

Abstract
Voltage-gated Kv7 (KCNQ) channels underlie important K+ currents, including the neuronal M current, and are thought to be sensitive to membrane phosphatidylinositol 4,5-bisphosphate (PIP2) and PIP2 depletion to underlie muscarinic receptor inhibition. We studied regulation of Kv7.2-7.4 channels by PIP2 in Chinese hamster ovary (CHO) cells using single-channel and whole-cell patch clamp and biochemical analysis. Maximal open probabilities (Po) of Kv7.2-Kv7.4 homomultimers and of Kv7.2/7.3 heteromultimers were found to be strongly dependent on the [diC8-PIP2] applied to inside-out patches, with differential apparent affinities that correlate with their maximal Po in on-cell mode. Unitary conductance was not affected by PIP2. Raising tonic [PIP2] by coexpression of phosphatidylinositol (4)5-kinase increased the maximal Po of both Kv7.2 and Kv7.2/7.3 channels studied in on-cell patches and increased whole-cell Kv7.2, but not Kv7.3, current amplitudes. In cells coexpressed with muscarinic M1 receptors, bath application of muscarinic agonist reduced the maximal Po of Kv7.2/7.3 channels isolated in on-cell patches. Coexpression of a PIP2 sequestering construct moderately reduced whole-cell Kv7.2/7.3 currents, and coexpression of a construct containing a PIP2 phosphatase nearly abolished them. Finally, biochemical analysis of anionic phospholipids in CHO cells stably expressing M1 receptors shows that PIP2 and PIP are nearly depleted 1 min after muscarinic stimulation, with an unexpected rebound after 10 min. These results strongly support the direct regulation of Kv7 channels by PIP2 and its depletion as the mechanism of muscarinic suppression of M channels. Divergent apparent affinities of Kv7.2-7.4 channels for PIP2 may underlie their highly differential maximal Po observed in cell-attached patches.
Keywords: lipid signaling; potassium channel; M current; phosphatidylinositol 4,5-bisphosphate; single channel; gating; patch clamp
Introduction
Many ion channels and transporters have been shown to be sensitive to the presence of phosphatidylinositol 4,5-bisphosphate (PIP2) in the membrane (Suh and Hille, 2005). Particularly well studied has been PIP2 regulation of the family of inward rectifier K+ (Kir) channels, for which differential sensitivity to membrane PIP2 has been suggested to underlie the highly divergent open probabilities of this channel family (Rohacs et al., 2003). In this scenario, PIP2 has differential apparent affinities for each channel protein, and a given channel with a high apparent affinity for PIP2 will display a high open probability (Po) at tonic PIP2 levels and be relatively insensitive to decreases in PIP2, whereas channels with a low apparent affinity display a much lower tonic Po and are very sensitive to depletion of PIP2, as may happen after strong activation of phospholipase C (PLC).
An emerging literature suggests that a similar permissive requirement for membrane PIP2 controls the activity of the Kv7 family of M-type voltage-gated K+ channels, made by Kv7.1-7.5 (KCNQ1-5) subunits (Delmas and Brown, 2005). Most M currents in neurons are made by homomultimers and heteromultimers of Kv7.2, Kv7.3, and Kv7.5 (Wang et al., 1998; Lerche et al., 2000; Cooper et al., 2001; Roche et al., 2002; Hadley et al., 2003); Kv7.4 makes important K+ currents in the inner ear (Kubisch et al., 1999), and Kv7.1 co-assembles with auxiliary KCNE subunits to make the cardiac IKs current (Sanguinetti et al., 1996). Wide agreement is appearing that M channels are sensitive to the abundance of membrane PIP2 and that activation of PLC, and subsequent PIP2 hydrolysis, can sufficiently deplete the membrane of PIP2 to cause the observed downregulation of M currents by stimulation of Gq/11-coupled M1 muscarinic acetylcholine receptors (mAChRs) (Suh and Hille, 2002; Ford et al., 2003; Zhang et al., 2003; Suh et al., 2004). Although this hypothesis is compelling, single-channel demonstration of the direct gating of M channels by PIP2 has been lacking.
We recently characterized the gating of Kv7.2-7.5 channels at the single-channel level and found them to have strikingly different open probabilities in cell-attached patches (Li et al., 2004). Whereas Kv7.3 has a maximal Po near unity, Kv7.2, Kv7.4, and Kv7.5 have much lower maximal Po values and, in particular, Kv7.4 has a maximal Po over 10-fold lower than Kv7.3. Using a chimeric strategy, the highly divergent Po values for Kv7.3 and Kv7.4 were found to be conferred by the cytoplasmic C terminal of the channel. Here, we sought to more directly test the putative PIP2 regulation of Kv7 channel gating at the single-channel and whole-cell level and by biochemical analysis. We find that Kv7 channels are indeed highly sensitive to the PIP2 concentration at the cytoplasmic face of the patch and that the different Kv7 channels display a highly differential apparent affinity for PIP2 that may underlie their differential Po in intact cells. Furthermore, changes in [PIP2] have the expected effect on channel activity consistent with the PIP2-regulation hypothesis. Finally, PIP2 and PIP levels are shown to dramatically decrease and then rebound partially in parallel with a continuing fall of phosphatidylinositol (PI) and rise of phosphatidic acid (PA).
Materials and Methods
cDNA constructs. Plasmids encoding human Kv7.2, rat Kv7.3, human Kv7.4, and human Kv7.5 (GenBank accession numbers AF110020, AF091247, AF105202, and AF249278, respectively) were kindly given to us by D. McKinnon (State University of New York, Stony Brook, NY; Kv7.2 and Kv7.3), T. Jentsch (Zentrum für Molekulare Neurobiologie, Hamburg, Germany; Kv7.4), and K. Steinmeyer (Aventis Pharma, Frankfurt am Main, Germany; Kv7.5). Kv7.2 and Kv7.3 were subcloned into pcDNA3 (Invitrogen, San Diego, CA) as described previously (Shapiro et al., 2000). Kv7.4 and Kv7.5 were subcloned into pcDNA3.1zeo+ and pcDNA3.1zeo-(Invitrogen) using XhoI/HindIII and XbaI/EcoRI, respectively. EGFP-PLCδ-PH, EGFP-Lyn-PH-PP, and EGFP-Akt-PH constructs were kind gifts from T. Meyer (Stanford University, Stanford, CA). Type 1α PI(4)P5-kinase [PI(4)5-kinase] was kindly given to us by L. Pott (Ruhr-University, Bochum, Germany). The Chinese hamster ovary (CHO) cell line stably expressing M1 muscarinic receptors was kindly given to us by Prof. D. A. Brown (University College London, London, UK). The human mAChR M1 clone was purchased from the Guthrie Foundation (Sayre, PA).
Cell culture and transfections. CHO cells were used for electrophysiological analysis as described recently (Gamper et al., 2005a). Cells were grown in 100 mm tissue culture dishes (Falcon; Becton Dickinson, Mountain View, CA) in DMEM with 10% heat-inactivated fetal bovine serum and 0.1% penicillin and streptomycin in a humidified incubator at 37°C (5% CO2) and passaged every 3-4 d. Cells were discarded after ∼30 passages. For transfection, cells were plated onto poly-l-lysine-coated coverslip chips and transfected 24 h later with Polyfect reagent (Qiagen, Hilden, Germany) according to the instructions of the manufacturer. For electrophysiological experiments, cells were used 48-96 h after transfection. As a marker for successfully transfected cells, cDNA encoding green fluorescent protein (GFP) was cotransfected together with the cDNAs of the genes of interest. We found that >95% of green-fluorescing cells expressed KCNQ currents in control experiments.
Cell-attached/inside-out patch/single-channel electrophysiology. The methods of recording and analysis were similar to those previously used for studying unitary Kv7 channels (Li et al., 2004). Channel activity was recorded 48-72 h after transfection in the cell-attached configuration. Pipettes had resistances of 7-15 MΩ when filled with a solution of the following composition (in mm): 150 NaCl, 5 KCl, 1 MgCl2, and 10 HEPES, pH 7.4 with NaOH. Cells were bath perfused with a solution of the following composition (in mm): 175 KCl, 4 MgCl2, and 10 HEPES, pH 7.4 with KOH. There was no Ca2+ nor Ca2+-chealator added to these solutions, and we assume free Ca2+ to be in the range of ∼50 μm. The resting membrane voltage was assumed to be 0 mV. Currents were recorded using an Axopatch 1-D amplifier (Molecular Devices, Union City, CA), sampled at 5 kHz, and filtered at 200-500 Hz (Kv7.4) or at 0.5-1 kHz (all others). The short-chain, water-soluble PIP2 analog diC8-PIP2 (Echelon Biosciences, Salt Lake City, UT) was dissolved in an aqueous stock solution at 10 mm and sonicated immediately before each use.
Single-channel data were analyzed using PulseFit and TAC (Bruxton, Seattle, WA) software. Open and closed events were analyzed by using the “50% threshold criterion.” All events were carefully checked visually before being accepted. Po histograms were generated using TACFit (Bruxton). The presence of only one channel in a patch was assumed if the Po was >0.25 for >1 min without superimposed openings, especially at strongly depolarized potentials at which Po is highest. At a given potential, the single-channel amplitude (i) was calculated by fitting all-point histograms with single or multi-Gaussian curves. The difference between the fitted “closed” and “open” peaks was taken as i. When superimposed openings were observed, the number of channels in the patch was estimated from the maximal number of superimposed openings. The apparent NPo was estimated as follows:
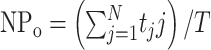 |
(1) |
where tj is the time spent at each current level corresponding to j = 0, 1, 2.. .N, T is the duration of the recording, and N is the number of current levels (minimum number of active channels).
Dose-response curves of channel Po versus [diC8-PIP2] were fit using IgorPro software (WaveMetrics, Lake Oswego, OR) by the following Hill equation:
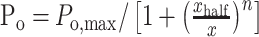 |
(2) |
where Po,max is set to 1, x is [diC8-PIP2], xhalf is the [diC8-PIP2] at which Po = 0.5, and n is the Hill coefficient.
Whole-cell electrophysiology. Whole-cell and perforated-patch experiments were performed as described previously (Gamper et al., 2003). To evaluate the amplitude of macroscopic Kv7.2/7.3 currents, CHO cells were held at 0 mV, and 800 ms hyperpolarizing steps to -60 mV, followed by 1 s pulses back to 0 mV, were applied. The amplitude of the current was usually defined as the difference between the holding current at 0 mV and the current at the beginning (after any capacity current as subsided) of the 1 s pulse back to 0 mV. In some cells, a more precise measurement was the XE991- or linopirdine-sensitive current at the holding potential of 0 mV. CHO cells have negligible endogenous macroscopic K+ currents under our experimental conditions, and 50 μm XE991 or linopirdine completely blocked the K+ current in Kv7-transfected CHO cells but had no effect on currents in nontransfected cells (Gamper et al., 2005a). All results are reported as mean ± SEM.
Phosphoinositide analysis. Anionic phospholipids were extracted from cells, deacylated, and quantified from the glycerol head groups by suppressed conductivity as described previously (Nasuhoglu et al., 2002).
Results
PIP2 stabilizes opening of unitary Kv7.2/7.3 channels and reactivates “run-down” channels
We first examined the sensitivity of Kv7.2/7.3 (KCNQ2/3) heteromultimers to PIP2, because these heteromeric channels comprise most “M-type” channels in sympathetic ganglia cells (Wang et al., 1998; Roche et al., 2002). Cloned Kv7.2+7.3 subunits were expressed in CHO cells and recordings from channels isolated in cell-attached patches, followed by excised inside-out patches. Heterologously expressed Kv7.2/7.3 channels produce voltagegated K+ currents very similar to native M currents in neurons (Wang et al., 1998; Selyanko et al., 2000; Shapiro et al., 2000). Because the muscarinic inhibition of M-type channels, thought to be attributable to depletion of PIP2 (Delmas and Brown, 2005), is voltage independent (Shapiro et al., 2000), we focus here on the Po of the channels at saturating voltages, which we refer to as their maximal Po. This value was measured usually at 0 mV, which is a saturating voltage for these channels (Li et al., 2004).
For channels sensitive to membrane PIP2 abundance, their activity in cell-attached patches should reflect the tonic level of PIP2, channel run-down during excision the loss of PIP2 in the patch by membrane-associated phosphatases, and channel activity in inside-out patches should be governed by the PIP2 concentration ([PIP2]) in the cytoplasmically facing bath solution. Because full-length PIP2 itself is water insoluble, making quantification of its concentration in aqueous solution uncertain, we used the short-chain, water-soluble analog dioctanoyl (diC8)-PIP2, as have other investigators studying the PIP2 sensitivity of these channels (Zhang et al., 2003). Figure 1 shows a representative experiment on a patch containing a single Kv7.2/7.3 channel examined at a saturating voltage of 0 mV, which is sufficiently positive to induce maximal Po (Li et al., 2004). In on-cell mode, the activity of the channel was present, but modest. After excision, the channel ran down considerably, displaying only rare open events. The channel activity, however, was strongly reactivated by the addition of diC8-PIP2 to the bath, resulting in channel activity much greater than that exhibited in on-cell mode. The modest channel activity in on-cell mode in this patch probably reflects a low tonic PIP2 abundance in this particular cell. At 50 μm diC8-PIP2, the channel Po was almost 0.5, and application of 200 μm diC8-PIP2 caused the channel to remain open almost all of the time. For Kv7.2/7.3 channels isolated in patches, the maximal Po in on-cell mode was 0.31 ± 0.04 (n = 41); after excision, the channels ran down to a Po of 0.14 ± 0.04 (n = 18), and the addition of 50 μm diC8-PIP2 reactivated the channels to a Po of 0.65 ± 0.09 (n = 8). An additional increase in diC8-PIP2 to 200 μm increased the Po at 0 mV to near unity (0.95; n = 2).
Figure 1.

DiC8-PIP2 stabilizes the opening of Kv7.2/7.3 channels. A, Current records from a patch containing a single Kv7.2/7.3 channel in cell-attached mode (On-cell) and in inside-out mode with various concentrations of diC8-PIP2 in the cytoplasmically facing bath solution. B, Each 3 s sweep during the experiment was analyzed for channel Po, and the time course of the Po during the experiment is plotted. The transition from on-cell to inside-out mode is indicated by the arrow, and the applications of different diC8-PIP2 concentrations are indicated by the bars.
Kv7.2-7.4 channels are sensitive to PIP2 with different apparent affinities
We then similarly investigated the PIP2 sensitivity of Kv7.2-7.4 homomultimers. We have previously showed Kv7.2-7.4 homomultimers to have a very different maximal Po (Li et al., 2004), raising the possibility that a differential apparent affinity for PIP2 may underlie these differences. Figure 2 shows representative experiments at a patch potential of 0 mV, which is a saturating voltage for these channels. A patch containing a single Kv7.2 channel (Fig. 2A) was excised and ran down to the marginal Po value of 0.01. Application of 3 μm diC8-PIP2 failed to significantly increase channel activity, but application of 100 μm had a robust effect, increasing channel Po to 0.35. Records from a patch containing a unitary Kv7.3 channel are shown in Figure 2B. In on-cell mode, channel activity was near unity, as is usual for this channel (Li et al., 2004). After excision, channel activity somewhat ran down, but remained substantial, with a Po value of 0.24. Application of only 3 μm diC8-PIP2 increased the Po to 0.72, and 30 μm diC8-PIP2 sufficed to restore channel Po to near its value in on-cell mode. A patch containing a single Kv7.4 channel, on the other hand, exhibited a negligible Po after excision to inside-out mode (0.001), and this value was only marginally increased by application of 20 μm diC8-PIP2 (Po = 0.005). However, the increased concentration in diC8-PIP2 to 150 μm resulted in a dramatic increase in channel Po, to 0.43.
Figure 2.

A-C, Individual sweeps from patches containing a single Kv7.2 (A), Kv7.3 (B), or Kv7.4 (C) channel in the presence of the indicated concentration of diC8-PIP2. The records were filtered at 500 Hz (A, B) and 200 Hz (C).
These types of experiments on homomeric Kv7.2-7.4 channels were performed over a range of diC8-PIP2 concentrations, and the data were summarized as a dose-response relationship as Po versus diC8-PIP2 concentration (Fig. 3A). For Kv7.2-7.4 homomultimers and Kv7.2/7.3 heteromultimers, channel Po was well described by such a dose-response relationship with diC8-PIP2 concentration, but with dramatically differential apparent affinities. For all of the channels, channel Po near unity could be achieved by application of diC8-PIP2, but at very different concentrations. For Kv7.2/7.3, Kv7.2, Kv7.3, and Kv7.4, the EC50 values were 40 ± 6 μm (n = 1-4 patches), 205 ± 36 μm (n = 1-8 patches), 2.6 ± 0.5 μm (n = 1-7 patches), and 215 ± 19 μm (n = 1-3 patches), respectively. Thus, there is a 100-fold difference in the apparent affinity of Kv7.3 versus Kv7.2 or Kv7.4 to diC8-PIP2, and we suppose also to full-length PIP2, and heteromeric Kv7.2/7.3 channels have an intermediate apparent affinity, as one would expect for a heteromeric channel composed of Kv7.2 and Kv7.3 subunits that both contribute to PIP2 binding, and to gating. For the four types of channels, the Hill coefficients were 1.24 ± 0.24, 1.33 ± 0.15, 0.97 ± 0.17, and 1.74 ± 0.15, respectively, very near that reported previously for diC8-PIP2 action on Kv7.2/7.3 (Zhang et al., 2003) and slightly lower than that modeled for full-length PIP2 action on Kv7.2/7.3 channels (Suh et al., 2004).
Figure 3.
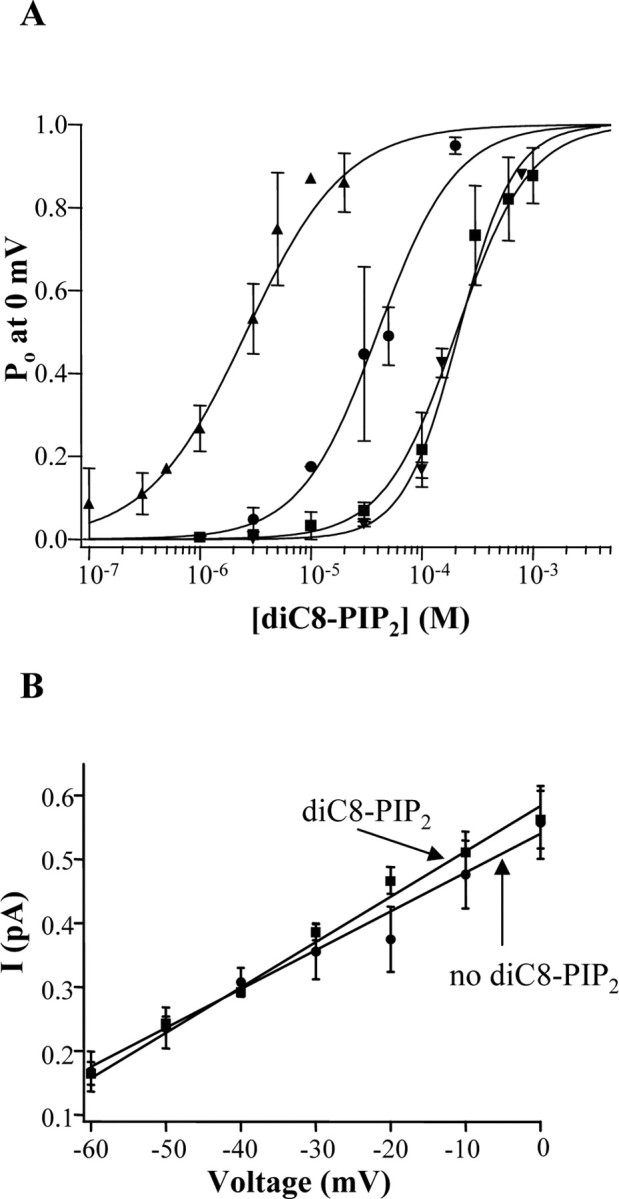
A, Dose-response relationship of the Po for Kv7.2 (squares), Kv7.3 (triangles), Kv7.4 (inverted triangles), and Kv7.2/7.3 (circles) channels as a function of diC8-PIP2 concentration in inside-out patches. The data were fit by a Hill equation (see Materials and Methods), with data errors taken into account by the fitting routine for all channels, except Kv7.2/7.3. The parameters of the fits are given in the text. B, The unitary current amplitudes for Kv7.2/7.3 channels over a range of voltages, either in the absence or presence of diC8-PIP2 (30 μm) in the bath, are plotted. The data were fit linearly, yielding single-channel conductances given in the text.
The differential apparent affinities of diC8-PIP2 for activation of Kv channels parallels their maximal Po in cell-attached patches, for which the rank order is Kv7.3 > Kv7.2/7.3 > Kv7.2 > Kv7.4 (Li et al., 2004), suggesting that the differential Po of channels in on-cell mode may be governed by their differential apparent affinity for plasma-membrane PIP2. By correlating the maximal Po of Kv7 channels in on-cell mode with their values as a function of diC8-PIP2 concentration, we can estimate what diC8-PIP2 concentration correlates with tonic [PIP2] in intact cells. In cell-attached patches, the maximal Po of Kv7.2/7.3, Kv7.2, Kv7.3, and Kv7.4 are 0.30-0.31, 0.04-0.17, 0.59-0.89, and 0.07 (Selyanko et al., 2001; Li et al., 2004; present study), respectively. From these values, we can estimate that an applied diC8-PIP2 concentration of ∼23 μm in inside-out patches corresponds to the tonic [PIP2] seen by channels in intact cells, because this concentration gives Po values similar to that seen in on-cell patches for all of the channels. We presume that the affinity of the channels to PIP2 differs from that for the shorter-chain diC8-PIP2 analog but that the relative apparent affinities for the different channels are likely similar for the two phosphoinositides. We also investigated whether PIP2 changes the single-channel conductance of Kv7 channels. Kv7.2/7.3 channels were recorded in inside-out patches over a range of potentials, both in the presence or absence of 30 μm diC8-PIP2. The presence of the phosphoinositide, although having a dramatic effect on channel Po, did not affect their unitary conductance, which was 6.1 ± 0.4 pS (n = 5) and 6.8 ± 0.4 pS (n = 5) in the absence or presence of 30 μm diC8-PIP2, respectively (Fig. 3B).
The molecule we call PIP2 is not the only doubly phosphorylated phosphoinositide used as a membrane signaling molecule; the other is phosphatidylinositol 3,4-bisphosphate [PI(3,4)P2]. The latter is produced by activation of phosphatidylinositol 3′-kinase (PI3-kinase), typically as a result of growth factor receptor activation, and is well known for activation of the kinase Akt/PKB. Although plasma-membrane PI(3,4)P2 concentrations are typically only a small fraction of [PIP2], even after strong PI3-kinase activation, the sensitivity of other PIP2-sensitive channels to [PI(3,4)P2] has been investigated as a probe of phosphoinositide binding (Rohacs et al., 1999). We tested whether bath application of PI(3,4)P2 to inside-out patches containing Kv7.2/7.3 channels would also stabilize channel activity. We found that at 10 μm, PI(3,4)P2 reactivated channels to a similar maximal Po observed in cell-attached patches, and at 20 μm, channel activity was further increased (data not shown). Thus, like certain types of Kir3 channels (Rohacs et al., 1999), Kv7.2/7.3 channels may have a nonspecific phosphoinositide activation profile, at least in terms of these two distinct moieties of doubly phosphorylated phosphoinositides.
Increasing tonic [PIP2] by overexpression of PI(4)5-kinase increases on-cell channel activity
Having ascertained that Kv7 channels are sensitive to plasma-membrane PIP2, we then manipulated [PIP2] in intact cells in several different ways and examined the effects on maximal Po. PIP2 is produced in the plasma membrane by sequential phosphorylation of PI by PI4-kinase and PI(4)5-kinase. Overexpression of PI(4)5-kinase in atrial myocytes or in sympathetic neurons has been shown to tonically increase membrane PIP2 and to dramatically reduce desensitization of Kir3 channels in the former case and muscarinic modulation of endogenous M current in the latter case (Bender et al., 2002; Winks et al., 2005). We thus reasoned that if Kv7 channels are indeed sensitive to membrane [PIP2], then its tonic increase by overexpression of PI(4)5-kinase should increase channel Po recorded in cell-attached patches. CHO cells were coexpressed with Kv7.2+Kv7.3 or only Kv7.2, either alone or together with PI(4)5-kinase. For both Kv7.2/7.3 and Kv7.2 channels, there was a dramatic increase in maximal Po in cells coexpressed with PI(4)5-kinase. In addition, the profound run-down in channel activity observed after excision of patches from control cells was sharply reduced in patches from PI(4)5-kinase-transfected cells (Fig. 4A). These data are summarized in Figure 4B. In on-cell patches containing Kv7.2 channels only, the Po at 0 mV was 0.035 ± 0.013 (n = 12), but in patches from cells coexpressing PI(4)5-kinase, the Po was 0.74 ± 0.09 (n = 10). In on-cell patches containing Kv7.2/7.3 channels only, the Po at 0 mV was 0.31 ± 0.04 (n = 41), but in patches from cells coexpressing PI(4)5-kinase, the Po was 0.95 ± 0.03 (n = 13). These results are in accord with recent work from the Brown laboratory, which showed that overexpression of PI(4)5-kinase increased [PIP2] nearly threefold in sympathetic neurons, resulting in strongly blunted muscarinic modulation of M currents (Winks et al., 2005). Although we cannot quantify the increase produced here, we estimate it to be at least that much, judging from the robust increase in on-cell channel Po and from the strongly reduced run-down of the channels after patch excision.
Figure 4.
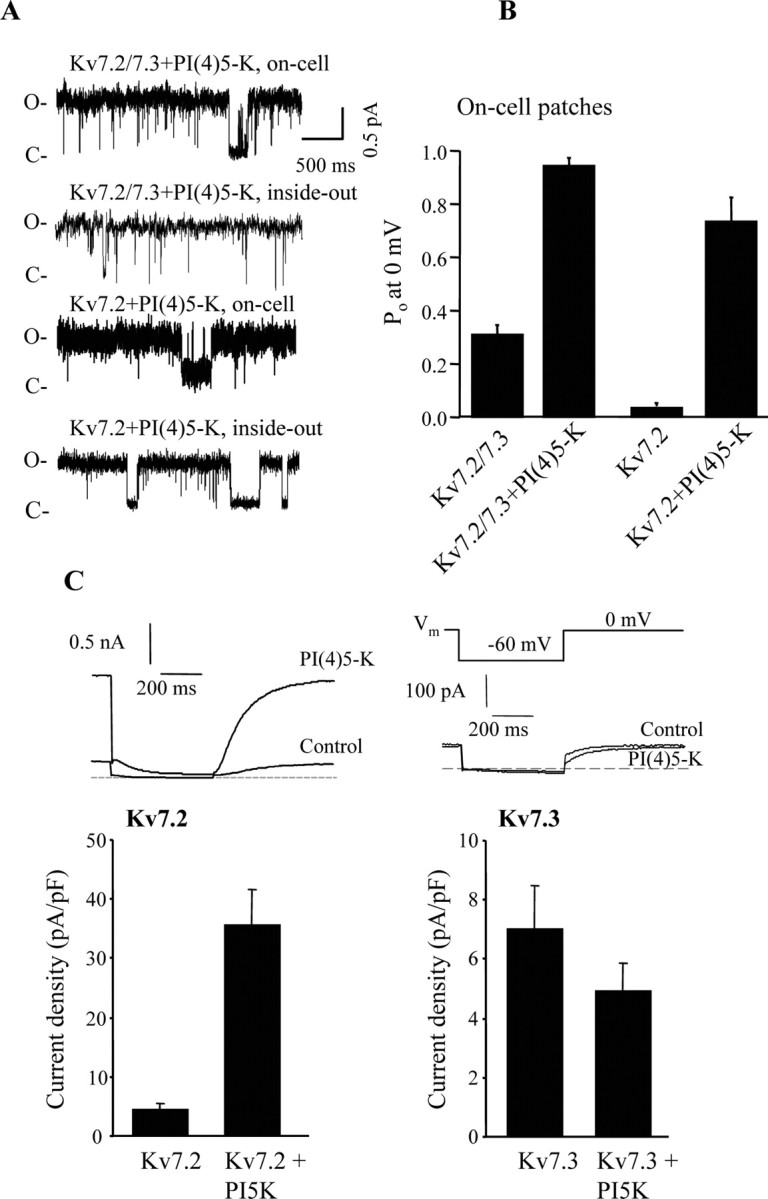
Overexpression of PI(4)5-kinase increases the Po of Kv7 channels in patches and increases whole-cell Kv7.2 but not Kv7.3, currents. A, Individual sweeps from a patch containing either a single Kv7.2/7.3 or a Kv7.2 channel from control CHO cells or those cotransfected with PI(4)5-kinase are shown. Recordings were first made in on-cell modes, followed by excision to inside-out modes. O, Open channel levels; C, closed channel levels. B, Bars show the mean Po at 0 mV for all of the on-cell patches. C, Bars show whole-cell current amplitudes from CHO cells transfected with Kv7.2 or Kv7.3 alone or together with PI(4)5-kinase. Traces from representative experiments are displayed above. The voltage protocol used is as shown.
Heterologously expressed homomeric Kv7.2 and 7.3 channels display whole-cell current amplitudes some 10-fold smaller than do Kv7.2/7.3 heteromultimers (Wang et al., 1998; Gamper et al., 2003; Maljevic et al., 2003; Schwake et al., 2003). Although it has been suggested that this difference is attributable to differential surface expression of the homomeric and heteromeric channels (Schwake et al., 2000), a number of factors are likely involved (Etxeberria et al., 2004). The dramatic increase in maximal Po of Kv7.2 seen in cell-attached patches from PI(4)5-kinase-expressing cells suggested that part of the reason for the low macroscopic expression of Kv7.2 channels might be as a result of its low apparent affinity for PIP2 and that such expression might be much larger in cells with higher tonic [PIP2] produced by overexpression of PI(4)5-kinase. Indeed, this turned out to be the case (Fig. 4C). Control cells displayed very low Kv7.2 current densities (4.5 ± 1.1 pA/pF; n = 10), but those coexpressed with PI(4)5-kinase displayed current densities some eightfold larger (34.7 ± 5.8 pA; n = 10; p < 0.001). On the other hand, the high maximal Po of Kv7.3 homomultimers (Li et al., 2004) and its high apparent affinity for diC8-PIP2 (present study) predicts that overexpression of PI(4)5-kinase should not increase macroscopic Kv7.3 currents, and this was indeed the case as well (Fig. 4C). In control cells, Kv7.3 current densities were 7.0 ± 1.5 pA/pF (n = 10), and in cells coexpressed with PI(4)5-kinase, they were still low, at 4.9 ± 1.0 pA/pF (n = 10). These data reinforce the conclusion that Kv7.2 and Kv7.3 channels have highly differential apparent affinities for PIP2 and furthermore suggest that part of the difference between Kv7.2 and Kv7.2/7.3 current amplitudes may be attributable to the lower apparent affinity of Kv7.2 for PIP2. However, the high affinity of Kv7.3 for diC8-PIP2 found in this study, reinforced by the lack of increase in whole-cell currents in cells overexpressing PI(4)5-kinase, suggests that the low macroscopic expression Kv7.3 is not attributable to weak binding to PIP2 and indicates a further difference between Kv7.2 and Kv7.3.
Cell-attached channel Po is suppressed by bath application of muscarinic agonists
To further explore PIP2 regulation of Kv7 channels, we reduced membrane PIP2 abundance in intact CHO cells by receptor stimulation and by tonic PIP2 depletion. The separation of types of G-protein-mediated modulation of channels between “direct” and “second-messenger-mediated” mechanisms is best observed using cell-attached patch experiments. In direct mechanisms involving interactions between G-protein and channels, bath application of receptor agonists does not modulate the activity of channels isolated in the patch membrane, presumably because activated G-protein subunits cannot diffuse in the plane of the membrane past its interface with the glass of the pipette tip. However, when second-messenger molecules underlie the modulation, as is the case for Gq/11 actions, channels in the patch can be modulated by bath-applied agonists. For the case of muscarinic modulation of N-type Ca2+ channels in SCG neurons, the rapid inhibition by G-protein βγ dimers, thought to underlie modulation by Go/i-coupled M2/4 receptors (Herlitze et al., 1996), is not seen in such cell-attached patch experiments, but the slower inhibition by Gq/11-coupled M1 receptors does affect channels in the patch, implicating an intracellular second messenger in the action (Hille, 1994). The latter pathway has been shown to be attributable to depletion of PIP2 in the membrane (Gamper et al., 2004). Likewise, the M1 receptor-mediated modulation of M channels that gives M current its name also reaches past the glass/membrane interface, suppressing M channels in the cell-attached patch (Selyanko et al., 1992). Finally, M1 receptor-mediated desensitization of GIRK (G-protein-activated inwardly rectifying K+; Kir3) channels, thought also to be attributable to depletion of membrane PIP2, is also evident on channels in the patch when M1 receptors in the rest of the cell are stimulated (Zhang et al., 2003). Thus, it seems likely that, unlike membrane proteins, membrane PIP2 can diffuse into and out of the membrane in the tips of patch pipettes and is the exemplar case of a type signal recently termed “diffusible membrane delimited” (Delmas et al., 2005).
We wanted to analyze the muscarinic suppression of Kv7.2/7.3 channels, mediated by depletion of PIP2, at the single-channel level, to compare it to regulation of the channels by diC8-PIP2 in inside-out patches. CHO cells transfected with Kv7.2, Kv7.3, and the human M1 receptor were studied in cell-attached patches, and unitary currents recorded before, during, and after application of the muscarinic agonist oxotremorine-M (oxo-M) (Fig. 5). Although such stimulation of heterologously expressed M1 receptors likely raises [Ca2+]i (Shapiro et al., 2000), we have shown Kv7.2/7.3 channels heterologously expressed in CHO cells to be mostly insensitive to rises in [Ca2+]i in the absence of coexpressed calmodulin (Gamper and Shapiro, 2003). In the experiment shown in Figure 5A, Po was 0.40 in control, slightly higher than that typically seen for Kv7.2/7.3 channels. Bath application of oxo-M (10 μm) strongly reduced channel Po within several minutes to 0.07, and channel activity was reversible to a Po of 0.24 several minutes after washout of the agonist. In all such patches tested, the Po in control was 0.22 ± 0.06; after application of oxo-M, the Po was sharply reduced by 73% to 0.060 ± 0.018, and after washout, it mostly recovered, to 0.19 ± 0.03 (n = 4). This action of bath-applied muscarinic agonists on unitary Kv7.2/7.3 channels isolated in the cell-attached patch corresponds very closely with the action on whole-cell currents seen previously (Selyanko et al., 2000; Shapiro et al., 2000). These data suggest that, although there are known differences in this signaling pathway between a heterologous expression system and real neurons, muscarinic modulation of neuronal M currents can be wholly accounted for by a modulation of channel Po. It also reinforces the conclusion that PIP2 can diffuse into and out of patches of membrane in the tips of patch pipettes. From similarly transfected CHO cells, we also used “macropatches,” containing many Kv7.2/7.3 channels to observe such inhibition from bath-applied agonist. These experiments also included the membrane-permeant IP3-receptor blocker 2-aminoethoxydiphenyl borate (Maruyama et al., 1997) to exclude the possibility that the inhibition was attributable to rises in [Ca2+]i. Bath application of oxo-M (10 μm) strongly and reversibly inhibited the Kv7.2/7.3 currents in the patches (data not shown), similar to previous results using such macropatch experiments (Selyanko et al., 2000).
Figure 5.
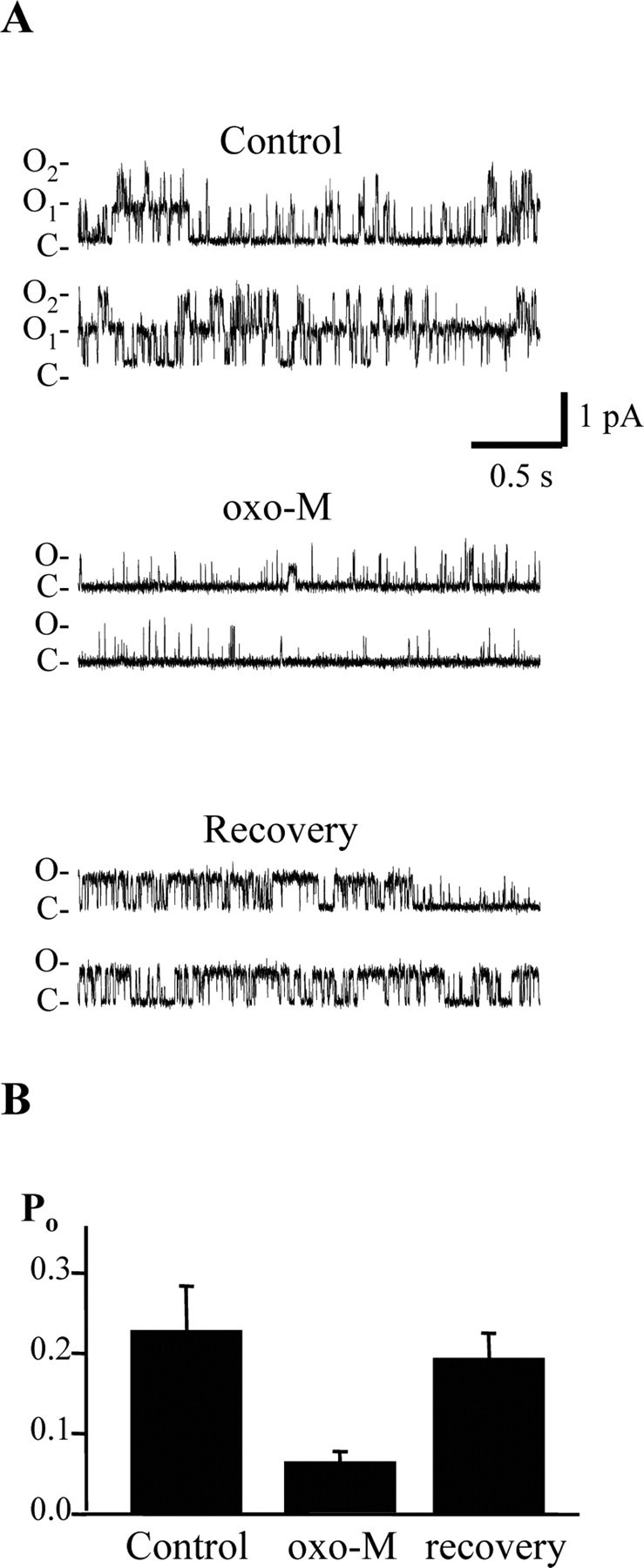
Stimulation of M1 receptors by bath application of agonist reduces the Po of channels in cell-attached patches. CHO cells were cotransfected with Kv7.2+7.3 channels and M1 receptors. A, Sweeps show channel activities from a patch containing two Kv7.2/3 channels before, during, and after bath application of oxo-M (10 μm). B, Bars show the mean unitary Po from four experiments such as in A. O, Open channel levels; C, closed channel levels.
Coexpression of PIP2 sequestering or degrading constructs reduces whole-cell Kv7.2/7.3 currents in CHO cells
We also assayed the PIP2 sensitivity of Kv7.2/7.3 channels by testing several enhanced GFP (EGFP)-tagged constructs that have been shown to bind and sequester PIP2 or to decrease its plasma membrane concentration. The first, PLCδ-PH, contains the “pleckstrin homology” (PH) domain of PLCδ that binds to PIP2 to achieve its plasma-membrane targeting (Shaw, 1996). Overexpression of PLCδ-PH has been shown to sequester PIP2 and thus make it unavailable to act as an intracellular effector (Stauffer et al., 1998; Raucher et al., 2000; Gamper et al., 2004). The second, Akt-PH, contains the PH domain of the serine/threonine kinase Akt. Akt binds to, and is activated by, PI3-kinase-generated phosphoinositol PI(3,4)P2 but not PI(4,5)P2 (the molecule in this work abbreviated as PIP2) (Franke et al., 1997), and so Akt-PH is a useful control for PLCδ-PH. The third, Lyn-PH-PP, codes for a PIP2-specific phospholipid, 5′-phosphatase (Stolz et al., 1998), that selectively reduces plasma membrane PIP2 concentrations but not other intracellular pools of PIP2 or other plasma membrane PI phosphates, expressed as a fusion protein to a myristoylation/palmitoylation sequence of the tyrosine kinase Lyn that achieves its plasma membrane localization (Raucher et al., 2000). Because all of these PIP2-testing constructs are linked to EGFP, we can use single-cell imaging to (1) confirm their presence in the cell and (2) localize their distribution within the cell (i.e., plasma membrane or cytoplasm).
CHO cells were cotransfected with Kv7.2/7.3 channels and only GFP, Akt-PH, PLCδ-PH, or Lyn-PH-PP. For all four, we observed robust fluorescence that was strongly membrane localized for the cases of PLCδ-PH and Lyn-PH-PP or diffusely cytoplasmic for the case of GFP and Akt-PH. We found that cotransfection of PLCδ-PH strongly reduced the amplitude of the Kv7.2/7.3 currents and Lyn-PH-PP nearly abolished the currents, but cotransfection of Akt-PH had little effect (Fig. 6). For these four groups of cells, the mean current densities were 58.3 ± 9.4 pA/pF (n = 12), 69.9 ± 14 pA/pF (n = 9), 22.4 ± 6.5 pA/pF (p < 0.05; n = 14), and 6.5 ± 1.0 pA/pF (p < 0.001; n = 19). We interpret the reduced currents in the PLCδ-PH cells as being attributable to the reduced abundance of free PIP2, resulting in a reduced tonic maximal Po of the channels, consistent with the inability of PLCδ PH to bind to channels and stabilize their opening. The Lyn-PH-PP-expressing cells are predicted to have a severely depleted PIP2 abundance from the strongly augmented PIP2 phosphatase activity and the strongly reduced current density to arise from a much lower Po of the channels. We verified that expression of the PIP2-binding constructs does not affect expression of channel protein using immunoblots of whole-cell lysates from cells expressed with myc-tagged Kv7.2+Kv7.3 together with each construct (data not shown). Assuming that the effects on current amplitudes are as a result of reduction in the Po, we can use the dose-response curve of unitary Kv7.2/7.3 Po versus [diC8-PIP2] (Fig. 3) to estimate changes in tonic membrane PIP2 caused by expression of the two PIP2 sequestering/degrading constructs. Assuming a normal maximal Kv7.2/7.3 Po of 0.31 from our data, that corresponds to a [diC8-PIP2] of 23 μm in our inside-out patch experiments. In cells expressing PLCδ-PH, the whole-cell currents were reduced by 69%, corresponding to a Po of 0.096 and a [diC8-PIP2] of 8.2 μm; in cells expressing Lyn-PH-PP, whole-cell currents were reduced by 91%, corresponding to a Po of 0.026 and a [diC8-PIP2] of 3.0 μm. Thus, under the assumption that changes in [diC8-PIP2] and in [PIP2] have similar effects, we estimate that expression of PLCδ-PH and Lyn-PH-PP reduced free PIP2 in CHO cells by 64 and 87%, respectively.
Figure 6.
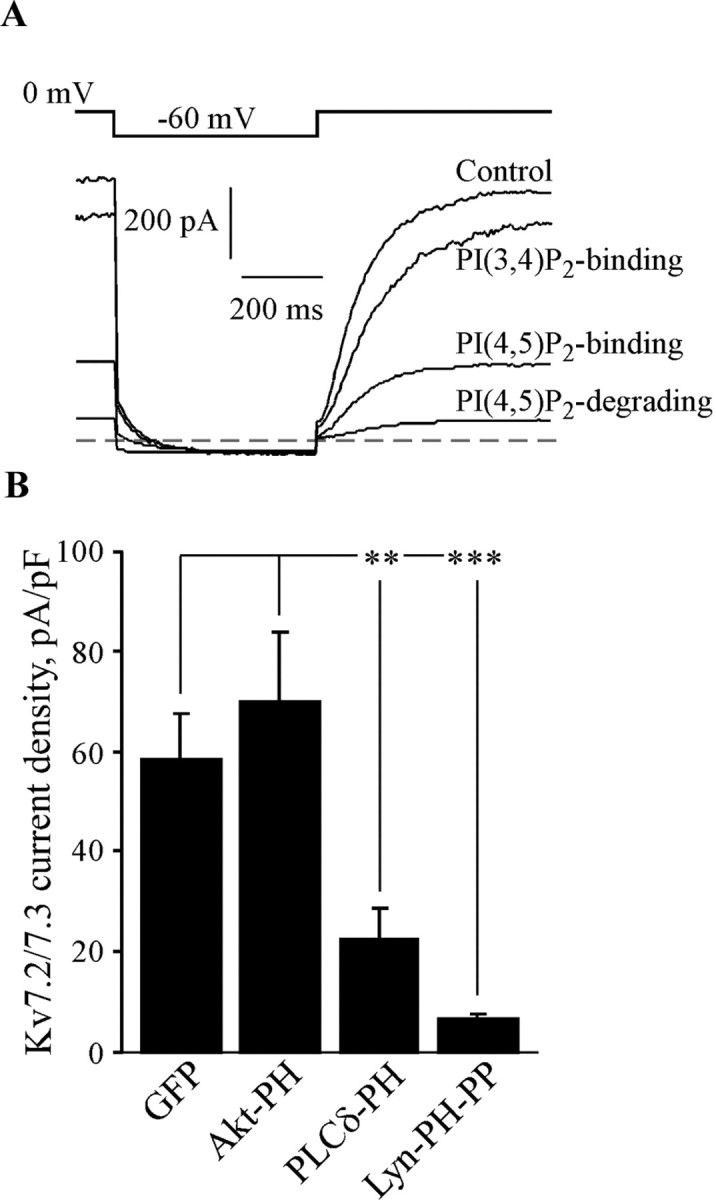
Expression of PIP2 sequestering or degrading constructs reduces whole-cell Kv7.2/7.3 current amplitudes. CHO cells were cotransfected with Kv7.2+Kv7.3 together with either GFP only (control) or the PLCδ PH, Lyn-PH-PP, or Akt-PH constructs. Currents were elicited using the indicated voltage protocol in whole-cell configuration. A, Plotted are superimposed current traces from four different cells transfected with each different construct. B, Bars show the mean whole-cell current amplitudes for these four groups of cells. **p < 0.01; ***p < 0.001.
Stimulation of Gq/11-coupled M1 receptors depletes PIP2 in CHO cells
As discussed previously, muscarinic suppression of M-type currents is thought to primarily involve depletion of membrane PIP2 by activation of PLC (Suh and Hille, 2002; Zhang et al., 2003; Suh et al., 2004). Our findings here suggest that the Po of these channels are highly sensitive to PIP2 abundance but that a strong depletion of PIP2 would be required to cause the profound (>70%) suppression of the currents that is seen. Because this hypothesis of how muscarinic stimulation suppresses M channels predicts a strong reduction in PIP2, we took advantage of a CHO cell line stably expressing M1 receptors to analytically measure changes in phosphoinositide abundance induced by muscarinic stimulation of these cells. Using HPLC analysis, we assayed the abundance of several different phosphoinositides, including PIP2, phosphatidylinositol 4-phosphate (PIP), PI, and PA, before and after exposure of the cells to the cholinergic agonist carbachol (100 μm) for 1 or 10 min (Fig. 7).
Figure 7.

Effects of muscarinic stimulation on anionic phospholipids in CHO cells expressing M1 receptors. Anionic phospholipids were determined from their deacylation products by anion exchange HPLC with suppressed conductivity detection. Data are compiled for application of the cholinergic agonist carbachol (100 μm) for 1 or 10 min. Bars show the mean PIP2, PIP, PI, or PA, expressed as a percentage of total membrane phosphoinositides. The left axis refers to %PIP2 and %PIP, and the right axis refers to %PI and %PA.
We found that muscarinic stimulation for 1 min indeed reduced PIP2 abundance. Results are given as percentages of total anionic phospholipids, a pool that includes the phosphatidylserine and cardiolipin moieties that are not involved in PLC activity. The %PIP2 was strongly reduced from 0.80 ± 0.02% (n = 7) before to 0.10 ± 0.04% (n = 5) after 1 min of carbachol exposure. Receptor stimulation also caused a strong decline in %PIP, from 0.99 ± 0.06% (n = 7) before to 0.19 ± 0.04% (n = 5) after 1 min of carbachol exposure. This parallel decline in %PIP2 and %PIP is similar to that measured in human neuroblastoma cells (Willars et al., 1998). Unexpectedly, the %PI also displayed a significant decline, falling from 49.5 ± 0.7% (n = 7) before to 46.5 ± 0.3% (n = 5) after 1 min of carbachol exposure. Because diacylglycerol (DAG), the membrane-bound byproduct of PIP2 hydrolysis, is rapidly converted to PA by DAG-kinase, the %PA increased after receptor stimulation, as expected. The %PA rose from 15.8 ± 1.1% (n = 7) before to 17.6 ± 0.9% (n = 5) after 1 min of carbachol exposure. Although muscarinic suppression of M/Kv7 channels does not noticeably desensitize in patch-clamp experiments, we found the abundance of PIP2, and to a lesser extent PIP, to slowly recover during maintained muscarinic stimulation. Thus, the %PIP2 and the %PIP recovered to 0.57 ± 0.15% (n = 5) and 0.40 ± 0.06% (n = 5) after 10 min of carbachol exposure, respectively. The %PI, however, continued to fall during maintained receptor stimulation, falling to 39.0 ± 1.1% (n = 5) after 10 min of carbachol exposure. The %PA also continued to rise during the maintained stimulation, rising further to 24.1 ± 1.4% (n = 5) after 10 min of carbachol exposure. Both the continued fall in %PI and the continued rise in %PA suggest that PIP2 hydrolysis continued unabated during the longer carbachol exposure, arguing against receptor desensitization as the reason for the partial recovery of %PIP2 and %PIP during the 10 min treatment. Perhaps this partial recovery is attributable to the agonist-mediated stimulation of PIP and PIP2 synthesis by PI4-kinase and PI(4)5-kinase, as has been shown recently for Gq/11-coupled bradykinin receptors in neuroblastoma cells (Xu et al., 2003) and suggested in sympathetic neurons (Gamper et al., 2004).
Discussion
The importance of phosphoinositides in cellular signals triggered by plasma-membrane receptors coupled to the Gq/11 family of G-proteins has been appreciated for decades. In this pathway, hormonal stimulation results in activation of PLC, which hydrolyzes PIP2 into membrane-bound DAG and soluble inositol trisphosphate, both potent intracellular signaling molecules. However, in the past decade, it has become clear that the PIP2 molecule functions as a second messenger in its own right. Much evidence has accumulated over the past several years suggesting that Kv7 channels are sensitive to the abundance of PIP2 in the plasma membrane (Delmas and Brown, 2005). Although Kv7 channels are voltage-gated K+ channels (unlike PIP2-sensitive Kir channels), the presence of PIP2 would act to stabilize opening by favoring the allosteric conformational change that opens the channel, such that Po is increased at all voltages. The physiological role of such PIP2 sensitivity is perhaps most clear for the case of M-type channels, which are thus regulated by depletions of membrane PIP2 produced by PLC activation downstream of Gq/11-coupled receptor stimulation. This mechanism predicts the maximal Po of Kv7 channels in intact cells to be governed by the tonic abundance of PIP2 in the membrane and increases or decreases in tonic PIP2 levels to increase or decrease maximal channel Po, respectively. Stimulation of other Gq/11-coupled receptors in sympathetic neurons also suppress M currents, however not by large changes in membrane PIP2 (Gamper et al., 2004) but by generation of intracellular Ca2+ signals (Cruzblanca et al., 1998; Bofill-Cardona et al., 2000), which may act in concert with calmodulin (Gamper and Shapiro, 2003; Gamper et al., 2005b) to alter the affinity of the channels for PIP2, such that tonic PIP2 abundance is insufficient to keep most channels open (Winks et al., 2005).
Here, we directly tested the PIP2-gating hypothesis by a variety of methods. The first was inside-out patch recording and a range of water-soluble diC8-PIP2 concentrations applied to the cytoplasmic face of the patch. We found that the maximal single-channel Po of Kv7.2-7.4 and Kv7.2/7.3 channels is highly governed by [diC8-PIP2], with a strongly differential apparent affinity of the channels for diC8-PIP2 that parallels the differential maximal Po in cell-attached patches (Li et al., 2004). In this study, we did not posit that the affinity of the channels for diC8-PIP2 is the same as for (full-length) PIP2, but did assume that relative differences in the apparent affinities of the different channels for diC8-PIP2 and full-length PIP2 do correspond. For all of the channels studied in inside-out patches, the Po could be increased well beyond their values normally seen in cell-attached patches by increasing the diC8-PIP2 concentration. Consistent with this result is the strong increase in maximal channel Po of Kv7.2/7.3 and Kv7.2 observed in cell-attached patches from cells overexpressing PI(4)5-kinase, resulting presumably from greater tonic PIP2 abundance in the membrane. The increase in macroscopic Kv7.2, but not Kv7.3, currents by overexpression of PI(4)5-kinase was particularly dramatic and reinforces both the conclusions of low apparent affinity of Kv7.2 for PIP2 and the highly differential maximal Po of Kv7.2 and Kv7.3 in cell-attached patches reported here and previously (Li et al., 2004). Conversely, maneuvers that tonically decreased the abundance of free membrane PIP2 strongly lowered tonic current amplitudes, almost certainly by lowering channel Po. A strong reduction in unitary channel Po by muscarinic stimulation was observed that correlates well with the suppression of macroscopic currents, and that resembles the changes in channel Po seen in the direct diC8-PIP2 applications. Finally, HPLC analysis confirms the necessary profound PIP2 depletion in cells heterologously expressing M1 receptors. There is a discrepancy at later times, in that PIP2 and PIP levels recover in part over a few minutes during receptor stimulation, whereas the inhibition of channel activity is usually stable over a similar time course. One explanation is that PIP2 is being generated in membrane domains from which PIP2 diffusion to M channels is in some way restricted. Despite this discrepancy, our work is strongly supportive of PIP2 regulation of Kv7 channels as being physiologically integral to G-protein channel signaling.
Although our data suggest that the highly divergent Po values of Kv7 channels are associated with very differential apparent affinities for PIP2, we have no biochemical data on their binding affinities. Indeed, although several investigators assume that PIP2-sensitive channels are regulated by direct binding of PIP2 molecules to the channel proteins in the membrane, clear evidence for this remains sparse. However, if there is an intermediate protein that mediates PIP2 regulation of Kv7 channels, it would have to be membrane associated because the regulation happens in excised inside-out patches and must be present both in the plasma membrane of mammalian cells (present study) and of oocytes (Zhang et al., 2003). For the case of inward rectifier K+ channels, the large effects on their PIP2 regulation by neutralizations of positively charged amino acids lends support to the idea of direct binding, as does a recent crystal structure (Pegan et al., 2005). Thus, direct binding to Kv7 channels is our working hypothesis. For both families of channels that contain members with divergent apparent affinities for PIP2, it seems as yet unknown whether their biochemical binding affinities are so divergent, or whether their intrinsic energy of opening diverges, resulting in a differential amount of stabilization energy coming from PIP2 binding being required to achieve the same level of channel opening. Because K+ channels are tetramers, and presumably each subunit can bind PIP2, it will be critical to learn the stoichiometry between PIP2 molecules and channel and the relationship between bound PIP2 molecules/channel and opening. Work from the Logothetis laboratory has also demonstrated direct gating of Kv7 channels by PIP2 in oocyte-excised macropatches (Zhang et al., 2003). Their EC50 value for activation of Kv7.2/7.3 channels by diC8-PIP2 was 87 μm, with a Hill slope of 1.35, in the range of that seen here for regulation of unitary channel Po. They also showed direct regulation of Kv7.1, Kv7.4, and Kv7.5 channels by PIP2, but they did not investigate any differential apparent affinities among the channels. The mild cooperativity for the binding of multiple PIP2 molecules to the channel modeled by Suh et al. (2004) seems in the correct range both for the slope of the dose-response curves and of muscarinic agonist versus M current seen by many laboratories and for diC8-PIP2 versus single-channel Po seen here (nHill = 1-1.8).
The maximal on-cell Po for Kv7.2/7.3 heteromultimers is seen here to be ∼0.3, an intermediate value between low-Po Kv7.2 and high-Po Kv7.3 (Li et al., 2004), and to be in the range of 0.13-0.3 by the Brown laboratory (Selyanko et al., 2001; Tatulian and Brown, 2003). Our inside-patch data show that the maximal Po continues to rise past this range of values as the applied [diC8-PIP2] is increased, and in on-cell patch, overexpression of PI(4)5-kinase likewise strongly raises maximal Po. Together, these data suggest that the tonic [PIP2] in intact cells is well under that necessary to saturate PIP2 binding to the channels. However, in SCG neurons, the M channels of which heavily consist of Kv7.2/7.3 heteromultimers, overexpression of PI(4)5-kinase did not increase tonic M-current amplitudes (Winks et al., 2005), and we found that its overexpression likewise did not significantly increase whole-cell Kv7.2/Kv7.3 currents expressed in CHO cells (our unpublished observations). Perhaps this discrepancy between single-channel and macroscopic effects is attributable to a secondary effect of increased [PIP2] in promoting endocytosis of membrane proteins, as has been described recently (Huang et al., 2004). Thus, an increase in current amplitudes by higher tonic [PIP2] increasing channel Po might be balanced by a decrease in tonic surface abundance of the channel proteins.
Left unanswered is the molecular mechanism by which PIP2 binding to the channel proteins stabilizes the allosteric conformational change that is opening. Because the “kink” in the sixth transmembrane segment (S6) thought to be the swivel point of the main gate of Kv channels (Jiang et al., 2002) is located in the middle of the membrane, and because the S6 helix is thought to end just after it emerges from the inner leaflet of the membrane, PIP2 should bind to the channel downstream of S6 (i.e., to the cytoplasmic tail). Indeed, mutagenesis (Zhang et al., 2003) and single-channel recording of Kv7.3/Kv7.4 chimeras (Li et al., 2004; Gamper et al., 2005b) indicate that this is where PIP2-mediated regulation of channel opening occurs. From that point, the PIP2/C-terminal interaction could result in some force that pulls the S6 helix away from the pore, opening it. Because Kv7 channels, like other Kv channels, are voltage gated, this must occur after the movement of the voltage sensors of the channels permits gating. Perhaps the PIP2 action is to anchor the post-S6 end of the channel in place, causing the voltage-induced flexing of the channel to splay the pore open. Alternatively, the hydrophobic part of the PIP2 molecule may cause it to favor being surrounded by lipid, generating a force away from the channel axis as the carboxy-tail is pulled from the rest of the protein. Such structural questions await more detailed knowledge of the molecular architecture of Kv7 channels and of how PIP2 binding to the protein alters its configuration in the plasma membrane.
Footnotes
This work was supported by National Institutes of Health Grants R01 NS043394 (M.S.S.) and R01 HL067942 (D.W.H.) and American Heart Association Postdoctoral Fellowship 0325120Y (N.G.). We thank Pamela Martin, Kathryn Boyd, and Chengcheng Shen for expert technical assistance and Brad Rothberg and James Stockand for helpful discussions.
Correspondence should be addressed to Dr. Mark S. Shapiro, Department of Physiology, MS 7756, University of Texas Health Science Center, 7703 Floyd Curl Drive, San Antonio, TX 78229. E-mail: shapirom@uthscsa.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259825-11$15.00/0
References
- Bender K, Wellner-Kienitz MC, Pott L (2002) Transfection of a phosphatidyl-4-phosphate 5-kinase gene into rat atrial myocytes removes inhibition of GIRK current by endothelin and alpha-adrenergic agonists. FEBS Lett 529: 356-360. [DOI] [PubMed] [Google Scholar]
- Bofill-Cardona E, Vartian N, Nanoff C, Freissmuth M, Boehm S (2000) Two different signaling mechanisms involved in the excitation of rat sympathetic neurons by uridine nucleotides. Mol Pharmacol 57: 1165-1172. [PubMed] [Google Scholar]
- Cooper EC, Harrington E, Jan YN, Jan LY (2001) M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci 21: 9529-9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruzblanca H, Koh DS, Hille B (1998) Bradykinin inhibits M current via phospholipase C and Ca2+ release from IP3-sensitive Ca2+ stores in rat sympathetic neurons. Proc Natl Acad Sci USA 95: 7151-7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Brown DA (2005) Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci, in press. [DOI] [PubMed]
- Delmas P, Coste B, Gamper N, Shapiro MS (2005) Phosphoinositide lipid second messengers: new paradigms for calcium channel modulation. Neuron 47: 179-182. [DOI] [PubMed] [Google Scholar]
- Etxeberria A, Santana-Castro I, Regalado MP, Aivar P, Villarroel A (2004) Three mechanisms underlie KCNQ2/3 heteromeric potassium M-channel potentiation. J Neurosci 24: 9146-9152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Stemkowski PL, Light PE, Smith PA (2003) Experiments to test the role of phosphatidylinositol 4,5-bisphosphate in neurotransmitter-induced M-channel closure in bullfrog sympathetic neurons. J Neurosci 23: 4931-4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC, Toker A (1997) Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 275: 665-668. [DOI] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS (2003) Calmodulin mediates Ca2+-dependent modulation of M-type K+ channels. J Gen Physiol 122: 17-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Stockand JD, Shapiro MS (2003) Subunit-specific modulation of KCNQ potassium channels by Src tyrosine kinase. J Neurosci 23: 84-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Reznikov V, Yamada Y, Yang J, Shapiro MS (2004) Phosphatidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J Neurosci 24: 10980-10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Stockand JD, Shapiro MS (2005a) The use of Chinese hamster ovary (CHO) cells in the study of ion channels. J Pharmacol Toxicol Methods 51: 177-185. [DOI] [PubMed] [Google Scholar]
- Gamper N, Li Y, Shapiro MS (2005b) Structural requirements for differential sensitivity of KCNQ K+ channels to modulation by Ca2+/calmodulin. Mol Biol Cell 16: 3538-3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley JK, Passmore GM, Tatulian L, Al-Qatari M, Ye F, Wickenden AD, Brown DA (2003) Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J Neurosci 23: 5012-5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA (1996) Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature 380: 258-262. [DOI] [PubMed] [Google Scholar]
- Hille B (1994) Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci 17: 531-536. [DOI] [PubMed] [Google Scholar]
- Huang S, Lifshitz L, Patki-Kamath V, Tuft R, Fogarty K, Czech MP (2004) Phosphatidylinositol-4,5-bisphosphate-rich plasma membrane patches organize active zones of endocytosis and ruffling in cultured adipocytes. Mol Cell Biol 24: 9102-9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R (2002) Crystal structure and mechanism of a calcium-gated potassium channel. Nature 417: 515-522. [DOI] [PubMed] [Google Scholar]
- Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ (1999) KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96: 437-446. [DOI] [PubMed] [Google Scholar]
- Lerche C, Scherer CR, Seebohm G, Derst C, Wei AD, Busch AE, Steinmeyer K (2000) Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J Biol Chem 275: 22395-22400. [DOI] [PubMed] [Google Scholar]
- Li Y, Gamper N, Shapiro MS (2004) Single-channel analysis of KCNQ K+ channels reveals the mechanism of augmentation by a cysteine-modifying reagent. J Neurosci 24: 5079-5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maljevic S, Lerche C, Seebohm G, Alekov AK, Busch AE, Lerche H (2003) C-terminal interaction of KCNQ2 and KCNQ3 K+ channels. J Physiol (Lond) 548: 353-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K (1997) 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem (Tokyo) 122: 498-505. [DOI] [PubMed] [Google Scholar]
- Nasuhoglu C, Feng S, Mao J, Yamamoto M, Yin HL, Earnest S, Barylko B, Albanesi JP, Hilgemann DW (2002) Nonradioactive analysis of phosphatidylinositides and other anionic phospholipids by anion-exchange high-performance liquid chromatography with suppressed conductivity detection. Anal Biochem 301: 243-254. [DOI] [PubMed] [Google Scholar]
- Pegan S, Arrabit C, Zhou W, Kwiatkowski W, Collins A, Slesinger PA, Choe S (2005) Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci 8: 279-287. [DOI] [PubMed] [Google Scholar]
- Raucher D, Stauffer T, Chen W, Shen K, Guo S, York JD, Sheetz MP, Meyer T (2000) Phosphatidylinositol 4,5-bisphosphate functions as a second messenger that regulates cytoskeleton-plasma membrane adhesion. Cell 100: 221-228. [DOI] [PubMed] [Google Scholar]
- Roche JP, Westenbroek R, Sorom AJ, Hille B, Mackie K, Shapiro MS (2002) Antibodies and a cysteine-modifying reagent show correspondence of M current in neurons to KCNQ2 and KCNQ3 K+ channels. Br J Pharmacol 137: 1173-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohacs T, Chen J, Prestwich GD, Logothetis DE (1999) Distinct specificities of inwardly rectifying K+ channels for phosphoinositides. J Biol Chem 274: 36065-36072. [DOI] [PubMed] [Google Scholar]
- Rohacs T, Lopes CM, Jin T, Ramdya PP, Molnar Z, Logothetis DE (2003) Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc Natl Acad Sci USA 100: 745-750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT (1996) Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 384: 80-83. [DOI] [PubMed] [Google Scholar]
- Schwake M, Pusch M, Kharkovets T, Jentsch TJ (2000) Surface expression and single channel properties of KCNQ2/KCNQ3, M-type K+ channels involved in epilepsy. J Biol Chem 275: 13343-13348. [DOI] [PubMed] [Google Scholar]
- Schwake M, Jentsch TJ, Friedrich T (2003) A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep 4: 76-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Stansfeld CE, Brown DA (1992) Closure of potassium M-channels by muscarinic acetylcholine-receptor stimulants requires a diffusible messenger. Proc R Soc Lond B Biol Sci 250: 119-125. [DOI] [PubMed] [Google Scholar]
- Selyanko AA, Hadley JK, Wood IC, Abogadie FC, Jentsch TJ, Brown DA (2000) Inhibition of KCNQ1-4 potassium channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J Physiol (Lond) 522: 349-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Hadley JK, Brown DA (2001) Properties of single M-type KCNQ2/KCNQ3 potassium channels expressed in mammalian cells. J Physiol (Lond) 534: 15-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B (2000) Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J Neurosci 20: 1710-1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw G (1996) The pleckstrin homology domain: an intriguing multifunctional protein module. BioEssays 18: 35-46. [DOI] [PubMed] [Google Scholar]
- Stauffer TP, Ahn S, Meyer T (1998) Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol 8: 343-346. [DOI] [PubMed] [Google Scholar]
- Stolz LE, Kuo WJ, Longchamps J, Sekhon MK, York JD (1998) INP51, a yeast inositol polyphosphate 5-phosphatase required for phosphatidylinositol 4,5-bisphosphate homeostasis and whose absence confers a cold-resistant phenotype. J Biol Chem 273: 11852-11861. [DOI] [PubMed] [Google Scholar]
- Suh B, Hille B (2002) Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35: 507-520. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B (2005) Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr Opin Neurobiol 15: 370-378. [DOI] [PubMed] [Google Scholar]
- Suh BC, Horowitz LF, Hirdes W, Mackie K, Hille B (2004) Regulation of KCNQ2/KCNQ3 current by G-protein cycling: the kinetics of receptor-mediated signaling by Gq J Gen Physiol 123: 663-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian L, Brown DA (2003) Effect of the KCNQ potassium channel opener retigabine on single KCNQ2/3 channels expressed in CHO cells. J Physiol (Lond) 549: 57-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McK-innon D (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282: 1890-1893. [DOI] [PubMed] [Google Scholar]
- Willars GB, Nahorski SR, Challiss RA (1998) Differential regulation of muscarinic acetylcholine receptor-sensitive polyphosphoinositide pools and consequences for signaling in human neuroblastoma cells. J Biol Chem 273: 5037-5046. [DOI] [PubMed] [Google Scholar]
- Winks JS, Hughes S, Filippov AK, Tatulian L, Abogadie FC, Brown DA, Marsh SJ (2005) Relationship between membrane phosphatidylinositol-4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels. J Neurosci 25: 3400-3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Watras J, Loew LM (2003) Kinetic analysis of receptor-activated phosphoinositide turnover. J Cell Biol 161: 779-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T, Logothetis DE (2003) PIP2 Activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37: 963-975. [DOI] [PubMed] [Google Scholar]