

Abstract
The expansion of a polyglutamine tract in the ubiquitously expressed huntingtin protein causes Huntington's disease (HD), a dominantly inherited neurodegenerative disease. We show that the activity of the cholesterol biosynthetic pathway is altered in HD. In particular, the transcription of key genes of the cholesterol biosynthetic pathway is severely affected in vivo in brain tissue from HD mice and in human postmortem striatal and cortical tissue; this molecular dysfunction is biologically relevant because cholesterol biosynthesis is reduced in cultured human HD cells, and total cholesterol mass is significantly decreased in the CNS of HD mice and in brain-derived ST14A cells in which the expression of mutant huntingtin has been turned on. The transcription of the genes of the cholesterol biosynthetic pathway is regulated via the activity of sterol regulatory element-binding proteins (SREBPs), and we found an ∼50% reduction in the amount of the active nuclear form of SREBP in HD cells and mouse brain tissue. As a consequence, mutant huntingtin reduces the transactivation of an SRE-luciferase construct even under conditions of SREBP overexpression or in the presence of an exogenous N-terminal active form of SREBP. Finally, the addition of exogenous cholesterol to striatal neurons expressing mutant huntingtin prevents their death in a dose-dependent manner. We conclude that the cholesterol biosynthetic pathway is impaired in HD cells, mice, and human subjects, and that the search for HD therapies should also consider cholesterol levels as both a potential target and disease biomarker.
Keywords: degeneration, GABAergic neurons, Huntington's disease, lipids, neuronal death, transcription factor
Introduction
Huntington's disease (HD) is a progressive, autosomal dominant, neurodegenerative disorder characterized by motor, cognitive, and behavioral dysfunctions. The mutation leading to HD is an expansion of a CAG repeat close to the 5′ end of the HD gene (The Huntington's Disease Collaborative Research Group, 1993) that leads to an expanded glutamine tract in huntingtin, a 348 kDa protein with individual neuroprotective activities (Rigamonti et al., 2000; Cattaneo et al., 2001; Zuccato et al., 2001). Although huntingtin is ubiquitously expressed, the brain is primarily affected in HD, with the striatal medium spiny neurons and cortical neurons being particularly vulnerable (Reiner et al., 1988). The underlying mechanism in HD is attributable to the increased toxicity of the mutant protein. In addition, there is evidence indicating that some disease phenotypes may be attributable to the loss of normal huntingtin prosurvival activities (Cattaneo et al., 2001).
The aim of this study was to test the hypothesis of a dysfunction in cholesterol biosynthesis in HD. Cholesterol is synthesized locally in the brain (Jurevics and Morell, 1995), in which it constitutes a major component of myelin. During development, the demand for cholesterol in the brain is particularly high, and cholesterol/sphingolipid depletion leads to a gradual loss of synapses and spines (Thiele et al., 2000; Hering et al., 2003). Cholesterol is also crucially involved in optimal neurotransmitter release (Mauch et al., 2001). In adult brain, the available cholesterol seems to derive from basal biosynthesis, mainly by astrocytes (Barres and Smith, 2001; Pfrieger, 2003a), and its accumulation during the early phases of life. Although little is known about the role of cholesterol in this tissue district, defects in cholesterol homeostasis have been linked to neurodegenerative diseases such as Niemann-Pick type C and Alzheimer's disease (Mahley and Huang, 1999; Battaile and Steiner, 2000; Dietschy and Turley, 2001), and our previous microchip studies of a cell model of HD (Sipione et al., 2002) showed decreased transcription of the genes encoding for the enzymes of the cholesterol biosynthetic pathway.
We show here that the cholesterol biosynthetic pathway is altered in HD cells and mouse and human brain. This dysfunction is attributable to a mutant huntingtin-dependent decrease in the amount of the active form of SREBP transcription factor translocating to the nucleus and a subsequent reduction in the transcription of key genes of cholesterol biosynthesis, a decrease in the end product (i.e., cholesterol level) and a smaller total cholesterol mass. We also found that the addition of exogenous cholesterol to striatal neurons expressing mutant huntingtin prevents their death in a dose-dependent manner.
Materials and Methods
Radioactive semiquantitative reverse transcription-PCR analyses. Total RNA was isolated from the tissues and cells using TRIZOL reagent (Invitrogen, Carlsbad, CA) and reverse-transcribed into single-stranded cDNA using Superscript II RNase H- Reverse Transcriptase (Invitrogen) as described by the manufacturer. Radioactive PCR was performed in a total volume of 50 μl containing cDNA, consisting of 0.25 μg of RNA, 20 mm Tris-HCl, pH 8.4, 50 mm KCl, 1.5 mm MgCl2, 0.2 mm dNTPs, 1.7 μCi of [α32P]dCTP, 0.4 μm of each primer, and 2 U of Taq polymerase (Invitrogen). All of the genes were amplified using multiple cycle numbers (20-25 cycles) to determine the appropriate conditions for obtaining semiquantitative differences in their expression. The cycle steps and primer sequences are available on request. The PCR products were separated by means of nondenaturing 6% PAGE and visualized by autoradiography.
Cell cultures. The human primary fibroblasts and huntingtin-inducible cells were cultured in 5% CO2 at, respectively, 37°C and 33°C in DMEM, supplemented as described previously (Rigamonti et al., 2000; Sipione et al., 2002), unless otherwise specified. Only one control fibroblast cell line was used, because, as reported previously (Corsini et al., 1991), cholesterol biosynthetic pathway parameters are well characterized and similar in multiple fibroblast cell lines. We tested 10 other control lines in the past, and the individual experiments have shown the same production of de novo cholesterol after lipoprotein-deprived serum (LPDS) exposure. The well characterized fibroblast control cell line, namely PF in Corsini et al. (1991), was used. Fibroblast HD cell lines were obtained according to standard procedures from symptomatic Italian HD patients and were provided by Prof. S. Di Donato (Carlo Besta Neurological Institute) and by Prof. M. Racchi (University of Pavia). LPDS was prepared as described previously (Corsini et al., 1986) and was used in the experiments evaluating cholesterol biosynthesis. Lipid-deprived serum was prepared as described previously (Hannah et al., 2001).
Evaluation of cholesterol biosynthesis in HD cells. Cultured human fibroblasts were incubated under normal growth conditions (with medium plus 10% FCS) or lipid-deprived conditions (medium plus 10% LPDS) on Barloword Scientific (Stone, UK) cell culture-treated plates. Cholesterol synthesis was measured by incorporating radioactive acetate into cellular sterols (Brown et al., 1978; Corsini et al., 1987) (supplemental material, available at www.jneurosci.org). Triplicate samples of each cell clone were tested in two independent experiments.
Measurement of total cholesterol in tissues and cells. To measure total cholesterol levels in tissue, the animals were perfused with saline, their brains were dissected, and the striatum and cortex were isolated. The lipid fraction was isolated by means of solvent extraction (supplemental material, available at www.jneurosci.org), and total cholesterol was assayed using the Cholesterol kit (Clonital, Lombardia, Italy) according to the instructions of the manufacturer and normalized by tissue weight and protein concentration, which was assayed using a BCA protein assay kit (Pierce, Rockford, IL). An adapted procedure was followed to evaluate cholesterol concentrations in cell cultures (supplemental material, available at www.jneurosci.org).
Preparation of membrane and nuclear extracts. The cells were plated at 70% confluence and, on day 1, doxycycline (1 μg/ml) was added to induce the expression of exogenous huntingtin. On day 2, the cells were exposed to 5% delipidated fetal calf serum containing medium and, on day 3, all of the cells received 25 μg/ml N-acetyl-leucine-leucine-norleucinal (Sigma, St. Louis, MO) 2 h before harvesting. The membrane and nuclear lysates were extracted as described in the supplemental material (available at www.jneurosci.org).
Western blotting and immunocytochemistry. The protein concentrations in the samples were measured using BCA reagent (Pierce), and aliquots (50 μg of protein) of the membrane and nuclear fractions underwent, respectively, 6.5 or 10% SDS-PAGE before being transferred to nitrocellulose membranes. The membranes were saturated with 5% non-fat milk and Tris-buffered saline with 1× Tween 0.1% and incubated at room temperature for 2 h with anti-SREBP1 mouse monoclonal antibody 2A4 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA) or mouse monoclonal anti-SREBP2 antibody 7D4 (a gift from Dr. T. F. Osborne, University of California, Irvine, CA), followed by 1 h at room temperature with a secondary antibody against mouse IgG labeled with horseradish peroxidase (Bio-Rad, Hercules, CA) diluted 1:3000. They were then developed by chemiluminescence using the SuperSignal West Pico Chemiluminescent Substrate (Pierce). Histone H1 rabbit polyclonal antibody (1:200; Santa Cruz Biotechnology) or calnexin rabbit polyclonal antibody (1:2000; Stressgen Biotechnologies, San Diego, CA) were used as internal controls. The Mab2166 (1:5000; Chemicon, Temecula, CA) and EM48 antibodies (1:200; Chemicon) were used to detect huntingtin and mutant huntingtin aggregates, respectively. The endogenous SREBPs were immunocytochemically analyzed, as described in the supplemental material (available at www.jneurosci.org).
DNA transfection and luciferase assays. The following constructs were used: SRE-luciferase reporter construct (pSynSRE), expressing the partial hamster hydroxy-methyl-glutaryl CoA (HMGCoA) synthase promoter cloned upstream of the luciferase reporter gene (Dooley et al., 1998); TK-HSV-SREBP1a, encoding herpes simplex virus (HSV) epitope-tagged human SREBP1a driven by the HSV thymidine kinase (TK) promoter (Sakai et al., 1996); CMV-nSREBP1, encoding the transcriptionally active fragment of the human SREBP1a protein (1-460 aa) driven by the cytomegalovirus (CMV) promoter (Sanchez et al., 1995). Electroporation and luciferase activity were evaluated as detailed in the supplemental material (available at www.jneurosci.org). Luciferase activity was normalized to protein content in each sample, as measured by a BCA Protein Assay kit (Pierce).
Assay of huntingtin-mediated cell death. This assay was established on the basis of previous reports (Saudou et al., 1998). Primary striatal neurons prepared from embryonic day 17 rats were electroporated with cDNA plasmids encoding the 480 N-terminal amino acids of human huntingtin containing an expanded (68) CAG repeat (Htt480.68) and a plasmid encoding green fluorescent protein (GFP). The expression of Htt480.68 and GFP in the electroporated cells was, respectively, confirmed by Western blotting 6 d after plating using the 1HU-4C8 antibody (Euromedex, Mundolsheim, France) and anti-GFP (clone 7.1 and 13.1; Roche Diagnostics, Indianapolis, IN). The cells were plated onto 96-well microplates and exposed to the test compounds or controls on the day of plating. BDNF was added to eight wells per plate at a final concentration of 5 ng/ml, as a positive control. Eight control wells received 0.5% DMSO. Survival was measured six d after plating by counting the number of GFP-positive cells as an indicator of Htt480.68 transgene expression using a Trophos (Marseilles, France) Flash Cytometer and associated software. Htt480.68 induces 50% cell death on postplating day 6, 80-100% of which is antagonized by adding BDNF in the culture on plating day 0.
Statistical analysis. The data were compared using Bonferroni's post hoc test and one-way ANOVA, as indicated.
Results
Microarray studies of an inducible cell model of HD showed reduced mRNA levels for a subset of genes involved in the cholesterol biosynthetic pathway (Sipione et al., 2002). We specifically tested whether mouse and human HD brain tissues are affected by analyzing three key genes of this pathway: HMGCoA reductase (HMG-CoAred), cytochrome P450 lanosterol 14 α-demethylase (Cyp51) and 7-dehydroxy-cholesterol reductase (7dhcred). Figure 1A shows that the levels of mRNA for all three were reduced by up to 62% in the striatum and cortex of 12-week-old R6/2 HD transgenic mice (Mangiarini et al., 1996) compared with controls. A less dramatic, but still significant decrease was also detected at early symptomatic stages (6 weeks) in most brain samples (Fig. 1B). The changes in mRNA levels were also confirmed in human postmortem striatal (Fig. 1C) and cortical tissues taken from grades I and II (Fig. 1D) and from grades III and IV (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) HD patients.
Figure 1.
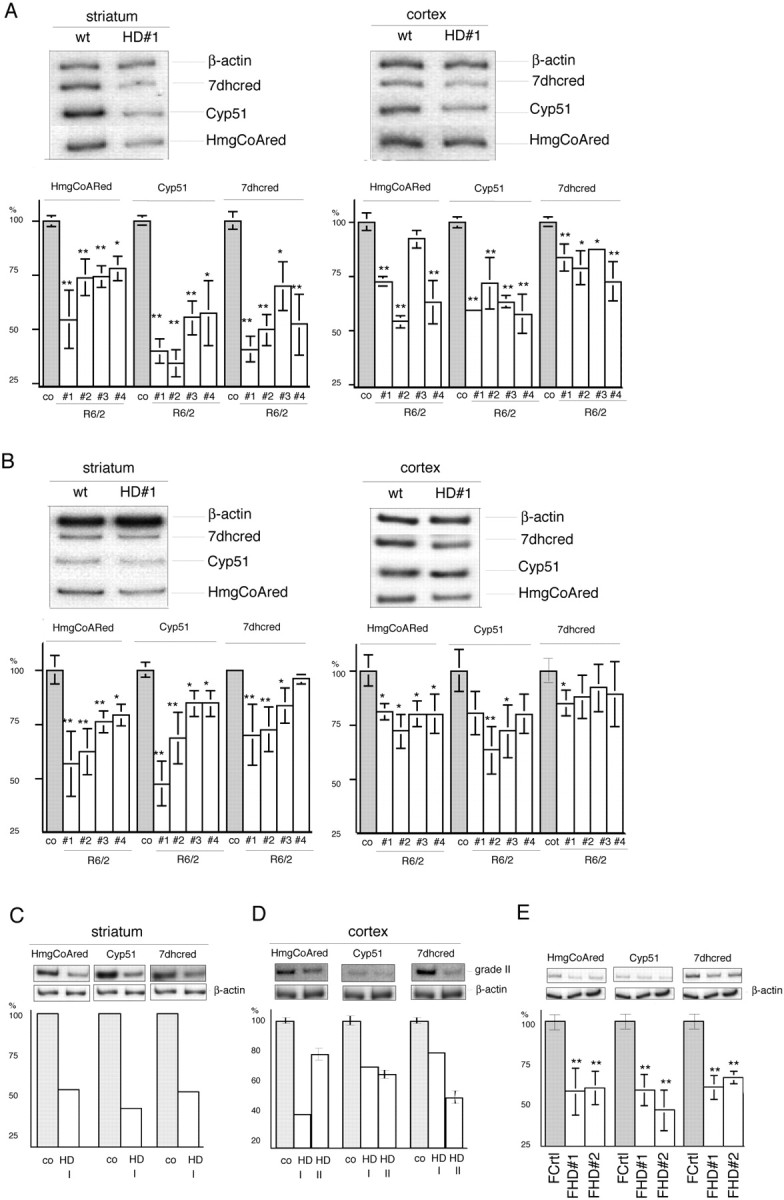
Reduced HMGCoAred, Cyp51, and 7dhcred gene transcription in mouse and human HD brain and peripheral cells. A, B, Semiquantitative radioactive RT-PCR analyses of striatum and cortex from individual symptomatic 12-week-old (A) and early symptomatic 6-week-old (B) R6/2 transgenic mice compared with the mean values ± SD of three age-matched controls (co). The tissue from each transgenic mouse was investigated by means of six independent RT-PCR analyses. wt, Wild type. C, Human postmortem striatum from grade I HD (HD I). D, Human postmortem cortical tissue from grade I (HD I) and grade II (HD II) versus control. E, Normal (FCrtl) and HD cultured human fibroblasts (FHD). Because of limitations in the available RNA, the grade I striatum and cortex data relate to a single RT-PCR, repeated twice; the data from the grade II samples were obtained by repeating the RT-PCR three times. The graphs show the quantitative analyses of HMGCoAred, Cyp51, and 7dhcred mRNA levels. The peak densitometric area was normalized over the peak densitometric area of the β-actin band. The data are expressed as percentages of control values. *p < 0.05 and **p < 0.01 versus wild-type littermates (co) (A, B), controls (C, D), or normal cultured fibroblasts (FCrtl) (E) (ANOVA). Error bars represent SEM.
Wild-type and mutant huntingtin are ubiquitously expressed in the CNS and peripheral tissue (Trottier et al., 1995), and thus, to test whether mutant huntingtin induces similar gene expression changes in peripheral cells, we analyzed the RNA extracted from primary fibroblasts taken from control and HD patients. Figure 1E shows that HD fibroblasts show a 35-45% reduction in HMGCoAred, Cyp51, and 7dhcred mRNA levels.
Together, these data indicate that there is a significant reduction in the mRNAs for HMGCoAred, Cyp51, and 7dhcred in HD mice and human subjects.
To evaluate the biological relevance of these alterations, we next analyzed the activity of the mevalonate pathway in cultured human fibroblasts from control, heterozygous, and homozygous HD subjects. It is known that control fibroblasts exposed to complete medium produce negligible levels of de novo synthesized cholesterol (William and Avigan, 1972; Brown et al., 1978), but their exposure to LPDS containing medium led to a 15-fold increase in cholesterol production, a well standardized value that is also reported in the literature (Corsini et al., 1991). Importantly, when the heterozygous and homozygous HD fibroblasts were exposed to LPDS, the increase was, respectively, only eightfold and fourfold (Fig. 2a). These data suggest that HD cells are less capable of upregulating the mevalonate pathway in response to low levels of cholesterol. However, as shown in Figure 2b, 25-OH-cholesterol, a cholesterol derivative that blocks cholesterol biosynthesis by increasing cholesterol concentration in the endo-plasmatic reticulum (ER) (Brown et al., 2002), equally repressed the cholesterol biosynthesis induced by LPDS in control and homozygous HD fibroblasts, and the same occurred when low-density lipoproteins were added (Fig. 2b). These data indicate that human HD cells appropriately suppress cholesterol biosynthesis in response to high levels of sterols but that their ability to activate the pathway is severely affected.
Figure 2.

Modulation of cholesterol biosynthesis in HD cells. a, The activity of the mevalonate pathway was evaluated by incorporating radioactive (14C) acetate into the cellular sterols of cells exposed to medium containing 10% FCS or 10% LPDS. No differences in cholesterol biosynthesis were observed between normal cultured fibroblasts (FCrtl) and HD fibroblasts (FHD) in serum containing medium (dark gray columns). When the cells were exposed to LPDS (light gray columns), cholesterol biosynthesis was significantly reduced in heterozygous and homozygous HD fibroblasts compared with control. **p < 0.01 versus FCrtl (ANOVA). b, Effect of 25-OH-cholesterol (25OHChol) and low-density lipoprotein (LDL) on cholesterol biosynthesis in FCrtl and HD fibroblasts exposed to LPDS. The basal level of cholesterol biosynthesis was restored by the addition of 25OHChol or LDL in both normal and HD cells. The results are normalized to protein content. Mean values ± SD of triplicate experiments. Error bars represent SEM.
Because these results may have been caused by excessive cholesterol accumulation, we measured total cholesterol levels in HD cells and tissues of HD mice. Figure 3 shows the behavior of brain-derived inducible cells expressing mutant huntingtin (Sipione et al., 2002) exposed to delipidated medium. The uninduced cells had higher total cholesterol levels, whereas exposure to the doxycycline inducer led to the expected expression of mutant huntingtin (data not shown) (Sipione et al., 2002) and a progressive reduction in total cholesterol content after 96 and 120 h (Fig. 3). This effect was specific to the delipidated condition and mutant huntingtin cells because there were no differences in serum containing medium (Fig. 3, shaded line) or in wild-type huntingtin-inducible cells (data not shown). We also measured total cholesterol content in the striatum and cortex of 10-week-old R6/2 HD transgenic mice and found a 32 ± 4.8% reduction compared with controls (supplemental Fig. 2, available at www.jneurosci.org as supplemental material); similar results were obtained in 5-week-old mice (data not shown). These data together demonstrate that the reduced cholesterol synthesis in HD cells is not caused by cholesterol accumulation and that the reduction in total cholesterol levels may be a biological consequence of altered cholesterol biosynthesis.
Figure 3.

Total cholesterol (tot. chol.) content in inducible HD cells during exposure to delipidated medium (delip). Total cholesterol levels remained unchanged in induced and uninduced cells cultured in serum containing medium [FCS, -doxy (doxycycline); FCS, +doxy; shaded lines]. Exposure to delipidated medium progressively decreased total cholesterol in the cells expressing mutant huntingtin (delip, +doxy) compared with the same cells under uninduced conditions (delip, -doxy). The results are normalized to protein (prot) content. Mean values ± SD of triplicate experiments. **p < 0.01 versus mutant huntingtin cells grown in the absence of doxy (delip, -doxy; ANOVA). Error bars represent SEM.
In the presence of low cholesterol levels, cholesterol biosynthesis is triggered by the SREBPs (Hua et al., 1993), which are known to translocate from the ER to the Golgi after activation of the sterol-sensing domain of SREBP cleavage activation protein (SCAP). In the Golgi, SREBPs are cleaved by two proteases (S1P and S2P) to obtain a 68 kDa NH2-terminal fragment that enters the nucleus and activates the transcription of SRE-controlled cholesterogenic genes. The three known SREBP isoforms each have a molecular mass of 120-125 kDa (Brown and Goldstein, 1997): SREBP1a and SREBP1c are derived from a single gene, whereas SREBP2 (which has 50% identity with SREBP1) is encoded by a different gene. SREBP1a is a potent activator of all SREBP-responsive genes, including those that mediate the synthesis of cholesterol, fatty acids, and triglycerides, but the roles of SREBP1c and SREBP2 are more restricted (Horton et al., 2002). Moreover, many SRE-controlled genes (including those studied by us) are similarly modulated by both SREBP1a and SREBP2, as has been shown by microarray and chromatine immunoprecipitation data (Horton et al., 2003; Bennett et al., 2004).
To test the hypothesis of impaired SREBP activity in HD cells, we evaluated the transactivation potential of an SRE element in huntingtin-inducible cells before and after the expression of mutant or wild-type huntingtin. After transiently transfecting huntingtin-inducible cells with an pSynSRE, we performed a luciferase assay under basal conditions or after incubation in delipidated medium 24 h after the addition of the doxycycline inducer of huntingtin expression. As shown in Figure 4a, no significant changes in SRE activity were detected when the different cultures were grown in the presence of FCS. However, in line with the data thus far reported, the exposure of the cells to delipidated medium led to a statistically significant 48 ± 10% decrease in SRE activity only in the cultures expressing mutant huntingtin (Fig. 4a). Because the activation of the SREBP1a isoform has broader consequences on lipid metabolism (Shimomura et al., 1997; Dooley et al., 1998), we performed a similar experiment by cotransfecting the pSynSRE vector and a construct constitutively expressing full-length SREBP1a (TK-HSV-SREBP1a). Figure 4b shows a similar reduction in SRE activity in mutant huntingtin-expressing cells, thus confirming suboptimal SRE activation in mutant cells even when SREBP is overexpressed. Finally, when a plasmid encoding the 68 kDa N terminus of the active form of SREBP1a was cotransfected with pSynSRE, transactivation of the SRE reporter gene was 24 ± 8% lower in the cells expressing mutant huntingtin than in those expressing wild-type huntingtin (Fig. 4c).
Figure 4.

Impaired SRE activity in HD cells. a, The HMGCoA synthase SRE promoter construct (PSynSRE) was transfected into parental, wild-type huntingtin (wt htt), or mutant huntingtin (mu htt) inducible cells, after which doxycycline (doxy) was added for 24 h and was followed by 16 h of exposure to FCS containing medium or lipid-deprived conditions (delip). Parental cells ± doxy were used as controls. b, The cells were cotransfected with PSynSRE and a construct encoding HSV epitope-tagged human SREBP1a driven by the HSV TK promoter and exposed to complete medium (with FCS) or delipidated conditions. c, Cotransfection of pSynSRE and a construct encoding the N-terminal active form of SREBP1a (CMV-SREBP1a Nt). In this experimental paradigm, 1 μg/ml 25-OH-cholesterol and 10 μg/ml cholesterol (suppressed conditions) were added to the complete medium to suppress the activation of endogenous SREBPs. In all of the luciferase assays, relative light units (RLU) were normalized to protein content, and the data were expressed as the percentages of RLU per microgram of protein from the same cells in the absence of doxy, under delipidated (a, b) or suppressed (c) conditions. Data are mean values of triplicate experiments, one of which is shown. **p < 0.01 versus each cell type grown in the absence of doxy (ANOVA). Error bars represent SEM.
To analyze whether the reduced SREBP-transactivating capability in HD was attributable to impaired translocation of active SREBPs into the nucleus, we next evaluated the distribution of these transcription factors in cells before and after the induction of mutant or wild-type huntingtin (Fig. 5a). We concentrated on SREBP1 (the available antibodies do not distinguish whether the endogenous protein is SREBP1a or SREBP1c) because it is highly expressed in cell cultures and brain (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), and first confirmed that the immunoreactive bands corresponding to the precursor of SREBP1 were equally present in controls and HD cells and mice (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Subsequently, we evaluated SREBP1 levels in membrane (inactive form) and nuclear preparations (active form) made of cells exposed to delipidated conditions, as described above. Figure 5b shows that the exposure of cells expressing wild-type huntingtin (or parental cells; data not shown) to delipidated medium did not affect the translocation of active SREBP1 into the nucleus, whereas the nuclear preparations of mutant huntingtin-expressing cells showed a 46% reduction in the amount of the 68 kDa active form of SREBP1 (Fig. 5b). Figure 5, b and c, also show that, under the same conditions, SREBP1 precursor protein and mRNA levels in the inducible HD cells were similar to controls at the same time points.
Figure 5.

Impaired translocation of endogenous SREBP in HD models. a, b, Western blot analyses of endogenous SREBP1 in nuclear and membrane extracts from parental, wild-type huntingtin (wt htt) (HD19), and mutant huntingtin (mu htt) (HD43) inducible cells in the absence or presence of the inducer [1 μg/ml doxycycline (doxy)] in delipidated growth medium. HD43 cells (Q105; mutant clone) and HD19 cells (Q26; normal clone), respectively, overexpress the N-548aa fragment of mutant or wild-type huntingtin. a, Induction of wild-type or mutant huntingtin in the cells after 40 h exposure to doxy. b, Localization of the 125 kDa inactive membrane form and the 68 kDa active nuclear form of SREBP1 in the absence or presence of wild-type or mutant huntingtin, after 16 h in delipidated medium. Calnexin and histone H1 were, respectively, used as loading controls in the membrane and nuclear fractions. The graphs show the quantitative densitometric analyses of the inactive and active forms of SREBP1 compared with calnexin and histone H1. The data are expressed as percentages of the wild-type or mutant cells without doxy. c, Semiquantitative radioactive RT-PCR analysis of mRNA levels of SREBP1 in HD43 after incubation in delipidated growth medium. There were no differences in SREBP1 mRNA levels in the absence or presence of mutant huntingtin. d, Immunocytochemistry for SREBP1 in HD19 and HD43 clones under the same conditions as those described above. The graph shows the mean percentage of nuclear immunoreactivity over the total cell number obtained by counting four microscope fields (10× magnification) from three replicates per experiment. The parental (P) cells (± doxy) behaved like the wild-type huntingtin cells (data not shown). a-d, *p < 0.05 and ** p < 0.01 versus each cell type grown in the absence of doxy (ANOVA). e, Localization of the 68 kDa-active form of SREBP1 in nuclear preparations from HD and control mice. Histone H1 was used as the loading control for the nuclear fraction. The graph shows the quantitative densitometric analysis of the nuclear form of SREBP1 compared with histone H1. The Western blot represents one of five experiments that gave similar results. The data are expressed as percentages of controls (Co); the SD in the graph was obtained by including all of the experiments. **p < 0.01 versus control tissues (ANOVA). Error bars represent SEM.
To investigate further the impairment in SREBP1 translocation, we exposed parental cells, and cells expressing wild-type or mutant huntingtin, to delipidated conditions, and analyzed them immunocytochemically and confocally 24 h after the addition of the inducer. Under these conditions, most (>80 ± 12%) of the cells expressing wild-type huntingtin showed the immunoreactive signal localized inside the nucleus, consistent with SREBP1 translocation (Fig. 5d), and the same was observed in the parental cell cultures (see graph); however, nuclear SREBP1 could be detected in only 50 ± 14% of the cells bearing mutant huntingtin.
Together, this suggests that both the nuclear translocation of SREBP1 and the activation of the SRE are compromised in HD cells. To extend these data in vivo, we evaluated the levels of SREBP1 in nuclear extracts of HD mouse brain. As shown in Figure 5e, the nuclear level SREBP1 was reduced in 10-week-old HD mice versus controls.
Because SREBP2 activity is also linked to cholesterol biosynthesis (Horton et al., 2002), we evaluated nuclear SREBP2 levels in extracts from HD and control samples and found that the levels of active SREBP2 were also reduced in HD cells (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Together, these results demonstrate that HD nuclei have reduced levels of both SREBP1 and SREBP2.
A pathological hallmark of Huntington's disease and other polyglutamine diseases is the presence of inclusions or aggregates of the expanded polyglutamine protein (Davies et al., 1997; Scherzinger et al., 1997). The results of previous studies suggest that polyglutamine inclusions may sequester polyglutamine-containing transcription factors and deplete their concentration in the nucleus, thus altering gene expression (Cha, 2000; Li and Li, 2004), and thus, to test whether the reduced level of nuclear SREBP in HD is attributable to its sequestration into mutant huntingtin aggregates, we used Western blots to analyze brain lysates from R6/2 transgenic mice and the inducible HD cell lines. Despite the absence of detectable aggregates (Sipione et al., 2002) (supplemental Fig. 5a, available at www.jneurosci.org as supplemental material), the latter showed reduced nuclear SREBP levels. The Western blots of nuclear and cytoplasmic lysates from the brain of R6/2 mice revealed the expected mutant huntingtin aggregates in the stacking gel, but no immunoreactive bands were codetected using the anti-SREBP antibody (supplemental Fig. 5b, available at www.jneurosci.org as supplemental material). We therefore conclude that the reduced nuclear SREBP levels in the HD samples were unlikely to be because of SREBP sequestration into aggregates.
To estimate the pathogenic impact of the dysfunction and the potential value of its rescue in HD, we investigated whether the addition of exogenous cholesterol may be beneficial to primary neurons undergoing cell death after the transfection of mutant huntingtin. Primary striatal neurons were coelectroporated with plasmids encoding the 480 N-terminal amino acids of human huntingtin containing an expanded CAG repeat (Htt480.68) and GFP cDNA (for the expression levels of Htt480.68 and GFP, see supplemental Fig. 6, available at www.jneurosci.org as supplemental material). On the basis of the number of GFP+ cells (Saudou et al., 1998), 50% of the cells expressing mutant huntingtin had died after 6 d; this cell death was specifically attributable to mutant huntingtin because no cell death was observed after transfection with wild-type huntingtin (data not shown). As demonstrated previously, the addition of BDNF to cultures prevents mutant huntingtin-induced cell death in a dose-dependent manner, with maximum effect after the addition of 5 ng/ml at the time of plating (Saudou et al., 1998; Zuccato et al., 2001) (Fig. 6a). Importantly, the addition of increasing cholesterol concentrations (from 0.3 to 10 μm) promoted cell survival in a dose-dependent manner and to an extent similar (although not identical) to the results obtained after BDNF. Figure 6b shows the combined data of single replicates as ratios to the effect of BDNF.
Figure 6.

Cholesterol rescues the cell death induced by the transfection of mutant huntingtin in primary neurons. a, Primary rat striatal neurons were prepared and electroporated with plasmids encoding Htt480.68 and GFP and then plated into 96-well microplates. The expression of Htt480.68 and GFP was investigated by means of Western blotting using 1HU-4C8 antibody (Euromedex) for huntingtin and anti-GFP antibody (clone 7.1 and 13.1; Roche Diagnostics) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). The quantities were normalized to those of β-tubulin revealed using anti-β-tubulin antibody (clone DM1A; Sigma). Eight wells per plate were untreated [negative control (NC)] or treated with 5 ng/ml BDNF or increasing concentrations of cholesterol (chol) dissolved in DMSO. BDNF was used as a positive control. All of the wells received the same final concentration of DMSO (0.5%). Six days after plating, the GFP-positive cells were counted using a fluorescence imaging plate reader (Trophos Flash Cytometer). One of two experiments is shown. b, The combined data from two experiments are expressed as ratios relating to the effect of BDNF. Bonferroni's post hoc analyses revealed significant differences between each tested concentration and the negative control (except for 0.3 μm, as shown in the graph), as well as between each tested concentration and the positive controls. Moreover, among the tested concentrations, significance was reached between 10 and 1 μm, 10 and 0.3 μm, and 3 and 0.3 μm, thus showing the progressive effect of cholesterol dosing. ANOVA also revealed significant differences between treatments (F(5,90) = 34.77; p < 0.0001) and no differences within each treatment group (F(7,40) = 0.048, p = 1 for experiment 1, and F(7,40) = 0.17, p = 0.99 for experiment 2). Error bars represent SEM.
Discussion
Cholesterol is a multifaceted molecule that acts as an essential membrane component, a cofactor for signaling molecules, and a precursor of steroid hormones (Pfrieger, 2003a,b). Although its role in brain is far from understood, adult neurons depend on cholesterol for most of their activities, and defects in cholesterol homeostasis have been linked with neurodegenerative diseases. We report, for the first time, that cholesterol biosynthesis is impaired in cells carrying the HD mutation and that this dysfunction is specific to mutant huntingtin because inducible cells expressing the wild-type protein behave like mock-untransfected cells. In particular, the levels of mRNA for the genes of the cholesterogenic pathway are severely reduced in rodent HD cell models, HD transgenic mice, and human postmortem brain tissues. The molecular mechanism underlying this dysfunction is a mutant huntingtin-dependent decrease in the amount of active SREBP, which translocates from the cytosolic compartment to the nucleus in which, under conditions of low cholesterol levels, it should bind to, and activate the transcription of, SRE-controlled genes. We found reduced SREBP translocation in vitro and in HD mouse brain tissue.
The reduced entry of SREBP into the nucleus leads to decreased cholesterol synthesis, a finding that was confirmed in fibroblasts from HD patients. It is known that the intermediates of cholesterol biosynthesis are critical for neurite outgrowth, synaptic activity and stability. Neurite loss is an early manifestation of various neurodegenerative disorders, including HD, in which morphological abnormalities of the brain and defects in synaptic activity have been documented (Li et al., 2003; Levine et al., 2004; Schulz et al., 2004).
Our data indicate that cholesterol dysfunction is linked to mutant huntingtin, presumably because of its toxic activity. It is also possible that the molecular network controlling cholesterol biosynthesis is already compromised during development, even in the absence of a detectable phenotype, and that the late-onset clinical signs may determine “compensatory” mechanisms, because gene mutation carriers show subtle molecular dysfunctions at presymptomatic stages, even when they show no detectable clinical signs (Varani et al., 2003; Rosas et al., 2003).
Importantly, cholesterol turnover is known to be very slow in adult brain and could emphasize the late-onset HD phenotype. If the supply of cholesterol to the adult brain is attributable to de novo synthesis during development and is sufficient to maintain neuronal functions in adulthood, it is likely that altered cholesterol biosynthesis appears later than during the first phases of life. Impaired cholesterol biosynthesis may contribute to the synaptic activity and remodeling, therefore possibly contributing to the neuronal dysfunction observed in HD. It has also been suggested that different neuronal populations and brain areas may require different amounts of cholesterol that may make them more vulnerable (Pfrieger, 2003a,b).
Mutant huntingtin protein may mechanistically interfere with SREBP activation at different levels: (1) during the SCAP-mediated transport of SREBP from the ER to the Golgi; (2) during SREBP cleavage by S1p and S2p proteases in the Golgi; or (3) during the translocation of active SREBP into the nucleus. The demonstration that an exogenous 68 kDa active form of SREBP is also unable to activate the SRE fully in the presence of mutant huntingtin supports the hypothesis that huntingtin acts down-stream of SREBP activation, at the level of its entry into the nucleus and that reduced SREBP nuclear translocation/activity may depend on a broad modification of the systems controlling the nucleocytoplasmic shuttling of transcription factors (Nakielny and Dreyfuss, 1999; Ossareh-Nazari et al., 2001). Alternatively, mutant huntingtin may affect the posttranslational modifications of SREBPs, whose SUMOylation subsequently reduces their own transcriptional activities (Hirano et al., 2003), and increased SUMO-1 (small ubiquitin-related modifier-1) activity has been reported in HD (Steffan et al., 2004).
Finally, our data also show that addition of cholesterol partially rescues neurons expressing mutant huntingtin from cell death, thus indicating that treatments aimed at restoring cholesterol levels may be beneficial in HD.
Dysfunctions such as those reported here could perhaps also be exploited as a means of monitoring disease evolution in peripheral cells. The changes in cholesterol biosynthesis observed in human primary HD fibroblasts imply that extraneural tissues are also affected by the mutation. The reduced cholesterol biosynthesis observed in fibroblasts anticipates the possibility that cholesterol measurement in these cells may be a good target in the search for biomarkers in HD (Varani et al., 2003; Mayeux, 2004).
Footnotes
This work was supported by Telethon (Italy), the Hereditary Disease Foundation, the Huntington's Disease Society of America, and Fondazione Cariplo (Italy). This study stems from a microchip analysis in our inducible HD cells (Sipione et al., 2002) performed in the context of the Huntington Disease Array Group (Hereditary Disease Foundation). We are grateful to T. F. Osborne, Nico Dantuma, R. Lehner, and the members of the laboratory for their very helpful comments and suggestions; to T. F. Osborne for the gift of the pSynSRE, HSV-TK-SREBP1a and anti-SREBP2 (7D4) antibody; to D. Vance for the CMV-SREBP1a Nt construct; to R. Faull for kindly providing the human HD brain specimens to J.M.O.; and to Erika Reitano and Francesca Zanin for their technical support. The animals were cared for in accordance with institutional guidelines.
Correspondence should be addressed to Prof. Elena Cattaneo, Department of Pharmacological Sciences, University of Milan, 20133 Milan, Italy. E-mail: elena.cattaneo@unimi.it.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259932-08$15.00/0
M.V. and D.R. contributed equally to this work.
D.G. and C.Z. contributed equally to this work.
References
- Barres BA, Smith SJ (2001) Cholesterol—making or breaking the synapse. Science 294: 1296-1297. [DOI] [PubMed] [Google Scholar]
- Battaile KP, Steiner RD (2000) Smith-Lemli-Opitz syndrome: the first mal-formation syndrome associated with defective cholesterol synthesis. Mol Genet Metab 71: 154-162. [DOI] [PubMed] [Google Scholar]
- Bennett MK, Toth JI, Osborne TF (2004) Selective association of sterol regulatory element-binding protein isoforms with target promoters in vivo. J Biol Chem 279: 37360-37367. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL (2002) Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol Cell 10: 237-245. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL (1997) The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89: 331-340. [DOI] [PubMed] [Google Scholar]
- Brown MS, Faust JR, Goldstein JL (1978) Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase. J Biol Chem 253: 1121-1128. [PubMed] [Google Scholar]
- Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S (2001) Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci 24: 182-187. [DOI] [PubMed] [Google Scholar]
- Cha JH (2000) Transcriptional dysregulation in Hungtington's disease. Trends Neurosci 9: 387-392. [DOI] [PubMed] [Google Scholar]
- Corsini A, Granata A, Fumagalli R, Paoletti R (1986) Calcium antagonists and low density lipoproteins metabolism by human fibroblasts and by human hepatoma cell line HEP G2. Pharmacol Res Commun 18: 1-16. [DOI] [PubMed] [Google Scholar]
- Corsini A, Bernini F, Cighetti G, Soma M, Galli G, Fumagalli R (1987) Lipophilic beta-adrenoceptor antagonists stimulate cholesterol biosynthesis in human skin fibroblasts. Biochem Pharmacol 15: 1901-1906. [DOI] [PubMed] [Google Scholar]
- Corsini A, McCarthy BJ, Granata A, Soria LF, Fantappiè S, Bernini, Romano C, Romano L, Fumagalli R, Catapano AL (1991) Familiar defective apo B-100, characterization of an italian family. Eur J Clin Invest 21: 389-397. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90: 537-548. [DOI] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD (2001) Cholesterol metabolism in the brain. Curr Opin Lipidol 12: 105-112. [DOI] [PubMed] [Google Scholar]
- Dooley KA, Millinder S, Osborne T (1998) Sterol regulation of 3-hydroxy-3-methylglutaryl coenzyme A synthase gene through a direct interaction between sterol regulatory element binding protein and the trimeric CCAAT-binding factor/nuclear factor Y. J Biol Chem 273: 1349-1356. [DOI] [PubMed] [Google Scholar]
- Hannah VC, Ou J, Luong A, Goldstein JL, Brown MS (2001) Unsaturated fatty acids down-regulate SREBP isoforms 1a and 1c by two mechanisms in HEK-293 cells. J Biol Chem 276: 4365-4372. [DOI] [PubMed] [Google Scholar]
- Hering HE, Lin C, Sheng M (2003) Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J Neurosci 23: 3262-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Murata S, Tanaka K, Shimizu M, Sato R (2003) Sterol regulatory element-binding proteins are negatively regulated through SUMO-1 modification independent of the ubiquitin/26 S proteasome pathway. J Biol Chem 278: 16809-16819. [DOI] [PubMed] [Google Scholar]
- Horton JD, Brown MS, Goldstein JL (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109: 1125-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shah N, Warrington J, Anderson N, Park S, Brown MS, Goldstein JL (2003) Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA 100: 12027-12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL, Wang X (1993) SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci USA 90: 11603-11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971-983. [DOI] [PubMed] [Google Scholar]
- Jurevics H, Morell P (1995) Cholesterol for synthesis of myelin is made locally, not imported into brain. J Neurochem 64: 895-901. [DOI] [PubMed] [Google Scholar]
- Levine MS, Cepeda C, Hickey MA, Fleming SM, Chesselet MF (2004) Genetic mouse models of Huntington's and Parkinson's diseases: illuminating but imperfect. Trends Neurosci 11: 691-697. [DOI] [PubMed] [Google Scholar]
- Li JY, Plomann M, Brundin P (2003) Huntington's disease: a synaptopathy? Trends Mol Med 10: 414-420. [DOI] [PubMed] [Google Scholar]
- Li SH, Li XJ (2004) Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet 20: 146-154. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Huang Y (1999) Apolipoprotein E: from atherosclerosis to Alzheimer's disease and beyond. Curr Opin Lipidol 10: 207-217. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87: 493-506. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nagler K, Schumacher S, Goritz C, Muller E, Otto A, Pfrieger FW (2001) CNS synaptogenesis promoted by glia-derived cholesterol. Science 294: 1354-1357. [DOI] [PubMed] [Google Scholar]
- Mayeux R (2004) Biomarkers: potential uses and limitations. NeuroRx 1: 182-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakielny S, Dreyfuss G (1999) Transport of proteins and RNAs in and out of the nucleus. Cell 99: 677-690. [DOI] [PubMed] [Google Scholar]
- Ossareh-Nazari B, Gwizdek C, Dargemont C (2001) Protein export from the nucleus. Traffic 2: 684-698. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW (2003a) Role of cholesterol in synapse formation and function. Biochim Biophys Acta 1610: 271-280. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW (2003b) Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci 60: 1158-1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D'amato CJ, Penney JB, Young AB (1988) Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA 85: 5733-5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti D, Bauer JH, De-Fraja C, Conti L, Sipione S, Sciorati C, Clementi E, Hackam A, Hayden MR, Li Y, Cooper JK, Ross CA, Govoni S, Vincenz C, Cattaneo E (2000) Wild-type huntingtin protects from apoptosis upstream of caspase-3. J Neurosci 20: 3705-3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME, Caplan K, Marek K, Seidman LJ, Makris N, Jenkins BG, Goldstein JM (2003) Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology 10: 1615-1620. [DOI] [PubMed] [Google Scholar]
- Sakai J, Elizabeth A, Duncan EA, Rawson RB, Hua X, Brown MS, Goldstein JL (1996) Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85: 1037-1046. [DOI] [PubMed] [Google Scholar]
- Sanchez HB, Yieh L, Osborne TF (1995) Cooperation by sterol regulatory element-binding protein and Sp1 in sterol regulation of low density lipoprotein receptor gene. J Biol Chem 270: 1161-1169. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95: 55-66. [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE (1997) Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90: 549-558. [DOI] [PubMed] [Google Scholar]
- Schulz JG, Bosel J, Stoeckel M, Megow D, Dirnagl U, Endres M (2004) HMG-CoA reductase inhibition causes neurite loss by interfering with geranylgeranylpyrophosphate synthesis. J Neurochem 1: 24-32. [DOI] [PubMed] [Google Scholar]
- Shimomura I, Bashmakov Y, Shimano H, Horton JD, Goldstein JL, Brown MS (1997) Cholesterol feeding reduces nuclear forms of sterol regulatory element binding proteins in hamster liver. Proc Natl Acad Sci USA 94: 12354-12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J, Kooperberg C, Olson JM, Cattaneo E (2002) Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum Mol Genet 11: 1953-1965. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL (2004) SUMO Modification of huntingtin and Huntington's disease pathology. Science 304: 100-104. [DOI] [PubMed] [Google Scholar]
- Thiele C, Hannah MJ, Fahrenholz F, Huttner WB (2000) Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicle. Nat Cell Biol 2: 42-49. [DOI] [PubMed] [Google Scholar]
- Trottier Y, Devys D, Imbert G, Saudou F, An I, Lutz Y, Weber C, Agid Y, Hirsch EC, Mandel JL (1995) Cellular localization of the Huntington's disease protein and discrimination of the normal and mutated form. Nat Genet 10: 104-110. [DOI] [PubMed] [Google Scholar]
- Varani K, Abbracchio MP, Cannella M, Cislaghi G, Giallonardo P, Mariotti C, Cattabriga E, Cattabeni F, Borea PA, Squitieri F, Cattaneo E (2003) Aberrant A2A receptor function in peripheral blood cells in Huntington's disease. FASEB J 17: 2148-2150. [DOI] [PubMed] [Google Scholar]
- William CD, Avigan J (1972) In vitro effects of serum proteins and lipids on lipid synthesis in human skin fibroblasts and leukocytes grown in culture. Biochim Biophys Acta 260: 413-423. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E (2001) Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science 293: 493-498. [DOI] [PubMed] [Google Scholar]