

Abstract
Reelin, a large protein that regulates neuronal migration during embryonic development, activates a conserved signaling pathway that requires its receptors, very low-density lipoprotein receptor and apolipoprotein E receptor 2, the cytoplasmic adaptor protein Disabled-1 (Dab1), and Src family kinases (SFK). Reelin also markedly enhances long-term potentiation in the adult hippocampus, suggesting that this developmental signaling pathway can physiologically modulate learning and behavior. Here, we show that Reelin can regulate NMDA-type glutamate receptor activity through a mechanism that requires SFKs and Dab1. Reelin mediates tyrosine phosphorylation of and potentiates calcium influx through NMDA receptors in primary wild-type cortical neurons but not in Dab1 knock-out neurons or in cells in which Reelin binding to its receptors is blocked by a receptor antagonist. Inhibition of SFK abolishes Reelin-induced and glutamate-dependent enhancement of calcium influx. We also show that Reelin-induced augmentation of Ca2+ entry through NMDA receptors increases phosphorylation and nuclear translocation of the transcription factor cAMP-response element binding protein. Thus, Reelin may physiologically modulate learning and memory by modulating NMDA receptor functions.
Keywords: neurotransmission, brain development, Disabled, Apoer2, Vldlr, LTP
Introduction
Reelin is a large secreted signaling protein with important roles in the developing nervous system as well as in the adult nervous system. During neuronal development, it is abundantly secreted by Cajal-Retzius cells at the surface of the neocortex (Alcantara et al., 1998; Drakew et al., 1998). Reelin binding to the very low-density lipoprotein (LDL) receptor (Vldlr) and to the apolipoprotein E receptor 2 (Apoer2) on migrating neurons induces tyrosine phosphorylation of Disabled-1 (Dab1), an adaptor protein that interacts with the cytoplasmic domains of both receptors (D'Arcangelo et al., 1999; Hiesberger et al., 1999). Activation of this Reelin signaling pathway is essential for the proper positioning of neurons and the lamination of the cortex and the cerebellum (D'Arcangelo et al., 1995; Howell et al., 1997; Trommsdorff et al., 1999).
Dab1 is a neuron-specific cytoplasmic protein that binds to the NPxY motif in the cytoplasmic tails of Vldlr and Apoer2 (Trommsdorff et al., 1999). Clustering of Vldlr and/or Apoer2 by Reelin binding results in tyrosine phosphorylation of Dab1 and activation of nonreceptor tyrosine kinases of the Src family (SFK) through a feedforward mechanism (Arnaud et al., 2003; Bock and Herz, 2003; Strasser et al., 2004).
We showed recently that Reelin can also modulate synaptic plasticity and learning in the adult mouse brain (Weeber et al., 2002). Reelin enhances long-term potentiation (LTP), and this function is abolished if either Vldlr or Apoer2 is absent. These findings raise the possibility of a functional cross-talk between the Reelin signaling complex and synaptic ion channels. In the CNS, synaptic strength and activity-dependent synaptic plasticity depend primarily on the function of NMDA receptors, which are mainly expressed on the postsynaptic side of excitatory synapses (Bliss and Collingridge, 1993). By controlling Ca2+ entry into the neurons, the NMDA receptor has been shown to regulate both synapse formation during development as well as synaptic plasticity in the adult brain (Ghosh and Greenberg, 1995). It is also required for the LTP in the CA1 region of the hippocampus (Malenka, 2003). NMDA receptor activity is partly regulated by phosphorylation and dephosphorylation on tyrosine residues in the cytoplasmic domains of the NMDA receptor NR2A or NR2B subunits by SFK (Yu et al., 1997), and phosphorylation increases ion conductance (Wang and Salter, 1994; Lau and Huganir, 1995; Kohr and Seeburg, 1996; Rostas et al., 1996).
Together, these findings raise the possibility of a role for Reelin in the regulation of the NMDA receptor and hence synaptic activity. In this study, we have quantitatively determined NMDA receptor-dependent and glutamate-stimulated Ca2+ flux in primary cortical neurons in response to Reelin. We find that Reelin is a potent enhancer of glutamate-stimulated Ca2+ influx through NMDA receptors. This Reelin-mediated modulation of NMDA receptor functions requires SFK and Dab1. These findings provide the first direct evidence for a physiological role of Reelin signaling in the regulation of NMDA receptor function and synaptic strength.
Materials and Methods
Preparation of primary neurons. All animal experiments were conducted according to the procedures approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center. Cortical neurons were prepared from embryonic day 18 (E18) embryos from timed pregnant Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) or E16 embryos from timed pregnant Dab1 heterozygous mice, as described previously (Bock and Herz, 2003). Neurons were plated on poly-d-lysine (Sigma, St. Louis, MO)-coated 12 mm round coverslips (Carolina Biological Supply Company, Burlington, NC) at a density of 5 × 104 per cm2. Neurons were cultured in Neurobasal medium (Invitrogen, Carlsbad, CA) supplemented with 2% B27 (Invitrogen), 1 mm glutamine, and penicillin–streptomycin at 37°C in a 5% CO2 environment.
Calcium imaging experiments. At 12–13 d in vitro (DIV), the cortical neurons were loaded with 5 μm fura-2 AM (Molecular Probes, Eugene, OR) for 30 min at 37°C in artificial CSF [ACSF; containing the following (in mm): 140 NaCl, 5 KCl, 2.5 CaCl2, 1.6 MgCl2, 10 HEPES, and 24 d-glucose, pH 7.3] supplemented with 1 μm tetrodotoxin (Sigma), 5 μm nimodipine (Sigma), and 40 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; Sigma). Calcium-free ACSF was prepared by omitting CaCl2 from ACSF and supplementing with 100 μm EGTA. Coverslips were mounted onto a recording/perfusion chamber (RC-26G; Warner Instruments, Hamden, CT), positioned on the movable stage of an Olympus (Melville, NY) IX-70 inverted microscope, and perfused with supplemented ACSF by gravity flow. Images at 340 and 380 nm excitation wavelengths were acquired every 5 s and shown as 340/380 image ratios as described previously (Tang et al., 2003). Neurons were stimulated with 40 μm glutamate (Sigma) after baseline measurements.
Data processing. Responses R(basal) and R(peak) indicate the fura-2 340/380 ratio values before and after glutamate application, respectively. R(peak) – R(basal) = ΔR indicates the change in the fura-2 340/380 ratios induced by glutamate in a certain cell. In the responding distribution chart, we defined the 0 value of ΔR as a response 0 <ΔR < 0.01, 0.01 as 0.01 <ΔR < 0.02, and so on. ΔR = 0.17 was defined as an upper cutoff value beyond which all cells were classified as ΔR > 0.17. The basal ratios R(basal) were determined as the average of the ratio values 35–15 s before glutamate application. The peak values were determined from maximal signals observed 25–60 s after glutamate application. For each coverslip, we calculated the mean value and SD of R(basal). Cells with R(basal) values more than or less than mean ± SD were excluded. Cells were thus categorized into 18 groups (from 0 to 0.17) by their ΔR values. The total number of cells in each group were plotted as histograms. To identify responding and nonresponding cell populations, we defined a threshold value in the valley between the peaks of the resulting bimodal distribution. This value was set to ΔR = 0.05, and cells with ΔR values ≥0.05 were defined as responders. A summary of the total data set is shown in Table 1.
Table 1.
Activation of the Reelin signaling pathway enhances the response of cortical neurons to glutamate
|
Treatment |
Percentage of responsive cells |
n (number of coverslips) |
|---|---|---|
| Group 1 | ||
| Control | 29.7 ± 1.6 | 5 |
| Reelin | 57.5 ± 4.4** | 5 |
| Reelin plus Ca2+-free ACSF | 2.50 ± 2.5 | 4 |
| Reelin plus d-APV | 5.50 ± 3.2 | 4 |
| Group 2 | ||
| Mock control | 10.4 ± 7.4 | 5 |
| GST plus Reelin | 44.6 ± 11.2* | 5 |
| GST—RAP plus Reelin | 15.3 ± 1.7 | 4 |
| Group 3 | ||
| Dab1 +/+, control | 24.6 ± 8.8 | 4 |
| Dab1 +/+, Reelin | 47.9 ± 6.6** | 7 |
| Dab1 -/-, control | 14.4 ± 4.5 | 10 |
| Dab1 -/-, Reelin | 11.7 ± 3.4 | 9 |
| Group 4 | ||
| PP2, control | 5.57 ± 3.8 | 7 |
| PP2, Reelin | 10.6 ± 3.3 | 5 |
| PP3, control | 27.4 ± 8.8 | 7 |
| PP3, Reelin |
48.1 ± 18.3* |
5 |
At 12-13 DIV, the cultured cortical neurons were subjected to different treatments and stimulated with glutamate in the presence of TTX, nimodipine, and CNQX. Percentage of responsive cells of each individual coverslip was calculated as described in Materials and Methods. Data are presented in the form of mean ± SE. *p < 0.05 and **p < 0.01 versus the first control in each group, one-way ANOVA test. The data in this table reflect the results of individual coverslips from all experiments examined over the course of this study to test for overall statistical significance. Figures 2, 3, 4, 5 reflect results from individual and internally controlled experimental sessions.
Reelin stimulation and inhibitors. Reelin- and mock-conditioned media were collected and concentrated as described previously (Beffert et al., 2002). Concentrated Reelin samples were purified by ion exchange on HiTrap Q HP (Amersham Biosciences, Piscataway, NJ) and then by gel filtration on Sephacryl S300 (Amersham Biosciences). Concentrated mock samples were subjected to the same purification procedure, and the according fractions were collected. Reelin (∼5nm) was applied to cortical neurons for 25 min before fluorescence measurements. Mock fractions were applied to neurons in exactly the same relative concentrations as Reelin samples. The glutathione S-transferase (GST) receptor-associated protein (RAP) fusion protein and GST protein were prepared as described previously (Herz et al., 1991). GST–RAP and GST were applied at 50 μg/ml for 50–60 min before and during Reelin stimulation. 2-Amino-5-phosphonovalerate (d-APV; Sigma) was applied at 10 μm to block NMDA receptors. 4-Amino-5-(4-chlorophenyl)-7-(t-butyl)-pyrazolo[3,4-d]pyrimidine (PP2) (Calbiochem, La Jolla, CA) and 4-amino-7-phenylpyrazol[3,4-d]pyrimidine (PP3) (Calbiochem) were applied at 10 μm for 60 min before and during Reelin stimulation or before acquiring images.
Transfections and immunoprecipitations. Human embryonic kidney 293 (HEK 293) cells were transfected by FuGene 6 (Roche Applied Science, Indianapolis, IN) with 1 μg of pCMV-myc-NR2B, 1 μg of pEF1α-mDab1, or 1 μg of pEF1α-m Dab1–5F and 0.5 μg of pCMV-cSrc. Dab1–5F bears the following mutations: Y185F, Y198F, Y200F, Y220F, and Y232F (Howell et al., 2000). After 48 h, cells were lysed as described previously (Bock and Herz, 2003). Cell lysates were analyzed by immunoblotting using a rabbit polyclonal phosphorylation-specific antibody against phosphorylated SFK (Y416; Cell Signaling Technology, Beverly, MA), a monoclonal Src antibody (GD11; Upstate Biotechnology, Lake Placid, NY), and a rabbit polyclonal murine Dab1 antibody (Trommsdorff et al., 1999). Myc-tagged NR2B was immunoprecipitated as described previously (Stolt et al., 2005) and analyzed by immunoblotting using a phosphotyrosine-specific antibody (4G10; Upstate Biotechnology) and a polyclonal goat NR2B antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
Immunoprecipitation of neuronal NR2B. Cortical neurons were exposed to mock-treated or Reelin-containing medium for the indicated times at 4°C and lysed as described previously (Bock and Herz, 2003). Lysates were precleared using goat nonimmune serum and incubated with 4 μg of goat anti-NR2B antibody at 4°C overnight. Immune complexes were precipitated using 40 μl of protein G-Sepharose slurry (Sigma). Precipitated proteins were analyzed by immunoblotting using a phosphotyrosine-specific antibody (4G10; Upstate Biotechnology) and a rabbit polyclonal antibody directed against NR2B (Upstate Biotechnology). Densitometric scans were quantitated using NIH ImageJ software.
Slice preparation. Transverse hippocampal brain slices (400 μm) were prepared from 12- to 21-d-old deeply anesthetized mice and incubated in oxygenated ACSF [containing the following (in mm): 124 NaCl, 5 KCl, 12 NaH2PO4, 26 NaHCO3, 10 d-glucose, 2 CaCl2, and 1 MgCl2] at 34°C for 30 min and thereafter at room temperature (24–27°C) for at least 45 min. After cutting between CA1 and CA3 regions, individual slices were transferred to the recording chamber mounted on a Nikon (Tokyo, Japan) E600FN microscope. Slices were submerged and perfused continuously with ACSF, which was continuously exchanged by through a gravity-driven superfusion system. Neurons were visualized using a water-immersion objective and Nomarski differential interference contrast optics. Images were detected with an infrared-sensitive video camera.
Whole-cell voltage-clamp recordings on CA1 pyramidal neurons. Patch electrodes (3–6 mΩ) were filled with the following (in mm): 110 K-gluconate, 20 KCl, 10 NaCl, 10 HEPES, 0.6 EGTA, 4 Mg-ATP, 0.3 GTP, and 10 N-ethyl bromide quaternary salt, pH 7.2. Series resistance (Rs) ranged from 5 to 15 MΩ. Experiments in which Rs varied ≥20% within a recording session were discarded. Recordings were obtained with an Axopatch-200B patch-clamp amplifier (Molecular Devices, Union City, CA) digitized at 10 kHz through Digidata 1322A (Molecular Devices). EPSCs were evoked by stimulation (duration, 200 μs; amplitude, 10–100 μA) of Schaeffer collateral commissural afferents using concentric bipolar electrodes through a stimulus isolation unit (A365; World Precision Instruments, Sarasota, FL) driven by a stimulator (Grass S88; Grass Instruments, Quincy, MA). I–V curves for NMDA currents were plotted in the presence of Reelin (estimated at ∼5 nm) or control media. To isolate NMDA currents, the AMPA receptor antagonist CNQX (10 μm; Sigma) and the GABA receptor antagonist picrotoxin (50 μm; Sigma) were added. The L-type Ca2+ channel antagonist nimodipine (5 μm; Sigma) was present where indicated. Statistical differences were established using the two-tailed Student's t test. All data are expressed as mean ± SEM.
Phospho-cAMP-response element binding protein immunoblotting. After treatment with concentrated mock-conditioned or Reelin-conditioned media for 20 min at 37°C as described (Bock et al., 2003), cortical neurons were lysed in SDS-PAGE sample buffer. Lysates were separated by SDS-PAGE. The gel was transferred to polyvinylidene difluoride membrane, and the membrane was immunoblotted with a phosphorylated cAMP-response element binding protein (CREB; Ser133) antibody (Cell Signaling Technology), a CREB antibody (Cell Signaling Technology), and a phosphotyrosine antibody (4G10; Upstate Biotechnology). Densitometric scans were quantitated using ImageJ software.
Immunofluorescent imaging. Cortical neurons were fixed using 4% paraformaldehyde. After rinsing in PBS, fixed cells were incubated in blocking buffer (PBS) containing 1% BSA and 5% normal goat serum for 1 h. Neurons were then incubated overnight with primary antibodies diluted in blocking solution at 4°C. Phosphorylated CREB (Ser133)-specific antibody (from rabbit; Cell Signaling Technology) and microtubule-associated protein 2 (MAP2) antibody (mouse monoclonal; Sigma) were used at final concentrations of 1:100 and 1:500, respectively. After washing with PBS containing 0.1% BSA, cells were incubated with the appropriate secondary antibodies (Molecular Probes) for 1 h at room temperature and mounted on microscope slides with 4′,6′-diamidino-2-phenylindole (DAPI)-containing mounting medium (Vectashield; Vector Laboratories, Burlingame, CA). Fluorescent images were acquired using a Zeiss (Oberkochen, Germany) Axioplan 2 imaging microscope and Aixo-Vision software. The experiment was repeated three times, and random fields were chosen and evaluated independently by two scorers blinded to the identity of the groups.
Results
Reelin enhances glutamate-stimulated Ca2+ influx in cortical neurons
We first investigated whether Reelin could modulate glutamate-stimulated Ca2+ influx in primary rat cortical neurons from E18 after 12 or 13 DIV. We incubated cortical neurons with 5 μm fura-2 AM, a calcium indicator dye. The ratio of fura-2 signals at 340 and 380 nm excitation wavelengths was used to quantitatively determine the concentration of intracellular Ca2+ ([Ca2+]i). [Ca2+]i increased dramatically after glutamate (40 μm) application, which shows that glutamate receptors were expressed and that cortical neurons responded normally under our culture conditions (data not shown). Glutamate evoked very little [Ca2+]i increase when neurons were perfused with 5 μm nimodipine (an inhibitor of L-type Ca2+ channels), 40 μm CNQX (an inhibitor of AMPA-type glutamate channels), and 1 μm tetrodotoxin, which blocks basal neuronal activity (Figs. 1, first row, 2A,B). The NMDA receptor cannot overcome the voltage-dependent Mg2+ block under these conditions. Cortical neurons were then preincubated for 25 min in the absence or presence of Reelin and then assessed for [Ca2+]i changes in response to glutamate stimulation under the same perfusion conditions described above. Reelin alone did not increase [Ca2+]i above baseline level, whereas glutamate stimulated a large increase in [Ca2+]i in Reelin-treated neurons (Figs. 1, second row, 2D,E).
Figure 1.

Representative images of Reelin-induced enhancement of Ca2+ influx in cortical neurons. Representative images of fura-2 AM 340/380 ratios of Ca2+ transients induced by 40 μm glutamate in cortical neurons are shown. A pseudocolored calibration scale for 340/380 ratios is shown on the right. All recordings were performed in ACSF supplemented with TTX, nimodipine, and CNQX. Wild-type neurons were recorded without (first row) and with (second row) Reelin treatment. Fura-2 AM 340/380 ratio images are shown 30 s before (first column) and 30 s after (second column), 1.5 min after (third column), and 3 min after (fourth column) application of 40 μm glutamate.
Figure 2.

Reelin enhances glutamate-stimulated Ca2+ influx through the NMDA receptor in wild-type cortical neurons. At 12–13 DIV, the cultured cortical neurons were stimulated with glutamate in the presence of TTX, nimodipine, and CNQX. A, Basal and peak 340/380 ratios are shown for individual cortical neurons. Basal ratios were determined as the average of the ratio values 35–15 s before glutamate application. Peak values were determined from maximal signals observed 25–60 s after glutamate application. Trace of 340/380 ratio for the representative cell is shown in B. Presence of glutamate in the buffer is indicated by the horizontal bar. Changes in 340/380 ratios at peak and basal were calculated for each cell as R(peak) – R(basal). Cells were categorized by their R(peak) – R(basal) values, and responding distribution of the cells was plotted in C (n = number of cells). Basal and peak 340/380 ratio, trace of representative 340/380 ratio, and distribution of Reelin responding cells are shown in D–F respectively. Basal and peak 340/380 ratios are shown for individual Reel in-treated cortical neurons perfused with Ca2+-free ACSF (G) and d-APV (J). Traces of 340/380 ratios for representative cells are shown in H and K. Presence of glutamate in the buffer is indicated by the horizontal bar. Responding distributions for each different treatment are shown in I and L (n = number of cells).
To better evaluate the effect of Reelin, we plotted the changes in [Ca2+]i in response to glutamate as a histogram depicting the distribution of the magnitude of the response of the analyzed cell population (Fig. 2). A cutoff value was defined as described in Materials and Methods and as indicated by the black vertical solid line in the histograms. By this definition, 18% of the neurons responded to glutamate in the absence of Reelin addition (Fig. 2C). In contrast, after Reelin application, 75% of the neurons responded to glutamate (Fig. 2F).
Reelin-induced enhancement of glutamate-stimulated Ca2+ influx requires NMDA receptors
To exclude the contribution of intracellular Ca2+ release to the observed effect of Reelin, we performed a control experiment in which we removed the Ca2+ ions from the perfusion buffer. Only 2% of the neurons treated with Reelin responded to glutamate in the absence of extracellular Ca2+ (Fig. 2G–I), indicating that Reelin stimulated Ca2+ influx from the extracellular space, as opposed to release from intracellular storage compartments. To further investigate whether this effect of Reelin on glutamate-stimulated Ca2+ influx requires NMDA receptors, we pretreated cortical neurons with the NMDA receptor antagonist d-APV before Reelin application. d-APV decreased the percentage of responding cells to 5.6% (Fig. 2 J–L), indicating that NMDA receptors are required for the observed Reelin-mediated increase in Ca2+ influx.
Reelin-mediated enhancement of glutamate-stimulated Ca2+ influx is receptor dependent
Reelin binding to the LDL receptor family members Apoer2 and Vldlr is required for transmission of the Reelin signal and the activation of kinases in the neuronal cytoplasm. To investigate whether Reelin-mediated modulation of NMDA receptor activity requires binding of this ligand to Apoer2 and Vldlr, we incubated cortical neurons with recombinant GST–RAP fusion protein before Reelin treatment. RAP is a universal competitor of ligand binding to LDL receptor family members and blocks Reelin binding to Apoer2 and Vldlr (Strickland et al., 1990; Herz et al., 1991). In the presence of GST–RAP, only 19% of Reelin-treated neurons responded to glutamate, whereas 64% of the neurons responded to Reelin and glutamate when GST control protein was used (Fig. 3D–I). Only 6% of mock-treated neurons responded to glutamate (Fig. 3A–C). These findings indicate that Reelin binding to LDL receptor family members, and thus activation of the signaling pathway, is required to enhance glutamate-stimulated Ca2+ currents.
Figure 3.

RAP, a ligand for LDL receptor family members, blocks Reelin-mediated enhancement of glutamate-stimulated Ca2+ influx. Basal and peak 340/380 ratios are shown for individual cortical neurons treated with mock media (A), GST and Reelin (D), and GST–RAP and Reelin (G). Traces of 340/380 ratios for representative cells are shown in B, E, and H. Presence of glutamate in the buffer is indicated by the horizontal bar. Responding distributions for each different treatment are shown in C, F, and I (n = number of cells).
Reelin-induced enhancement of glutamate-stimulated Ca2+ influx requires Dab1
Dab1 is a cytoplasmic adaptor protein that interacts with Apoer2 and Vldlr. Binding of Reelin to Apoer2 or Vldlr leads to phosphorylation of Dab1 by SFK, resulting in additional feedback of tyrosine kinase activation (Bock and Herz, 2003). To determine the role of Dab1 in the Reelin-mediated modulation of NMDA receptor activity, we compared the response of neurons from wild-type and Dab1-deficient mouse embryos to Reelin (Fig. 4). After glutamate application alone, the relative response of Dab1+/+ and Dab1–/– neurons was low and comparable (7.7 and 0%, respectively) (Fig. 4A–C, G–I). However, after Reelin treatment, 62.5% of the Dab1+/+ neurons (Fig. 4D–F) responded to glutamate, whereas only 4.8% of the Dab1–/– neurons (Fig. 4J–L) showed any increase in intracellular Ca2+ levels.
Figure 4.

Dab1 is required for Reelin-induced enhancement of Ca2+ influx. Basal and peak 340/380 ratios are shown for individual Dab1+/+ cortical neurons without treatment (A), Dab1+/+ neurons treated with Reelin (D), Dab1–/– neurons without treatment (G), and Dab1–/– neurons treated with Reelin (J). Traces of 340/380 ratio for representative cells are shown in B, E, H, and K. Presence of glutamate in the buffer is indicated by the horizontal bar. Responding distributions for each different treatment are shown in C, F, I, and L (n = number of cells).
Reelin-induced enhancement of glutamate-stimulated Ca2+ influx requires Src family tyrosine kinases
Reelin binding to Apoer2 and Vldlr activates SFK (Bock and Herz, 2003), and tyrosine phosphorylation is known to modulate NMDA receptor activity (Yu et al., 1997). To investigate the role of SFK in the Reelin-induced enhancement of glutamate-stimulated Ca2+ influx, we used a commonly used inhibitor of SFK, PP2, and its inactive structural analog, PP3, in our assay. Pretreatment of neurons with PP2 almost completely abolished the Reelin response, with only 10.8% of neurons responding (Fig. 5D–F), whereas 67.7% of the neurons responded to Reelin and glutamate in the presence of PP3 (Fig. 5J–L). PP2 or PP3 alone did not affect the basal response rate, with only 17.4% of the neurons treated with PP2 and 10.5% of the neurons treated with PP3 responding to glutamate.
Figure 5.

Src family tyrosine kinases are required for Reelin-induced enhancement of Ca2+ influx. Basal and peak 340/380 ratios are shown for individual cortical neurons treated with PP2 (A), PP2 and Reelin (D), PP3 (G), and PP3 and Reelin (J). Traces of 340/380 ratio for representative cells are shown in B, E, H, and K. Presence of glutamate in the buffer is indicated by the horizontal bar. Responding distributions for each different treatment are shown in C, F, I, and L (n = number of cells).
Reelin increases tyrosine phosphorylation of and ion flux through NMDA receptors
SFK can modulate NMDA receptor activity through tyrosine phosphorylation of NR2 subunits (Yu et al., 1997), and Reelin signaling activates SFK in a Dab1-dependent manner (Bock and Herz, 2003). To determine whether Reelin can induce tyrosine phosphorylation of NMDA receptors, we tested the effect of Reelin- and Dab1-dependent activation of SFK on tyrosine phosphorylation of NR2B, the predominant subunit in embryonic neurons. In a first experiment, we reconstituted the essential intracellular components of the Reelin signaling complex by transfection in a heterologous cell system. Myc-tagged NR2B and c-Src were transfected into HEK 293 cells in the absence or presence of wild-type Dab1 or Dab1–5F (Howell et al., 2000), a nonphosphorylatable mutant form that cannot activate SFK (Fig. 6A). In this cell-culture model system, overexpression of Src and Dab1 mimics the clustering of these two proteins that is physiologically induced by Reelin at the cell surface. Tyrosine phosphorylation of immunoprecipitated NR2B was detected by immunoblotting using 4G10 as well as an antibody that specifically detects SFK phosphorylated at the active site residue Y416 (Bock and Herz, 2003). Tyrosine phosphorylation of NR2B was increased slightly by c-Src alone (lane 4) but increased dramatically when wild-type Dab1 was also present (lane 5). The inactive Dab1–5F failed to stimulate Src-mediated phopshorylation of NR2B (lane 6). Thus, Dab1 can mediate tyrosine phosphorylation of NR2 subunits in an SFK-dependent manner.
Figure 6.

Stimulation of NR2B tyrosine phosphorylation. A, HEK 293 cells were transfected with the indicated expression vectors, and whole-cell lysates (WCL) were analyzed by immunoblotting for SFK tyrosine-phosphorylated at Y416, total cellular Src, and Dab1. Immunoprecipitated (IP) myc-tagged NR2B was probed for tyrosine phosphorylation [pY (4G10)]. Total amounts of immunoprecipitated NR2B are shown below (NR2B). B, Cortical neuronal cultures were treated in the absence (M) or presence (R) of Reelin at 4°C for the indicated times. NR2B was immunoprecipitated from the lysates and analyzed for tyrosine phosphorylation [pY (4G10)]. Total amounts of immunoprecipitated NR2B are shown below. Densitometric scanning of a total of 16 sample lanes from three independent experiments revealed a 20-fold average increase in detectable NR2B phosphotyrosine levels in response to Reelin (SEM ± 10; p < 0.05; Student's paired t test). Values were normalized to total NR2B levels.
In another experiment, we investigated whether Reelin can stimulate tyrosine phosphorylation of endogenous NR2B in neuronal cultures in vitro. Because of high background levels of Reelin-independent tyrosine phosphorylation of NMDA receptor subunits at 37°C, we performed this experiment at 4°C, where neuronal activity is suspended, cells are quiescent, and background tyrosine phosphorylation levels are very low. At this temperature, any activation-induced dynamic turnover through internalization, dephosphorylation, degradation, and recycling of the NMDA receptor is reduced or prevented. Neurons were treated in the absence or presence of Reelin at 4°C for 2–4 h. Figure 6B demonstrates that Reelin potently increased tyrosine phosphorylation of NR2B under these conditions.
To test whether Reelin could alter the electrophysiological properties of NMDA receptor conductance in neurons, we performed whole-cell patch-clamp experiments in hippocampal slices (Fig. 7). Synaptically evoked NMDA currents were recorded under holding potentials ranging from –70 to +70 mV in the absence or presence of nimodipine. Pretreatment of the slices with Reelin for 10 min significantly increased NMDA currents over the entire voltage range. As expected, nimodipine had no effect, indicating that Reelin does not affect L-type Ca2+ channels. These findings directly support our fura-2 data and show that Reelin functions by increasing the ion flux through NMDA receptors by mediating tyrosine phosphorylation of receptor subunits.
Figure 7.

Reelin potentiates synaptically evoked NMDA currents. A, Responses were recorded using whole-cell voltage clamp at potentials varying between –70 and +70 mV. I–V curves of residual NMDA currents recorded in the presence of GABAA and AMPA receptor antagonists, with (triangles) or without (circles) nimodipine, are shown in either the presence (open symbols) or absence (filled symbols) of Reelin. Values were obtained from seven (circles) and four (triangles) different CA1 pyramidal neurons, respectively. I–V curves were recorded 10 min after Reelin application. B, Representative responses recorded at holding potentials between –70 and +70 mV (steps of 10 mV) after application of either Reelin or control fractions in the absence and presence of nimodipine. Reelin treatment significantly increased the amplitudes of NMDA currents over the entire voltage range.
Reelin increases glutamate-induced CREB Ser133 phosphorylation
CREB is a transcription factor that is activated by increases in intracellular calcium concentration and that contributes to NMDA-receptor-dependent gene transcription and expression (Shaywitz and Greenberg, 1999). Phosphorylation of CREB at Ser133 is required for CREB-mediated gene transcription. To test whether Reelin-mediated modulation of the NMDA receptor can activate CREB, we determined the effect of Reelin on glutamate-stimulated CREB Ser133 phosphorylation in cortical neurons (Fig. 8A). Cellular depolarization with KCl was used as a positive control to elicit Reelin and NMDA-receptor-independent stimulation of CREB phosphorylation (lane 6). Activation of the Reelin signaling pathway under the indicated conditions was monitored by determining tyrosine phosphorylation of Dab1 (Fig. 8A, bottom). In the absence of Reelin and glutamate (lane 1), CREB phosphorylation was virtually undetectable. In the presence of Reelin alone, CREB phosphorylation increased slightly but noticeably (lane 2), likely through Ca2+-independent activation of other intracellular kinases by Reelin (Beffert et al., 2002; Arnaud et al., 2003; Bock et al., 2003). Addition of glutamate alone, without Reelin pretreatment, induced CREB phosphorylation, consistent with the small number of neurons that responded to glutamate alone in the fura-2 experiments. However, after pretreatment with Reelin, glutamate induced strong CREB Ser133 phosphorylation (lane 4), which was reduced to baseline levels (i.e., glutamate alone) by the NMDA receptor blocker d-APV (lane 5). Thus, all of the observed Reelin-mediated increase in glutamate-induced CREB Ser133 phosphorylation was mediated by the effect of Reelin on the NMDA receptor.
Figure 8.
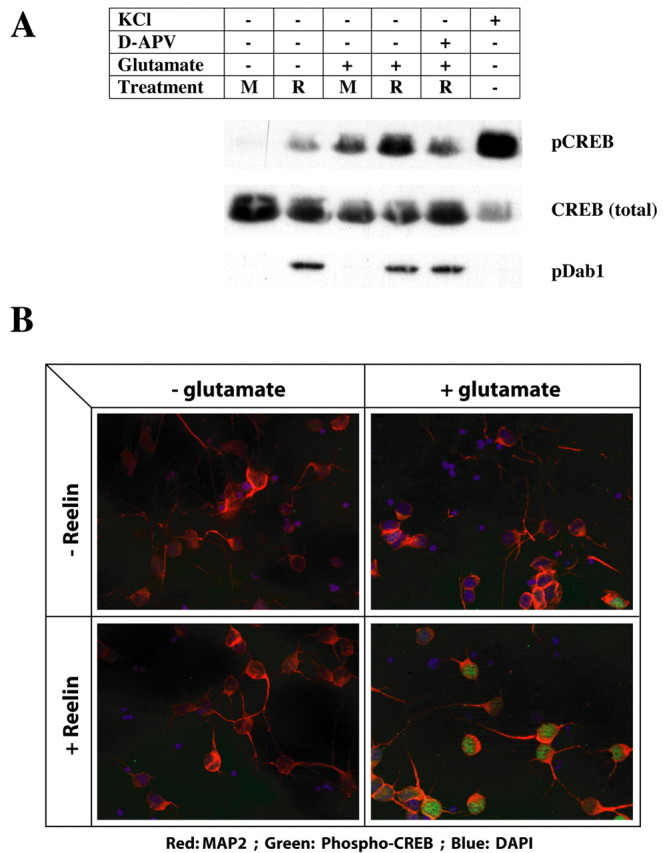
Reelin increases glutamate-induced CREB Ser133 phosphorylation. A, Primary cortical neurons at 7 DIV were treated in the absence (M) or presence (R) of Reelin in ACSF supplemented with TTX, nimodipine, and CNQX. d-APV (100 μm) was applied to block NMDA receptors. Cells were stimulated with 10 μm glutamate for 8 min, and lysates were analyzed by Western blotting for levels of CREB, phosphorylated at Ser133(pCREB; top), total CREB (middle), and tyrosine-phosphorylated Dab1 (pDab1; bottom). The latter serves as an indicator of activation of the Reelin signaling pathway. KCl (55 mm) was applied in the absence of inhibitors as a positive control to depolarize neurons and induce calcium entry through voltage-gated channels. Densitometric scanning of a total of 15 sample lanes from three independent experiments revealed a statistically significant (p < 10–7) average 2.3 ± 0.14-fold increase in phospho-CREB levels in response to Reelin and glutamate compared with glutamate alone. The average increase in Reelin treatment over mock treatment in the absence of glutamate was sevenfold (SEM ± 2; p < 0.01). The density of phospho-CREB signals was normalized to the corresponding total CREB signals from the same blot. Student's paired t test was used. B, Mixed neuronal cultures were treated as described in A and analyzed by immunocytochemistry for the presence of Ser133-phosphorylated CREB in the nucleus (green). The neuronal cell population was identified with a MAP2-specific antibody (red). Nuclei were visualized using DAPI (blue). In the absence of Reelin and glutamate, two of a total of 69 scored nuclei from MAP2-expressing neurons stained positive for phospho-CREB (2 of 69), in the presence of Reelin alone (7 of 67), in the presence of glutamate alone (9 of 64), and in the presence of Reelin and glutamate (59 of 78). The number of phospho-CREB-positive nuclei after glutamate exposure was thus significantly increased in the presence of Reelin (p < 0.000001).
To further confirm that Reelin can enhance glutamate-stimulated CREB Ser133 phosphorylation and nuclear localization, immunofluorescent staining was performed on cultured cortical neurons (Fig. 8B). MAP2 was used to identify the neuronal cell population. Consistent with the results shown in Figure 8A, Reelin pretreatment dramatically increased the amounts of phosphorylated CREB in the nuclei of neurons during glutamate stimulation, whereas glutamate or Reelin alone had little effect.
Discussion
The role of the Reelin signaling pathway in the regulation of neuronal migration during brain development has been studied extensively (Tissir and Goffinet, 2003). In a previous study, we showed that this pathway is also involved in the modulation of synaptic activity (i.e., LTP, memory, and learning) (Weeber et al., 2002). In the current study, we have now investigated the effect of Reelin signaling on the function of NMDA receptors by measuring NMDA receptor-dependent calcium influx in primary cortical neurons. We show that Reelin can potently enhance NMDA receptor activity during glutamate stimulation in wild-type cortical neurons (Fig. 2) but not in Dab1-deficient neurons (Fig. 4), which are unable to respond to Reelin. Inhibition of either activity of SFK (Fig. 5) or of Reelin binding to its receptors Apoer2 and Vldlr by RAP (Fig. 3) abolished this modulatory effect of Reelin. Tyrosine phosphorylation of NMDA receptor subunits was increased by Reelin (Fig. 6), resulting in a marked potentiation of NMDA receptor-mediated ion currents (Fig. 7). Reelin signaling also stimulated phosphorylation of the transcription factor CREB through Ca2+-dependent and -independent mechanisms (Fig. 8). Together, these data implicate the Reelin signaling complex, consisting of the lipoprotein receptors Apoer2 and Vldlr, their ligand Reelin, the adaptor protein Dab1, and SFK in the physiological regulation of neuronal NMDA receptor activity, memory, and learning.
We showed recently a role for Reelin in synaptic plasticity. Specifically, we showed that Reelin and its receptors, Apoer2 and Vldlr, can positively regulate LTP in mouse hippocampal slices (Weeber et al., 2002). SFK, which are activated by Reelin, are also essential for LTP induction (Bock and Herz, 2003) as well as for NMDA receptor regulation (Wang and Salter, 1994; Kohr and Seeburg, 1996). Blockade of Reelin signaling on any level, either by disrupting binding of Reelin to its receptors (Fig. 3), by genetic inactivation of Dab1 (Fig. 4), or by pharmacological inhibition of SFK (Fig. 5), prevented the Reelin-enhanced NMDA-receptor-mediated Ca2+ entry in response to glutamate. Activation of the Reelin pathway resulted in tyrosine phosphorylation of NMDA receptor subunits (Fig. 6) as well as increased NMDA-receptor-mediated currents in hippocampal neurons (Fig. 7). Together, these findings suggest that Reelin can physiologically modulate LTP through regulation of NMDA receptor activity.
Increases in intracellular calcium concentrations in the postsynaptic neuron trigger a series of downstream signals that can induce long-term changes in synaptic strength. The transcription factor CREB, which is activated by phosphorylation by calmodulin-dependent kinases in response to an NMDA receptor-mediated rise in cytoplasmic Ca2+ levels, is part of this cascade. Reelin was able to potently stimulate CREB phosphorylation, and thus activation and nuclear translocation, through an NMDA-receptor-dependent mechanism (Fig. 8). This raises the possibility that Reelin may physiologically modulate activity-dependent gene transcription in vivo.
NMDA-receptor-mediated calcium influx can also trigger signaling pathways and the expression of genes that participate in neurite outgrowth and dendrite development. For example, Reelin has been shown to promote the branching of entorhinocortical–hippocampal projections (Del Rio et al., 1997), and Niu et al. (2004) showed that Reelin can promote dendrite development in the hippocampus through an Apoer2/Vldlr-Dab1-dependent pathway. Reelin-mediated sensitization of NMDA receptors to the stimulation of glutamate could therefore also contribute to the activity-dependent regulation of neurite outgrowth, dendritogenesis, and synapse formation.
We propose a model in which Reelin binding to its receptors results in the activation of SFK. These kinases in turn can enhance NMDA receptor activity through phosphorylation of tyrosine residues in the cytoplasmic domains of NR2 subunits or possibly by modifying other components of the NMDA receptor complex that alter the gating of the channel. In addition, activity-mediated regulation of NMDA receptor trafficking (Chung et al., 2004) might also be modulated by SFK-dependent mechanisms. The NMDA receptor is crucial for regulating the formation and strength of synapses, and thus for learning and memory, by controlling the flow of calcium ions during synaptic signal transmission. The present findings raise the possibility that Reelin might serve a function as a novel physiological signaling molecule that can selectively modulate synaptic plasticity and activity-dependent gene transcription by gating NMDA receptor-mediated calcium currents in mature synapses. Thus, the same fundamental signaling pathway that is required for the regulation of neuronal migration and positioning during brain development can also modulate synaptic plasticity in the mature brain.
Footnotes
This work was supported by grants from the National Institutes of Health (HL20948, HL63762, NS39552, and NS43408), the Perot Family Foundation, and the Humboldt Foundation.
Correspondence should be addressed to Joachim Herz, Department of Molecular Genetics, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX 75390-9046. E-mail: joachim.herz@utsouthwestern.edu.
DOI:10.1523/JNEUROSCI.1951-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258209-08$15.00/0
References
- Alcantara S, Ruiz M, D'Arcangelo G, Ezan F, de Lecea L, Curran T, Sotelo C, Soriano E (1998) Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J Neurosci 18: 7779–7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud L, Ballif BA, Forster E, Cooper JA (2003) Fyn tyrosine kinase is a critical regulator of disabled-1 during brain development. Curr Biol 13: 9–17. [DOI] [PubMed] [Google Scholar]
- Beffert U, Morfini G, Bock HH, Reyna H, Brady ST, Herz J (2002) Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3beta. J Biol Chem 277: 49958–49964. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31–39. [DOI] [PubMed] [Google Scholar]
- Bock HH, Herz J (2003) Reelin activates SRC family tyrosine kinases in neurons. Curr Biol 13: 18–26. [DOI] [PubMed] [Google Scholar]
- Bock HH, Jossin Y, Liu P, Forster E, May P, Goffinet AM, Herz J (2003) Phosphatidylinositol 3-kinase interacts with the adaptor protein Dab1 in response to Reelin signaling and is required for normal cortical lamination. J Biol Chem 278: 38772–38779. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Huang YH, Lau LF, Huganir RL (2004) Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J Neurosci 24: 10248–10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T (1995) A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374: 719–723. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T (1999) Reelin is a ligand for lipoprotein receptors. Neuron 24: 471–479. [DOI] [PubMed] [Google Scholar]
- Del Rio JA, Heimrich B, Borrell V, Forster E, Drakew A, Alcantara S, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Derer P, Frotscher M, Soriano E (1997) A role for Cajal-Retzius cells and reelin in the development of hippocampal connections. Nature 385: 70–74. [DOI] [PubMed] [Google Scholar]
- Drakew A, Frotscher M, Deller T, Ogawa M, Heimrich B (1998) Developmental distribution of a reeler gene-related antigen in the rat hippocampal formation visualized by CR-50 immunocytochemistry. Neuroscience 82: 1079–1086. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME (1995) Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 268: 239–247. [DOI] [PubMed] [Google Scholar]
- Herz J, Goldstein JL, Strickland DK, Ho YK, Brown MS (1991) 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor. J Biol Chem 266: 21232–21238. [PubMed] [Google Scholar]
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J (1999) Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24: 481–489. [DOI] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P, Cooper JA (1997) Neuronal position in the developing brain is regulated by mouse disabled-1. Nature 389: 733–737. [DOI] [PubMed] [Google Scholar]
- Howell BW, Herrick TM, Hildebrand JD, Zhang Y, Cooper JA (2000) Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr Biol 10: 877–885. [DOI] [PubMed] [Google Scholar]
- Kohr G, Seeburg PH (1996) Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J Physiol (Lond) 492: 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau LF, Huganir RL (1995) Differential tyrosine phosphorylation of N-methyl-d-aspartate receptor subunits. J Biol Chem 270: 20036–20041. [DOI] [PubMed] [Google Scholar]
- Malenka RC (2003) The long-term potential of LTP. Nat Rev Neurosci 4: 923–926. [DOI] [PubMed] [Google Scholar]
- Niu S, Renfro A, Quattrocchi CC, Sheldon M, D'Arcangelo G (2004) Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron 41: 71–84. [DOI] [PubMed] [Google Scholar]
- Rostas JA, Brent VA, Voss K, Errington ML, Bliss TV, Gurd JW (1996) Enhanced tyrosine phosphorylation of the 2B subunit of the N-methyl-d-aspartate receptor in long-term potentiation. Proc Natl Acad Sci USA 93: 10452–10456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68: 821–861. [DOI] [PubMed] [Google Scholar]
- Stolt PC, Chen Y, Liu P, Bock HH, Blacklow SC, Herz J (2005) Phosphoinositide binding by the disabled-1 PTB domain is necessary for membrane localization and Reelin signal transduction. J Biol Chem 280: 9671–9677. [DOI] [PubMed] [Google Scholar]
- Strasser V, Fasching D, Hauser C, Mayer H, Bock HH, Hiesberger T, Herz J, Weeber EJ, Sweatt JD, Pramatarova A, Howell B, Schneider WJ, Nimpf J (2004) Receptor clustering is involved in Reelin signaling. Mol Cell Biol 24: 1378–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland DK, Ashcom JD, Williams S, Burgess WH, Migliorini M, Argraves WS (1990) Sequence identity between the alpha 2-macroglobulin receptor and low density lipoprotein receptor-related protein suggests that this molecule is a multifunctional receptor. J Biol Chem 265: 17401–17404. [PubMed] [Google Scholar]
- Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I (2003) Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39: 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM (2003) Reelin and brain development. Nat Rev Neurosci 4: 496–505. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J (1999) Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97: 689–701. [DOI] [PubMed] [Google Scholar]
- Wang YT, Salter MW (1994) Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature 369: 233–235. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Beffert U, Jones C, Christian JM, Forster E, Sweatt JD, Herz J (2002) Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem 277: 39944–39952. [DOI] [PubMed] [Google Scholar]
- Yu XM, Askalan R, Keil II GJ, Salter MW (1997) NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science 275: 674–678. [DOI] [PubMed] [Google Scholar]