

Abstract
Microglia undergo a phenotypic activation in response to fibrillar β-amyloid (fAβ) deposition in the brains of Alzheimer's disease (AD) patients, resulting in their elaboration of inflammatory molecules. Despite the presence of abundant plaque-associated microglia in the brains of AD patients and in animal models of the disease, microglia fail to efficiently clear fAβ deposits. However, they can be induced to do so during Aβ vaccination therapy attributable to anti-Aβ antibody stimulation of IgG receptor (FcR)-mediated phagocytic clearance of Aβ plaques.
We report that proinflammatory cytokines attenuate microglial phagocytosis stimulated by fAβ or complement receptor 3 and argue that this may, in part, underlie the accumulation of fAβ-containing plaques within the AD brain. The proinflammatory suppression of fAβ-elicited phagocytosis is dependent on nuclear factor κB activation. Significantly, the proinflammatory cytokines do not inhibit phagocytosis elicited by antibody-mediated activation of FcR, which may contribute to the efficiency of Aβ vaccination-based therapy. Importantly, the proinflammatory suppression of fAβ phagocytosis can be relieved by the coincubation with anti-inflammatory cytokines, cyclooxygenase inhibitors, ibuprofen, or an E prostanoid receptor antagonist, suggesting that proinflammatory cytokines induce the production of prostaglandins, leading to an E prostanoid receptor-dependent inhibition of phagocytosis. These findings support anti-inflammatory therapies for the treatment of AD.
Keywords: phagocytosis, β-amyloid, Alzheimer's disease, NSAIDs, inflammation, cytokines
Introduction
The Alzheimer's disease (AD) brain is characterized by the presence of senile plaques, invariantly surrounded by abundant activated microglia. One of the central enigmas in AD is why microglia, despite their “activated” state, fail to effectively mount a phagocytic response to ingest and degrade the insoluble fibrillar β-amyloid (fAβ) deposits. Vaccination studies have demonstrated that phagocytic removal of amyloid plaques can result from the addition of antibodies to Aβ directly to the brain (Bacskai et al., 2002) or after immunization in animal models of AD (Schenk et al., 1999; Bard et al., 2000; Wilcock et al., 2003). Similarly, it has been argued that this mechanism may be responsible for reduction of plaque burden in humans immunized with fAβ. These studies argue that plaque removal results from stimulation of microglial phagocytosis during engagement of IgG (FcR) receptors by anti-Aβ antibodies bound to the plaques (Schenk et al., 1999; Bard et al., 2000; Wilcock et al., 2003, 2004), although other mechanisms may act in parallel (Wilcock et al., 2003, 2004; Wyss-Coray et al., 2003; Koistinaho et al., 2004).
Microglia interact with fAβ through an ensemble of cell surface receptors (Bamberger et al., 2003), leading to the activation of signal transduction pathways that result in phenotypic activation of these cells (Combs et al., 1999, 2001). The secretion of cytokines, chemokines, and a diverse array of other proinflammatory molecules, including prostaglandins (PGs), accompany microglial activation. Indeed, the AD brain and animal models of the disease exhibit elevated levels of these molecules (Akiyama et al., 2000). This chronic proinflammatory environment is postulated to contribute to the cognitive deficits and neuronal loss that characterize the disease (Akiyama et al., 2000).
Microglia are typically characterized as “quiescent” or “activated,” and it has only recently been appreciated that these cells exhibit an array of phenotypes, governed by their local environment (Goerdt and Orfanos, 1999; Gordon, 2003). Cells exhibiting these nonclassical phenotypes have recently been termed “alternatively activated” (Goerdt and Orfanos, 1999; Mantovani et al., 2002; Duffield, 2003; Gordon, 2003). Microglia may display an activated proinflammatory phenotype yet be unable to initiate a phagocytic response. We have shown recently that microglia respond to fAβ by stimulation of a robust β1-integrin-dependent phagocytic process (Koenigsknecht and Landreth, 2004). We now question why this response is not evident in the AD brain.
We report that proinflammatory cytokines inhibit phagocytosis elicited by engagement of the Aβ receptor complex and complement receptor 3 (CR3). This effect may cause the accumulation of amyloid plaques within the AD brain by suppression of endogenous fAβ peptide clearance mechanisms. Importantly, antibody-stimulated phagocytosis mediated through FcR was unaffected by proinflammatory cytokines and argue that this effect may underlie the efficacy of vaccination-based therapies in reducing Aβ plaque burden. We also report that incubation of cells with anti-inflammatory cytokines or cyclooxygenase (COX) inhibitors, including ibuprofen, blocks the ability of proinflammatory cytokines to suppress fAβ-elicited phagocytosis through the inhibition of prostaglandin E2 (PGE2) production or its signaling pathways. These findings support the use of anti-inflammatory therapies in the treatment of AD.
Materials and Methods
Materials. Aβ25–35 and Aβ1–42 were purchased from American Peptide Company (Sunnyvale, CA). Aβ peptides corresponding to amino acids 25–35 and 1–42 were dissolved in sterile water and incubated for 5 d at 37°C to allow for fibrilization (Burdick et al., 1992; Lorenzo and Yankner, 1994; Terzi et al., 1994). We cannot rule out that the Aβ1–42 preparation may contain some oligomers. Zymosan was purchased from Sigma (St. Louis, MO) and was opsonized with complement by incubating with mouse serum for 30 min at 37°C, followed by 10 washes with 0.85% NaCl before resuspension in PBS. Immune IgG was prepared from goat anti-rabbit IgG from Sigma and rabbit anti-mouse IgG from Cappel (Aurora, OH) in a 5:1 ratio. Parthenolide, butaprost, and AH6809 were also purchased from Sigma. Interleukin-4 (IL-4) and IL-10 were purchased from Biosource (Camarillo, CA). IL-13, interferon γ (IFNγ), IL-1β, tumor necrosis factor α (TNFα), and monocyte chemoattractant protein (MCP-1) were from R & D Systems (Minneapolis, MN). CD40L was purchased from Alexis (San Diego, CA). Nile red fluorospheres (1 μm microspheres) and AlexaFluor 488-phalloidin were purchased from Molecular Probes (Eugene, OR). TGFβ was purchased from Promega (Madison, WI), lipopolysaccharide (LPS) was from List Biologic (Campbell, CA), SN50 and SN50M were from Biomol (Plymouth Meeting, PA), and CAY10404 and SC-560 were from Cayman Chemicals (Ann Arbor, MI).
Tissue culture. The immortalized murine microglia cell line BV-2 (Blasi et al., 1990) was grown and maintained in DMEM containing 2% fetal bovine serum and gentamycin in 5% CO2. BV-2 cells used in these experiments were between passage numbers 20 and 40. No differences in results were observed between different passage numbers. Primary microglia were derived from postnatal 1–2 d mouse brains (C57BL/6J) as described previously (McDonald et al., 1997).
Phagocytosis assay. Phagocytosis was assayed as described previously (Koenigsknecht and Landreth, 2004). Briefly, BV-2 cells were collected, and 1 × 106 cells were plated in serum-free medium in 35 mm plates and then incubated in the presence or absence of proinflammatory or anti-inflammatory cytokines overnight. The cells were then treated for 30 min in the absence or presence of 60 μm (63.6 μg/ml) fAβ25–35 peptide, 5 μm (22.6 μg/ml) fAβ1–42 peptide, 1 mg/ml complement-opsonized zymosan, or 1 mg/ml immune IgG. Fluorescent microspheres were washed in PBS containing 1 mg/ml BSA and then added to the cells for 30 min. The fluorescent microspheres were used as a marker of fluid phase phagocytosis. Cells were then fixed with 2% paraformaldehyde, and three random fields of cells (>100 cells) were counted on an inverted fluorescent microscope.
Phalloidin staining. BV-2 cells were collected and then plated on coverslips (150,000 BV-2 cells per coverslip) resting in a 24-well plate overnight in serum-free medium in the presence or absence of cytokines. The cells were then incubated in the absence or presence of 60 μm (63.6 μg/ml) fAβ25–35 peptide for 30 min. The cells were rinsed with PBS before being fixed in 3.6% paraformaldehyde and washed again in PBS. The cells were then incubated at room temperature with 0.1% Triton X-100 buffer for 5 min and washed again in PBS. Finally, AlexaFluor 488-phalloidin (1.6 U diluted in PBS) was added to the coverslips and incubated at room temperature protected from light for 25 min. The coverslips were mounted on glass slides.
Results
Proinflammatory cytokines inhibit fAβ and opsonized zymosan-stimulated phagocytosis but do not inhibit immune IgG-mediated phagocytosis
We established a phagocytosis assay that measures ligand-stimulated phagocytosis through the bulk phase uptake of fluorescent microspheres to examine the phagocytic response in microglia (Koenigsknecht and Landreth, 2004). BV-2 microglial cells were used because they have been proven to faithfully mimic behaviors of primary microglial cells (Bocchini et al., 1992) as well as exhibit a robust phagocytic response (Kopec and Carroll, 1998; Koenigsknecht and Landreth, 2004). We wanted to test the hypothesis that a proinflammatory environment may act to suppress the capacity of microglia to mount a phagocytic response. We have shown previously that microglia exhibit both classical (types I and II) phagocytic responses to immune IgG stimulation of the FcR and complement-opsonized zymosan stimulation of CR3, respectively. However, microglia respond to fAβ through the fAβ receptor complex via a novel and mechanistically distinct β1-integrin-dependent phagocytic response (Koenigsknecht and Landreth, 2004). To test the effect of a proinflammatory environment on the various types of phagocytosis, BV-2 cells were treated with LPS for 6 h. LPS elicits the production of proinflammatory cytokines from microglia and macrophages (Zielasek and Hartung, 1996). Moreover, LPS treatment has been demonstrated to increase neuroinflammation and accelerate fAβ deposition in animal models of AD attributable to microglial cytokine production (Qiao et al., 2001). After LPS treatment, the cells were incubated with phagocytic-stimulating ligands, and phagocytosis was quantified. LPS incubation before stimulation of the cells with fAβ1–42, fAβ25–35, or mouse complement-opsonized zymosan suppressed phagocytosis, indicating that proinflammatory stimulation of microglia abrogates the capacity of the cells to mount a phagocytic response during stimulation of the fAβ receptor complex or CR3 (Fig. 1A). Remarkably, immune IgG-elicited phagocytosis was unaffected, demonstrating that the FcR still effectively functions to stimulate phagocytosis in the presence of inflammatory stimuli.
Figure 1.

Proinflammatory cytokines inhibit fAβ and opsonized zymosan-stimulated phagocytosis but do not affect immune IgG phagocytosis. A, BV-2 cells were exposed to LPS (1μg/ml) for 6 h to produce a proinflammatory environment. The cells were then stimulated with fAβ (60 μm fAβ25–35 and 5 μm fAβ1–42), mouse complement-opsonized zymosan (mOZ; 1 mg/ml), and immune IgG (1 mg/ml) for 30 min, followed by 30 min of incubation with fluorescent microspheres. The fraction of phagocytically active cells was then determined. B, BV-2 cells were incubated with proinflammatory cytokines (15 ng/ml IL-1β, 10 ng/ml TNFα, and 100 ng/ml IFNγ), CD40L (1 μg/ml), or MCP-1 (20 ng/ml) overnight before stimulation with phagocytic-stimulating ligands for 30 min. Fluorescent microspheres were then added to the cells for 30 min. The fraction of phagocytically active cells was then determined. *p < 0.01, **p < 0.001, and #p < 0.05 compared with control. This experiment is representative of three independent experiments.
The observation that a difference in modulation of phagocytosis was shown among the three types of phagocytosis by LPS treatment led us to evaluate the effects of specific proinflammatory cytokines and chemokines. BV-2 cells were treated overnight with IL-1β, TNFα, IFNγ, MCP-1, and CD40L before stimulation with phagocytic ligands. MCP-1 is a microglial-derived chemokine found in the AD brain associated with senile plaques (Ishizuka et al., 1997). CD40 is a transmembrane receptor that binds CD40L (CD154), stimulating the production of proinflammatory cytokines, and has been postulated to participate in disease pathophysiology (Tan et al., 1999, 2002; Wesemann et al., 2002). Microglia express low amounts of CD40, but CD40 expression is upregulated after stimulation with proinflammatory stimuli. Microglia associated with senile plaques express increased levels of the proinflammatory cytokines, IL-1β, and TNFα (Hoozemans et al., 2001). Every proinflammatory agent tested in the phagocytosis assay demonstrated the same inhibition of fAβ and opsonized zymosan-stimulated phagocytosis but had no affect on immune IgG-mediated phagocytosis, identical to the effect observed in LPS-treated cells (Fig. 1B). These proinflammatory agents, acting alone, did not affect phagocytosis.
We next examined in detail the ability of IL-1β to suppress fAβ-elicited phagocytosis in BV-2 cells. A time course demonstrated that the cells required an exposure to IL-1β for 3 h or more before fAβ stimulation to attenuate fAβ-stimulated phagocytosis (Fig. 2A). Phagocytosis stimulated by fAβ was not affected by the presence of IL-1β for 1 h or less, suggesting that proinflammatory inhibition of fAβ-stimulated phagocytosis might be transcriptionally regulated.
Figure 2.
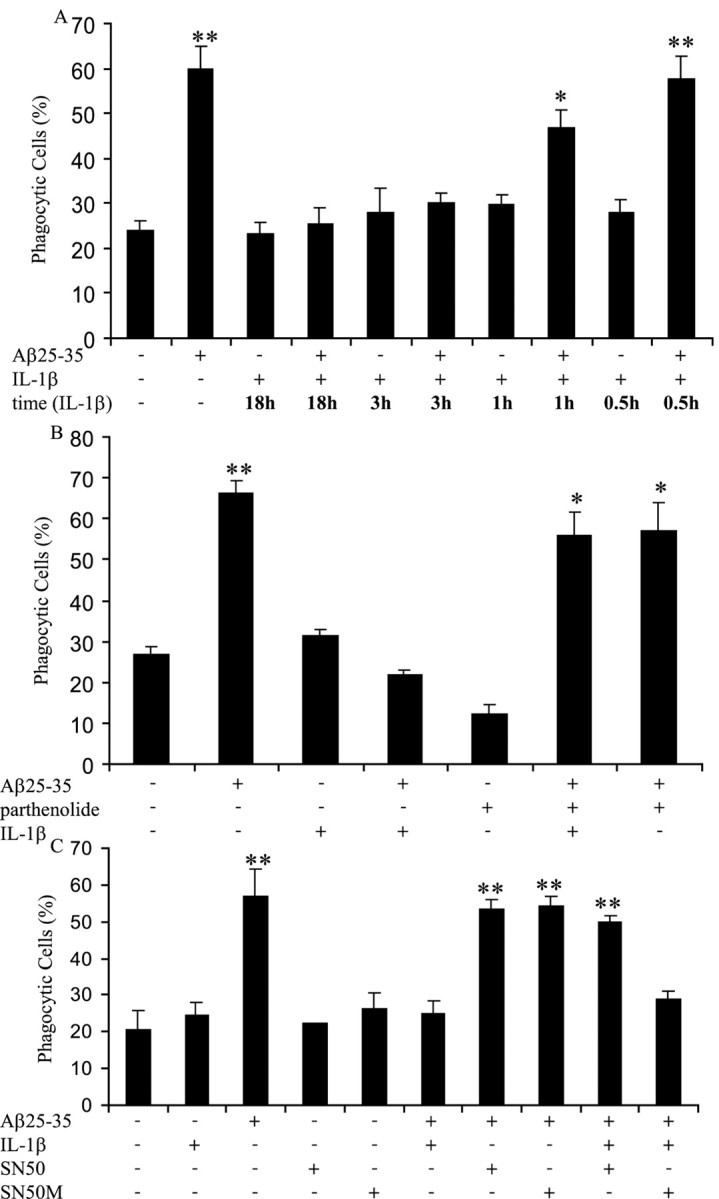
Proinflammatory cytokines inhibit fAβ-elicited phagocytosis by a transcriptionally dependent mechanism. A, BV-2 cells were incubated with IL-1β (15 ng/ml) for indicated time periods before fAβ25–35 (60 μm) stimulation for 30 min and the addition of fluorescent microspheres for 30 min. BV-2 cells were incubated with parthenolide (15μm)(B), SN50 (100μg/ml) (C), or SN50M (100 μg/ml) (C) for 3 h before stimulation with IL-1β (15 ng/ml) for 3 h. The cells were then incubated with fAβ25–35 (60μm) for 30 min, followed by the addition of fluorescent microspheres for 30 min. The fraction of phagocytically active cells was then determined. *p < 0.01 and **p < 0.001 compared with control. The data are representative of three independent experiments.
We further evaluated a role for transcriptional regulation in IL-1β-inhibited fAβ-stimulated phagocytosis. Inhibitors of nuclear factor κB (NFκB) activation were used. NFκB has been proven to play an essential role in the regulation of inflammation (Baldwin, 1996; Combs et al., 2001; Tak and Firestein, 2001). Parthenolide specifically targets the inhibitor of κB kinase and therefore inhibits NFκB activation (Hehner et al., 1999). Treatment of BV-2 cells with parthenolide for 3 h, followed by a 3 h incubation with IL-1β, restored phagocytosis in fAβ-treated cells (Fig. 2B). To verify the involvement of NFκB, we also used SN50 to inhibit NFκB. SN50 has been shown to inhibit the translocation of NFκB from the cytoplasm to the nucleus (Lin et al., 1995). SN50 also rescued phagocytosis elicited by fAβ in the presence of IL-1β (Fig. 2C). Furthermore, SN50M, an inactive form of SN50, did not restore phagocytosis of fAβ in a proinflammatory environment (Fig. 2C). From these data, we concluded that NFκB-dependent transcription of proinflammatory genes was essential for the suppression of fAβ-elicited phagocytosis in a proinflammatory environment.
Anti-inflammatory cytokines do not directly stimulate or inhibit phagocytosis
We next examined the effect of anti-inflammatory cytokines on phagocytosis by overnight preincubation of BV-2 cells with IL-4, IL-10, IL-13, or TGFβ before stimulation of the cells with fAβ, opsonized zymosan, or immune IgG. These cytokines were tested for their effects on microglial phagocytosis because they have been shown to promote alternative activation of microglia (Goerdt and Orfanos, 1999; Mantovani et al., 2002; Duffield, 2003). The anti-inflammatory cytokines neither stimulated nor inhibited phagocytosis elicited by any of the phagocytic ligands (Fig. 3).
Figure 3.

Anti-inflammatory cytokines do not stimulate or inhibit phagocytosis. BV-2 cells were exposed to anti-inflammatory cytokines IL-4, IL-10, IL-13 (each at 10 ng/ml), or TGFβ (5 ng/ml) overnight before stimulation with fAβ (60 μm fAβ25–35 and 5 μm fAβ1–42), opsonized zymosan (mOZ; 1 mg/ml), or immune IgG (1 mg/ml) for 30 min. Fluorescent microspheres were then added to the cells for 30 min. The fraction of phagocytically active cells was then determined. **p < 0.001 compared with control. The experiment is representative of three independent experiments.
fAβ1–42 elicits phagocytosis in primary microglia in an anti-inflammatory, but not a proinflammatory, milieu
Primary murine microglia were evaluated to determine whether proinflammatory environments could also regulate their capacity to stimulate phagocytosis. Microglia were treated with LPS for 6 h to induce a proinflammatory environment as was demonstrated previously with BV-2 cells. LPS treatment before stimulation with fAβ1–42 suppressed phagocytosis in primary microglial cells (Fig. 4A). Specific proinflammatory and anti-inflammatory cytokines were also evaluated for their ability to regulate microglial phagocytosis. The proinflammatory cytokine IL-1β suppressed fAβ-mediated phagocytosis (Fig. 4B). However, the fAβ-stimulated phagocytic mechanism was unaffected by the presence of IL-4, an anti-inflammatory cytokine (Fig. 4B), and IL-4 had no affect when added alone. We concluded that proinflammatory cytokines regulated microglial phagocytosis in primary microglial cells in a similar manner as in BV-2 cells.
Figure 4.

fAβ1–42 elicits phagocytosis in primary microglia in an anti-inflammatory, not a proinflammatory, milieu. A, Primary microglial cells were treated with LPS (1 μg/ml) for 6 h before stimulation with fAβ1–42 (5μm) for 30 min. Fluorescent microspheres were then added to the cells, and the fraction of phagocytically active cells was determined. B, Primary microglial cells were incubated overnight in the presence or absence of IL-1β (15ng/ml) or IL-4 (10ng/ml) before a 30 min treatment with fAβ1–42 (5μm). Fluorescent microspheres were then added to the cells. The fraction of phagocytically active cells was then determined. **p < 0.001 compared with control. The data are representative of three independent experiments.
Proinflammatory and anti-inflammatory treated cells exhibit morphological differences
Treatment of microglia with fAβ elicits a β1-integrin dependent change in cellular morphology similar to that observed in a type I phagocytic response (Koenigsknecht and Landreth, 2004). We next asked whether the morphology of the phagocytically active cells was different between the anti-inflammatory and the proinflammatory cytokine-treated cells. BV-2 cells were treated in the presence or absence of cytokines overnight before fAβ stimulation. The cells were then stained with phalloidin to label cytoskeletal filamentous actin (F-actin). The cells treated with fAβ alone or in combination with IL-4 extended processes and contained phagocytosed microspheres (Fig. 5). In contrast, the cells that were treated with IL-1β before incubation with fAβ were similar to untreated cells, demonstrating that proinflammatory cytokines prevented cytoskeletal reorganization necessary for phagocytosis.
Figure 5.

Proinflammatory and anti-inflammatory treated cells exhibit morphological differences. BV-2 cells were treated with IL-1β (15 ng/ml) or IL-4 (10 ng/ml) overnight before stimulation with fAβ25–35 (60μm). Fluorescent microspheres were then added for 30 min. The cells were stained with phalloidin to visualize F-actin. Images of the cells were taken at 100×. Scale bar, 5 μm. This experiment is representative of three independent studies.
Anti-inflammatory cytokines restore phagocytic activity
It has been demonstrated that anti-inflammatory and proinflammatory cytokines exhibit antagonistic actions in macrophages (Hamilton et al., 1999; Duffield, 2003). We examined cells incubated simultaneously with anti-inflammatory and proinflammatory cytokines to determine whether anti-inflammatory cytokines could suppress the inhibitory actions of proinflammatory cytokines and restore phagocytosis in fAβ-treated cells. First, we incubated BV-2 cells with IL-1β (15 ng/ml) and IL-4 (5, 10, and 15 ng/ml) overnight before stimulation with fAβ. IL-4 treatments restored fAβ-elicited phagocytosis in a dose-dependent manner in the presence of IL-1β (Fig. 6A). We also demonstrated that IL-4 treatment prevented the ability of IFNγ to inhibit fAβ-mediated phagocytosis (Fig. 6B). Similarly, the LPS-mediated suppression of fAβ-stimulated phagocytosis was blocked by the action of the anti-inflammatory cytokine IL-10 (Fig. 6C). In each case, the anti-inflammatory cytokine restored phagocytosis in the presence of proinflammatory cytokines.
Figure 6.
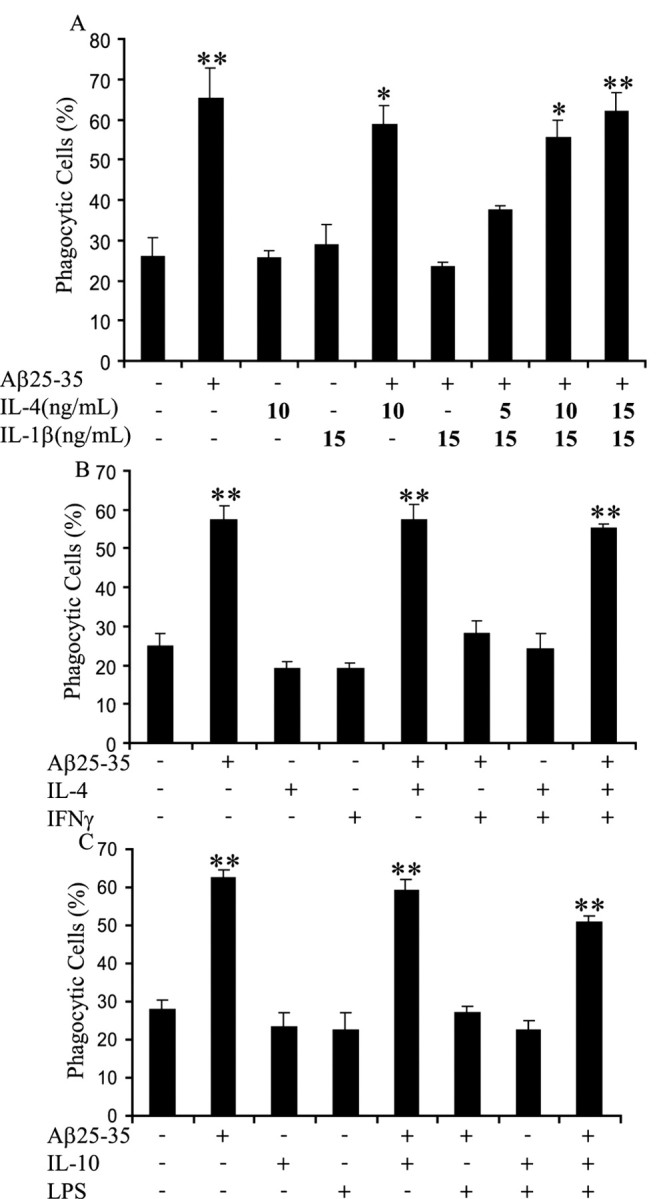
Anti-inflammatory cytokines restore phagocytic activity. A, BV-2 cells were incubated with 15 ng/ml IL-1β and indicated doses of IL-4 overnight before a 30 min stimulation with 60 μm fAβ25–35, followed by incubation with fluorescent microspheres for 30 min. The fraction of phagocytically active cells was then determined. B, BV-2 cells were incubated with 10 ng/ml IL-4 and 100 ng/ml IFNγ overnight before a 30 min stimulation with 60μm fAβ25–35 and incubation with fluorescent microspheres for 30 min. The phagocytically active cells were then measured. C, BV-2 cells were incubated with 10 ng/ml IL-10 overnight beforea 6 h stimulation with 1μg/ml LPS. The cells were then stimulated with 60μm fAβ25–35 for 30 min and incubated with fluorescent microspheres for 30 min. The fraction of phagocytically active cells was then determined. **p < 0.001 compared with control. These data are representative of three independent experiments.
Ibuprofen rescues fAβ-elicited phagocytosis in a proinflammatory milieu
The observation that anti-inflammatory cytokines were able to restore fAβ-elicited phagocytosis in a proinflammatory environment led us to evaluate whether the nonsteroidal anti-inflammatory drug (NSAID) ibuprofen would elicit similar effects in a proinflammatory milieu. BV-2 cells were stimulated with IL-1β and racemic (R and S enantiomers) ibuprofen overnight before incubation with fAβ. Ibuprofen was able to restore phagocytosis stimulated by fAβ dose dependently in the presence of IL-1β (Fig. 7A).
Figure 7.
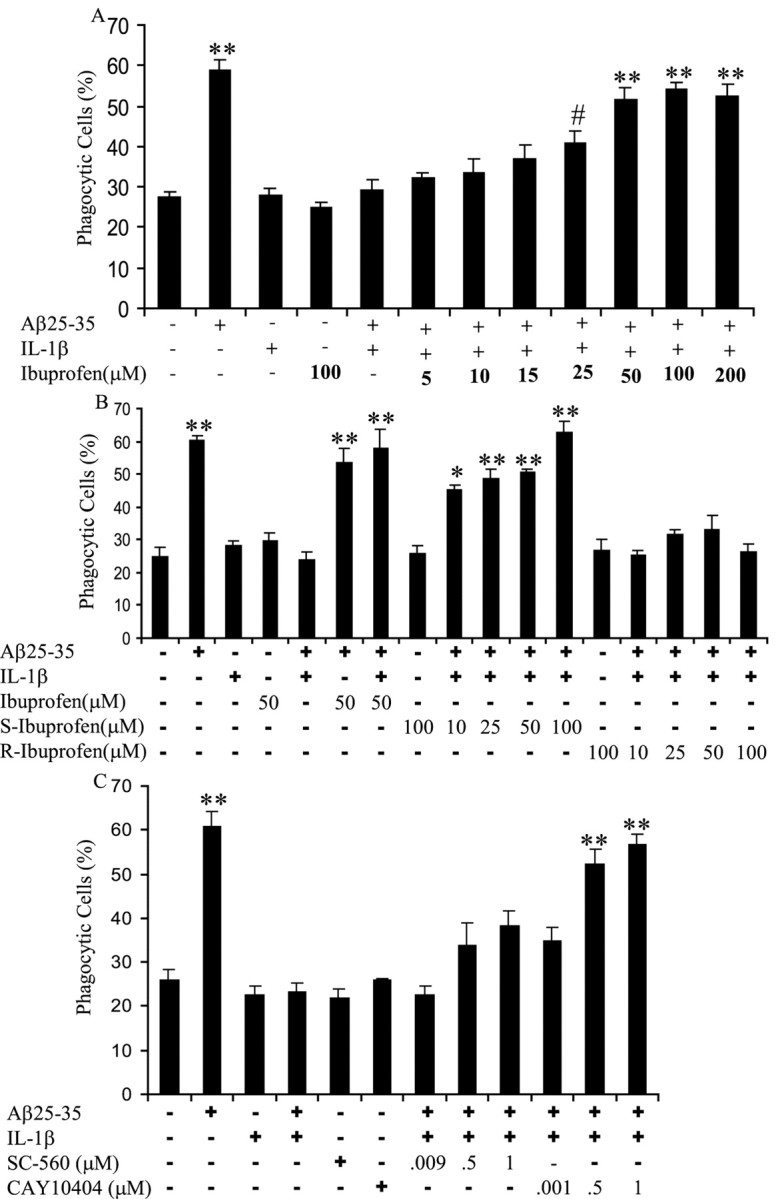
Ibuprofen rescues fAβ-elicited phagocytosis in a proinflammatory milieu. A, BV-2 cells were treated with racemic ibuprofen at indicated doses and IL-1β (15 ng/ml) overnight before stimulation with fAβ25–35 (60μm) for 30 min, followed by a 30 min incubation with fluorescent microspheres. The fraction of phagocytically active cells was then determined. B, BV-2 cells were treated with racemic ibuprofen, S-ibuprofen, or R-ibuprofen at indicated doses together with IL-1β (15 ng/ml) overnight. Cells were then stimulated with fAβ25–35 (60 μm) for 30 min before incubation with fluorescent microspheres for 30 min. The phagocytically active cells were then measured. C, BV-2 cells were treated with IL-1β (15 ng/ml) overnight beforea 1 h incubation with CAY10404, a COX-2 inhibitor, at indicated doses or SC-560, a COX-1 inhibitor, at indicated doses. Cells were then stimulated with fAβ25–35 (60μm) for 30 min before the addition of fluorescent microspheres for 30 min. The fraction of phagocytically active cells was then determined. *p < 0.01, **p < 0.001, and #p < 0.05 compared with control. These data are representative of three independent experiments.
We compared the ability of the two enantiomers of ibuprofen, S-ibuprofen and R-ibuprofen, to restore phagocytosis. The S-ibuprofen is the active enantiomer, whereas the R-enantiomer is inactive, with respect to COX inhibition. The S-ibuprofen was able to rescue phagocytosis in IL-1β-treated cells, but R-ibuprofen was without effect (Fig. 7B). These data argue that COX activity may be involved in the suppression of phagocytosis by IL-1β and other proinflammatory cytokines.
To determine which of the two COX isoforms may play a role in the regulation of fAβ-stimulated phagocytosis, we used two specific COX inhibitors. SC-560 specifically inhibits COX-1 (Smith et al., 1998), whereas CAY10404 is a highly specific inhibitor of COX-2 (Parashar et al., 2005). COX-1 is constitutively expressed, whereas COX-2 expression is induced by inflammatory stimulation of microglia and macrophages (Vane et al., 1998; Akiyama et al., 2000). BV-2 cells were incubated with IL-1β overnight before a 1 h incubation with the COX inhibitors. Cells were then stimulated with fAβ, and the percentage of phagocytic cells was determined. The inhibition of COX-2 restored fAβ phagocytosis to a level similar to cells stimulated with only fAβ (Fig. 7C). The COX-1 inhibitor was unable to rescue phagocytosis driven by fAβ in a proinflammatory environment (Fig. 7C). We concluded from this experiment that COX-2 is involved in the suppression of fAβ-elicited phagocytosis in a proinflammatory environment.
Prostaglandin E2 suppresses fAβ-stimulated phagocytosis
The principal action of COX is to catalyze the synthesis of PGs from arachidonic acid. COX-2 is responsible for the production of PGE2. PGE2 binds and activates G-protein-coupled E prostanoid receptors (Narumiya et al., 1999) and has been shown to modulate phagocytosis of macrophages (Hutchison and Myers, 1987; Rossi et al., 1998; Aronoff et al., 2004). Shie et al. (2005) have shown recently that microglia in which the E prostanoid 2 (EP2) receptor gene was knocked out demonstrated increased phagocytosis of Aβ compared with wild-type microglia. To determine whether PGE2 was involved in the suppression of fAβ-elicited phagocytosis in a proinflammatory milieu, we used the specific EP2 receptor agonist butaprost and an EP receptor antagonist, AH6809 (Aronoff et al., 2004; Pooler et al., 2004). Treatment of microglia with butaprost before fAβ stimulation inhibited fAβ-elicited phagocytosis, mimicking the effect that was seen in cells treated with IL-1β before stimulation with fAβ (Fig. 8A). Importantly, treatment with AH6809 after IL-1β incubation restored the ability of fAβ to elicit a phagocytic response (Fig. 8B). These data are consistent with a mechanism in which proinflammatory stimuli induce COX-2 expression and PGE2 production, which then likely signals through the EP2 receptor to inhibit fAβ-driven phagocytosis.
Figure 8.

Prostaglandin E2 regulates the suppression of fAβ-stimulated phagocytosis in a proinflammatory milieu. A, BV-2 cells were treated with IL-1β (15 ng/ml) overnight before the addition of butaprost, an EP2 receptor agonist, at indicated doses for 1 h before stimulation with fAβ25–35 (60 μm) for 30 min. Cells were then incubated with fluorescent microspheres for 30 min. The percentage of phagocytically active cells was then determined. B, BV-2 cells were treated with IL-1β (15 ng/ml) overnight before the addition of AH6809, an EP receptor antagonist, at indicated doses for 1 h. Cells were then stimulated with fAβ25–35 (60 μm) for 30 min, followed by a 30 min incubation with fluorescent microspheres. The percentage of phagocytically active cells was then determined. *p < 0.01 and **p < 0.001 compared with control. These data are representative of three independent experiments.
Discussion
We have reported previously the characterization of a microglial fAβ cell surface receptor complex (Bamberger et al., 2003) whose engagement leads to stimulation of phagocytosis (Koenigsknecht and Landreth, 2004). This receptor complex, whose principle constituents are CD36, CD47 (also termed integrin-associated protein), and the α6β1-integrin, recognizes and binds fAβ and elicits a novel phagocytic response that is reliant on the β1-integrin and is mechanistically distinct from classical type I or type II phagocytosis (Koenigsknecht and Landreth, 2004). It remains unclear why activated microglia that physically associate with amyloid plaques are unable to effectively phagocytose fAβ deposits in the AD brain or its animal models (Stalder et al., 2001).
The mechanisms subserving the removal of fAβ from the brain are complex, and microglial phagocytosis is clearly only one of several mechanisms that operate in parallel to effect the elimination of the amyloid peptides and plaques from the brain. These include the phagocytic uptake of Aβ through complement-mediated mechanisms (Wyss-Coray et al., 2002) and through the fAβ receptor complex (Koenigsknecht and Landreth, 2004), as well as trafficking of Aβ into the peripheral circulation through apolipoprotein-mediated uptake by vasculature (Zlokovic, 2004). We postulated that chronic inflammation in the AD brain suppresses activation of phagocytic machinery and affects the ability of microglial cells to mount a phagocytic response, thereby inhibiting normal microglial clearance of fAβ and senile plaques. The brains of AD patients exhibit elevated levels of cytokines, chemokines, immune cell surface proteins, and complement proteins, which results in a feedforward process with a progressive upregulation of inflammation over the course of the disease (Akiyama et al., 2000). It has been demonstrated that inflammatory stimuli, such as CD40, upregulates expression of proinflammatory cytokines, as well as COX-2 and PGE2 levels (Tan et al., 1999, 2002; Inoue et al., 2004). There is clear evidence that microglia routinely survey the brain parenchyma (Nimmerjahn et al., 2005) and act normally through complement-dependent processes to remove Aβ (Wyss-Coray et al., 2002) but are unable to prevent plaque formation in the AD brain. We found that microglia stimulated through the CR3 or the fAβ receptor complex are incapable of mounting a phagocytic response in the presence of proinflammatory cytokines, consistent with a very recent report by Townsend et al. (2005). Thus, inflammation inhibits both endogenous fAβ phagocytic mechanisms and reduces fAβ clearance and may explain, in part, the development and accumulation of senile plaques in the AD brain. The present study has demonstrated these effects in vitro with acute stimulation of phagocytes, and it remains possible that the chronic in vivo situation may be different.
We demonstrated that proinflammatory cytokines act selectively to regulate different types of microglial phagocytosis, because these cytokines had no effect on immune IgG-elicited phagocytosis. The molecular basis for this effect is unknown but suggests that elements of the phagocyte machinery are not subject to inhibition by cAMP. Previous studies that characterized the classical types of microglial phagocytosis demonstrated that type I or FcR-mediated phagocytosis occurred in the presence of inflammation, whereas type II phagocytosis occurred in the absence of inflammatory signals (Caron and Hall, 1998). Importantly, we conclude from these experiments that microglial FcRs are able to maintain their ability to stimulate phagocytosis despite the chronic inflammatory environment found in the AD brain. This is significant because FcR-mediated phagocytosis of senile plaques induced by antibodies directed against fAβ are unaffected by the inflammatory status of the brain, and this effect may underlie the efficacy of Aβ vaccination therapy in murine models of AD (Schenk et al., 1999; Bard et al., 2000; Wilcock et al., 2003, 2004).
The efficacy of Aβ vaccination-based therapy in preventing amyloid deposition, removal of existing plaques, and behavioral improvement is well documented in animal models of AD (Morgan et al., 2000; Arendash et al., 2001; Jensen et al., 2005). Indeed, there is suggestive evidence of analogous effects in human clinical trials of Aβ vaccination therapy (Hock et al., 2003; Orgogozo et al., 2003; Bayer et al., 2005). However, the mechanisms underlying the reduction of plaque burden are controversial. Wilcock et al. (2003, 2004) have elegantly demonstrated that attenuation of AD-like pathology in murine models of AD after Aβ vaccination arises from both microglial phagocytosis and nonphagocytic mechanisms (Aβ trafficking) that act in parallel. Aβ peptide levels initially fall through microglia-independent mechanisms, whereas later reduction of Aβ peptide levels and plaque burden is the result of phagocytic removal by microglia.
There is strong epidemiological evidence that long-term NSAID therapy has beneficial effects by reducing AD risk by 60–80% (McGeer et al., 1996; Stewart et al., 1997; in t'Veld et al., 2001), delaying onset, reducing the severity, and slowing the progression of AD (Rogers et al., 1993; Rich et al., 1995). Importantly, NSAID treatment is associated with a reduced number of plaque-associated microglia in the AD brain (Mackenzie and Munoz, 2001). The effect of proinflammatory stimulation of microglia is the rapid induction of COX-2 expression and the subsequent production of PGs (Akiyama et al., 2000). Indeed, COX-2 protein levels have been shown to be elevated in AD brains (Pasinetti and Aisen, 1998; Ho et al., 1999; Kitamura et al., 1999). The effect of anti-inflammatory cytokines is to suppress the NFκB-dependent stimulation of COX-2 expression and PGE2 production in CD40-stimulated monocytes (Inoue et al., 2004), and we argue that this effect underlies the antagonist actions of these anti-inflammatory cytokines in restoring phagocytic activity in the presence of inflammatory stimuli. TGFβ overexpression in an animal model of AD resulted in a dramatic reduction in Aβ plaque burden that was associated with microglial activation and enhanced phagocytosis (Wyss-Coray et al., 2001).
The primary effect of NSAID action is the inhibition of COXs and the subsequent suppression of PG synthesis (Vane and Botting, 1998). We provide data that ibuprofen acts via this mechanism to counteract the ability of proinflammatory cytokines to inhibit phagocytosis elicited by engagement of the fAβ receptor complex. Ibuprofen has been implicated in the prevention and treatment of AD. Treatment of AD murine models with ibuprofen resulted in a reduction of plaque burden and Aβ peptide levels that was not observed in untreated mice (Lim et al., 2000; Jantzen et al., 2002; Yan et al., 2003). Ibuprofen-treated animals exhibited a reduction in the number of reactive microglia and their proinflammatory products, as well as improved behavior (Lim et al., 2000). We demonstrated that anti-inflammatory cytokines and ibuprofen were able to restore fAβ-elicited phagocytosis in a proinflammatory milieu. This rescue effect was mediated by the inhibition of COX-2, reducing PGE2 levels, and signaling through the EP2 receptor. Importantly, this conclusion was validated by a recent report by Shie et al. (2005). Our results also define a mechanism that may explain, in part, the animal data in which administration of ibuprofen results in a reduction of fAβ plaques. Our finding that ibuprofen acts to antagonize the action of inflammatory stimuli and restore phagocytosis provides a new mechanism whereby NSAIDs act to reduce AD risk.
There has been considerable controversy over the efficacy of anti-inflammatory therapies in reducing AD risk, as well as in the treatment of patients with the disease. The epidemiological studies have led to clinical trials of steroids (Aisen et al., 2000), COX-2-specific inhibitors, and naproxen in patient populations with mild to moderate AD (Aisen et al., 2003). These trials failed to demonstrate a salutary effect on AD progression. This outcome likely reflects the requirement for drug intervention at earlier stages of the disease because the epidemiological studies revealed that only long-term (at least 2 years) drug treatment provided the protection against AD (Stewart et al., 1997; in t'Veld et al., 2001), suggesting that these agents cannot functionally intervene in the disease process after it has become clinically evident. From a mechanistic prospective, the action of NSAIDs relative to disease pathophysiology has generated substantial controversy with the newly recognized ability of a subset of these drugs, including ibuprofen, to have anti-inflammatory actions through peroxisome proliferator-activated receptor γ activation (Lehmann et al., 1997; Jiang et al., 1998; Ricote et al., 1998; Combs et al., 2000) and to act on neurons to alter the specificity of amyloid precursor protein processing by directly effecting γ-secretase specificity, although the effects are only observed at high drug concentrations (Weggen et al., 2001; Sagi et al., 2003; Kukar et al., 2005). The effects observed in the present study were elicited at ibuprofen concentrations consistent with its action on COX and not through alternate mechanisms (Tegeder et al., 2001).
In summary, the present study supports the use of anti-inflammatory therapies in patient populations at risk for AD. Specifically, we have shown that ibuprofen, acting at therapeutically relevant concentrations on its canonical targets, the COXs, facilitate the normal clearance of fAβ by phagocytosis. Moreover, the action of anti-inflammatory cytokines in facilitating the clearance of fAβ validate this therapeutic approach. The present data also illustrate the phenotypic complexity of “reactive microglia” and argue that it may be possible to selectively target specific actions of these cells. These findings also have direct therapeutic relevance to AD through the recognition that vaccination-based removal of fAβ from the AD brain is unaffected by its inflammatory status.
Footnotes
This work was supported by National Institutes of Health Grant AG20202 and by the Blanchette Hooker Rockefeller Foundation.
Correspondence should be addressed to Gary E. Landreth, Alzheimer Research Laboratory, E504, Case Western Reserve University School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106. E-mail:gel2@cwru.edu.
DOI:10.1523/JNEUROSCI.1808-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258240-10$15.00/0
References
- Aisen PS, Davis KL, Berg JD, Schafer K, Campbell K, Thomas RG, Weiner MF, Farlow MR, Sano M, Grundman M, Thal LJ (2000) A randomized controlled trial of prednisone in Alzheimer's disease. Alzheimer's Disease Cooperative Study. Neurology 54: 588–593. [DOI] [PubMed] [Google Scholar]
- Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ (2003) Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289: 2819–2826. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendash GW, Gordon MN, Diamond DM, Austin LA, Hatcher JM, Jantzen P, DiCarlo G, Wilcock D, Morgan D (2001) Behavioral assessment of Alzheimer's transgenic mice following long-term Abeta vaccination: task specificity and correlations between Abeta deposition and spatial memory. DNA Cell Biol 20: 737–744. [DOI] [PubMed] [Google Scholar]
- Aronoff DM, Canetti C, Peters-Golden M (2004) Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J Immunol 173: 559–565. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT (2002) Non-Fc-mediated mechanisms are involved in clearance of amyloid-β in vivo by immunotherapy. J Neurosci 22: 7873–7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin Jr AS (1996) The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol 14: 649–683. [DOI] [PubMed] [Google Scholar]
- Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE (2003) A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J Neurosci 23: 2665–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. (2000) Peripherally administered antibodies against amyloid betapeptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6: 916–919. [DOI] [PubMed] [Google Scholar]
- Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, Millais SB, Donoghue S (2005) Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology 64: 94–101. [DOI] [PubMed] [Google Scholar]
- Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F (1990) Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol 27: 229–237. [DOI] [PubMed] [Google Scholar]
- Bocchini V, Mazzolla R, Barluzzi R, Blasi E, Sick P, Kettenmann H (1992) An immortalized cell line expresses properties of activated microglial cells. J Neurosci Res 31: 616–621. [DOI] [PubMed] [Google Scholar]
- Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C (1992) Assembly and aggregation properties of synthetic Alzheimer's A4/beta amyloid peptide analogs. J Biol Chem 267: 546–554. [PubMed] [Google Scholar]
- Caron E, Hall A (1998) Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science 282: 1717–1721. [DOI] [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Cannady SB, Lehman TM, Landreth GE (1999) Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of β-amyloid and prion proteins. J Neurosci 19: 928–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE (2000) Inflammatory mechanisms in Alzheimer's disease: inhibition of β-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARγ agonists. J Neurosci 20: 558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CK, Karlo JC, Kao SC, Landreth GE (2001) β-Amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci 21: 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield JS (2003) The inflammatory macrophage: a story of Jekyll and Hyde. Clin Sci (Lond) 104: 27–38. [DOI] [PubMed] [Google Scholar]
- Goerdt S, Orfanos CE (1999) Other functions, other genes: alternative activation of antigen-presenting cells. Immunity 10: 137–142. [DOI] [PubMed] [Google Scholar]
- Gordon S (2003) Alternative activation of macrophages. Nat Rev Immunol 3: 23–35. [DOI] [PubMed] [Google Scholar]
- Hamilton TA, Ohmori Y, Tebo JM, Kishore R (1999) Regulation of macrophage gene expression by pro- and anti-inflammatory cytokines. Pathobiology 67: 241–244. [DOI] [PubMed] [Google Scholar]
- Hehner SP, Hofmann TG, Droge W, Schmitz ML (1999) The antiinflammatory sesquiterpene lactone parthenolide inhibits NF-kappa B by targeting the I kappa B kinase complex. J Immunol 163: 5617–5623. [PubMed] [Google Scholar]
- Ho L, Pieroni C, Winger D, Purohit DP, Aisen PS, Pasinetti GM (1999) Regional distribution of cyclooxygenase-2 in the hippocampal formation in Alzheimer's disease. J Neurosci Res 57: 295–303. [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, deq Uervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM (2003) Antibodies against beta-amyloid slow cognitive decline in Alzheimer's disease. Neuron 38: 547–554. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, Rozemuller AJ, Veerhuis R, Eikelenboom P (2001) Immunological aspects of alzheimer's disease: therapeutic implications. Bio-Drugs 15: 325–337. [DOI] [PubMed] [Google Scholar]
- Hutchison DL, Myers RL (1987) Prostaglandin-mediated suppression of macrophage phagocytosis of Listeria monocytogenes. Cell Immunol 110: 68–76. [DOI] [PubMed] [Google Scholar]
- Inoue Y, Otsuka T, Niiro H, Nagano S, Arinobu Y, Ogami E, Akahoshi M, Miyake K, Ninomiya I, Shimizu S, Nakashima H, Harada M (2004) Novel regulatory mechanisms of CD40-induced prostanoid synthesis by IL-4 and IL-10 in human monocytes. J Immunol 172: 2147–2154. [DOI] [PubMed] [Google Scholar]
- in t'Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH (2001) Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med 345: 1515–1521. [DOI] [PubMed] [Google Scholar]
- Ishizuka K, Kimura T, Igata-yi R, Katsuragi S, Takamatsu J, Miyakawa T (1997) Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer's disease. Psychiatry Clin Neurosci 51: 135–138. [DOI] [PubMed] [Google Scholar]
- Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN (2002) Microglial activation and β-amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci 22: 2246–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MT, Mottin MD, Cracchiolo JR, Leighty RE, Arendash GW (2005) Lifelong immunization with human beta-amyloid (1–42) protects Alzheimer's transgenic mice against cognitive impairment throughout aging. Neuroscience 130: 667–684. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B (1998) PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 391: 82–86. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Shimohama S, Koike H, Kakimura J, Matsuoka Y, Nomura Y, Gebicke-Haerter PJ, Taniguchi T (1999) Increased expression of cyclooxygenases and peroxisome proliferator-activated receptor-gamma in Alzheimer's disease brains. Biochem Biophys Res Commun 254: 582–586. [DOI] [PubMed] [Google Scholar]
- Koenigsknecht J, Landreth G (2004) Microglial phagocytosis of fibrillar β-amyloid through a β1 integrin-dependent mechanism. J Neurosci 24: 9838–9846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM (2004) Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med 10: 719–726. [DOI] [PubMed] [Google Scholar]
- Kopec KK, Carroll RT (1998) Alzheimer's beta-amyloid peptide 1–42 induces a phagocytic response in murine microglia. J Neurochem 71: 2123–2131. [DOI] [PubMed] [Google Scholar]
- Kukar T, Murphy MP, Eriksen JL, Sagi SA, Weggen S, Smith TE, Ladd T, Khan MA, Kache R, Beard J, Dodson M, Merit S, Ozols VV, Anastasiadis PZ, Das P, Fauq A, Koo EH, Golde TE (2005) Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Abeta42 production. Nat Med 11: 545–550. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA (1997) Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem 272: 3406–3410. [DOI] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM (2000) Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci 20: 5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J (1995) Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem 270: 14255–14258. [DOI] [PubMed] [Google Scholar]
- Lorenzo A, Yankner BA (1994) Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci USA 91: 12243–12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Munoz DG (2001) Effect of anti-inflammatory medications on neuropathological findings in Alzheimer disease. Arch Neurol 58: 517–519. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23: 549–555. [DOI] [PubMed] [Google Scholar]
- McDonald DR, Brunden KR, Landreth GE (1997) Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci 17: 2284–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Schulzer M, McGeer EG (1996) Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology 47: 425–432. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW (2000) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408: 982–985. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto Y, Ushikubi F (1999) Prostanoid receptors: structures, properties, and functions. Physiol Rev 79: 1193–1226. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308: 1314–1318. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C (2003) Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 61: 46–54. [DOI] [PubMed] [Google Scholar]
- Parashar B, Latha Shankar S, O'Guin K, Butler J, Vikram B, Shafit-Zagardo B (2005) Inhibition of human neuroblastoma cell growth by CAY10404, a highly selective Cox-2 inhibitor. J Neurooncol 71: 141–148. [DOI] [PubMed] [Google Scholar]
- Pasinetti GM, Aisen PS (1998) Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer's disease brain. Neuroscience 87: 319–324. [DOI] [PubMed] [Google Scholar]
- Pooler AM, Arjona AA, Lee RK, Wurtman RJ (2004) Prostaglandin E2 regulates amyloid precursor protein expression via the EP2 receptor in cultured rat microglia. Neurosci Lett 362: 127–130. [DOI] [PubMed] [Google Scholar]
- Qiao X, Cummins DJ, Paul SM (2001) Neuroinflammation-induced acceleration of amyloid deposition in the APPV717F transgenic mouse. Eur J Neurosci 14: 474–482. [DOI] [PubMed] [Google Scholar]
- Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J (1995) Nonsteroidal anti-inflammatory drugs in Alzheimer's disease. Neurology 45: 51–55. [DOI] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK (1998) The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391: 79–82. [DOI] [PubMed] [Google Scholar]
- Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, Zalinski J, Cofield M, Mansukhani L, Willson P (1993) Clinical trial of indomethacin in Alzheimer's disease. Neurology 43: 1609–1611. [DOI] [PubMed] [Google Scholar]
- Rossi AG, McCutcheon JC, Roy N, Chilvers ER, Haslett C, Dransfield I (1998) Regulation of macrophage phagocytosis of apoptotic cells by cAMP. J Immunol 160: 3562–3568. [PubMed] [Google Scholar]
- Sagi SA, Weggen S, Eriksen J, Golde TE, Koo EH (2003) The non-cyclooxygenase targets of non-steroidal anti-inflammatory drugs, lipoxygenases, peroxisome proliferator-activated receptor, inhibitor of kappa B kinase, and NF kappa B, do not reduce amyloid beta 42 production. J Biol Chem 278: 31825–31830. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, et al. (1999) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400: 173–177. [DOI] [PubMed] [Google Scholar]
- Shie FS, Breyer RM, Montine TJ (2005) Microglia lacking E prostanoid receptor subtype 2 have enhanced Abeta phagocytosis yet lack Abeta-activated neurotoxicity. Am J Pathol 166: 1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Zhang Y, Koboldt CM, Muhammad J, Zweifel BS, Shaffer A, Talley JJ, Masferrer JL, Seibert K, Isakson PC (1998) Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc Natl Acad Sci USA 95: 13313–13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder M, Deller T, Staufenbiel M, Jucker M (2001) 3D-Reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol Aging 22: 427–434. [DOI] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ (1997) Risk of Alzheimer's disease and duration of NSAID use. Neurology 48: 626–632. [DOI] [PubMed] [Google Scholar]
- Tak PP, Firestein GS (2001) NF-kappaB: a key role in inflammatory diseases. J Clin Invest 107: 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M (1999) Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science 286: 2352–2355. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Mullan M (2002) CD40-CD40L interaction in Alzheimer's disease. Curr Opin Pharmacol 2: 445–451. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Pfeilschifter J, Geisslinger G (2001) Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J 15: 2057–2072. [DOI] [PubMed] [Google Scholar]
- Terzi E, Holzemann G, Seelig J (1994) Alzheimer beta-amyloid peptide 25–35: electrostatic interactions with phospholipid membranes. Biochemistry 33: 7434–7441. [DOI] [PubMed] [Google Scholar]
- Townsend KP, Town T, Mori T, Lue LF, Shytle D, Sanberg PR, Morgan D, Fernandez F, Flavell RA, Tan J (2005) CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid beta-peptide. Eur J Immunol 35: 901–910. [DOI] [PubMed] [Google Scholar]
- Vane JR, Botting RM (1998) Mechanism of action of antiinflammatory drugs. Int J Tissue React 20: 3–15. [PubMed] [Google Scholar]
- Vane JR, Bakhle YS, Botting RM (1998) Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol 38: 97–120. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH (2001) A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414: 212–216. [DOI] [PubMed] [Google Scholar]
- Wesemann DR, Dong Y, O'Keefe GM, Nguyen VT, Benveniste EN (2002) Suppressor of cytokine signaling 1 inhibits cytokine induction of CD40 expression in macrophages. J Immunol 169: 2354–2360. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D (2003) Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci 23: 3745–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock D, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman M, Gordon M, Morgan D (2004) Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci 24: 6144–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L (2001) TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med 7: 612–618. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E (2002) Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci USA 99: 10837–10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J (2003) Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 9: 453–457. [DOI] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G (2003) Anti-inflammatory drug therapy alters β-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci 23: 7504–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielasek J, Hartung HP (1996) Molecular mechanisms of microglial activation. Adv Neuroimmunol 6: 191–222. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV (2004) Clearing amyloid through the blood-brain barrier. J Neurochem 89: 807–811. [DOI] [PubMed] [Google Scholar]