

Abstract
Current knowledge about the molecular mechanisms of NMDA receptor (NMDAR)-independent long-term potentiation (LTP) in the hippocampus and its function for memory formation in the behaving animal is limited. NMDAR-independent LTP in the CA1 region is thought to require activity of postsynaptic L-type voltage-dependent Ca2+ channels (Cav1.x), but the underlying channel isoform remains unknown. We evaluated the function of the Cav1.2 L-type Ca2+ channel for spatial learning, synaptic plasticity, and triggering of learning-associated biochemical processes using a mouse line with an inactivation of the CACNA1C (Cav1.2) gene in the hippocampus and neocortex (Cav1.2HCKO). This model shows (1) a selective loss of protein synthesis-dependent NMDAR-independent Schaffer collateral/CA1 late-phase LTP (L-LTP), (2) a severe impairment of hippocampus-dependent spatial memory, and (3) decreased activation of the mitogen-activated protein kinase (MAPK) pathway and reduced cAMP response element (CRE)-dependent transcription in CA1 pyramidal neurons. Our results provide strong evidence for a role of L-type Ca2+ channel-dependent, NMDAR-independent hippocampal L-LTP in the formation of spatial memory in the behaving animal and for a function of the MAPK/CREB (CRE-binding protein) signaling cascade in linking Cav1.2 channel-mediated Ca2+ influx to either process.
Keywords: calcium channels, learning, memory, NMDA receptor independent, Cav1.2, synaptic plasticity
Introduction
Glutamatergic synapses in the CNS can undergo activity-dependent long-lasting changes of synaptic efficacy such as long-term potentiation (LTP) (Bliss and Lomo, 1973). The mechanisms involved in these synaptic modifications are thought to give insight into the molecular processes underlying memory in the behaving animal and, ultimately, in humans (Bliss and Collingridge, 1993; Kandel, 2001; Martin and Morris, 2002; Malenka and Bear, 2004). Depending on the pattern of neuronal stimulation, LTP consists of an early phase (E-LTP) that requires covalent protein modifications and a late phase (L-LTP) that is protein synthesis dependent (Frey et al., 1988). Specifically, L-LTP is believed to be a cellular equivalent of long-lasting memory (Kandel, 2001). In line with this view, inhibition of protein synthesis not only blocks hippocampal L-LTP but also impairs hippocampus-dependent learning. Both hippocampal synaptic plasticity and various forms of learning-related behavior critically depend on the mitogen-activated protein kinase (MAPK)/extracellular signal-related protein kinase (ERK) cascade and the resulting stimulation of gene transcription and de novo protein synthesis [e.g., via cAMP response element-binding protein (CREB)] (English and Sweatt, 1997; Atkins et al., 1998; Hardingham et al., 2001; Kandel, 2001; Wu et al., 2001; Pittenger et al., 2002; Thomas and Huganir, 2004). Induction of L-LTP at Schaffer collateral/CA1 synapses, as well as activation of the ERK/CREB pathway in hippocampal CA1 neurons, requires an increase in the postsynaptic intracellular Ca2+ concentration (Shaywitz and Greenberg, 1999; Kandel, 2001). Ca2+ influx via L-type Ca2+ (Cav1.x) channels can specifically trigger the transcription of Ca2+-regulated genes (e.g., Zif/268) and brain-derived neurotrophic factor (BDNF), which play a major role in learning (Murphy et al., 1991; West et al., 2001). Ca2+ influx via postsynaptic Cav1.x channels can also support a form of NMDA receptor (NMDAR)-independent LTP (Grover and Teyler, 1990; Grover, 1998; Morgan and Teyler, 1999) and sustained CREB phosphorylation with subsequent activation of cAMP response element (CRE)-dependent gene expression in hippocampal neurons (Impey et al., 1996; Dolmetsch et al., 2001). However, the functional significance of these findings for memory formation remains unclear, because compelling evidence for a role of L-type Ca2+ channel-dependent, NMDAR-independent synaptic plasticity in the behaving animal is missing.
Hippocampal pyramidal neurons express predominantly the Cav1.2 channel and only rather low levels of the Cav1.3 isoform (Hell et al., 1993; Davare et al., 2001; Sinnegger-Brauns et al., 2004). Accordingly, a knock-out mouse model lacking the Cav1.3 channel showed neither a defect in hippocampus-dependent learning nor a defect in hippocampal LTP (Clark et al., 2003). To investigate their role in hippocampal LTP and memory formation, we generated a mouse line (Cav1.2HCKO) with an inactivation of the CACNA1C (Cav1.2) gene, mainly in the hippocampus and neocortex.
Here, we report that Cav1.2HCKO mice show a defect in protein synthesis-dependent, NMDAR-independent LTP in the CA1 region that is paralleled by a deficit in spatial learning and an impairment of CREB activation. These findings demonstrate that Cav1.2 L-type Ca2+ channels serve a critical function in hippocampus-dependent spatial memory by coupling NMDAR-independent synaptic activity to transcriptional events, which are thought to be molecular prerequisites for L-LTP and learning.
Materials and Methods
Conditional inactivation of the Cav1.2 gene. The generation of mice lacking Cav1.2 channels is described in supplemental Methods (available at www.jneurosci.org as supplemental material).
Behavior. Animals were maintained on a 12 h light/dark cycle and had access to water and food ad libitum. Litter-matched male mice of each genotype at the age of 8-15 weeks were used in all experiments. Experiments were performed with the investigator unaware of the genotype. Behavioral analysis was done as described previously (Kleppisch et al., 2003) and is delineated in detail in supplemental Methods (available at www.jneurosci.org as supplemental material).
Field EPSP recordings. Field EPSPs (fEPSPs) and whole-cell currents from CA1 pyramidal neurons were recorded in transverse hippocampal slices using the Axoclamp 2B amplifier (Molecular Devices, Union City, CA), and an EPC9 and the Pulse software (HEKA, Lambrecht/Pfalz, Germany), respectively, and a submersion-type recording chamber as described previously (Kleppisch et al., 2003). Experiments are delineated in detail in supplemental Methods (available at www.jneurosci.org as supplemental material).
Western blot analysis. Western blotting was performed using standard methods. Briefly, brain homogenates were separated on an SDS-PAGE, blotted on a polyvinylidene difluoride membrane (Millipore, Bedford, MA), and probed with Cav1.2- and Cav1.3-specific antibodies (Moosmang et al., 2003), antibodies against CREB and phospho-CREB (pCREB) Ser133 (Upstate Biotechnology, Lake Placid, NY), and ERK1/2 (Upstate Biotechnology) and phospho-ERK (pERK) Thr202/Tyr204 (Cell Signaling, Beverly, MA, and Promega, Madison, WI), respectively. Equal loading of slots was ascertained by the use of a polyclonal anti-cGMP-dependent kinase I antibody. Antibodies were visualized by the ECL system (NEN, Boston, MA). For densitometry, blots were digitized and analyzed using Quantity One software (Bio-Rad, Hercules, CA).
Agonist treatment of perfused hippocampal slices. Transverse 400 μm hippocampal slices were continuously perfused with oxygenated artificial CSF (aCSF) at 32°C for 2-3 h before the addition of tetraethylammonium chloride (TEA-Cl) (Sigma, St. Louis, MO). d-APV (50 μm; Sigma) or 1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)-butadiene (U0126) (40 μm; Tocris, Ballwin, MO) were added to the perfusate 1 h before and during the stimulation.
Immunocytochemistry. Hippocampal slices were fixed in 4% paraformaldehyde in PBS, paraffin-embedded, and sectioned at 8 μm intervals. Immunocytochemistry for β-galactosidase was performed using standard procedures with a rabbit anti-β-galactosidase antibody at 1:1000 dilution (Cappel, Cochranville, PA) and Cy3 goat-anti-rabbit IgG at 2 μg/ml (Jackson Immunochemicals, West Grove, PA). For immunocytochemistry of phospho-CREB and phospho-ERK1/2, slices were fixed in ice-cold PBS containing 4% paraformaldehyde for 30 min after the stimulation. A dual-label procedure was used: antibodies were used at 1:1000 dilution and the DNA stain/nuclear marker Hoechst 33342 was used at 200 μg/ml (Sigma). Images were captured on a Zeiss (Oberkochen, Germany) LSM 510 laser scanning confocal microscope.
Quantification of immunocytochemistry. Quantification was performed with Zeiss image analysis software. Briefly, integrated pixel intensity was measured in the desired region. A constant pixel area was used for 12 independent measurements, which were averaged. The averaged integrated pixel intensity for groups of slices was statistically analyzed. Imaging and quantitation were conducted in a blind manner.
Statistics. Data are expressed as mean + SEM. Statistical evaluations were performed by using ANOVA (repeated measures), and t tests were used to assess differences among individual time points.
Results
Regional inactivation of the CACNA1C gene in the murine hippocampus
We used the Cre recombinase system, using Nex-Cre transgenic mice (Schwab et al., 2000), to create a mouse line (Cav1.2HCKO mice) with an inactivation of the CACNA1C gene in the cerebral cortex and hippocampus (see supplemental Results and supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
CA1 pyramidal cells in Cav1.2HCKO mice lack Cav1.2 L-type Ca2+ currents
CA1 pyramidal cells of hippocampal slices from adult control and Cav1.2HCKO mice showed robust whole-cell Ca2+ inward currents at test potentials positive to -40 mV (Fig. 1a). To demonstrate the loss of functional Cav1.2 channels, we assessed the dihydropyridine (DHP)-sensitive Ca2+ inward currents evoked during voltage-clamp ramps (Fig. 1d-f). When the DHP isradipine (10 μm) was added to the bath perfusion after reaching a stable baseline current, we observed a prominent decrease of the Ca2+ current amplitude in CA1 neurons from control mice. In sharp contrast, isradipine had virtually no effect on Ca2+ currents in CA1 neurons of Cav1.2HCKO mice. On average, the portion of the peak Ca2+ inward current inhibited by isradipine was 22.3 ± 2.8% (control; n = 14) and 4.4 ± 1.8% (Cav1.2HCKO; n = 9). This is equivalent to >80% reduction of the DHP-sensitive current in CA1 neurons of the mutant mice (p < 0.001). The tiny residual DHP-sensitive current is likely caused by the Cav1.3 L-type channel. Magee et al. (1996) have suggested that a population of DHP-sensitive Ca2+ channels in CA1 pyramidal cells may be active under physiological conditions at potentials as hyperpolarized as -70 mV. Therefore, we tested for a possible impact of the Cav1.2 channel knock-out on resting membrane potential (RP) and input resistance (RN) at RP. Neither parameter was significantly altered in CA1 pyramidal cells of mutant mice (Fig. 1b,c). Control and Cav1.2HCKO mice exhibited an RP of -70.8 ± 0.9 mV (n = 13) and -69.9 ± 1.0 mV (n = 10), respectively. Corresponding RN were 92 ± 8 MΩ (n = 8) and 107 ± 12 MΩ (n = 6), respectively.
Figure 1.

CA1 pyramidal cells of mice with an inactivation of the CACNA1C gene (in Cav1.2HCKO) in the hippocampus display a nearly complete loss of L-type Ca2+ currents without changes in RP and RN. a, Ca2+ channel currents evoked by voltage-clamp steps in pyramidal neurons of hippocampal slices from control (ctr) and Cav1.2HCKO (ko) mice. The traces illustrated correspond to test potentials of -30, -20, and -10 mV. The holding potential was -65 mV, and 2 mm Ca2+ was used as a charge carrier. Calibration: 50 ms, 100 pA. b, Average RP measured in CA1 neurons of hippocampal slices from control (▪) (n = 13 cells from 6 mice) and Cav1.2HCKO (□) (n = 10 cells from 6 mice) mice in the current-clamp mode. Means + SEM are shown. c, Average RN of hippocampal CA1 neurons from control (▪) (n = 8 cells from 5 mice) and Cav1.2HCKO (□) (n = 6 cells, 5 mice) mice determined by injecting hyperpolarizing currents from a membrane potential of -70 mV. Means + SEM are shown. d, Representative examples of whole-cell Ca2+ currents (ICa) evoked by voltage-clamp ramps from -80 to +80 mV (0.5 ms/mV) in hippocampal CA1 neurons from ctr and ko mice before (-isr) and after (+isr) application of the L-type channel blocker isradipine (10 μm). The holding potential was -65 mV, and 2 mm Ca2+ was used as a charge carrier. e, Fraction of the total voltage-dependent Ca2+ inward current blocked by isradipine for the representative examples shown in d. Fitted I-V curves of the DHP-sensitive current component in control (filled in black) and Cav1.2HCKO (filled in white) mice, normalized to the corresponding peak inward current in the absence of isradipine (for details on fit function, see Materials and Methods), are represented. f, Average component of the total voltage-dependent Ca2+ current block by isradipine. The reduction of the peak inward current in hippocampal CA1 neurons from control (▪) (n = 14 cells from 6 mice) and Cav1.2HCKO (□) (n = 9 from 5 mice) mice, shown in percentage, is represented. Data are means ± SEM (***p < 0.001).
In summary, the data above demonstrate an efficient deletion of the CACNA1C gene in hippocampal CA1 neurons of Cav1.2HCKO mice without affecting RP and RN at rest.
NMDAR-independent hippocampal synaptic plasticity critically depends on the function of the Cav1.2 Ca2+ channel
To evaluate the role of Cav1.2 channels in synaptic plasticity, we studied NMDAR-dependent and NMDAR-independent forms of LTP. Initially, we compared basal synaptic transmission in hippocampal slices from control and Cav1.2HCKO mice. We observed no difference between the two genotypes in the dependency of the fEPSP slope on the stimulus intensity (Fig. 2a) and the fiber volley amplitude (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Paired-pulse facilitation (PPF), a form of short-term plasticity thought to rely on presynaptic mechanisms, was also normal in the Cav1.2HCKO mice (Fig. 2b). Moreover, Cav1.2HCKO mice showed normal LTP in response to a weak theta burst (Fig. 2c), which is known to induce NMDAR-dependent LTP (Bliss and Collingridge, 1993; Kleppisch et al., 1999). Collectively, these findings show that mutant mice lack a general defect in basal synaptic transmission and have normal NMDAR-dependent LTP.
Figure 2.
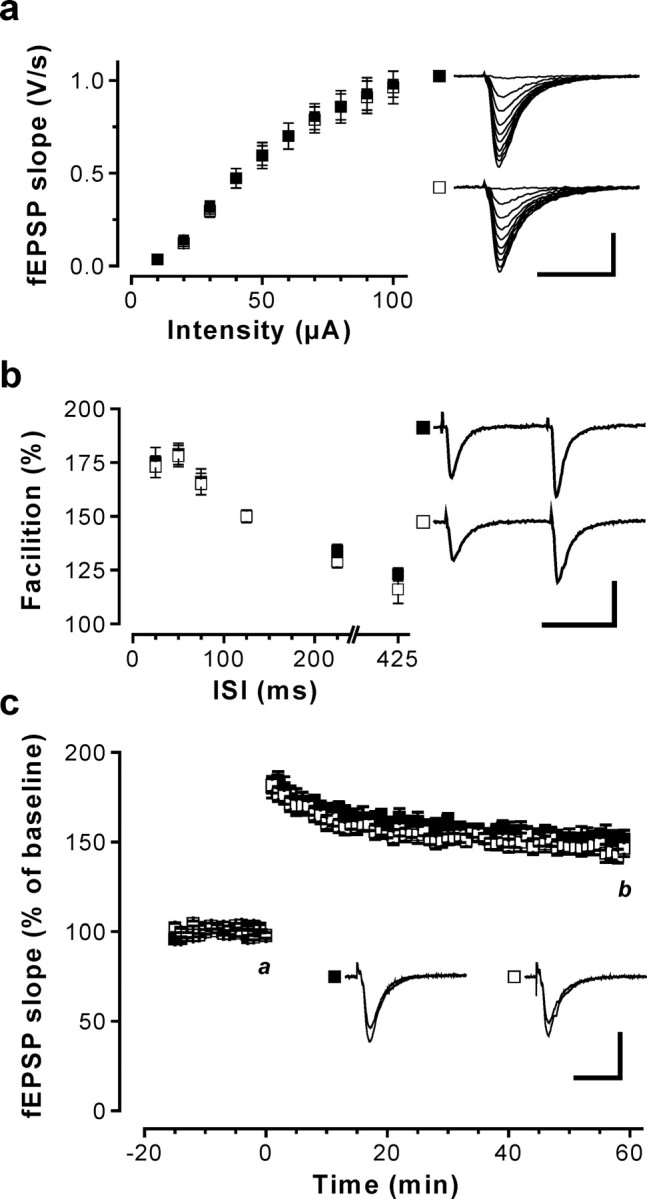
Basal synaptic transmission and NMDAR-dependent synaptic plasticity in the hippocampal CA1 region of Cav1.2HCKO mice. fEPSPs in response to stimulation of the Schaffer collaterals were recorded in slices from control (▪) and Cav1.2HCKO (□) mice. a, Input-output relationship curves obtained from control (37 slices from 16 mice) and Cav1.2HCKO mice (41 slices from 21 mice) were not different. Representative recordings are shown on the right (calibration: 20 ms, 1 mV). b, Short-term plasticity was assessed by measuring PPF at interstimulus intervals (ISIs) of 25, 50, 75, 125, 225, and 425 ms. PPF was not altered in control (n = 27 slices from 12 mice) compared with Cav1.2HCKO mice (n = 25 slices from 13 mice). Representative PPF recordings for the 75 ms interval are shown on the right (calibration: 50 ms, 1 mV). c, NMDAR-dependent Schaffer collateral/CA1 LTP was induced by theta-burst stimulation (12 times 4 pulses at 100 Hz; 200 ms pause) applied at time 0. The time course of the fEPSP slope (mean + SEM) was obtained from control (8 slices from 3 mice) and Cav1.2HCKO (9 slices from 4 mice) slices. Representative fEPSPs recorded at the times indicated (a, b) are shown in the corresponding inset. Calibration: 20 ms, 1 mV. Error bars indicate SE.
Contrary to the situation in NMDAR-dependent synaptic plasticity, the Cav1.2 Ca2+ channel might be critical for NMDAR-independent synaptic plasticity. Robust NMDAR-independent LTP can be induced using 200 Hz tetanus (Grover and Teyler, 1990). Taking into account their postsynaptic localization, a recent report suggests that activation of Cav1.x channels by 200 Hz tetanic stimulation triggers a retrograde signal, leading to presynaptic BDNF release and, ultimately, the induction of a presynaptic component of LTP (Zakharenko et al., 2003). Indeed, after a 200 Hz tetanus, Cav1.2HCKO mice showed significantly reduced LTP compared with control mice (factor genotype, F = 4.72; p = 0.048; ANOVA); ∼2 h after the tetanus, fEPSP slope was 136.5 ± 6.5 (n = 7) versus 167.6 ± 10.5 (n = 10). To explore whether this decrease in LTP was caused by a reduction of the NMDAR-independent component, we further examined 200 Hz LTP in the presence of the NMDAR antagonist APV (50 μm). Under these conditions, again, Cav1.2HCKO showed significantly decreased LTP (factor genotype, F = 7.55; p = 0.018; ANOVA) (Fig. 3a). Moreover, the extent of the reduction in LTP in the presence of APV was nearly identical to that without APV, arguing for a selective defect in NMDAR-independent LTP in Cav1.2HCKO mice. To further substantiate the evidence for a specific role of Cav1.2 channels in NMDAR-independent LTP, we used multiple 100 Hz tetanic stimulation (Impey et al., 1996) and examined L-LTP in the presence of APV for up to 4 h after the tetanus. Corroborating the results presented above, significant L-LTP was observed exclusively in the control but not in Cav1.2HCKO mice (factor genotype, F = 6.28; p = 0.02; ANOVA) (Fig. 3b).
Figure 3.

NMDAR-independent LTP in the hippocampal CA1 region of Cav1.2HCKO mice. fEPSPs in response to stimulation of the Schaffer collaterals were recorded in slices from control (▪) and Cav1.2HCKO (□) mice. a, LTP was induced at time 0 by a 200 Hz tetanic stimulation with the NMDAR antagonist APV (50 μm) present throughout the experiment. The time course of the fEPSP slope represents mean + SEM of experiments in eight slices from five mice for each genotype. Representative fEPSPs recorded at the times indicated (a, b) are shown in the corresponding inset. b, LTP was induced at time 0 by multiple strong 100 Hz tetani with the NMDAR antagonist APV (50 μm) present throughout the experiment. The time course of the fEPSP slope represents the mean + SEM in control (10 slices from 6 mice) and Cav1.2HCKO (14 slices from 6 mice) slices. Representative fEPSPs recorded at the times indicated (a, b) are shown in the corresponding inset. c, LTPK was induced by superfusing hippocampal slices with aCSF containing 25 mm potassium channel blocker TEA for 15 min (indicated by the bar). The NMDAR antagonist, APV (50 μm), was present throughout the experiment. The time course of the fEPSP slope represents the mean ± SEM in control (15 slices from 12 mice) and Cav1.2HCKO mice (11 slices from 7 mice). Representative fEPSPs recorded at the times indicated (a, b) are shown in the corresponding inset. d, Effect of the protein synthesis inhibitor anisomycin (20 μm) and the MEK1/2 inhibitor U0126 (40 μm) on L-LTPK in control and Cav1.2HCKO mice. ▪, The magnitude of LTPK 2 h after treatment with TEA in control mice in normal aCSF, in the presence of anisomycin (n = 8 slices from 5 mice) and U0126 (n = 7 slices from 5 mice) is illustrated. □, The magnitude of LTPK 2 h after treatment with TEA in control mice in normal aCSF and in the presence of anisomycin (n = 11 slices from 7 mice), respectively, is illustrated. *Significant difference (p < 0.05). Calibrations: 20 ms, 1 mV. Error bars indicate SE.
An alternative test for NMDAR-independent synaptic plasticity is to study LTPK [i.e., LTP induced by brief superfusion of hippocampal slices with the potassium channel blocker TEA-Cl (25 mm)]. Importantly, LTPK is thought to depend mostly on L-type channel-mediated Ca2+ influx (Aniksztejn and Ben-Ari, 1991; Zakharenko et al., 2001). We compared LTP in control and Cav1.2HCKO mice after 15 min of superfusion with a modified TEA-containing aCSF and APV (50 μm) present throughout the experiment. Under these conditions, control mice exhibited robust LTP, even 2 h after washout of TEA (147.4 ± 11.3; n = 15) (Fig. 3c). In sharp contrast, mutant mice showed only marginal LTP 2 h after treatment with TEA (113.1 ± 4.47; n = 11; p < 0.05) (Fig. 3c). LTP in control mice 2 h after superfusion with TEA might represent the so-called late-phase LTP, a hallmark of which is the dependence on de novo protein synthesis (Frey et al., 1988; Kandel, 2001). To test for this obligatory feature, we performed additional experiments with hippocampal slices from control and mutant mice in which the inhibitor of protein synthesis, anisomycin (20 μm), was present throughout the experiment. Anisomycin significantly reduced hippocampal LTP observed 2 h after treatment with TEA in slices from control mice (121.8 ± 8.3; n = 8; p < 0.05) compared with LTP without anisomycin, but had no effect in slices from Cav1.2HCKO mice (114.4 ± 12.9; n = 8) (Fig. 3d). Together, these findings clearly demonstrate that NMDAR-independent, protein synthesis-dependent L-LTP in the hippocampal CA1 region depends mostly on Cav1.2 L-type Ca2+ channels.
Cav1.2HCKO mice have normal basic motor functions, exploratory behavior, anxiety, and visual acuity
Does the lack of NMDAR-independent L-LTP in Cav1.2HCKO mice correlate with altered spatial memory? The performance of Cav1.2HCKO mice in learning tasks might, in principle, be affected by defects in other behavioral traits. Therefore, we initially performed a series of control tests. Arguing against nonspecific effects of the CACNA1C gene inactivation on exploratory behavior and anxiety, control and Cav1.2HCKO mice (n = 10 for both groups) showed no significant differences in the open field test. The total number of fields entered by control mice (119 ± 20 on day 1 and 89 ± 9 on day 4) matched that of Cav1.2HCKO mice (112 ± 19 on day 1 and 70 ± 18 on day 4). For both genotypes, the vast majority were entries into outer fields, amounting to 114 ± 18 (day 1) and 85 ± 7 (day 4) in control mice, and 105 ± 19 (day 1) and 70 ± 15 (day 4) in the Cav1.2HCKO mice. The following findings rule out a deficit in general motor ability in the Cav1.2 knock-out. First, control and Cav1.2HCKO mice (n = 10 for both groups) performed equally in a rotarod test. The mean duration that mice of both genotypes managed to keep balance on the accelerating rotarod increased similarly to a maximal level at day 3 and remained constant throughout the following 2 d of examination. Second, swim speed in a water maze pool was the same in knock-out and control groups (control mice, 0.35 ± 0.06 m/s; Cav1.2HCKO mice, 0.36 ± 0.03 m/s) (Fig. 4a).
Figure 4.

Spatial memory defects in Cav1.2HCKO mice. a, Correct-choice rate of mice trained to find the correct platform in a visual acuity task by using the only a visible landmark in a dimly lit room. There were no differences between the Cav1.2HCKO (□) animals (n = 7) and the control (▪) mice (n = 7) in the percentage of correct choices during the trial days (10 trials per day). Inset, Swim speed. Cav1.2HCKO (□) animals (n = 10) present the same swim-speed rate (meters/second) compared with control (▪) mice (n = 10). Data are expressed as mean + SEM. b, c, The performance of Cav1.2HCKO mice in the discriminatory water-maze task. b, The graph represents the correct-choice rate of mice trained to find the correct platform in a discriminatory water maze by using the distal cues surrounding it. The Cav1.2HCKO (□) animals (n = 10) display a significantly (p < 0.05) lower correct-choice rate from day 3 (10 trials per day) than the control (▪) mice (n = 10). During a control reversal trial (Rv), the percentage of correct choices made by control mice dropped by a similar extent to values near chance level, indicating that spatial searching strategies wereused. c, The graph represents the escape latencies of mice that found the correct platform. The Cav1.2HCKO (□) animals (n = 10) displayed a longer latency, starting from day 3 (10 trials per day) than the control (▪) mice (n = 10). d, Spatial learning in a labyrinth maze made of transparent, brightly lit glass tubes, with a single correct way leading to a dark box. Performance was assessed by the number of tubes traversed by control (▪) and Cav1.2HCKO (□) mice until they reached the dark box. Two trials were performed on the first 3 d (n = 11 animals for both groups) and one trial on days 10 and 17 (n = 5 animals of both groups). Data are expressed as mean + SEM. *p < 0.05; **p < 0.01.
It is a general problem when studying mutants in a task using distal cues (like the two learning tasks used in this study) to rule out a deficit based on an inability to properly see them. Therefore, we trained mice in a visual acuity, discriminatory water-maze task, in which they were required to identify the correct platform, kept in a permanent position and marked by a single large, distal, well lit landmark in a dimly lit room with no additional distal cues (for details, see Materials and Methods). In this test, again, there was no difference in the performance of control and Cav1.2HCKO mice (Fig. 4a). Collectively, these findings allow us to separate a learning defect from other kinds of behavioral changes (e.g., perceptual, motivational, or attentional) in Cav1.2HCKO mice.
Cav1.2HCKO mice display severely impaired hippocampus-dependent spatial memory
Next, we chose two mouse-friendly learning tasks that tax spatial reference memory but require different sets of sensory and motor abilities. Initially, we examined hippocampus-dependent spatial learning in a well established discriminatory water-maze task (Arns et al., 1999; Steckler et al., 1999; Kleppisch et al., 2003), adapted from the paradigm originally described by Morris et al. (1986b) for rats. This task has been proven to be hippocampus dependent using excitotoxic lesions (Arns et al., 1999). The water-maze spatial-discrimination task differs from the more frequently used place-navigation paradigm in several aspects. First, we used visible instead of hidden platforms with the consequence that animals are not required to first learn that there is a platform at all. Second, escape latencies are relatively short, providing the advantage of greatly reduced swim stress. Third, the setup with two platforms gives the opportunity to assess spatial learning using a probability parameter, the frequency of correct choices (Fig. 4b), that is less likely to be affected by changes in other behavioral, perceptional, or motor functions than a time (e.g., the water escape latency) or distance (e.g., the path length in the pool) parameter (Fig. 4c).
Cav1.2HCKO and control mice were trained to identify a fixed platform visually identical to a second sinkable platform. Both groups showed the same initial rate of correct choices and an equivalent increase of this parameter from day 1 to day 2. However, we observed a dramatic difference in the performance of control and mutant mice beyond trial day 2 (Fig. 4b). Whereas Cav1.2HCKO mice did not further improve their correct choice score from the value at day 2 (∼65%), the performance of their control siblings significantly improved to >80%. In addition, when knock-out animals found the correct platform, their water escape latency was significantly increased (Fig. 4c). These findings suggest a severe impairment of spatial learning in the Cav1.2HCKO mice.
To test for the possibility that mice used salient cues provided by the platforms for discrimination, the correct platform was moved to the opposite quadrant (reversal) and animals were tested again. During this reversal trial, the percentage of correct choices made by control mice dropped to values near chance level (Fig. 4b). This rules out that the two platforms were discriminated merely based on their appearance but confirms that control animals used spatial cues in the environment for successful navigation.
To further substantiate that Cav1.2-mediated, NMDAR-independent synaptic plasticity is crucial for spatial memory formation, we sought after an alternative learning task preferably differing from the water maze in various aspects, such as task complexity, response requirement, and the nature of the rein-forcer. Therefore, we further examined mice in a validated spatial-learning labyrinth paradigm that has been shown to be well suited to resolve alterations in spatial memory [e.g., using NR2B (NMDAR 2B)-overexpressing transgenic mice] (Tang et al., 1999; Adelsberger et al., 2005). The task consists of the repeated walk through a labyrinth made of clear, transparent acrylic glass tubes with a length of 50 cm each to a familiar dark box in a laboratory environment presenting distal cues (for details, see Materials and Methods). The test is primarily based on the positive motivation of the animals and avoids the escape from forced swimming associated with water-maze tests (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
During the initial trial, the number of tubes traversed (which represents the path length) before reaching the dark box was not significantly different for control and Cav1.2HCKO mice (Fig. 4d). This indicates equally effective searching patterns in naive animals of either genotype. The number of tubes traversed is expected to decline during training to an extent depending on the efficiency of spatial learning strategies and the formation of spatial memories. In nice agreement with the differences observed between the two genotypes in the discriminatory water maze, Cav1.2HCKO mice showed significantly longer path lengths than the control mice during all subsequent trial days (Fig. 4d). The coincidence of a defect in NMDAR-independent protein synthesis-dependent L-LTP with a defect in two spatial learning tasks strongly suggests a functional link between these processes.
Activation of the MAPK pathway is impaired in Cav1.2HCKO mice
There is comprehensive evidence supporting the view that the MAPK signaling cascade is involved in synaptic plasticity in the CA1 area of the mammalian hippocampus and in various forms of long-term memory (e.g., spatial learning and fear conditioning) (Thomas and Huganir, 2004). Experiments with neuronal cell cultures have shown that Ca2+ influx can efficiently activate ERK/MAPK (Rosen et al., 1994; Dolmetsch et al., 2001; Hardingham et al., 2001). To assess the function of Cav1.2 L-type Ca2+ channels for the activation of ERK1/2, we asked whether phosphorylation and nuclear translocation of ERK occurs in postsynaptic CA1 pyramidal neurons after treatment with TEA (i.e., under experimental conditions resulting in robust L-LTPK in Schaffer collateral/CA1 synapses) (compare Fig. 3c). As visualized by confocal immunofluorescence, superfusion with TEA caused a strong increase in phosphorylated, activated ERK in the postsynaptic dendrites (Fig. 5a). In addition, we were able to detect phospho-ERK-positive nuclei exclusively in TEA-treated slices, but not in untreated control slices. Remarkably, neither nuclear translocation nor phosphorylation of ERK1/2 in CA1 pyramidal cells of control mice was affected by addition of the NMDAR antagonist APV. In contrast to the findings in control mice, TEA-treated hippocampal CA1 pyramidal cells of Cav1.2HCKO mice showed only a delicate dendritic phospho-ERK signal and only a negligible number of phospho-ERK-positive nuclei. To test whether the defect of TEA-induced NMDAR-independent L-LTP observed in Cav1.2HCKO mice results from impaired MAPK cascade signaling, we examined the effect of an inhibitor of the MAPK pathway, U0126, on this form of synaptic plasticity in hippocampal slices of control mice. Supporting our hypothesis, U0126 (40 μm) mimicked the effect of the protein synthesis inhibitor anisomycin in these mice (Fig. 3d). We therefore concluded that ERK activity is required for NMDAR-independent, protein synthesisdependent L-LTP in hippocampal CA1 neurons.
Figure 5.
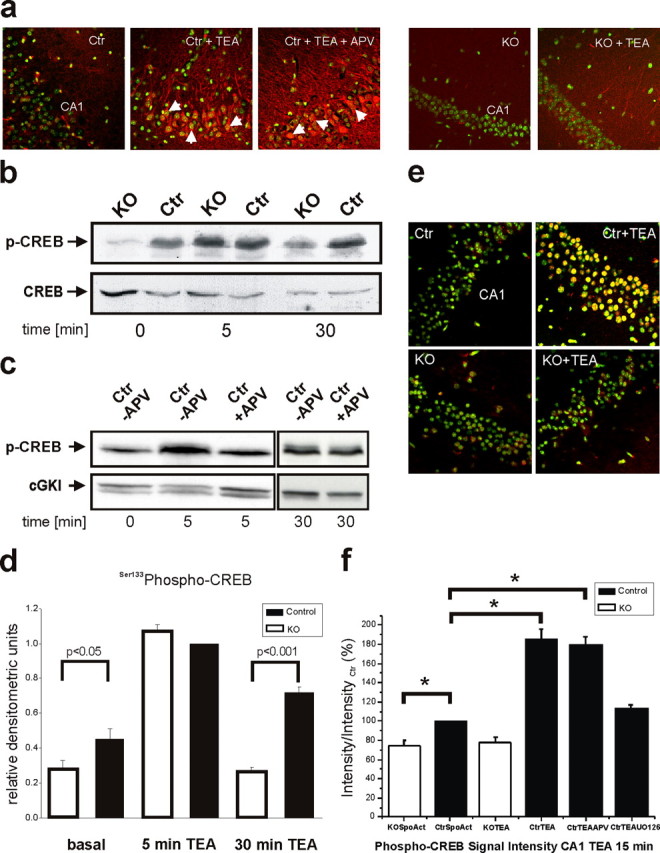
Disturbed MAPK activation and impaired phosphorylation of CREB by stimulus paradigms that generate L-LTP in area CA1 of Cav1.2HCKO mice. a, Representative examples of phospho-ERK1/2 immunocytochemistry in a untreated control (Ctr), in slices that were treated with a stimulus paradigm (TEA) that generates L-LTP in the presence or absence of the NMDAR antagonist APV, and in untreated and TEA treated knock-out (KO) slices. Green, Hoechst 33258 nuclear marker; red, phospho-ERK1/2; yellow, overlay. Arrowheads, Nuclear localization of ERK1/2. Images are at 400× (original) magnification. b, Western blot of the time course of CREB Ser133 phosphorylation in hippocampal neurons depolarized with TEA in KO or Ctr mice. Ca2+ influx through Cav1.2 L-type Ca2+ channels results in sustained CREB phosphorylation (30 min). Blots (n = 6 independent experiments) were stripped and reprobed with anti-CREB. c, Western blot of the time course of CREB Ser133 phosphorylation in hippocampal neurons depolarized with TEA in (Ctr) mice in the presence (+APV) or absence (-APV) of the NMDAR antagonist APV. NMDAR activity contributes to the early phase (5 min) but not to the late phase of CREB phosphorylation (30 min). Blots (n = 6 independent experiments) were stripped and reprobed with anti-cGMP-dependent protein kinase I (anti-cGKI). d, Densitometric analysis of Western blots for CREB Ser133 phosphorylation. Data are normalized phospho-CREB Ser133 values in control mice after 5 min of TEA treatment. e, Representative examples of phospho-CREB immunofluorescence in untreated control and knock-out slices, and in slices that were treated with a stimulus paradigm (TEA) that generates L-LTP. Images are at 400× (original) magnification. Green, Hoechst 33258 nuclear marker; red, phospho-CREB Ser133; yellow, overlay. f, Fold increase in phospho-CREB immunocytochemistry compared with untreated slices [spontaneous activity (SpoAct)]. Slices were stimulated with a paradigm (TEA) that generates L-LTP in the presence and absence of APV. Slices (n = 12 per condition) were fixed 30 min after tetanus. *Significant differences (p < 0.05). Error bars indicate SE.
Impaired sustained CREB-Ser133 phosphorylation and CRE-dependent transcription in Cav1.2HCKO mice
Based on the above conclusion, we next asked whether effectors of ERK1/2 associated with synaptic plasticity and memory are affected in Cav1.2HCKO mice. Activation of ERK/MAPK may contribute to memory formation by stimulating many different events, including transcription of memory-related genes (Sweatt, 2004). A major role in these processes has been assigned to the downstream effector of ERK1/2, CREB, a transcription factor related to synaptic plasticity and essential for various forms of memory (Silva et al., 1998; Lonze and Ginty, 2002). CRE-dependent gene expression critically depends on CREB-Ser133 phosphorylation (Brindle and Montminy, 1992), which provides a duration-sensitive switch (i.e., only a sustained pCREB-Ser133 signal is sufficient to activate CRE-mediated transcription) (Bito et al., 1996). Therefore, we next asked whether decreased ERK activity in hippocampal neurons of Cav1.2HCKO mice was accompanied by a suppression of the prolonged activation of CREB. Hippocampal extracts obtained from tissue before and at various times after treatment with TEA (25 mm) were blotted with an antibody specifically binding pCREB-Ser133. Hippocampal tissue from Cav1.2HCKO mice showed a reduced basal pCREB-Ser133 level [0.28 ± 0.05 relative densitometric units (rdu)] compared with that in tissue from control mice (0.45 ± 0.06 rdu; p < 0.05). This difference is likely caused by spontaneous activity of Cav1.2 channels. In hippocampi from control mice, TEA treatment induced a strong CREB phosphorylation, detectable at 5 min and lasting at least 30 min (Fig. 5b,d). Interestingly, initial TEA-induced CREB phosphorylation (5 min) was also detected in hippocampi from Cav1.2HCKO mice. But, in striking contrast with the findings in control animals, CREB phosphorylation was completely reversed within 30 min (control mice, 0.72 ± 0.03 rdu; Cav1.2HCKO mice, 0.27 ± 0.02 rdu; p < 0.001). Thus, hippocampal neurons of Cav1.2HCKO mice specifically lack sustained CREB phosphorylation under these conditions. Additional analysis of TEA-induced CREB activation in hippocampal tissue from control mice in the presence of the NMDAR antagonist APV (Fig. 5c) revealed that the early phase (5 min) was caused by an NMDAR-mediated Ca2+ influx. Importantly, APV did not affect CREB phosphorylation at 30 min, confirming the role of an NMDAR-independent mechanism for sustained CREB activation.
In the next series of experiments, we used confocal immunofluorescence to visualize nuclear phospho-CREB localization and CRE-dependent transcription in CA1 neurons under conditions leading to L-LTPK. CRE-dependent transcriptional activity was assessed in control and Cav1.2HCKO mice additionally carrying a transgene for a CRE-regulated reporter construct (Impey et al., 1996). TEA treatment caused a strong increase in nuclear pCREB (Fig. 5e,f) and CRE-dependent gene transcription (Fig. 6a,b) exclusively in hippocampi from control mice. Hippocampal pyramidal cells in Cav1.2HCKO mice lacked such effects (Fig. 6a,b). Again, neither nuclear translocation nor phosphorylation of CREB (Fig. 5e,f) was affected by adding APV. Importantly, the inhibitor of the ERK pathway, U0126 (40 μm), abolished nuclear pCREB-Ser133 signals (Fig. 5f) and blocked activation of CRE-mediated transcription after treatment with TEA (Fig. 6a,b). These data indicate that both L-LTPK and the increases in CRE-mediated gene expression are associated with increased ERK1/2 activity.
Figure 6.

Increases in Cre-LacZ expression by stimulus paradigms that generate L-LTP at Schaffer collateral/CA1 synapses requires Cav1.2 L-type Ca2+ channel activity.a, Representative examples of CRE-LacZ expression in control (Ctr) and knock-out slices (KO) that were untreated, treated with TEA, or treated with TEA and U0126. All experiments were performed in the presence of APV. Green, β-Galactosidase reporter protein. b, Percentage of increase in β-galactosidase immunocytochemistry compared with untreated slices. Slices (n = 12 per condition) were fixed 240 min after the LTP induction. *Significant difference (p < 0.05). Error bars indicate SE.
Discussion
Numerous studies have provided pharmacological and genetic evidence, both loss of function and gain of function, linking NMDAR-dependent LTP in the hippocampal CA1 region to certain types of learning (Morris et al., 1986a; McHugh et al., 1996; Tsien et al., 1996; Tang et al., 1999). However, a description of hippocampal synaptic plasticity and memory merely based on NMDAR-mediated processes falls short in various aspects, as shown in this study and by others (Impey et al., 1996; Borroni et al., 2000; Rampon and Tsien, 2000; Woodside et al., 2004). Thus, LTP can also be induced in a Cav1.x channel-dependent, NMDAR-independent manner in the CA1 region (Grover and Teyler, 1990; Aniksztejn and Ben-Ari, 1991; Cavus and Teyler, 1996; Morgan and Teyler, 1999). In particular, activity of Cav1.x channels has been shown to be necessary for induction of L-LTP by strong stimulus protocols (Impey et al., 1996; Raymond and Redman, 2002). In addition, biochemical processes, like the activation of ERK and the subsequent phosphorylation of CREB, have been linked to Cav1.x channel activity (Impey et al., 1996; Mermelstein et al., 2000; West et al., 2001; Wu et al., 2001). Therefore, we hypothesized that NMDAR-independent, L-type Ca2+ channel-dependent synaptic plasticity might, in parallel to NMDAR-activated processes, be necessary for spatial learning in the behaving animal.
Although the pharmacological analysis of cultured cells and slice preparations has provided much insight into the regulation and function of L-type Ca2+ channels in neurons (for review, see Hofmann et al., 1999; Striessnig, 1999; Moosmang et al., 2005), their significance in vivo is far less clear because of several problems associated with the currently available pharmacological tools. Most importantly, all L-type Ca2+ channel blockers affect the function of the cardiovascular system to some extent, which may have substantial input on the function of the CNS. In fact, a number of investigations demonstrate that pharmacological block of L-type calcium channels may enhance learning through changes in cerebral blood flow (Deyo et al., 1989; Deyo and Hittner, 1995). Conversely, other studies show that the pharmacological block of neuronal L-type Ca2+ channels impairs memory (e.g., amygdala-dependent conditioned fear memory) (Bauer et al., 2002). Moreover, available pharmacological tools do not allow separation of the individual function from the various members of the L-type Ca2+ channel family (e.g., Cav1.2 and Cav1.3). The interpretation of in vivo data are further complicated because all known classes of Ca2+ channel blockers can also block other voltage-gated channels at higher concentrations (Galper and Catterall, 1979; Ragsdale et al., 1991; Rampe et al., 1993; Diochot et al., 1995; Ishibashi et al., 1995).
Therefore, we decided to use a genetic approach to analyze the role of NMDAR-independent hippocampal synaptic plasticity in vivo. We created a second-generation knock-out mouse strain using the Cre/loxP technique and Nex-Cre mice (Schwab et al., 2000). This approach yielded a mouse line (Cav1.2HCKO mice) deficient in the Cav1.2 protein, mainly in the hippocampal formation and cerebral cortex. The deletion of the CACNA1C gene became effective only after the first weeks of postnatal development; there was a normal level of hippocampal Cav1.2 protein in 2- to 3-week-old Cav1.2HCKO mice, whereas the protein was absent in adult mice. Hippocampal pyramidal cells undergo maximal dendritic growth during this period, and their cellular organization and synaptic connections are principally established at this age (Wong and Ghosh, 2002). Thus, Cav1.2HCKO mice provide a model that should be devoid of developmental disturbances in the brain architecture (Redmond et al., 2002; Aizawa et al., 2004). This assumption is supported by our findings that control and Cav1.2HCKO mice show no differences in the anatomical properties of the brain and in their characteristics of basal synaptic transmission in the hippocampus.
Our combined electrophysiological, biochemical, and behavioral analysis of the Cav1.2HCKO mice provides strong evidence that NMDAR-independent synaptic plasticity in the hippocampal CA1 region is functionally linked to hippocampus-dependent spatial learning. The following findings support this conclusion. First, NMDAR-dependent LTP was not altered in the hippocampal CA1 region of Cav1.2HCKO mice (i.e., defects observed in other forms of synaptic plasticity were selective). Second, NMDAR-independent L-LTP induced by various paradigms was defective in the Schaffer collateral/CA1 pathway of Cav1.2HCKO mice. Furthermore, the protein synthesis inhibitor anisomycin decreased NMDAR-independent LTPK 2 h after treatment with TEA in the control mice to the level observed in the mutants. These results clearly suggest that Cav1.2HCKO mice are deficient in protein synthesis-dependent L-LTP, which has been explicitly linked to long-term memory (Kandel, 2001; Martin and Morris, 2002). Third, the defect of protein synthesis-dependent synaptic plasticity in the Cav1.2HCKO mice was accompanied by reduced activation of MAPK cascade signaling. Fourth, this impairment of the MAPK pathway was paralleled by diminished CRE-dependent transcription in the CA1 region after L-LTP induction. Fifth, as a consequence, the mutant mice were severely impaired in two different spatial learning tasks. They showed inferior performance in a well established discriminatory water-maze task (Arns et al., 1999; Steckler et al., 1999; Kleppisch et al., 2003) and in a more complex labyrinth-maze task (Adelsberger et al., 2005).
The gene inactivation in Cav1.2HCKO mice was not limited strictly to the hippocampus but occurred also in the cerebral cortex. We cannot completely rule out an impact of the loss of Cav1.2 protein in cortical areas on behavioral functions. However, differences between control and Cav1.2HCKO mice were detected exclusively in tests designed to reveal defects in learning and memory associated with the hippocampus (e.g., the water maze task) (Arns et al., 1999; Morris, 2003). However, both genotypes performed equally in a series of behavioral control tests that are supposed to depend at least partially on a normal function of the cortex.
How might Cav1.2 channels support NMDAR-independent LTP and spatial learning? We found that Cav1.2 channel-mediated Ca2+ influx activates the protein kinase ERK and CREB-dependent transcription under conditions leading to L-LTP. It has been reported that LTP-inducing high-frequency stimulation activates ERK in the CA1 area, and that the MAPK kinase (MEK) inhibitor PD98059 [2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one] blocks both ERK activation and NMDAR-dependent LTP (English and Sweatt, 1996, 1997). ERK activity is also required for LTP in the dentate gyrus (Coogan et al., 1999), the amygdala (Schafe et al., 2000), and NMDAR-independent forms of LTP in the CA1 area (Kanterewicz et al., 2000). The role of ERK for long-term memory in behaving animals has been characterized using spatial-learning and fear-conditioning (Schafe et al., 2000) paradigms. Training in the Morris water maze leads to activation of ERK in the hippocampus of the corresponding animals (Blum et al., 1999). Moreover, intrahippocampal infusion of ERK inhibitors impairs the performance in this task (Selcher et al., 1999). Studies in invertebrates and rodents indicate that CREB-dependent transcription is essential for both L-LTP (Bourtchuladze et al., 1994; Deisseroth et al., 1996; Barco et al., 2002) and many forms of learning and memory (for review, see Lonze and Ginty, 2002). Phosphorylation of CREB at Ser133 leads to recruitment of other transcription machinery to CREs to regulate gene transcription (Lonze and Ginty, 2002). As such, it is thought that CREB phosphorylation at Ser133 is an important step in the induction of gene expression critical for memory. We show that the prolonged phosphorylation of CREB at Ser133 is impaired in Cav1.2HCKO mice. This phosphorylation can also be prevented by inhibitors of the ERK cascade (Wu et al., 2001). In conclusion, we propose that the defects of LTP and memory in the Cav1.2HKO mice are the consequence of impaired activation of ERK and defective CREB-dependent transcription, which are normally coupled specifically toaCav1.2 channel-mediated intracellular signal.
The outstanding role of NMDARs for synaptic plasticity and memory formation has been explained with their unique property: a voltage-dependent block by Mg2+ allows them to function as Hebbian coincidence detectors (Bliss and Collingridge, 1993). Can Cav1.2 channels serve a similar function? Cav1.2 channel-mediated dendritic Ca2+ influx has been proposed to arise from strong depolarization caused by backpropagating action potentials (APs) (Spruston et al., 1995; Yuste and Denk, 1995; Magee and Johnston, 1997) that are likely to occur during the induction of NMDAR-independent LTP. In this scenario, the Cav1.2 channel might facilitate the invasion of antidromic APs into distal dendritic structures in an activity-dependent manner (Magee and Johnston, 1997) (i.e., they could also serve as coincidence detectors). However, electrophysiological characteristics of Cav1.x channels favor Ca2+ entry associated with EPSPs as opposed to APs, which is reflected in a much stronger L-type Ca2+ channel-dependent CREB phosphorylation in response to synaptic activity (Mermelstein et al., 2000). In line with these findings, Cav1.2 may support synaptic potentiation, independent of postsynaptic action potential firing, by contributing to regenerative local dendritic spikes (Golding et al., 2002). Future studies (e.g., dendritic Ca2+ imaging in Cav1.2HCKO mice) will elucidate the precise way in which Cav1.2 can function as a Hebbian coincidence detector.
In conclusion, our findings show that NMDA receptor-independent, Cav1.2 L-type Ca2+ channel-dependent synaptic plasticity significantly contributes to spatial learning in the behaving mouse.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft, Fond der Chemischen Industrie, and the Volkswagen-Stiftung. We thank Susanne Paparisto for excellent technical assistance.
Correspondence should be addressed to Dr. Sven Moosmang, Institut für Pharmakologie und Toxikologie der Technischen Universität München, Biedersteiner Strasse 29, 80802 München, Germany. E-mail: moosmang@ipt.med.tu-muenchen.de.
H. Adelsberger's present address: Institut für Physiologie, Ludwig-Maximilian-Universität München, Pettenkoferstraβe 12, 80336 München, Germany.
N. Klugbauer's present address: Institut für Experimentelle und Klinische, Pharmakologie und Toxikologie, Albert-Ludwigs-Universität Freiburg, Albertstraβe 25, 79104 Freiburg im Breisgau, Germany.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259883-10$15.00/0
References
- Adelsberger H, Tsien J, Konnerth A (2005) Superior learning abilities of NR2B transgenic mice in a new labyrinth task. In: Proceedings of the 6th Meeting of the German Neuroscience Society (Krieglstein K, ed), p 244B. Berlin: Neurowissenschaftliche Gesellschaft.
- Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A (2004) Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science 303: 197-202. [DOI] [PubMed] [Google Scholar]
- Aniksztejn L, Ben-Ari Y (1991) Novel form of long-term potentiation produced by a K+ channel blocker in the hippocampus. Nature 349: 67-69. [DOI] [PubMed] [Google Scholar]
- Arns M, Sauvage M, Steckler T (1999) Excitotoxic hippocampal lesions disrupt allocentric spatial learning in mice: effects of strain and task demands. Behav Brain Res 106: 151-164. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD (1998) The MAPK cascade is required for mammalian associative learning. Nat Neurosci 1: 602-609. [DOI] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER (2002) Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell 108: 689-703. [DOI] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE (2002) NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci 22: 5239-5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW (1996) CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell 87: 1203-1214. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31-39. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T (1973) Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 232: 331-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum S, Moore AN, Adams F, Dash PK (1999) A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J Neurosci 19: 3535-3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroni AM, Fichtenholtz H, Woodside BL, Teyler TJ (2000) Role of voltage-dependent calcium channel long-term potentiation (LTP) and NMDA LTP in spatial memory. J Neurosci 20: 9272-9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ (1994) Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79: 59-68. [DOI] [PubMed] [Google Scholar]
- Brindle PK, Montminy MR (1992) The CREB family of transcription activators. Curr Opin Genet Dev 2: 199-204. [DOI] [PubMed] [Google Scholar]
- Cavus I, Teyler T (1996) Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. J Neurophysiol 76: 3038-3047. [DOI] [PubMed] [Google Scholar]
- Clark NC, Nagano N, Kuenzi FM, Jarolimek W, Huber I, Walter D, Wietzorrek G, Boyce S, Kullmann DM, Striessnig J, Seabrook GR (2003) Neurological phenotype and synaptic function in mice lacking the CaV1.3 alpha subunit of neuronal L-type voltage-dependent Ca2+ channels. Neuroscience 120: 435-442. [DOI] [PubMed] [Google Scholar]
- Coogan AN, O'Leary DM, O'Connor JJ (1999) P42/44 MAP kinase inhibitor PD98059 attenuates multiple forms of synaptic plasticity in rat dentate gyrus in vitro. J Neurophysiol 81: 103-110. [DOI] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW (2001) A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science 293: 98-101. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, Tsien RW (1996) Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron 16: 89-101. [DOI] [PubMed] [Google Scholar]
- Deyo RA, Hittner JM (1995) Effects of the Ca2+ channel antagonist flunarizine on visual discrimination learning. Neurobiol Learn Mem 64: 10-16. [DOI] [PubMed] [Google Scholar]
- Deyo RA, Straube KT, Disterhoft JF (1989) Nimodipine facilitates associative learning in aging rabbits. Science 243: 809-811. [DOI] [PubMed] [Google Scholar]
- Diochot S, Richard S, Baldy-Moulinier M, Nargeot J, Valmier J (1995) Dihydropyridines, phenylalkylamines and benzothiazepines block N-, P/Q- and R-type calcium currents. Pflügers Arch 431: 10-19. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME (2001) Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294: 333-339. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD (1996) Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem 271: 24329-24332. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD (1997) A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem 272: 19103-19106. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H (1988) Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res 452: 57-65. [DOI] [PubMed] [Google Scholar]
- Galper JB, Catterall WA (1979) Inhibition of sodium channels by D600. Mol Pharmacol 15: 174-178. [PubMed] [Google Scholar]
- Golding NL, Staff NP, Spruston N (2002) Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature 418: 326-331. [DOI] [PubMed] [Google Scholar]
- Grover LM (1998) Evidence for postsynaptic induction and expression of NMDA receptor independent LTP. J Neurophysiol 79: 1167-1182. [DOI] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ (1990) Two components of long-term potentiation induced by different patterns of afferent activation. Nature 347: 477-479. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H (2001) A calcium microdomain near NMDA receptors: on switch for ERK-dependent synapse-to-nucleus communication. Nat Neurosci 4: 565-566. [DOI] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA (1993) Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol 123: 949-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F, Lacinova L, Klugbauer N (1999) Voltage-dependent calcium channels: from structure to function. Rev Physiol Biochem Pharmacol 139: 33-87. [DOI] [PubMed] [Google Scholar]
- Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR (1996) Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron 16: 973-982. [DOI] [PubMed] [Google Scholar]
- Ishibashi H, Yatani A, Akaike N (1995) Block of P-type Ca2+ channels in freshly dissociated rat cerebellar Purkinje neurons by diltiazem and vera-pamil. Brain Res 695: 88-91. [DOI] [PubMed] [Google Scholar]
- Kandel ER (2001) The molecular biology of memory storage: a dialogue between genes and synapses. Science 294: 1030-1038. [DOI] [PubMed] [Google Scholar]
- Kanterewicz BI, Urban NN, McMahon DB, Norman ED, Giffen LJ, Favata MF, Scherle PA, Trzskos JM, Barrionuevo G, Klann E (2000) The extracellular signal-regulated kinase cascade is required for NMDA receptor-independent LTP in area CA1 but not area CA3 of the hippocampus. J Neurosci 20: 3057-3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleppisch T, Pfeifer A, Klatt P, Ruth P, Montkowski A, Fassler R, Hofmann F (1999) Long-term potentiation in the hippocampal CA1 region of mice lacking cGMP-dependent kinases is normal and susceptible to inhibition of nitric oxide synthase. J Neurosci 19: 48-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleppisch T, Wolfsgruber W, Feil S, Allmann R, Wotjak CT, Goebbels S, Nave KA, Hofmann F, Feil R (2003) Hippocampal cGMP-dependent protein kinase I supports an age- and protein synthesis-dependent component of long-term potentiation but is not essential for spatial reference and contextual memory. J Neurosci 23: 6005-6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35: 605-623. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D (1997) A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275: 209-213. [DOI] [PubMed] [Google Scholar]
- Magee JC, Avery RB, Christie BR, Johnston D (1996) Dihydropyridine-sensitive, voltage-gated Ca2+ channels contribute to the resting intracellular Ca2+ concentration of hippocampal CA1 pyramidal neurons. J Neurophysiol 76: 3460-3470. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44: 5-21. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Morris RG (2002) New life in an old idea: the synaptic plasticity and memory hypothesis revisited. Hippocampus 12: 609-636. [DOI] [PubMed] [Google Scholar]
- McHugh TJ, Blum KI, Tsien JZ, Tonegawa S, Wilson MA (1996) Impaired hippocampal representation of space in CA1-specific NMDAR1 knock-out mice. Cell 87: 1339-1349. [DOI] [PubMed] [Google Scholar]
- Mermelstein PG, Bito H, Deisseroth K, Tsien RW (2000) Critical dependence of cAMP response element-binding protein phosphorylation on L-type calcium channels supports a selective response to EPSPs in preference to action potentials. J Neurosci 20: 266-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosmang S, Schulla V, Welling A, Feil R, Feil S, Wegener JW, Hofmann F, Klugbauer N (2003) Dominant role of smooth muscle L-type calcium channel Cav1.2 for blood pressure regulation. EMBO J 22: 6027-6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosmang S, Lenhardt P, Haider N, Hofmann F, Wegener JW (2005) Mouse models to study L-type calcium channel function. Pharmacol Ther 106: 347-355. [DOI] [PubMed] [Google Scholar]
- Morgan SL, Teyler TJ (1999) VDCCs and NMDARs underlie two forms of LTP in CA1 hippocampus in vivo. J Neurophysiol 82: 736-740. [DOI] [PubMed] [Google Scholar]
- Morris RG (2003) Long-term potentiation and memory. Philos Trans R Soc Lond B Biol Sci 358: 643-647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M (1986a) Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature 319: 774-776. [DOI] [PubMed] [Google Scholar]
- Morris RG, Hagan JJ, Rawlins JN (1986b) Allocentric spatial learning by hippocampectomised rats: a further test of the “spatial mapping” and “working memory” theories of hippocampal function. Q J Exp Psychol B 38: 365-395. [PubMed] [Google Scholar]
- Murphy TH, Worley PF, Nakabeppu Y, Christy B, Gastel J, Baraban JM (1991) Synaptic regulation of immediate early gene expression in primary cultures of cortical neurons. J Neurochem 57: 1862-1872. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S, Kandel ER (2002) Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron 34: 447-462. [DOI] [PubMed] [Google Scholar]
- Ragsdale DS, Scheuer T, Catterall WA (1991) Frequency and voltage-dependent inhibition of type IIA Na+ channels, expressed in a mammalian cell line, by local anesthetic, antiarrhythmic, and anticonvulsant drugs. Mol Pharmacol 40: 756-765. [PubMed] [Google Scholar]
- Rampe D, Wible B, Fedida D, Dage RC, Brown AM (1993) Verapamil blocks a rapidly activating delayed rectifier K+ channel cloned from human heart. Mol Pharmacol 44: 642-648. [PubMed] [Google Scholar]
- Rampon C, Tsien JZ (2000) Genetic analysis of learning behavior-induced structural plasticity. Hippocampus 10: 605-609. [DOI] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ (2002) Different calcium sources are narrowly tuned to the induction of different forms of LTP. J Neurophysiol 88: 249-255. [DOI] [PubMed] [Google Scholar]
- Redmond L, Kashani AH, Ghosh A (2002) Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 34: 999-1010. [DOI] [PubMed] [Google Scholar]
- Rosen LB, Ginty DD, Weber MJ, Greenberg ME (1994) Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron 12: 1207-1221. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE (2000) Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J Neurosci 20: 8177-8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab MH, Bartholomae A, Heimrich B, Feldmeyer D, Druffel-Augustin S, Goebbels S, Naya FJ, Zhao S, Frotscher M, Tsai MJ, Nave KA (2000) Neuronal basic helix-loop-helix proteins (NEX and BETA2/Neuro D) regulate terminal granule cell differentiation in the hippocampus. J Neurosci 20: 3714-3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcher JC, Atkins CM, Trzaskos JM, Paylor R, Sweatt JD (1999) A necessity for MAP kinase activation in mammalian spatial learning. Learn Mem 6: 478-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68: 821-861. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S (1998) CREB and memory. Annu Rev Neurosci 21: 127-148. [DOI] [PubMed] [Google Scholar]
- Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renstrom E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschutz R, Hering S, Bova S, Rorsman P, Pongs O, Singewald N, Striessnig JJ (2004) Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca2+ channels. J Clin Invest 113: 1430-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, Schiller Y, Stuart G, Sakmann B (1995) Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science 268: 297-300. [DOI] [PubMed] [Google Scholar]
- Steckler T, Weis C, Sauvage M, Mederer A, Holsboer F (1999) Disrupted allocentric but preserved egocentric spatial learning in transgenic mice with impaired glucocorticoid receptor function. Behav Brain Res 100: 77-89. [DOI] [PubMed] [Google Scholar]
- Striessnig J (1999) Pharmacology, structure and function of cardiac L-type Ca2+ channels. Cell Physiol Biochem 9: 242-269. [DOI] [PubMed] [Google Scholar]
- Sweatt JD (2004) Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14: 311-317. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ (1999) Genetic enhancement of learning and memory in mice. Nature 401: 63-69. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL (2004) MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5: 173-183. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S (1996) The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87: 1327-1338. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME (2001) Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA 98: 11024-11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RO, Ghosh A (2002) Activity-dependent regulation of dendritic growth and patterning. Nat Rev Neurosci 3: 803-812. [DOI] [PubMed] [Google Scholar]
- Woodside BL, Borroni AM, Hammonds MD, Teyler TJ (2004) NMDA receptors and voltage-dependent calcium channels mediate different aspects of acquisition and retention of a spatial memory task. Neurobiol Learn Mem 81: 105-114. [DOI] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW (2001) Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA 98: 2808-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Denk W (1995) Dendritic spines as basic functional units of neuronal integration. Nature 375: 682-684. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA (2001) Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci 4: 711-717. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A (2003) Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron 39: 975-990. [DOI] [PubMed] [Google Scholar]