

Abstract
During cortical neurogenesis, cell proliferation and cell cycle exit are carefully regulated to ensure that the appropriate numbers of cells are produced. The antiproliferative agent transforming growth factor β1 (TGFβ1) and its receptors are endogenously expressed in proliferative zones of the developing cerebral cortex, thus implicating the growth factor in cell cycle regulation. The present study tested the hypothesis that TGFβ1 promotes cell cycle exit in the cortical ventricular zone (VZ) through modulation of cell cycle protein expression, in particular cyclin D1 and the cyclin-dependent kinase inhibitors p27 and p21. Although it did not affect the length of the cell cycle, TGFβ1 decreased the fraction of VZ-cycling cells by 21% and increased the number of VZ cells exiting the cell cycle a commensurate 24%. TGFβ1 selectively increased the expression of p21 in the VZ. In addition, high p21 expression levels were observed in VZ cells as they exited the cell cycle, and TGFβ1 increased the number p21-positive cells exiting the cell cycle. Collectively, these data show the following: (1) TGFβ1 promotes cell cycle exit, (2) p21 upregulation is correlated with cell cycle exit, and (3) TGFβ1-induced cell cycle exit is mediated by p21.
Keywords: BrdU, corticogenesis, cyclin D1, Ki-67, p27, neurogenesis, ventricular zone, cell proliferation
Introduction
The generation of most cortical neurons occurs within the ventricular neuroepithelium or ventricular zone (VZ) during an intense, but relatively brief, period of proliferative activity (Sauer, 1935; Sidman et al., 1959; Caviness et al., 2000; Nowakowski et al., 2002). Two features of the proliferative population that are tightly regulated are the propagation of the cell cycle and exit from the cell cycle. The balance of these two activities defines both neuronal production and maintenance of a cycling population.
Temporal expression of cell cycle proteins is central to cell cycle progression and exit. Advancement through the cell cycle is governed by stage-specific protein complexes composed of a cyclin and a cyclin-dependent kinase (cdk) (Sherr and Roberts, 2004). Cell cycle exit is facilitated through inhibition of cyclin-cdk complexes by cyclin-dependent kinase inhibitors (CKIs) (Sherr and Roberts, 1999). The cip/kip family of CKIs includes p21 and p27, proteins that affect multiple cyclin-cdk complexes during G1 and S (Harper et al., 1993; Xiong et al., 1993).
CKIs p21 and p27 have been linked to cell cycle exit during corticogenesis. A notable increase in p21 expression is associated with a substantial loss of cycling progenitors in the forebrain of foxg1 knock-out mice (Seoane et al., 2004). Genetic deletion of p27 reduces cell cycle exit during corticogenesis, resulting in an increase in the size of the adult cortex (Fero et al., 1996; Kiyokawa et al., 1996; Nakayama et al., 1996). In contrast, an increase in p27 expression in VZ progenitors increases cell cycle exit but does not increase of the length of G1 (Tarui et al., 2005). In addition, the temporal increase in p27 closely correlates with an increase in cell cycle exit and the length of the cell cycle in the mouse cortical VZ (Delalle et al., 1999; Caviness et al., 2003).
Transforming growth factor β1 (TGFβ1) inhibits cell proliferation in non-neural cells during development and in adults. TGFβ1 reduces cell proliferation through transcriptional suppression of the cyclins, including cyclin D1 (Ko et al., 1998) and through transcriptional upregulation of CKIs. Decreases in cell proliferation have been tied to TGFβ1-mediated increases in both p21 (Elbendary et al., 1994; Datto et al., 1995; Li et al., 1995; Yoo et al., 1999; Wolfraim et al., 2004) and p27 (Polyak et al., 1994; Bouchard et al., 1997; Kamesaki et al., 1998).
The role of TGFβ1 in the cortical VZ has not been elucidated. That said, it is known that TGFβ ligands and receptors are expressed in the VZ during cortical development (Miller, 2003). Furthermore, the antiproliferative action of TGFβ1 can be modeled using neuroblastoma cells and fetal cortical neurons (Luo and Miller, 1999; Miller and Luo, 2002a,b). To investigate the role of TGFβ1 in situ, organotypic slice cultures from fetal rat brains were used. Three main hypothesis were tested: (1) TGFβ1 is an antiproliferative signal in the VZ, (2) TGFβ1 promotes cell cycle exit in the VZ, and (3) TGFβ1 decreases cell proliferation in the VZ through modulation of cell cycle proteins.
Materials and Methods
Organotypic slice cultures. Slice cultures were prepared from fetuses of timed pregnant Sprague Dawley rats (Taconic, Germantown, NY) on gestational day 17 (G17) (Siegenthaler and Miller, 2004). Briefly, fetal brains were cut coronally into 300 μm sections using a MacIlwain Tissue Chopper (Mickle Laboratory Engineering, Gomshell, UK). Slices were cultured on Millicell filter inserts with 0.40 μm pores (Millipore, Bedford MA) in Minimal Essential Medium with 20% fetal calf serum (FCS), Hanks salts, 200 mm glutamine (Invitrogen, Carlsbad CA), 25 mm HEPES, 100 mm dextrose, 25 mm KCl, 100 μm penicillin/streptomycin (Invitrogen), and 12.5 μm fungizone (Invitrogen). Cultures were incubated at 37°C with 6.0% CO2. Slices were kept in medium containing FCS for 3 h, after which the slices were incubated in an FCS-free medium containing TGFβ1 (0, 2.5, 5, 10, 20, or 40 ng/ml; Sigma, St. Louis, MO).
Determination of cell cycle kinetics. A cumulative labeling method was used to determine the total length of the cell cycle (Tc), the length of S (Ts), and the proportion of cells that were actively cycling, the growth fraction (GF) (Nowakowski et al., 1989; Jacobs and Miller, 2000). The principles of this method are as follows: (1) 5-bromo-2-deoxyuridine (BrdU) is a thymidine analog that is incorporated into cells during the S phase of the cell cycle, (2) continuously available BrdU is incorporated into previously unlabeled cycling cells as they pass from G1 to S, and (3) consequently, the proportion of BrdU-positive (BrdU+) cells or labeling index (LI) increases over time. The maximum LI or growth faction is attained when all of the cycling cells have passed into S, Tc-Ts.
BrdU (0.0040% in dH20; Sigma) was added to the medium after 10 h of incubation in untreated or TGFβ1-containing medium. The time of BrdU addition was designated as time 0 h, or t0. Slices were fixed with 4.0% paraformaldehyde in 0.10 m phosphate buffer for 30 min at room temperature at t3, t6, t9, t12, t14, t17, and t24. After fixation and a 30 min rinse in PBS, slices were cryosectioned into 15 μm sections.
Incorporated BrdU was detected immunohistochemically. Sections were incubated in 2.0N HCl for 30 min followed by a 1 min rinse in dH2O. The primary antibody was a mouse anti-BrdU antibody (Becton Dickinson, San Diego, CA) diluted 1:30 in PBS, 10% goat serum, and 0.75% Triton X-100 followed by incubation in biotinylated goat anti-mouse secondary (Vector Laboratories, Burlingame, CA) diluted 1:400 in PBS. Elite Vectastain ABC and DAB kits (Vector Laboratories) were used to detect BrdU-labeled cells, and the sections were counterstained with methyl green.
BrdU-immunolabeled sections were analyzed using the Bioquant Image Analysis System (R&M Biometrics, Nashville, TN). The analysis focused on the VZ. Briefly, the VZ was composed of a pseudostratified columnar epithelium in which the mitotic figures were located at the ventricular surface, and cells acutely labeled with BrdU were located at the ab-ventricular limit (Miller, 1989). This contrasted with the SZ in which no organization was detectable as M- and S-phase cells were intermingled. It was determined that a counting box 70 μm in height consistently accommodated the full width of the VZ. To determine the LI in the VZ, the number of BrdU+ cells and the total number of cells within a 100 μm width of the VZ were counted. The LI was calculated as the quotient of these two numbers. The LI for each time point represented the mean of counts from three slices collected from each of five different animals (n = 5). The LIs for untreated and TGFβ1-treated slices were best fit to a line(s) by regression analysis to determine the rate of increase in LI (slope) and whether the proliferating cells cycled as a single population. As in the past, the data were best fit to a single population model (Nowakowski et al., 1989; Jacobs and Miller, 2000). The slope and GF were used in formulas to determine Tc and Ts.
Assay for cell cycle exit. To label a cohort of cycling cells, pregnant dams received an intraperitoneal injection of BrdU (12 mg/kg of body weight) on G17. The time of injection was designated as time 0 h postinjection (pi) or t0pi. One hour later, fetuses were harvested and slices were generated and treated with TGFβ1 (0 or 40 ng/ml) as described above (see above, Organotypic slice cultures). Twenty-four hours after the BrdU injection (t24pi), slices were fixed and sectioned for immunohistochemistry as described above. Sections were processed for BrdU and Ki-67 immunoreactivity. BrdU immunolabeling was performed as described above, except that a goat anti-mouse, FITC-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA) was used. Immunolabeling of BrdU was immediately followed by serial incubation in rabbit anti-Ki-67 (Labvision, Fremont, CA) diluted 1:150 in PBS with 0.75% Triton X-100 and 10% goat serum for 2 h followed by rhodamine-conjugated goat anti-rabbit secondary (Jackson ImmunoResearch) for 1 h.
To obtain an index of cell cycle exit, sections were examined for BrdU and Ki-67 expression. The numbers of two groups of immunopositive cells in a defined area of the cerebral wall were counted: (1) cells that contained BrdU immunoreactivity (BrdU+) and (2) cells that were BrdU positive but immunonegative for Ki-67 (BrdU+/Ki-67–). The proportion of cells that exited the cell cycle was calculated by dividing the number of BrdU+/Ki-67– cells by the total number of BrdU+ cells.
Cell cycle protein immunohistochemistry. Sections were processed immunohistochemically for the expression of specific cell cycle proteins. Primary antibodies (and dilutions in PBS, 10% goat serum, and 0.75% Triton X-100) were rabbit monoclonal anti-cyclin D1 (1:100; Labvision), mouse monoclonal anti-p27 antibody (1:1000; PharMingen, San Diego, CA), rabbit monoclonal anti-Ki-67 (1:150; Labvision), and antibody mouse monoclonal anti-p21 antibody (1:100; PharMingen). Incubations with primary antibodies were done for 2 h at room temperature. Subsequently, sections were incubated with a biotinylated goat anti-rabbit or goat anti-mouse secondary (Vector Laboratories) diluted 1:400 in PBS for 1 h at room temperature. Elite Vectastain ABC and DAB kits (Vector Laboratories) were used to detect immunolabeled cells, and the sections were counterstained with cresyl violet.
After immunohistochemistry for cyclin D1, p27, Ki-67, or p21, the percentage of VZ cells expressing each cell cycle protein was determined using Bioquant software. We used the same size counting box (70 × 100 μm) that was used for BrdU analysis. The number of immunolabeled cells as well as the total number of cells was determined using a minimum of three slices collected from three different litters (n = 3).
Triple immunolabeling for p21 and cell cycle-related markers. A cohort of cycling cells was labeled with BrdU as described in the cell cycle exit assay and slices cultured for 18, 24, or 30 h after BrdU injection (i.e., t18pi, t24pi, or t30pi, respectively). Various combinations of triple antibody labeling were performed for the following antigens: p21, BrdU, Ki-67, cyclin D1, and p27. p21 protein was detected using the Tyramide Signal Amplification kit (Molecular Probes, Eugene, OR) in conjunction with the anti-p21 antibody (1:1000; PharMingen). After labeling of p21, sections were processed for BrdU, Ki-67, cyclin D1, or p27 immunofluorescence as described previously with the following exceptions: (1) before detection of p21, sections were steamed in 0.010 m citric acid for 15 min in lieu of the 2.0N HCl wash; and (2) rabbit monoclonal anti-Ki-67 and rabbit monoclonal anti-cyclin D1 were detected with cyanine 5-conjugated donkey anti-rabbit secondary (Jackson ImmunoResearch).
For triple-labeled sections, immunofluorescence was detected using a Leica (Nussloch, Germany) microscope outfitted with a confocal laser (Bio-Rad, Hercules, CA). For each section, a z-series of seven 1.0-μm-deep images was collected for each fluorophore. Colocalization of p21 with BrdU, p27, and/or Ki-67 was quantified from at least three slices collected from four different dams (n = 4).
Cerebral wall collection and immunoblotting. Quantitative changes in the concentration of cell cycle proteins were determined using a quantitative immunoblotting method (Mooney and Miller, 2001). Slices were prepared and treated as described above (Organotypic slice cultures) and collected at t3. The cerebral wall was isolated from the slice and collected in lysis buffer [1.0% Nonidet P-40, 0.50% deoxycholic acid, 0.010% SDS, and Complete Mini protease inhibitor cocktail tablets (one tablet per 10 ml of buffer; Roche, Indianapolis, IN) in PBS], sonicated, and spun at 10,000 rpm for 10 min. The protein content of the supernatant was determined.
Samples of the supernatant (50 μg), molecular weight markers (Amersham Biosciences, Piscataway, NJ), and an internal standard (lysate from the pooled cortices of 19-d-old fetuses; 50 μg) were loaded on 15% SDS-PAGE, separated by electrophoresis, and transferred to nitrocellulose membranes. After blocking in 5.0% nonfat-dehydrated milk in 0.10% Tween 20 and PBS, blots were probed overnight at 4°C with one of the following: mouse monoclonal anti-cyclin D1 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), mouse-monoclonal anti-p27 antibody (1:1000; PharMingen), and mouse monoclonal anti-p21 antibody (1:500; PharMingen). After washing, blots were incubated with horseradish peroxidase-linked anti-mouse (1:1000; Amersham Biosciences) for 30 min. Immunotagged protein bands were visualized using a chemiluminescent detection reagent (Amersham Biosciences). Membranes were then stripped of immunolabel and reprobed with a mouse monoclonal anti-actin antibody (1:5000; Sigma). The amount of actin protein was used as a loading control.
Densitometric analysis of the immunoblots was performed using an Image Station (Kodak, Rochester, NY). Variations in total protein content were corrected within each blot using the amount of actin expression as a standard. Interblot variation was addressed by normalizing the data against the internal control and the untreated condition.
Statistics. Differences among multiple treatment groups and/or across time points were assessed with ANOVA. In cases in which statistically significant (p < 0.05) differences were detected, specific differences (e.g., in time or treatment) were examined using post hoc analysis. For paired analyses, t tests were used. Error values in this study represent SEM.
Results
Cell proliferation in the VZ of organotypic slice cultures: effect of TGFβ1 on cell cycle kinetics
The challenge of studying cortical proliferation in vitro is that the model systems commonly used (e.g., dissociated cell cultures) lack the organization of cell proliferation exhibited in the intact fetal VZ or are composed of cells that fail to stop proliferating (e.g., cell lines derived from neural cancers). Additionally, it is difficult to study the effects of exogenous agents on cortical cell proliferation in systems that do not mirror the complexity of the cortical VZ. For this reason, organotypic slice cultures of the cerebral wall are advantageous for studying cortical cell proliferation as the integrity of the VZ is preserved.
TGFβ1 may alter proliferation in the VZ via changes in the kinetics of cell cycle progression. A BrdU cumulative labeling method was used to determine the total length of the cell cycle (Tc) and the length of S phase (Ts). BrdU is incorporated exclusively into S-phase cells. As more cells pass into S, the proportion of BrdU+ cells increases until all cells are labeled (Fig. 1B). To ensure that cycling cells in the subventricular zone (SZ) were not included in the analysis, the VZ was delineated from the SZ by four criteria: (1) the VZ contained many radially elongated nuclei, indicative of interkinetic nuclear migration; (2) after 1 h of BrdU exposure, labeled cells were confined to the outer third of the VZ; (3) the SZ contained cells with no obvious orientation; and (4) the SZ had mitotic figures and BrdU+ cells randomly distributed throughout its depth (Fig. 1C).
Figure 1.

BrdU cumulative labeling to determine cell cycle kinetics. A, Time line depicting the course of the BrdU cumulative labeling experiment. B, The schematic figures depict the principles of the cumulative labeling method. BrdU is incorporated into cells passing through S at the time of BrdU addition (t0). BrdU-labeled and unlabeled cells are represented by filled and open ovals, respectively. As time progresses, more cells pass into S and incorporate the BrdU (t9) until all cycling cells are BrdU+ (t17). C, The micrograph depicts the delineation between the two proliferative zones (dotted line) in an untreated slice after 3 h of continuous BrdU exposure (t3). Elongated BrdU+3 nuclei perpendicular to the ventricular surface are found only in the VZ (straight arrow), whereas ablumenal mitotic figures are restricted to the SZ (crossed arrow). D, For untreated slices and those treated with TGFβ1 (40 ng/ml), the temporal change in the proportion of BrdU-labeled cells in the VZ was plotted. The initial linear increase in BrdU labeling in both treatment conditions was similar. TGFβ1 treatment did not significantly alter the total length of the cell cycle (Tc) or the length of S (Ts). In contrast, the maximum BrdU labeling attained in the TGFβ1 treatment condition was less than that in the untreated condition. Consequently, TGFβ1 significantly (p < 0.05) reduced the growth fraction. The asterisk indicates a statistically significant difference relative to the untreated condition (p < 0.05). Scale bar, 20 μm.
To determine the rate of increase in BrdU labeling, the BrdU LI in the VZ was calculated for untreated and TGFβ1-treated slices collected at t3, t6, t9, t12, t14, t17, and t24 and the values plotted against time (Fig. 1A,D). TGFβ1 (40 ng/ml) significantly (F(11,35) = 16.668; p < 0.001) decreased the LI at each time point examined (Fig. 1D). The regression analysis of the linear increase in LI yielded r ≥ 0.92. Cells in the untreated slices took 23.9 ± 2.0 h to pass through a cell cycle, and 10.2 ± 1.7 h of that time was spent in S (Fig. 1D). TGFβ1 (40 ng/ml) treatment did not significantly alter Tc or Ts.
A second index of cell proliferation is the proportion of cells in the VZ that are actively cycling, also referred to as the growth fraction. The GF was calculated as the mean of LIs after Tc-Ts. In the untreated and TGFβ1-treated slices, the GFs were attained at 13.7 and 12.6 h, respectively. In untreated slices, 70.5 ± 1.5% of the cells in the VZ were cycling. This concurs with the frequency of cells (69.3 ± 1.5%) that were immunopositive for a marker of cycling cells, Ki-67. Treatment with TGFβ1 (40 ng/ml) significantly reduced the GF (p < 0.05), causing a 21% decrease (to 55.0 ± 2.0%) in the fraction of cycling cells in the VZ (Fig. 1C). The frequency of Ki-67 immunolabeling in the VZ was 55.6 ± 1.1% after treatment with TGFβ1 (40 ng/ml).
Concentration-dependent effect of TGFβ1on cell proliferation
To determine whether the effect of TGFβ1 is concentration dependent, TGFβ1-treated (0, 2.5, 5, 10, 20, or 40 ng/ml) slices were exposed to BrdU for 14 h (t0 → t14) and immunolabeled using a BrdU antibody to determine the LI. Most cells in the VZ of untreated slices were BrdU+, and slices treated with 2.5 and 10 ng/ml TGFβ1 had similar proportions of BrdU+ cells in the VZ as untreated slices (Fig. 2A–C). In contrast, treatment with a higher concentration of TGFβ1 (40 ng/ml) reduced the number of BrdU+ cells (Fig. 3D). Indeed, quantification of LIs in slices treated with a broad range of TGFβ1 reveal that only the highest concentration (40 ng/ml) significantly (F(5,22) = 26.584; p < 0.01) reduced the proportion of BrdU+ cells in the VZ (Fig. 2E).
Figure 2.
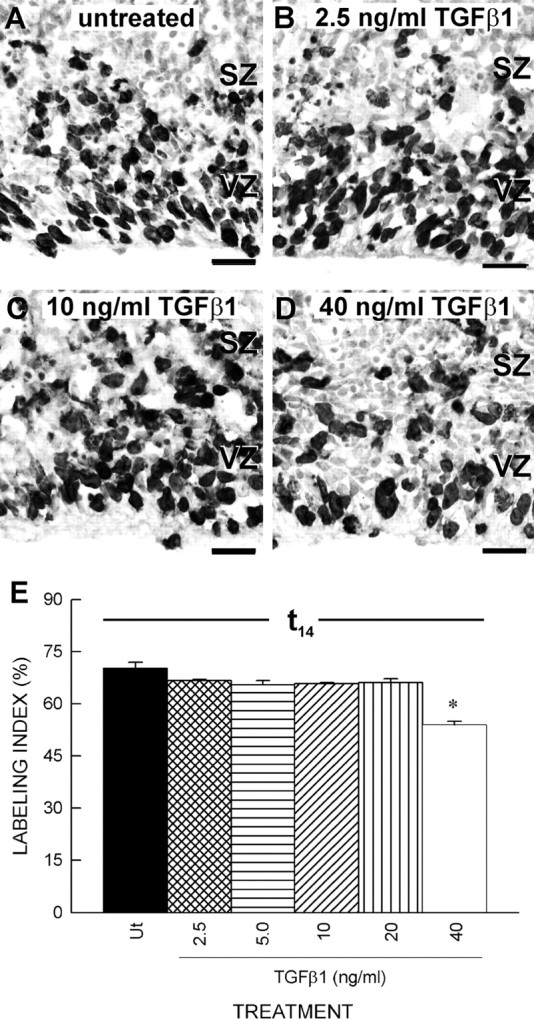
Concentration-dependent effect of TGFβ1 on proliferation of the VZ cells. A, In the untreated slices at t14, BrdU+ cells filled the VZ but were less prominent in the SZ. B, C, The density of BrdU+ VZ cells in slices treated with low concentrations of TGFβ1 (2.5 and 10 ng/ml, respectively) was similar to cells in untreated slices. D, Fewer BrdU+ VZ cells were found in slices treated with a high concentration of TGFβ1 (40 ng/ml). E, The LI at t14 was determined for slices treated with TGFβ1 (0, 2.5, 5.0, 10, 20, or 40 ng/ml). Only the highest tested concentration of TGFβ1 significantly (p < 0.05) reduced the LI. Scale bars, 20μm. The asterisk indicates a statistically significant difference relative to the untreated condition (p < 0.05)
Figure 3.

Cell cycle exit. A, Depiction of the experimental time line of the cell cycle exit assay. Injection of a G17 pregnant dam at 0 h after injection (t0pi) labels all cells in S with BrdU. At t0pi, all BrdU+ cells are in the cell cycle and colabel with the cell proliferation marker Ki-67 (BrdU+/Ki-67+; yellow ovals). Cells in the other phases of the cell cycle are not labeled with BrdU but are Ki-67 positive (BrdU–/Ki-67+; green ovals). Slices were generated 1 h after injection (t1pi). At t24pi, the BrdU+ cohort has completed one full cell cycle. A fraction of the BrdU+ cells stay in the cell cycle (BrdU+/Ki-67+; yellow ovals), and the remaining cells exit the cell cycle. The exited BrdU+ cells no longer express Ki-67 (BrdU+/Ki-67–; red ovals). The proportion of BrdU+/Ki-67– cells is an index of cell cycle exit. B, C, At t24pi, three types of immunopositive cells were identified in the untreated (B) and TGFβ1-treated (40 ng/ml; C) slices: (1) BrdU–/Ki-67+ cells (green arrow), (2) BrdU+/Ki-67+ cells (yellow arrow), and (3) BrdU+/Ki-67– cells (red arrow). Cell counts showed that TGFβ1 treatment significantly (p > 0.05) increased cell cycle exit by 24% (from a frequency of 0.40 ± 0.02 to 0.50 ± 0.01). Scale bars, 20 μm.
How does the effective concentration of exogenous TGFβ1 (40 ng/ml) compare with that of endogenous ligand? The concentration of endogenous TGFβ1 in the cerebral wall of slices from 17-d-old fetuses is ∼0.4 ng/ml; however, the total concentration of TGFβ1 in the cerebral wall is not indicative of regional concentrations of TGFβ1. Immunohistochemical analyses show that TGFβ1 is not uniformly distributed in the fetal rat cerebral wall (Miller, 2003). Indeed, TGFβ1 immunolabeling is concentrated in the VZ and CP. Thus, cycling cells in the VZ may reside in a much richer, endogenous TGFβ1 environment than indicated by studies of total TGFβ1 content.
TGFβ1 promotes cell cycle exit
The hypothesis that TGFβ1 reduces the fraction of VZ cycling cells by inducing cells to exit the cell cycle was tested using BrdU and Ki-67 colocalization (Chenn and Walsh, 2002). With this method, the presence of Ki-67 in the nuclei is used to assess the “proliferation status” of cells pulse-labeled with BrdU. Ki-67 is a nuclear protein that is expressed exclusively by proliferating cells during all stages of the cell cycle and is downregulated after cell cycle exit in G1 (du Manoir et al., 1991; Scholzen and Gerdes, 2000). The exclusive expression of Ki-67 was confirmed in the slice culture system by comparing GFs measured by BrdU cumulative labeling (70%) and Ki-67 immunolabeling (69%) in untreated slices. The consistency between the two measures indicates that Ki-67 is found in all cycling cells in the VZ but not in postmitotic cells.
A cohort of cycling cells was labeled with BrdU in utero, and slices were generated 1 h after injection (t1pi) (Fig. 3A). Tc in untreated and TGFβ1-treated slices was calculated at ∼24 h. Thus, slices were cultured for 24 h after injection (t24pi) to allow all cells labeled with BrdU to complete one full cell cycle, in particular the entirety of G1 (Fig. 3A). After fixation at t24pi, Ki-67 immunofluorescence was used to assess whether a BrdU+ cell was actively cycling. At the time of injection (t0pi), it was expected that all BrdU+ cells would colabel with Ki-67 (Fig. 3A). Indeed, slices fixed immediately after generation (t1pi) and analyzed for BrdU–Ki-67 colocalization revealed that <1% of the BrdU-labeled cells did not colocalize with Ki-67 (data not shown). Twenty-four hours after injection, after which the BrdU+ cells made the “decision” to remain in or exit the cell cycle, it was expected that BrdU+ cells that exited the cell cycle would no longer express Ki-67 (Fig. 3A). Thus, the proportion of BrdU+/Ki-67-negative (Ki-67–) cells was used as an estimation of cell cycle exit in the VZ.
In both untreated and TGFβ1-treated slices collected at t24pi, Ki-67-positive (Ki-67+) cells filled the VZ and select cells in the SZ (Fig. 3B,C). Many Ki-67+ cells co-labeled with BrdU, indicating that these cells remained in the cell cycle. The remaining BrdU+ cells did not colabel with Ki-67, presumably because the cells exited the cell cycle. The proportion of cells that exited the cell cycle, defined as the fraction of the total BrdU+ population immunonegative for Ki-67, was determined for untreated and TGFβ1 (40 ng/ml)-treated slices. In untreated slices, 40.0 ± 0.9% of the BrdU pulse-labeled cells exited the cell cycle 24 h after BrdU injection. TGFβ1 treatment significantly (p < 0.05) increased the fraction of cells that exited the cell cycle, increasing cell cycle exit to 50 ± 0.5%.
Effect of TGFβ1 on cell cycle proteins
TGFβ1 may induce cell cycle exit via downregulation of cyclin proteins, in particular cyclin D1, and/or upregulation of specific CKIs of the cip/kip families (e.g., p21 and p27). Immunoblotting and immunohistochemistry on slices were used to determine the effects of TGFβ1 on expression of cell cycle-related proteins in the cerebral wall. Slices were fixed for immunohistochemistry or collected for immunoblotting at a time point corresponding to t3 of the BrdU cumulative labeling time line (Fig. 1A).
Cyclin D1 immunoreactivity was expressed in the nuclei of select VZ and SZ cells and was essentially absent from cells in regions where cycling cells are rare, specifically the intermediate zone (IZ), cortical plate (CP), and marginal zone (MZ) (Fig. 4D–F). BrdU pulse-chase studies indicate that cyclin D1-positive (cyclin D1+) VZ cells are in G1 (data not shown). This is consistent with the notion that cyclin D1 is upregulated exclusively during G1 (Stacey, 2003). Immunoblots for cyclin D1 indicate that TGFβ1 treatment (2.5, 10, and 40 ng/ml) did not significantly alter cyclin D1 expression in the cerebral wall (Fig. 4A). Based on data from the study by Siegenthaler and Miller (2005), immunoblots show that longer exposure (e.g., slices collected at t24) to TGFβ1 (40 ng/ml) does not alter cyclin D1 expression.
Figure 4.

Cell cycle protein expression. A–C, Cortical slices treated with TGFβ1 (0, 2.5, 10, or 40 ng/ml) were collected for quantitative immunoblotting. Immunoblots were probed with antibodies directed against cyclin D1, p27, p21, or actin. TGFβ1 treatment did not significantly alter protein expression of cyclin D1 (A) and p27 (B). In contrast, p21 expression (C) was significantly (p < 0.05) increased in slices treated with a high concentration of TGFβ1 (40 ng/ml). Ut, Untreated. D, Organotypic slices were collected and processed for cyclin D1 immunohistochemistry. The compartments of the cerebral wall (VZ, SZ, IZ, CP, and MZ) were identifiable at low power. Virtually all cyclin D1+ cells were confined to the proliferative zones. E, Indeed, cyclin D1 expression in untreated slices was limited to the nuclei of select VZ and SZ cells. F, TGFβ1 (40 ng/ml) treatment did not alter the pattern or intensity of cyclin D1 expression but did decrease the number of cyclin D1-positive cells. G, p27 immunolabeling was distributed in all compartments of the cerebral wall. H, Some cells in the proliferative zones were p27 positive, although a number of p27-negative cells were present. I, Changes in p27 immunoreactivity were not evident in a TGFβ1 (40 ng/ml)-treated slice. J, K, Unlike p27, p21-positive nuclei were limited to the proliferative zones of untreated slices. L, Consistent with the p21 immunoblots, treatment with TGFβ1 (40 ng/ml) increased the number of p21-positive nuclei. The asterisk indicates a statistically significant difference relative to the untreated condition (p < 0.05). Scale bars, 20 μm.
Sections immunolabeled for cyclin D1 were analyzed to determine the proportion of VZ cells that expressed cyclin D1. In the untreated slices, 40.2 ± 1.1% of VZ cells expressed cyclin D1. Treatment with TGFβ1 (40 ng/ml) significantly (p < 0.05) reduced the proportion of VZ cells expressing cyclin D1, decreasing the proportion to 33.7 ± 0.9%. Qualitatively, the intensity of cyclin D1 staining did not appear different in slices treated with TGFβ1 (40 ng/ml). The decrease in VZ cyclin D1-positive cells caused by TGFβ1 treatment likely resulted from a decrease in VZ cell proliferation. Indeed, the difference in VZ cell proliferation (21%) between untreated and TGFβ1 was similar to the treatment-induced difference (18%) in cyclin D1-expressing cells.
p27-positive (p27+) cells were distributed in all layers of the cerebral wall, and almost all cells in the IZ and CP were p27+ (Fig. 4G). In the untreated slice, a conspicuous number of cells in the VZ were p27 negative (Fig. 4H). Analysis of the proportion of p27+ cells in the VZ revealed that in untreated slices, 66.0 ± 3.2% of VZ cells expressed p27. TGFβ1 (40 ng/ml) treatment did not significantly alter the proportion of p27+ cells in the VZ (72.0 ± 2.8%). This was consistent with immunoblots for p27 (Fig. 4B). Therefore, TGFβ1 treatment did not significantly change total p27 expression in the cerebral wall. As with cyclin D1, p27 expression does not change significantly between t3 and t24 (Siegenthaler and Miller, 2005).
Like cyclin D1, p21 was localized to the nucleus of cells in the proliferative zones, the VZ and SZ, but was essentially absent from cells in the IZ or CP (Fig. 4J–L). The small size of p21+ nuclei indicated that the nuclei likely contained 2N DNA and, consequently, were not in G2 or in late S. In addition, the p21+ cells did not localize to the ventricular surface, where mitotic cells are distributed (Sauer, 1935; Sidman et al., 1959). Therefore, it appears that p21+ cells are in G1 or G0.
TGFβ1 increased p21 expression in a concentration-dependent manner. Lower concentrations of TGFβ1 (2.5 and 10 ng/ml) did not significantly alter p21 expression (Fig. 4C). Incidentally, these two concentrations did not alter cell proliferation in the VZ. The concentration of TGFβ1, which affected cell proliferation (40 ng/ml) significantly (p < 0.05) increased p21 protein expression. Immunohistochemistry for p21 indicated that the increase in total p21 protein expression paralleled an increase in the number of p21+ cells (Fig. 4L). Analysis of p21-immunolabeled sections revealed that 10.9 ± 0.4% of cells in the VZ express p21. Treatment with TGFβ1 (40 ng/ml) significantly (p < 0.05) increased the percentage of p21+ cells in the VZ to 16.7 ± 0.6%.
p21 is associated with cell cycle exit
The confined distribution of p21+ cells in the cortical proliferative zones implies that this CKI participates in cell cycle regulation. If p21 is specifically involved in cell cycle exit, the expectation is that p21 should be expressed in a manner to facilitate this event, including the following: (1) upregulation in G1, (2) colocalization with other CKIs, and (3) expression in cells that are exiting the cell cycle.
To ascertain the timing of p21 expression in the cell cycle (e.g., G1 and G0), a cohort of proliferating cells in the VZ was labeled with BrdU during a 1.0 h pulse in vivo, and slices were fixed 18 h (t18pi), 24 h (t24pi), and 30 h (t30pi) after the BrdU injection (Fig. 5A). This method exploits the idea that the cohort of BrdU+ cells progress through the cell cycle in the intervening hours between the BrdU injection and the time of fixation. By collecting slices before one full cell cycle, at the end of one cell cycle, and after a full cell cycle, the p21 immunoreactivity of BrdU+ cells both in and out of the cell cycle can be established. Colocalization of p21 with phase-specific cell cycle proteins and other CKIs further defines the timing of p21 expression and determines colocalization with other CKIs (e.g., p27).
Figure 5.

p21 colocalization with cyclin D1 and p27. A, Time line of the triple-labeling experiment. A cohort of cells in S was labeled with BrdU at t0pi, and slices were generated from the BrdU-exposed embryos at t1pi and fixed at t18pi, t24pi, and t30pi for assessment triple immunoreactivity of a variety of cell cycle proteins. B–D, Slices fixed 18 h after the BrdU injection (t18pi) were triple immunolabeled for p21, cyclin D1, and BrdU. p21 was upregulated in G1, as indicated by the p21+ nuclei that coexpressed the G1 marker cyclin D1 and BrdU (white arrows). p21+, BrdU+, and cyclin D1– were also apparent (yellow arrows); evidence that p21 expression persisted in cells after they exit from the cell cycle is shown. E–G, To determine whether p21 and p27 are co-upregulated in cycling cells, t18pi slices were triple immunolabeled for p21, p27, and Ki-67. p21+ cells coexpressed p27 whether they were in the cell cycle (Ki-67+; white arrows) or out of the cell cycle (Ki-67–; yellow arrows). Cells that were p27+/Ki-67+, but did not express p21, were also present (pink arrows), indicating that p21 and p27 were not always co-upregulated. Scale bars, 20 μm.
To determine whether p21 protein expression increased in G1, triple immunofluorescence for p21, BrdU, and cyclin D1 was performed on untreated slices collected at t18pi. Some p21+/BrdU+ cells coexpressed cyclin D1 (Fig. 5B–D), indicating that p21 was upregulated during G1. Cells that were p21+ and BrdU+, but did not express cyclin D1, were also present. It is likely that these cells progressed out of G1 into G0 and continued to express p21.
Studies of p21, p27, and Ki-67 triple immunofluorescence were performed on untreated slices collected at t18pi to determine whether p21 and p27 were upregulated in tandem in cycling cells. In general, p21+ cells coexpressed p27 (Fig. 5E–G). Analysis of the colocalization revealed that 81.8 ± 2.1% of p21+ cells coexpress p27. Cells that coexpressed p21 and p27 in the VZ were either in the cell cycle (i.e., also Ki-67+) or out of the cell cycle (Ki-67–). The implication of this is that p21 and p27 are upregulated in G1 in preparation for cell cycle exit and continue to be expressed in G0. It was noteworthy that some p27+ cells in the cell cycle did not express p21, indicating that coinduction of p21 and p27 in G1 may not be compulsory.
To determine whether p21 is upregulated during cell cycle exit, cycling cells were labeled with BrdU for 1 h in vivo and then cultured for 18 h (t18pi), 24 h (t24pi), and 30 h (t30pi) after the BrdU injection and triple immunolabeled for p21, BrdU, and Ki-67 (Fig. 5B). Taking into account that p21 is upregulated in G1, we predicted that, at a time before one full cell cycle (e.g., at t18pi), more BrdU+/p21+ cells would be in the cell cycle (Ki-67+) than out of the cell cycle (Ki-67–) (Fig. 6A). Additionally, if p21 is pushing cells out of the cell cycle, the number of BrdU+/p21+ cells in the cell cycle should decrease over time (e.g., t18pi → t24pi), and the number of BrdU+/p21+ cells out of the cell cycle should increase (Fig. 6A).
Figure 6.

p21 upregulation and cell cycle exit. A, To determine whether p21 upregulation correlated with cell cycle exit, a model of p21 colocalization with BrdU and Ki-67 at t0pi, t18pi, t24pi, and t30pi is proposed. This model was built on the idea that p21 is upregulated in G1 and persists after cell cycle exit. At a time before one full cycle (t18pi), more p21+/BrdU+ should be in the cell cycle (Ki-67+) than out of the cell cycle (Ki-67–). As the BrdU+ cells progress through G1, cells that upregulate p21 should exit the cell cycle and downregulate Ki-67 expression. B, C, Triple-labeled nuclei (white arrows) were readily distinguishable from cells labeled with p21 and BrdU only (yellow arrows; B). The number of p21+, BrdU+, Ki-67+, and p21+, BrdU+, and Ki-67– per field was counted in untreated slices. As predicted, a decrease in the number of triple-labeled cells over time was countered by an increase in p21+, BrdU+, and Ki-67– cells (D). Scale bar, 20 μm.
Cells labeled with only p21 and BrdU were readily distinguishable from cells labeled with all three antibodies (Fig. 6B). To test the proposed model, slices were fixed at t18pi, t24pi, and t30pi and analyzed for the number of p21+/BrdU+ cells in the VZ that were in the cell cycle (Ki-67+) or out of the cell cycle (Ki-67–). At t18pi, the number of p21+/BrdU+ cells in the cell cycle (Ki-67+) was greater than the number of cells that had exited. From t18pi to t24pi, that trend was reversed, because p21+/BrdU+ cells presumably exited the cell cycle and stopped expressing Ki-67 (Fig. 6C). Little change was seen in either category of immunolabeling between t24pi and t30pi. This is consistent with the idea that the vast majority of BrdU+ cells that will exit have done so by t24pi. A similar pattern was observed in slices treated with TGFβ1 (data not shown). These data support the hypothesis that p21 is upregulated in G1 and that an increase in p21 is strongly linked with cell cycle exit.
TGFβ1 increases cell cycle exit through p21
Does a TGFβ1-dependent increase in p21 expression translate to an increase in cell cycle exit? To answer this question, the number of p21+/BrdU+/Ki-672 cells within a defined area of the VZ was determined for untreated and TGFβ1-treated slices at the three time points (t18pi, t24pi, and t30pi). At t24pi, more p21+/BrdU+/Ki-67– cells were evident in TGFβ1-treated slices (Fig. 7A,B). Indeed, at each time point examined, TGFβ1 (40 ng/ml) significantly (p < 0.05) increased the number of p21+/BrdU+/Ki-67– cells (Fig. 7C). From this evidence, a model emerges in which TGFβ1 upregulates p21 in VZ cells that would not normally have expressed it, thus catalyzing their premature exit from the cell cycle (Fig. 7D).
Figure 7.

TGFβ1 increases p21-mediated cell cycle exit. After collection at 1 h after BrdU administration (t1pi), slices were treated with TGFβ1 (0 or 40 ng/ml), collected at t18pi, t24pi, and t30pi, and triple immunolabeled for p21, BrdU, and Ki-67. A, B, Qualitatively, the number of p21+, BrdU+, and Ki-67– cells at t24pi was increased in the slice treated with 40 ng/ml TGFβ1 compared with untreated. C, The number of p21+, BrdU+, and Ki-67– cells in a field was determined for untreated and TGFβ1-treated slices. At each time point, TGFβ1 increased the number of p21+, BrdU+, and Ki-67– cells. D, The diagram illustrates the effect of TGFβ1 on p21-mediated cell cycle exit. Asterisks indicate a statistically significant (p < 0.05) difference from untreated. Scale bars, 20 μm.
Discussion
Proliferation in organotypic slice cultures
Tc and Ts in the slices from the rat cerebral wall are 30 and 6% longer, respectively, than in vivo calculations of Tc and Ts at similar stages of rat cortical development (Miller and Nowakowski, 1991; Vaccarino et al., 1999). In contrast, the present data are similar to those for the Tc and Ts in mouse organotypic slices (Haydar et al., 1999; Takahashi, 1999). Loss of projections from the thalamus, a byproduct of the slice preparation, may explain the lengthening of Tc. Loss of thalamic afferents leads to an increase in Tc in organotypic slice cultures (Dehay et al., 2001). The increase in Tc is potentially caused by a loss of diffusible factors, specifically basic fibroblast growth factor, secreted by the thalamic afferents.
TGFβ1 decreases the GF and increases cell cycle exit
TGFβ1 is intimately involved in growth control in a variety of cell types, including hematopoietic cells, epithelial cells, endothelial cells, and lymphocytes (Roberts and Sporn, 1990). Neural cells are also sensitive to TGFβ1. TGFβ1 inhibits the proliferation of B104 neuroblastoma cells (Luo and Miller, 1999) and cultured cortical neurons (Miller and Luo, 2002b). Thus, it is not unexpected that TGFβ1 has an antiproliferative effect on cycling VZ cells.
The concentration needed to inhibit cell proliferation (40 ng/ml) is greater than that needed to increase neural cell adhesion molecule and integrin protein expression in postmitotic, migrating cortical neurons (2.5 and 10 ng/ml) (Siegenthaler and Miller, 2004). One explanation may lie in the spatial distribution of foxg1, a transcription factor that inhibits TGFβ signal transduction. Foxg1 expression is high in the forebrain neuroepithelium and low in postmitotic neurons during corticogenesis (Tao and Lai, 1992). A large amount of TGFβ1 may be required to overcome inhibition by foxg1 expressed in VZ cells. Indeed, in the absence of foxg1, dissociated cortical progenitors are sensitive to lower concentrations (2.5 ng/ml) of TGFβ1 (Dou et al., 2000).
TGFβ1 decreases the number of actively proliferating cells in the VZ. A plausible mechanism for a TGFβ1-induced reduction in the GF is an increase in the number of cells exiting the cell cycle. The present assay of cell cycle exit indicates that the number of cells lost from the cycling population (21%) is equivalent to the TGFβ1-induced increase in the number of cells entering G0 [i.e., the number exiting the cell cycle (25%)]. Thus, TGFβ1-initiated cell cycle exit accounts for the decrease in the growth fraction. An alternative mechanism for the decrease in the GF is a TGFβ1-dependent increase in the death of cycling cells. TGFβ1 has been implicated in neuronal cell death; however, it appears to be neuroprotective (Scorziello et al., 1997; Sometani et al., 2001; Zhu et al., 2001; Brionne et al., 2003). Although cell death was not directly assayed in the present study, qualitatively, no notable increase in the number of pyknotic or condensed nuclei was observed after TGFβ1 treatment.
Although this study specifically examines p21, p27, and cyclin D1, a number of other cell cycle proteins can regulate cortical cell proliferation. For example, alterations in cyclin E expression are implicated in the alteration of cell cycle length during corticogenesis (Delalle et al., 1999). Unlike cyclin D1, however, suppression of cyclin E transcription has not been implicated in TGFβ1-mediated growth inhibition. Another example is the CKI p57. The expression of p57 transcripts is high in developing cortex, specifically in the cortical plate (van Lookeren Campagne and Gill, 1998). Upregulation of p57 by TGFβ1, however, has not been tied to growth arrest.
p21 and cell cycle exit
The present study provides strong correlative evidence that upregulation of p21 in G1 leads to cell cycle exit in the developing cerebral cortex. A role for p21 in cortical development is supported by in situ studies that show elevated p21 mRNA throughout corticogenesis in both the VZ and SZ (Delalle et al., 1999). An increase in p21, caused by the absence of foxg1, correlates with a substantial increase in cell cycle exit in the VZ (Seoane et al., 2004). Furthermore, an excess of the transcription factor Runx1 promotes cortical neural progenitor proliferation through repression of p21 transcription (Theriault et al., 2005).
Upregulation of p21 in G1 and its potential role in cell cycle exit is consistent with evidence that the decision to remain in the cell cycle or become postmitotic neuron occurs in G1 (Caviness et al., 1999). During mouse corticogenesis, the length of G1 increases as the neurogenic interval progresses and more cells enter the postmitotic population (Takahashi et al., 1995). The lengthening of G1 is coordinate with changes in cell cycle protein transcript levels, in particular p27 and cyclin E (Delalle et al., 1999). The current study provides additional evidence that G1 cell cycle proteins are critical for the precise regulation of cell cycle exit in the developing cerebral cortex.
Coexpression of p21 with p27 during the G1 → G0 transition suggests that the two CKIs work in tandem to promote cell cycle exit. In the VZ, p21 and p27 are coexpressed in cells before cell cycle exit. Both CKIs remained upregulated after cell cycle exit: p21 for a short period that ends before the commencement of migration, and p27 for a much longer time. This idea is supported by the observation that p27 is expressed by cycling cells in the proliferative zones as well as postmitotic cells in the IZ and CP, whereas p21 is only expressed by cells in the proliferative zones. This pattern of expression suggests that both p27 and p21 are involved in the G1 → G0 transition, whereas p27 alone is required for continued suppression of cycling activity in migrating and differentiating neurons. p27 and p21 often cooperate to move cells out of the cell cycle. For example, both p21 and p27 are upregulated during cell cycle exit in intestinal epithelial cells (Tian and Quaroni, 1999). The increase in p21 expression is transient, whereas increased p27 expression continues well after cell cycle exit, a pattern similar to that observed in this study.
Despite the presence of cells that expressed both p21 and p27, some cells express only p27 before and after cell cycle exit. This indicates that p21 upregulation is not required for all VZ progenitors exiting the cell cycle. Taking into account the numbers of p21+ cells in the VZ and cell cycle exit, it can be estimated that, at the minimum, approximately one-third of exiting cells upregulate p21. It is possible that VZ cells that require both p21 and p27 to exit the cell cycle represent a specific subset of neuronal progenitors. Indeed, recent work examining neuronal production in the VZ and SZ reveals that neurons are derived directly from asymmetric divisions in the VZ as well as from symmetric divisions of intermediate precursor cells in the SZ (Miyata et al., 2004; Noctor et al., 2004). The intermediate precursor cells in the SZ are generated from asymmetric divisions in the VZ, after which the cells divide again in the SZ to produce two postmitotic neurons. It is possible that permanent exit from the cell cycle in the VZ requires upregulation of multiple CKIs (e.g., p21 and p27), whereas the transient cell cycle exit seen in the intermediate precursor cells requires only one CKI.
TGFβ1 and cell cycle exit
Regulation of the expression of cell cycle proteins is the primary mechanism underlying growth control by TGFβ1. The present study shows that TGFβ1 specifically increases the expression of p21, and upregulation of this CKI is linked with an increase in cell cycle exit. Collectively, these observations provide strong correlative evidence that TGFβ1 promotes cell cycle exit through upregulation of p21. Additionally, these data support a model in which endogenous growth factors, such as TGFβ1, cooperate with intracellular factors to coordinate the transition of cycling cells to postmitotic neurons.
Support for the role of TGFβ1 in the regulation of cortical proliferation comes from the proliferation phenotype of the foxg1 knock-out mouse. In the absence of foxg1, untimely upregulation of p21 is correlated with premature cell cycle exit (Seoane et al., 2004). The antigrowth signaling by endogenous TGFβ1 is inhibited by the forebrain-specific transcription factor foxg1 (Dou et al., 2000; Rodriguez et al., 2001). It could be argued that the decrease in cell proliferation observed in foxg1 knock-out mice is a result of unchecked TGFβ1 signaling resulting in inappropriate upregulation of p21 in VZ cells.
Although the experiments presented here use exogenous TGFβ1, the endogenous TGFβ system is strategically expressed in a spatiotemporal pattern that is consistent with a role for TGFβ in cell proliferation (Miller, 2003). Combined with previous studies implicating TGFβ in cortical cell migration (Luo and Miller, 1999; Miller and Luo, 2002; Siegenthaler and Miller, 2004), the evidence presented here implicates the TGFβ system as an active participant in the developmental “conveyer belt” in which cortical progenitors are prompted to exit the cell cycle and migrate to the appropriate place in the emerging cerebral cortex.
Footnotes
This work was supported by National Institute for Alcohol Abuse and Alcoholism Grants AA06916 and AA07568 and by the Department of Veterans Affairs. We thank Brad Pawlikowski for constructive comments on this manuscript.
Correspondence should be addressed to Michael W. Miller, Department of Neuroscience and Physiology, State University of New York, Upstate Medical University, 750 East Adams Street, Syracuse, NY 13210. E-mail: millermw@upstate.edu.
DOI:10.1523/JNEUROSCI.1876-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258627-10$15.00/0
References
- Bouchard C, Fridman WH, Sautes C (1997) Effect of TGFβ1 on cell cycle regulatory proteins in LPS-stimulated normal mouse B lymphocytes. J Immunol 159: 4155–4164. [PubMed] [Google Scholar]
- Brionne TC, Tesseur I, Masliah E, Wyss-Coray T (2003) Loss of TGFβ1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 40: 1133–1145. [DOI] [PubMed] [Google Scholar]
- Caviness Jr VS, Takahashi T, Nowakowski RS (1999) The G1 restriction point as critical regulator of neocortical neuronogenesis. Neurochem Res 24: 497–506. [DOI] [PubMed] [Google Scholar]
- Caviness Jr VS, Takahashi T, Nowakowski RS (2000) Neuronogenesis and the early events of neocortical histogenesis. Results Prob Cell Differ 30: 107–143. [DOI] [PubMed] [Google Scholar]
- Caviness Jr VS, Goto T, Tarui T, Takahashi T, Bhide PG, Nowakowski RS (2003) Cell output, cell cycle duration and neuronal specification: a model of integrated mechanisms of the neocortical proliferative process. Cereb Cortex 13: 592–598. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA (2002) Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297: 365–369. [DOI] [PubMed] [Google Scholar]
- Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF (1995) Transforming growth factor β induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci USA 92: 5545–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay C, Savatier P, Cortay V, Kennedy H (2001) Cell cycle kinetics of neocortical precursors are influenced by embryonic thalamic axons. J Neurosci 21: 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delalle I, Takahashi T, Nowakowski RS, Tsai LH, Caviness Jr VS (1999) Cyclin E-p27 opposition and regulation of the G1 phase of the cell cycle in the murine neocortical PVE: a quantitative analysis of mRNA in situ hybridization. Cereb Cortex 9: 824–832. [DOI] [PubMed] [Google Scholar]
- Dou C, Lee J, Liu B, Liu F, Massagué J, Xuan S, Lai E (2000) BF-1 interferes with transforming growth factor β signaling by associating with Smad partners. Mol Cell Biol 20: 6201–6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Manoir S, Guillaud P, Camus E, Seigneurin D, Brugal G (1991) Ki-67 labeling in postmitotic cells defines different Ki-67 pathways within the 2c compartment. Cytometry 12: 455–463. [DOI] [PubMed] [Google Scholar]
- Elbendary A, Berchuck A, Davis P, Havrilesky L, Bast Jr RC, Iglehart JD, Marks JR (1994) Transforming growth factor β1 can induce CIP1/WAF1 expression independent of the p53 pathway in ovarian cancer cells. Cell Growth Differ 5: 1301–1307. [PubMed] [Google Scholar]
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, Kaushansky K, Roberts JM (1996) A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27Kip1-deficient mice. Cell 85: 733–744. [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75: 805–816. [DOI] [PubMed] [Google Scholar]
- Haydar TF, Bambrick LL, Krueger BK, Rakic P (1999) Organotypic slice cultures for analysis of proliferation, cell death, and migration in the embryonic neocortex. Brain Res Protoc 4: 425–437. [DOI] [PubMed] [Google Scholar]
- Jacobs JS, Miller MW (2000) Cell cycle kinetics and immunohistochemical characterization of dissociated fetal neocortical cultures: evidence that differentiated neurons have mitotic capacity. Dev Brain Res 122: 67–80. [DOI] [PubMed] [Google Scholar]
- Kamesaki H, Nishizawa K, Michaud GY, Cossman J, Kiyono T (1998) TGFβ1 induces the cyclin-dependent kinase inhibitor p27Kip1 mRNA and protein in murine B cells. J Immunol 160: 770–777. [PubMed] [Google Scholar]
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A (1996) Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27Kip1 Cell 85: 721–732. [DOI] [PubMed] [Google Scholar]
- Ko TC, Yu W, Sakai T, Sheng H, Shao J, Beauchamp RD, Thompson EA (1998) TGFβ1 effects on proliferation of rat intestinal epithelial cells are due to inhibition of cyclin D1 expression. Oncogene 16: 3445–3454. [DOI] [PubMed] [Google Scholar]
- Li CY, Suardet L, Little JB (1995) Potential role of Waf1/Cip1/p21 as a mediator of TGF-β cytoinhibitory effect. J Biol Chem 270: 4971–4974. [DOI] [PubMed] [Google Scholar]
- Luo J, Miller MW (1999) Transforming growth factor β1-regulated cell proliferation and expression of neural cell adhesion molecule in B104 neuroblastoma cells: differential effects of ethanol. J Neurochem 72: 2286–2293. [DOI] [PubMed] [Google Scholar]
- Miller MW (1989) Effects of prenatal exposure to ethanol on neocortical development: II. Cell proliferation in the ventricular and subventricular zones of the rat. J Comp Neurol 287: 326–338. [DOI] [PubMed] [Google Scholar]
- Miller MW (2003) Expression of transforming growth factor β in developing rat cerebral cortex: effects of prenatal exposure to ethanol. J Comp Neurol 460: 410–424. [DOI] [PubMed] [Google Scholar]
- Miller MW, Luo J (2002a) Effects of ethanol and basic fibroblast growth factor on the transforming growth factor β1 regulated proliferation of cortical astrocytes and C6 astrocytoma cells. Alcohol Clin Exp Res 26: 671–676. [PubMed] [Google Scholar]
- Miller MW, Luo J (2002b) Effects of ethanol and transforming growth factor β1 (TGFβ1) on neuronal proliferation and nCAM expression. Alcohol Clin Exp Res 26: 1281–1285. [DOI] [PubMed] [Google Scholar]
- Miller MW, Nowakowski RS (1991) Effect of prenatal exposure to ethanol on the cell cycle kinetics and growth fraction in the proliferative zones of fetal rat cerebral cortex. Alcohol Clin Exp Res 15: 229–232. [DOI] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Saito K, Kawano M, Muto T, Ogawa M (2004) Asymmetric production of surface-dividing and non-surface-dividing cortical progenitor cells. Development 131: 3133–3145. [DOI] [PubMed] [Google Scholar]
- Mooney SM, Miller MW (2001) Effects of prenatal exposure to ethanol on the expression of bcl-2, bax and caspase 3 in the developing rat cerebral cortex and thalamus. Brain Res 911: 71–81. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY (1996) Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 85: 707–720. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR (2004) Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci 7: 136–144. [DOI] [PubMed] [Google Scholar]
- Nowakowski RS, Lewin SB, Miller MW (1989) Bromodeoxyuridine immunohistochemical determination of the lengths of the cell cycle and the DNA-synthetic phase for an anatomically defined population. J Neurocytol 18: 311–318. [DOI] [PubMed] [Google Scholar]
- Nowakowski RS, Caviness Jr VS, Takahashi T, Hayes NL (2002) Population dynamics during cell proliferation and neuronogenesis in the developing murine neocortex. Results Prob Cell Differ 39: 1–25. [DOI] [PubMed] [Google Scholar]
- Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massagué J, Roberts JM, Koff A (1994) p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor β and contact inhibition to cell cycle arrest. Genes Dev 8: 9–22. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Sporn MB (1990) The transforming growth factors βs. In: Peptide growth factors and their receptors. Handbook of experimental pharmacology, Vol 95, pp 419–472. Berlin: Springer Verlag. [Google Scholar]
- Rodriguez C, Huang LJ, Son JK, McKee A, Xiao Z, Lodish HF (2001) Functional cloning of the proto-oncogene brain factor-1 (BF-1) as a Smad-binding antagonist of transforming growth factor-β signaling. J Biol Chem 276: 30224–30230. [DOI] [PubMed] [Google Scholar]
- Sauer F (1935) Mitosis in the neural tube. J Comp Neurol 62: 377–405. [Google Scholar]
- Scholzen T, Gerdes J (2000) The Ki-67 protein: from the known and the unknown. J Cell Physiol 182: 311–322. [DOI] [PubMed] [Google Scholar]
- Scorziello A, Florio T, Bajetto A, Thellung S, Schettini G (1997) TGFβ1 prevents gp120-induced impairment of Ca2+ homeostasis and rescues cortical neurons from apoptotic death. J Neurosci Res 49: 600–607. [DOI] [PubMed] [Google Scholar]
- Seoane J, Le HV, Shen L, Anderson SA, Massagué J (2004) Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 117: 211–223. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13: 1501–1512. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM (2004) Living with or without cyclins and cyclin-dependent kinases. Genes Dev 18: 2699–2711. [DOI] [PubMed] [Google Scholar]
- Sidman R, Miale I, and Feder N (1959) Cell proliferation and migration in the primitive ependymal zone: an autoradiographic study of histogenesis in the nervous system. Exp Neurol 1: 322–333. [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Miller MW (2004) Transforming growth factor β1 modulates cell migration in rat cortex: effects of ethanol. Cereb Cortex 14: 791–802. [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Miller MW (2005) Ethanol disrupts cell cycle regulation in the developing rat cortex: interaction with transforming growth factor β1. J Neurochem, in press. [DOI] [PubMed]
- Sometani A, Kataoka H, Nitta A, Fukumitsu H, Nomoto H, Furukawa S (2001) Transforming growth factor β1 enhances expression of brain-derived neurotrophic factor and its receptor, TrkB, in neurons cultured from rat cerebral cortex. J Neurosci Res 66: 369–376. [DOI] [PubMed] [Google Scholar]
- Stacey DW (2003) Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr Opin Cell Biol 15: 158–163. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness Jr VS (1995) The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J Neurosci 15: 6046–6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Bhide PG, Goto T, Miyama S, Caviness Jr VS (1999) Proliferative behavior of the murine cerebral wall in tissue culture: cell cycle kinetics and checkpoints. Exp Neurol 156: 407–417. [DOI] [PubMed] [Google Scholar]
- Tao W, Lai E (1992) Telencephalon-restricted expression of BF-1, a new member of the HNF-3/forkhead gene family, in the developing rat brain. Neuron 8: 957–966. [DOI] [PubMed] [Google Scholar]
- Tarui T, Takahashi T, Nowakowski RS, Hayes NL, Bhide PG, Caviness VS (2005) Overexpression of p27Kip1, probability of cell cycle exit, and laminar destination of neocortical neurons. Cereb Cortex 15: 1343–1355. [DOI] [PubMed] [Google Scholar]
- Theriault FM, Nuthall HN, Dong Z, Lo R, Barnabe-Heider F, Miller FD, Stifani S (2005) Role for Runx1 in the proliferation and neuronal differentiation of selected progenitor cells in the mammalian nervous system. J Neurosci 25: 2050–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian JQ, Quaroni A (1999) Involvement of p21WAF1/Cip1 and p27Kip1 in intestinal epithelial cell differentiation. Am J Physiol 276: C1245–C1258. [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Nilsen J, Rhee J, Zhou M, Doetschman T, Coffin JD, Wyland JJ, Hung YE (1999) Changes in cerebral cortical size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci 2: 246–253. [DOI] [PubMed] [Google Scholar]
- van Lookeren Campagne M, Gill R (1998) Tumor-suppressor p53 is expressed in proliferating and newly formed neurons of the embryonic and postnatal rat brain: comparison with expression of the cell cycle regulators p21Waf1/Cip1, p27Kip1, p57Kip2, p16Ink4a, cyclin G1, and the proto-oncogene Bax. J Comp Neurol 397: 181–198. [DOI] [PubMed] [Google Scholar]
- Wolfraim LA, Walz TM, James Z, Fernandez T, Letterio JJ (2004) p21Cip1 and p27Kip1 act in synergy to alter the sensitivity of naive T cells to TGFβ-mediated G1 arrest through modulation of IL-2 responsiveness. J Immunol 173: 3093–3102. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D (1993) p21 is a universal inhibitor of cyclin kinases. Nature 366: 701–704. [DOI] [PubMed] [Google Scholar]
- Yoo YD, Choi JY, Lee SJ, Kim JS, Min BR, Lee YI, Kang YK (1999) TGFβ-induced cell cycle arrest through the p21WAF1/CIP1-G1 cyclin/Cdks-p130 pathway in gastric-carcinoma cells. Int J Cancer 83: 512–517. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ahlemeyer B, Bauerbach E, Krieglstein J (2001) TGFβ1 inhibits caspase-3 activation and neuronal apoptosis in rat hippocampal cultures. Neurochem Int 38: 227–235. [DOI] [PubMed] [Google Scholar]