, Fath S, Lee VM, Milla ME (2001) Neuronal localization of the TNFalpha converting enzyme (TACE) in brain tissue and its correlation to amyloid plaques. J Neurobiol 49: 40–46. [DOI] [PubMed] [Google Scholar]
- Slack BE, Ma LK, Seah CC (2001) Constitutive shedding of the amyloid precursor protein ectodomain is up-regulated by tumour necrosis factor-alpha converting enzyme. Biochem J 357: 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Duff K, Capell A, Romig H, Grim MG, Lincoln S, Hardy J, Yu X, Picciano M, Fechteler K, Citron M, Kopan R, Pesold B, Keck S, Baader M, Tomita T, Iwatsubo T, Baumeister R, Haass C (1999) A loss of function mutation of presenilin-2 interferes with amyloid beta-peptide production and notch signaling. J Biol Chem 274: 28669–28673. [DOI] [PubMed] [Google Scholar]
- Suganuma M, Sueoka E, Sueoka N, Okabe S, Fujiki H (2000) Mechanisms of cancer prevention by tea polyphenols based on inhibition of TNF-alpha expression. Biofactors 13: 67–72. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Crawford F, Mori T, DelleDonne A, Crescentini R, Obregon D, Flavell RA, Mullan MJ (2002) Role of CD40 ligand in amyloidosis in transgenic Alzheimer's mice. Nat Neurosci 5: 1288–1293. [DOI] [PubMed] [Google Scholar]
- Ullmann U, Haller J, Decourt JP, Girault N, Girault J, Richard-Caudron AS, Pineau B, Weber P (2003) A single ascending dose study of epigallocatechin gallate in healthy volunteers. J Int Med Res 31: 88–101. [DOI] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME (1999) Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature 402: 533–537. [DOI] [PubMed] [Google Scholar]