

Abstract
Prion diseases are induced by pathologically misfolded prion protein (PrPSc), which recruit normal sialoglycoprotein PrPC by a template-directed process. In this study, we investigated the expression of PrPC in a rat model of cerebral ischemia to more fully understand its physiological role. Immunohistochemical analysis demonstrated that PrPC-immunoreactive cells increased significantly in the penumbra of ischemic rat brain compared with the untreated brain. Western blot analysis showed that PrPC protein expression increased in ischemic brain tissue in a time-dependent manner. In addition, PrPC protein expression was seen to colocalize with neuron, glial, and vascular endothelial cells in the penumbric region of the ischemic brain. Overexpression of PrPC by injection of rAd (replication-defective recombinant adenoviral)-PGK (phosphoglycerate kinase)-PrPC-Flag into ischemic rat brain improved neurological behavior and reduced the volume of cerebral infarction, which is supportive of a role for PrPC in the neuroprotective adaptive cellular response to ischemic lesions. Concomitant upregulation of PrPC and activated extracellular signal-regulated kinase (ERK1/2) under hypoxia–reoxygenation in primary cortical cultures was shown to be dependent on ERK1/2 phosphorylation. During hypoxia–reoxygenation, mouse neuroblastoma cell line N18 cells transfected with luciferase rat PrPC promoter reporter constructs, containing the heat shock element (HSE), expressed higher luciferase activities (3- to 10-fold) than those cells transfected with constructs not containing HSE. We propose that HSTF-1 (hypoxia-activated transcription factor), phosphorylated by ERK1/2, may in turn interact with HSE in the promoter of PrPC resulting in gene expression of the prion gene. In summary, we conclude that upregulation of PrPC expression after cerebral ischemia and hypoxia exerts a neuroprotective effect on injured neural tissue. This study suggests that PrPC has physiological relevance to cerebral ischemic injury and could be useful as a therapeutic target for the treatment of cerebral ischemia.
Keywords: prion protein (PrPC), stress protein, cerebral ischemia animal model, hypoxia–reoxygenation, primary cortical neurons, neuroprotection
Introduction
Prion protein (PrPC) is a cell-surface copper-binding glycoprotein expressed by neurons (Kretzschmar et al., 1986), glial (Moser et al., 1995; Brown et al., 1998c), and other cells (Brown et al., 1998a). An abnormal isoform of this protein (PrPSc) (Bolton et al., 1982), which is proteinase resistant, is involved in prion diseases such as Creutzfeldt-Jakob (White et al., 2003). The main features of prion disease are neurodegeneration, gliosis, and accumulation of PrPSc in the CNS (Prusiner, 1991). Although accumulation of PrPSc may damage cells in the CNS (Bueler et al., 1993), it is clear that the generation of spongiform encephalopathy requires the presence of both PrPC and PrPSC (Bueler et al., 1993). PrPSc is thought to recruit PrPC through a transconformation process causing the accumulation of PrPSc in brain in the form of plaques and fibrils (Prusiner et al., 1998; Caughey and Chesebro, 2001). Thus, studying differences in conformation between these two protein isoforms is essential for understanding how prion diseases occur. Moreover, it is also necessary to study the biological function of PrPC to understand why alteration of PrPC leads to disease occurrence.
PrPC is encoded by a single gene corresponding to ∼250 amino acids and is widely expressed in the CNS and the lymphoreticular system. Although PrPC is a highly conserved protein in vertebrates, its actual physiological role remains elusive. Recent work has shown that PrPC plays pleiotrophic roles in neuronal and non-neuronal cells, such as cellular trafficking, and also participates in copper uptake (Lee et al., 2001; Negro et al., 2001; Perera and Hooper, 2001), cell adhesion/differentiation (Luckenbill-Edds, 1997), cell signaling (Martins et al., 1997), and neuronal survival (Bounhar et al., 2001). Interestingly, some reports have shown that PrPC also has the ability to protect neural tissue against oxidative stress (Brown et al., 1999, 2002).
Some stress proteins, such as heat shock protein 70 (Hsp70) (Chen et al., 1996), are expressed in both neurons and glial cells after ischemia. In vivo chronic hypoxic injury also enhances the expression of Hsp70 (Rafiee et al., 2003). Previously, a close correlation between PrPC and Hsp70 was demonstrated (Jung et al., 2000, 2002). We recently found two heat shock elements located in the promoter of the PrPC gene that result in the upregulation of of PrPC expression under heat shock conditions in neuronal culture (Shyu et al., 2000, 2002; Hung et al., 2001). Therefore, PrPC expression may be regulated by ischemia/hypoxia, as is the case with Hsp70. However, the molecular mechanism and cell distribution of PrPC expression after ischemia/hypoxia has not been studied to date.
Ischemia/hypoxia has been shown to activate some mitogen-related protein kinases, including extracellular signal-related kinase (ERK1/2) and c-Jun N-terminal kinase (JNK), which directly respond to stress (Mishra et al., 2004). In a recent study, ERK-mediated phosphorylation of heat shock transcriptional factor 1 (HSTF-1) was shown to regulate molecular chaperone gene transcription (Wang et al., 2003). Thus, a central role for ERK regulation in heat shock protein expression in response to cells under stress is generally accepted (Mishra et al., 2004). Because of the similarity in regulatory patterns between Hsp70 and PrPC (Shyu et al., 2000, 2002; Hung et al., 2001), we hypothesized that ischemia/hypoxia treatment might induce the expression of PrPC through the activation of the ERK signaling pathway.
In this study, we investigated whether ischemia/hypoxia could upregulate the expression of PrPC. It was found that PrPC had a neuroprotective effect on primary cortical neurons. In addition, overexpression of PrPC transduced by adenovirus-mediated gene transfer could reduce ischemic injury and improve neurological dysfunction after cerebral ischemia in rats. Subsequently, we examined the molecular mechanism governing the regulation of PrPC expression by ischemia/hypoxia.
Materials and Methods
In vivo ischemia/reperfusion brain model. Adult male Sprague Dawley rats (weight, >300 g) were used in this study. All surgical procedures were performed using sterile/aseptic techniques in accordance with institutional guidelines. Rats were anesthetized with chloral hydrate (0.4 g/kg, i.p.) and subjected to cerebral ischemia. Ligation of the right middle cerebral artery (MCA) and bilateral common carotid arteries (CCAs) was performed by methods described previously (Chen et al., 1986). The CCAs were clamped with nontraumatic arterial clips. The right MCA was ligated with a l0-0 nylone suture. After 90 min of ischemia, the suture on the MCA and arterial clips on CCAs were removed to allow reperfusion. Core body temperature was monitored with a thermistor probe and maintained at 37°C with a heating pad during anesthesia. After recovery from anesthesia, rat body temperature was maintained at 37°C with a heat lamp.
Total protein extraction of brain tissue and Western blotting. Experimental animals were decapitated at 4 h, 12 h, 3 d, and 7 d after reperfusion with 90 min MCA ligation. Three rats without MCA ligation were used as normal controls. Samples of ischemic cerebral cortex were taken from the peripheral region of infarcted brains (penumbra area). Western blot analysis of PrPC was performed on these samples. Briefly, ischemic brain tissue was homogenized and lysed in a buffer containing 320 mm sucrose, 5 mm HEPES, 1 μg/ml leupeptin, and 1 μg/ml aprotinin. Lysates were centrifuged at 13,000 × g for 15 min. The resulting pellet was resuspended in sample buffer (62.5 mm Tris-HCl, 10% glycerol, 2% SDS, 0.1% bromophenol blue, and 50 mm DTT) and subjected to SDS-PAGE (4–12%). Then, the gel was transferred to a Hybond-P nylon membrane. This was followed by incubation with appropriately diluted antibodies of PrPC (dilution, 1:200; M-20; Santa Cruz Biotechnology, Santa Cruz, CA) and β-actin (dilution, 1:2000; Santa Cruz Biotechnology). Membrane blocking, primary and secondary antibody incubations, and chemiluminescence reactions were conducted individually for each antibody according to the manufacturer's protocol. The intensity of each band was measured using a Kodak Digital Science 1D Image Analysis System (Eastman Kodak, Rochester, NY). The ratio of band intensity of PrPC in Western blots compared with the internal control was calculated. Results were expressed as the mean value of the ratio ± SEM for n preparations.
Immunohistochemical analysis of rat brain. Experimental rats were reanesthetized with chloral hydrate (0.4 g/kg, i.p.) and were decapitated at 4 h, 12 h, 3 d, and 7 d after cerebral ischemia. Three rats without MCA ligation were used as normal controls. Rat brains were fixed by transcardial perfusion with saline, followed by perfusion and immersion in 4% paraformaldehyde, and embedded in paraffin. A series of adjacent 6-μm-thick sections were cut from each tissue block in the coronal plane, stained with hematoxylin and eosin (H&E), and analyzed by light microscopy [Nikon (Tokyo, Japan) E600]. Immunostaining was performed using the labeled streptavidin–biotin (LSAB) method (Dako LASB-2 kit, peroxidase; DakoCytomation, Carpinteria, CA). Briefly, tissue on a saline-coated slide was placed twice in boiling citrate buffer, pH 6 (ChemMate; DakoCytomation), for 5 min in a microwave oven at 750 W after depariffinization and rehydration, as described previously (Shyu et al., 1996). This was followed by incubation with appropriate diluted antibodies to PrPC (dilution, 1:200; M-20; Santa Cruz Biotechnology) at room temperature for 1 h. After washing with Tris-buffered saline containing 0.1% Tween 20 (TBS-T), the specimens were sequentially incubated for 10–30 min with biotinylated anti-rabbit and anti-mouse Igs and peroxidase-labeled streptavidine. Staining was performed after a 10 min incubation with a freshly prepared substrate-chromogen solution containing 3% 3-amino-9-ethylcarbazole and hydrogen peroxide. Finally, the slides were lightly counterstained with hematoxylin, washed with water, and then mounted. The extent of PrPC cell immunoreactivity was measured as the number of cells per square minimeter.
Laser-scanning confocal microscopy with double immunofluorescence. To identify the expression of cell type-specific markers in PrPC+ cells, double immunofluorescence was performed. Expression of glial fibrillary acidic protein (GFAP), von Willebrand factor (vWF), microtubule-associated protein 2 (MAP-2) and neuronal nuclei (Neu-N) were tested for using PrPC-GFAP, PrPC-vWF, PrPC-MAP-2, and PrPC-Neu-N double immunofluorescence under the manufacturer's instructions (Boehringer Mannheim, Mannheim, Germany). Each coronal section was first stained with primary PrPC antibody conjugated with cyanine 3 (Cy3; 1:500; Jackson ImmunoResearch, West Grove, PA), followed by treatment with cell-specific antibodies conjugated with FITC (1:500; Jackson ImmunoResearch): GFAP for astrocyte (1:400; Sigma, St. Louis, MO), vWF for endothelial cell (1:400; Sigma), Neu-N for neuronal nuclei (1: 200; Chemicon, Temecula, CA), MAP-2 for neuronal dendrites (1:200; Boehringer Mannheim). The tissue sections were analyzed with a Zeiss (Oberkochen, Germany) LSM 510 laser-scanning confocal microscope. For immunofluorescence-labeled slides, green (FITC) and red (Cy3) fluorochromes on the slides were excited by laser beam at 488 and 543 nm, respectively. The PrPC (labeled by red-Cy3 fluorochromes) and cell-type-specific markers, including Neu-N, MAP-2, vWF, and GFAP (labeled by green-FITC fluorochromes) were double immunostained to demonstrate their colocalization in one cell under laser-scanning confocal microscopy.
Construction of adenoviral vector carrying PrPC. The procedure was described previously (Zoldhelyi et al., 1996; Shyue et al., 2001). We constructed, in a replication-defective recombinant adenoviral (rAd) vector, a human phosphoglycerate kinase (PGK) promoter to drive human PrPC-Flag expression (rAd-PGK-PrPC-Flag) and a PGK-enhanced green fluorescent protein (eGFP) to serve as a control (rAd-PGK-eGFP). Replication-defective rAd vectors were generated by homologous recombination and amplified in 293 cells. A large-scale production of hightiter rAd was performed as described previously (Zoldhelyi et al., 1996; Shyue et al., 2001). The rAd stocks were prepared by CsCl gradient centrifugation, aliquoted, and stored at –80°C. Viral titers were determined by a plaque assay (Shyue et al., 2001).
Cerebral ischemic animal model treated with recombinant Ad-PGK-PrPC-Flag. The cerebral ischemic animal model was established as described above. Experimental rats were injected intracerebrally with 1 × 1010 virus particles of rAd-PGK-PrPC-Flag or rAd-PGK-eGFP (5 μl) 30 min after MCA ligation through a 26 gauge Hamilton (Reno, NV) syringe into three cortical areas adjacent to the right MCA, 3.0–5.0 mm below the dura. The approximate coordinates for these sites were l.0–2.0 mm anterior to the bregma and 2.5–3.0 mm lateral to the midline, 0.5-l.5 mm posterior to the bregma and 3.5–4.0 mm lateral to the midline, and 3.0–4.0 mm posterior to the bregma and 4.5–5.0 mm lateral to the midline. The needle was retained in place for 5 min after each injection, and a piece of bone wax was applied to the skull defects to prevent leakage of the injected solution. The CCAs were ligated with nontraumatic arterial clips, and the right MCA was ligated with a l0-0 nylon suture. After 90 min of ischemia, the suture on the MCA and arterial clips on CCAs were removed to allow reperfusion. Core body temperature was monitored with a thermistor probe and maintained at 37°C with a heating pad during anesthesia.
Triphenyltetrazolium chloride staining, immunofluorescent colocalized study, and Western blot. Three days after cerebral ischemia, animals receiving intracrebral administration of rAd-PGK-PrPC-Flag or rAd-PGK-eGFP were intracardially perfused with saline. The brain tissue was removed, immersed in cold saline for 5 min, and sliced into 2.0-mm-thick sections (seven slices per rat). The brain slices were incubated in 20 g/L triphenyltetrazolium chloride (TTC; Research Organics, Cleveland, OH), dissolved in saline for 30 min at 37°C, and then transferred into a 5% formaldehyde solution for fixation. The area of infarction in each slice was measured with a digital scanner, as described previously (Wang et al., 1997). The volume of infarction was obtained from the product of average slice thickness (2 mm) and by examining infarcted areas in all brain slices. To minimize any artifacts induced by postischemic edema in the infarcted tissue, the area of infarction was also calculated as described previously (Lin et al., 1993). To measure the infarcted area in the right cortex, we subtracted the noninfarcted area in the right cortex from the total cortical area of the left hemisphere.
Immunofluorescent colocalization analysis, using the same method as described above with a specific antibody against Flag (1:200; Sigma) conjugated to FITC (1:300; Jackson ImmunoResearch), was performed to demonstrate successful expression of PrPC at either cell type of ischemic brain after intracerebral injection of rAd-PGK-PrPC-Flag. Furthermore, the time course of PrPC-Flag expression after intracerebral injection of rAd-PGK-PrPC-Flag in the ipsilateral ischemic brain was examined by Western blot using Flag (1:400; Sigma) antibody as described in the procedure above.
To demonstrate that the addition of 2-(2-amino-3-methyoxyphenyl)-4 H-1-benzopyran-4-one (PD98059) (20 μm) to the ischemic brain would inhibit the protective effect of rAd-PGK-PrPC-Flag, experimental rats receiving either rAd-PGK-PrPC-Flag plus PD98059 or rAd-PGK-eGFP 90 min after cerebral ischemia were evaluated for infract size using TTC and H&E staining.
Neurological behavioral measurements. Behavioral assessments were performed 3 d before cerebral ischemia and 72 h after cerebral ischemia. The tests measured (1) body asymmetry and (2) locomotor activity. The baseline test scores were recorded to normalize those taken after cerebral ischemia. (1) The elevated body swing test was used to assess body asymmetry after MCA ligation and evaluated quantitatively as described previously (Borlongan et al., 1998). Initially, animals were examined for lateral movement on their bodies while being suspended by their tails. The frequency of initial head swing contralateral to the ischemic side was counted in 20 continuous tests and was normalized as follows: % recovery = [1 – (lateral swings in 20 tests – 10)/10 × 100%]. (2) Locomotor activity: rats were subjected to VersaMax animal activity monitoring (Accuscan Instruments, Columbus, OH) for ∼2 h for behavioral recording. The VersaMax animal activity monitoring contained 16 horizontal and 8 vertical infrared sensors spaced 87 cm apart. The vertical sensors were situated 10 cm from the floor of the chamber. Motor activity was counted as the number of beams broken by a rat's movement in the chamber. Three vertical parameters defined in the menu option of the manufacturer were calculated over 2 h at night: (1) vertical activity, (2) vertical time, and (3) number of vertical movements.
Blood pressure, heart rate, blood glucose, and blood gas measurement. Physiological parameters were measured in 14 rats after adenoviral delivery, by a procedure described previously (Lin et al., 1999).
In vitro primary cortical culture preparation and hypoxia-reoxygenation treatment. Primary cortical cells were prepared from the cerebral cortex of gestation day 17 embryos from Sprague Dawley rats as described previously (Murphy et al., 1990). Four days after isolation, the cultures were replenished with minimum essential medium (Invitrogen, Carlsbad, CA) containing 0.5 g/L BSA plus N-2 supplement, 0.5 × 10–3 mol/L pyruvate and antibiotics. Finally, the culture medium was changed to serum-free minimum essential medium containing 1 × 10–3 mol/L pyruvate, 1 × 10–3 mol/L glutamate, 0.5 g/L BSA, 0.3 × 10–3 mol/L KCl, and antibiotics on the seventh day. For hypoxia-reoxygenation treatment, the cells were placed for 12 h within a hypoxic chamber (Bug Box; Ruskinn Technology, London, UK), continuously flushed with 95% N2 and 5% CO2 at 37°C to maintain a gas phase PO2 of <1 mmHg (OM-14 oxygen monitor; SensorMedics, Yorba Linda, CA). After each hypoxic treatment, the cells were returned to a 37°C normoxic incubator (95% air and 5% CO2) for different time periods (1, 3, 10, and 24 h) of reoxygenation. The cells were then collected at each time point and stored at –80°C for protein extraction.
Immunocytochemical analysis of primary cortical cultures. For hypoxiareoxygenation treatment, primary cortical cultures were placed within a hypoxic chamber for 12 h, as described previously. After each hypoxic treatment, the cells were returned to a 37°C normoxic incubator (95% air and 5% CO2) at different time periods (1, 3, 10, and 24 h) for reoxygenation. For PrPC immunostaining at each time point, cell cultures were washed with PBS and fixed for 30 min at room temperature in 4% paraformaldehyde. After being washed with PBS, the fixed cultured cells were treated for 30 min with blocking solution (10 g/L BSA, 0.03% Triton X-100, and 4% serum in PBS). Cells were incubated overnight at 4°C with an antibody against PrPC (1:200; M-20; Santa Cruz Biotechnology) and then rinsed three times in PBS. After washing with TBS-T, the cells were sequentially incubated for 10–30 min with biotinylated anti-rabbit and anti-mouse Igs and peroxidase-labeled streptavidine. Staining was performed after a 10 min incubation with a freshly prepared substrate-chromogen solution containing 3% 3-amino-9-ethylcarbazole and hydrogen peroxide. Finally, the slides were lightly counterstained with hematoxylin, washed with water, and then mounted. The extent of PrPC cell immunoreactivity was measured as the number of cells per square minimeter.
Total protein extraction and Western blotting assay. Western blot analyses of PrPC, ERK1/2, p38, and JNK expression from primary cortical culture were performed after hypoxic-reoxygenation treatment. Briefly, primary cortical cells were lysed in a buffer containing 320 mm sucrose, 5 mm HEPES, 1 μg/ml leupeptin, and 1 μg/ml aprotinin. Lysates were centrifuged at 13,000 × g for 15 min. The resulting pellet was resuspended in sample buffer (62.5 mm Tris-HCl, 10% glycerol, 2% SDS, 0.1% bromophenol blue, and 50 mm DTT) and subjected to SDS-PAGE (4–12%). The gel was then transferred to a Hybond-P nylon membrane. This was followed by incubation with appropriately diluted antibodies to PrPC (dilution, 1:200; Santa Cruz Biotechnology), activated ERK1/2 (dilution, 1:200; Santa Cruz Biotechnology), activated p38 (dilution, 1:200; Santa Cruz Biotechnology), activated JNK (dilution, 1:200; Santa Cruz Biotechnology), and β-actin (dilution, 1:2000, Santa Cruz Biotechnology). Specific ERK1/2 pathway inhibitor PD98059 (10 μm) (Cell Signaling Technology, Beverly, MA), which was used to pretreat cells, was applied to the primary cortical culture to suppress enzyme binding to block the transcriptional signal of ERK1/2. Membrane blocking, primary and secondary antibody incubations, and chemiluminescence reactions were conducted individually for each antibody according to the protocol of the manufacturer. The intensity of each band was measured using a Kodak Digital Science 1D Image Analysis System.
Measurement of caspase-3 activity and lactate dehydrogenase activity analysis. To evaluate the antiapoptotic effect of PrPC, the adenoviral constructs of rAd-PGK-PrPC-Flag and rAd-PGK-eGFP were used to infect mouse neuroblastoma cell line (N18), after which the cells were subjected to hypoxia–reoxygenation. Fluorometric assays of caspase-3 activity were performed on the infected cells that received 12 h hypoxia incubation using commercial kits (Bio-Rad, Hercules, CA) following the instructions of the manufacturer. In addition, infected cells were subjected to hypoxia for up to 12 or 24 h and then assayed for lactate dehydrogenase (LDH) activity using the commercial kit CytoTox 96 (Promega, Madison, WI) according to the instructions of the manufacturer, as described previously (Chu and Ng, 2003).
Construction of reporter plasmids for heat shock element functional analysis. RaPrP promoter-luciferase reporter plasmids were constructed as described previously (Saeki et al., 1996), including praPrP(-2831)-luc, praPrP(-1026)-luc, praPrP(-514)-luc, and pGL2-control (Promega). The other plasmid, praPrP(-2831M)-luc, was constructed by site-directed mutagenesis of the heat shock element 1 (HSE1) in the rat prion promoter of praPrP(-2831)-luc, as described previously (Shyu et al., 2002). These previous reports show that praPrP(-2831)-luc contains HSE1 and HSE2 and that praPrP(-1026)-luc and praPrP(-2831M)-luc contain HSE1 and HSE2, respectively. These constructs were used to transfect N18 cells to evaluate the bioactivity of HSE in regulating the expression of PrPC. Cells were plated at a density of 1 × 106 in 60 mm plates and grown overnight in a 37°C incubator. They were then rinsed with serum-free medium and transiently transfected with 6 μg of super-coiled construct DNA (including 4 μg of promoter-luciferase plasmid and 2 μg of pCMVβ-gal) using lipofectamine (Invitrogen). Briefly, the lipofectamine-DNA solution was left at room temperature for 30 min, mixed with serum-free medium and added to cells. The cells were incubated for 5 h at 37°C and 5% CO2, after which the medium was aspirated and replaced with serum-containing medium. After transfection, cells were placed in a hypoxic chamber for 12 h and allowed to reoxygenate for up to 24 h. They were then harvested and lysed, and luciferase activity was determined using a luminometer (Lumat LB 9501; Berthold, Bad Wild-bad, Germany). Luciferase activity was measured in triplicate after normalization by β-gal assay.
Statistical analysis. All measurements in this study were performed blindly. Results are expressed as mean ± SEM. The behavioral scores have been evaluated for normality. Student's t tests were used to evaluate mean differences between the control and the treated group.
Results
Cerebral ischemia increases the immunoreactivity of PrPC
To determine whether cerebral ischemia increases the immunoreactivity of PrPC, experimental rats (n = 16) receiving cerebral ischemia were killed at different time points for PrPC immunostaining. Four hours (n = 4) after cerebral ischemia, greater numbers of PrPC-immunoreactive cells were detected, mainly in the ipsilateral cortex near the infarcted boundary and subventricular region of the ischemic rat brains (Fig. 1A) when compared with nonischemic rats (n = 4). Furthermore, increased numbers of PrPC-immunoreactive cells were found around the lumen of varying calibers in the perivascular portion of the blood vessels (and also in the vessel wall of endothelial cells; data not shown) when compared with nonischemic rats. The increase in PrPC expression was time dependent (Fig. 1B–F) and reached its peak level 3 d (n = 4) after cerebral ischemia compared with the nonischemic rats (Fig. 1G).
Figure 1.

Cerebral ischemia induces an increase in PrPC immunoreactivity. A, The square-mark (white square) in the representative figure of cerebral ischemia (black area) was selected to show the immunoreactivity of PrPC at different time points after cerebral ischemia. B, Rats not receiving cerebral ischemia are indicated as controls. C–F, Experimental rats received cerebral ischemia and were killed at different time points (4 h, 12 h, 3 d, and 7 d) for PrPC immunostaining. Scale bar, 40 μm. G, Results of quantitative analysis of PrPC-immunoreactive cells. The pattern of increased immunoreactivity of PrPC was time dependent and reached its peak level 3 d after cerebral ischemia compared with nonischemic rats, which are indicated as “C.” The mean ± SEM is shown. *p < 0.05 versus control.
Cerebral ischemia induced expression of PrPC in vivo
To determine the expression level of PrPC around the penumbra, experimental rats receiving cerebral ischemia (n = 8) for 90 min were killed at different time points for protein extraction of ischemic brain tissue. Three rats without MCA ligation were used as normal controls. Samples of ischemic cerebral cortex were taken from the peripheral region of the infarcted brains of test rats (penumbra area) for Western blot analysis. It was found that cerebral ischemia caused an increase in the expression of PrPC (Fig. 2A), which was compatible with our immunohistochemical findings. The ratio of PrPC/actin reached a peak level of approximately a twofold increase in ischemic rats compared with nonischemic rats 3 d after cerebral ischemia (Fig. 2B).
Figure 2.

Cerebral ischemia induces expression of PrPC in vivo. A, Experimental rats receiving cerebral ischemia for 90 min were killed at different time points (4 h, 12 h, 3 d, and 7 d) for protein extraction of ischemic brain tissue. Samples of ischemic cerebral cortex were taken from the peripheral region of the infarcted brains of test rats (penumbric area) for Western blot analysis. B, Result of densitometric analysis of Western blots; the ratio of PrPC/actin was taken as 1 for nonischemic rats, which represents the control (C). The mean ± SEM is shown. *p < 0.05 versus control. Time-dependent changes in PrPC expression are depicted.
Imunoreactivity of PrPC colocalized to neuron, glial, and endothelial cells
To identify which type of cerebral cells expressed PrPC after cerebral ischemia, double immunofluorescence with laser scanning confocal microscopy was performed on brain specimens of ischemic rats. The ischemic cortical areas of the rats revealed an increase in PrPC+ cells coexpressing the neuronal phenotype of Neu-N+ cells (Fig. 3A–D) and the glial phenotype GFAP+ (Fig. 3E–H). Some PrPC+ cells showing vascular phenotypes (vWF+ cells) were also found around the perivascular and endothelial regions (Fig. 3I–L) of the ischemic hemispheres.
Figure 3.

PrPC immunoreactivity is colocalized with neuronal, glial, and endothelial cells. To identify which type of cerebral cells expressed PrPC after cerebral ischemia, double immunofluorescence with laser-scanning confocal microscopy was performed on rat brain slices. A–H, Ischemic cortical areas of rats revealed an increase in PrPC+ coexpression with the neuronal phenotypes of Neu-N+ (A–D) and with the glial phenotype of GFAP+ cells (E–H). I–L, Some PrPC + cells showing vascular phenotypes (vWF+ cells) were also found around the perivascular and endothelial regions of the ischemic hemispheres. Scale bar, 50 μm. DAPI, 4′,6′-Diamidino-2-phenylindole dihydrochloride.
Intracerebral rAd-PGK-PrPC-Flag administration improves neurological behavior after cerebral ischemia
To evaluate the neuroprotective effect of intracerebral administration of rAd-PGK-PrPC-Flag in rats receiving cerebral ischemia, body asymmetry trials and locomotor activity tests were used to assess neurological deficits in rAd-PGK-PrPC-Flag-treated (n = 8) and control (rAd-PGK-eGFP) rats (n = 8). Rats intracerebrally treated with rAd-PGK-PrPC-Flag exhibited significantly reduced body asymmetry 3 d after cerebral ischemia compared with that of controls (Fig. 4A). The measured locomotor activities (i.e., vertical activity, vertical movement time, and number of vertical movements) increased significantly in rats receiving rAd-PGK-PrPC-Flag treatment compared with control animals 3 d after cerebral ischemia (Fig. 4B–D).
Figure 4.
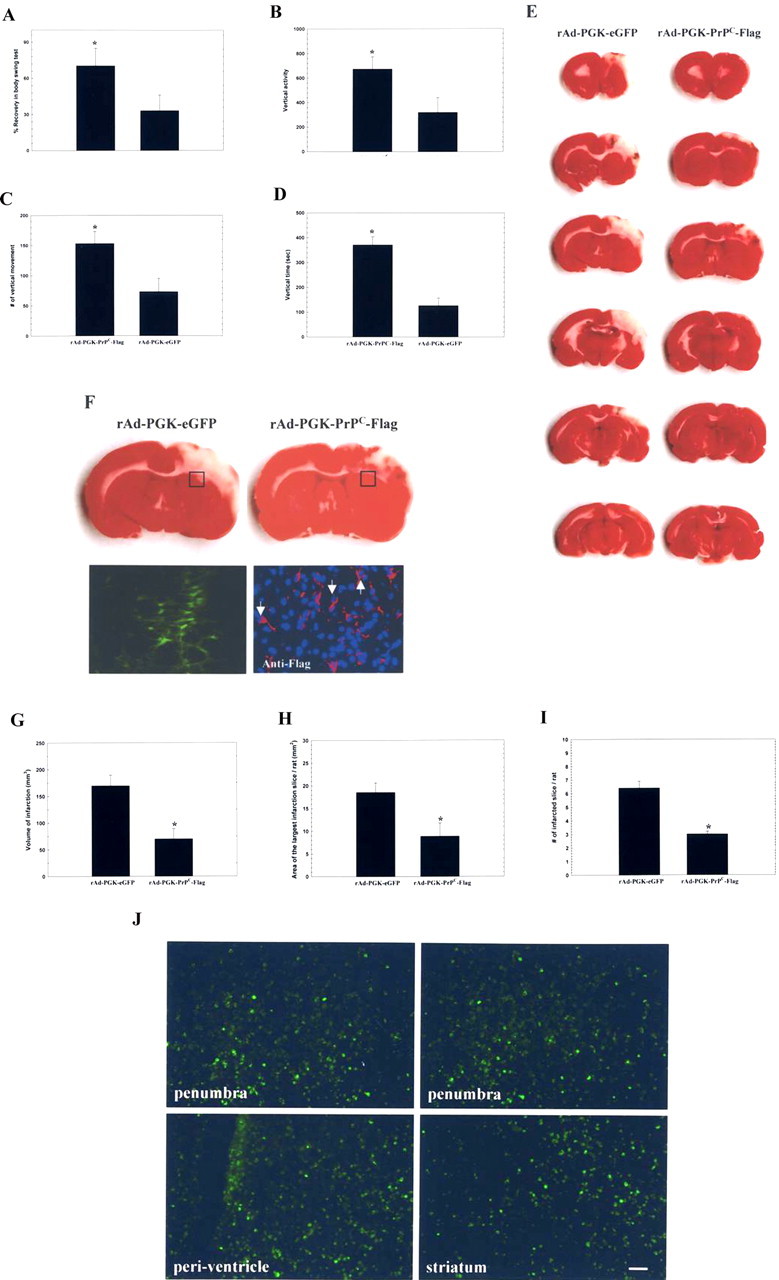
Intracerebral injection of rAd-PGK-PrPC-Flag improves neurological dysfunction and reduces infarcted volume in stroke rats. A–D, Three days after cerebral ischemia, body asymmetry trials, and locomotor activities, three vertical parameters were examined in each experimental rat. These showed significant improvement in rAd-PGK-PrPC-Flag-treated rats compared with rAd-PGK-eGFP-treated control rats. E, Injection of rAd-PGK-PrPC-Flag significantly reduced cortical infarction compared with rAd-PGK-eGFP-treated rats. The white area represents the infarcted zone in the right cerebral cortex by TTC staining. F, Under immunofluorescence, rats receiving rAd-PGK-PrPC-Flag showed expression of Flag [red-Cy3 and blue-4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI)] in the representative square area (□), compared with rats receiving rAd-PGK-eGFP, showing expression of eGFP (green-eGFP) in the square area. G, The volume of infarction was significantly reduced in rats treated with rAd-PGK-PrPC-Flag compared with the control (rAd-PGK-eGFP) group. H, The area with the largest infarction in a section from a given rat was significantly diminished by rAd-PGK-PrPC-Flag treatment. I, Finally, the number of infarcted sections per rat was also reduced by rAd-PGK-PrPC-Flag treatment. J, The results of fluorescent microscopy of rAd-PGK-PrPC-Flag-infected cells in ischemic brains were shown to be widely distributed in the peri-infarcted region, striatum, and periventricular area.
Intracerebral administration of rAd-PGK-PrPC-Flag reduces infarct volume after cerebral infarction
To investigate whether there was any effect from rAd-PGK-PrPC-Flag on infarct volumes, rats (n = 16) were killed for TTC staining 3 d after cerebral ischemia. Rats (n = 8) receiving rAd-PGK-PrPC-Flag treatment showed significantly decreased infarct volumes 3 d after cerebral ischemia compared with controls (rAd-PGK-eGFP) (Fig. 4E). Quantitative measurement of the infarct volume showed that it was significantly reduced from an average of 169 ± 15 mm3 in rAd-PGK-eGFP-treated controls (n = 8) to 70 ± 16 mm3 in rAd-PGK-PrPC-Flag-treated animals (n = 8) (Fig. 4G). The area of the largest infarct decreased significantly from 18.6 ± 2.5 mm2 in the controls (rAd-PGK-eGFP) to 8.9 ± 3.5 mm2 in the treated rats (Fig. 4H), and infarcted slices were also significantly reduced from 6.4 ± 0.5 slices/rat in the control animals to 3.0 ± 0.2 slices/rat in the rAd-PGK-PrPC-Flag-treated rats (Fig. 4I).
Intracerebral administration of rAd-PGK-PrPC-Flag could induce PrPC expression in neuron, glia, and endothelial cells
To demonstrate the successful induction of PrPC expression at which type of brain cells, immunofluorescent cololcalization analysis was performed after intracerebral injection of rAd-PGK-PrPC-Flag. For fluorescent microscopy, the results of spatial distribution of rAd-PGK-PrPC-Flag-infected cells were shown to be widely spread among the peri-infarcted region, striatum, and periventricular areas (Fig. 4J), etc. As shown by the laser-scanning confocal microscope, the PrPC-Flag+ cells coexpressed with the neuronal phenotype of MAP-2+, nestin+, and the glial phenotype of GFAP+ cells (Fig. 5A–C). In addition, PrPC-Flag expression was also colocalized with the endothelial phenotype of vWF expression (Fig. 5D). After semiquantitative measurement of specific cell phenotypes over the total PrPC-Flag+ cells in the colocalization study, the results showed that percentages of total PrPC-Flag+ cells colocalizing with specific markers of MAP-2 (neurons), GFAP (glial cells), and vWF (endothelial cells) were ≈19, ≈48, and ≈33%, respectively.
Figure 5.

Intracerebral injection of rAd-PGK-PrPC-Flag increases PrPC-Flag expression in the ischemic brain. A–D, The PrPC-Flag+ cells coexpressed with the MAP-2+, nestin+, Neu-N+, GFAP+, and vWF+ cells. E, Western blot analysis of the temporal expression of PrPC-Flag after intracerebral injection of either adenoviral constructs. F, Densitometric results indicate a significant increase in expression of PrPC-Flag, peaking 2 d after intracerebral injection of rAd-PGK-PrPC-Flag compared with rAd-PGK-eGFP (Cg). G, Representative H&E staining of the ischemic brains of three test groups of rAd-PGK-eGFP, rAd-PGK-PrPC-Flag, and PD98059 plus rAd-PGK-PrPC-Flag. H, Results of infarct measurement are shown in these three groups. The mean ± SEM is shown. *p < 0.05. Scale bar, 50 μm.
Intracerebral administration of rAd-PGK-PrPC-Flag increases PrPC-Flag expression in the ischemic brain
To confirm the neuroprotective effect of rAd-PGK-PrPC-Flag via the overexpression of PrPC in the ischemic brain, the time course (6 h, 12 h, 24 h, 2 d, and 5 d; n = 4 each) of PrPC-Flag expression in the ischemic brain after intracerebral injection of either adenoviral constructs was examined using Western blot analysis with a specific antibody against Flag (Fig. 5E). The results indicated a greater increase in expression of PrPC-Flag peaking 2 d after intracerebral injection of rAd-PGK-PrPC-Flag compared with rAd-PGK-eGFP (Fig. 5F).
Intracerebral addition of PD98059 exerts the reverse effect of rAd-PGK-PrPC-Flag
To clarify that addition of PD98059 (20 μm) to the ischemic brain would inhibit the protective effect of rAd-PGK-PrPC-Flag, infarct size was evaluated by H&E staining in three test groups of rAd-PGK-eGFP (n = 4), rAd-PGK-PrPC-Flag (n = 4), and PD98059 plus rAd-PGK-PrPC-Flag (n = 4) (Fig. 5G). The results revealed a significantly increased infarct size in PD98059 plus rAd-PGK-PrPC-Flag-treated rats compared with rAd-PGK-PrPC-Flag-treated rats. The infarct size of PD98059 plus rAd-PGK-PrPC-Flag-treated rats was similar to that found in rAd-PGK-eGFP-treated rats (Fig. 5H).
Intracerebral administration of rAd-PGK-PrPC-Flag does not influence the physiological properity of rats
To demonstrate that the neuroprotective effect of rAd-PGK-PrPC-Flag did not occur as a result of changes to other physiological parameters, systemic physiological parameters were analyzed in 14 experimental rats. Compared with rAd-PGK-eGFP (n = 7), intracerebral administration of rAd-PGK-PrPC-Flag (n = 7) did not alter systemic blood pressure, blood gases, blood glucose, or serum electrolyte levels (Table 1).
Table 1.
Physiological parameters not altered by rAd-PGK-PrPC-Flag treatment
|
Items |
rAd-PGK-PrPC-Flag (n = 7) |
rAd-PGK-eGFP (n = 7) |
pa |
|---|---|---|---|
| pH | 7.34 ± 0.016 | 7.32 ± 0.012 | 0.911 |
| PaCO2 (mmHg) | 47.22 ± 1.37 | 49.33 ± 2.6 | 0.230 |
| PaO2 (mmHg) | 93.51 ± 3.1 | 92.47 ± 3.4 | 0.220 |
| HCO3- (10-3 mol/L) | 27.9 ± 1.22 | 26.47 ± 1.14 | 0.414 |
| Hematocrit (%) | 43.62 ± 2.61 | 42.8 ± 3.78 | 0.285 |
| Hemoglobin (10 g/L) | 13.9 ± 0.38 | 14.69 ± 0.86 | 0.229 |
| Na+ (10-3 mol/L) | 140.1 ± 4.4 | 144.11 ± 2.4 | 0.642 |
| K+ (10-3 mol/L) | 4.53 ± 0.31 | 4.36 ± 0.16 | 0.819 |
| Ca+ (10-2 g/L) | 4.13 ± 0.29 | 3.76 ± 1.03 | 0.577 |
| Glucose (10-2 g/L) | 144.9 ± 27.2 | 146.91 ± 15.2 | 0.584 |
| MBP (mmHg) | 78.6 ± 8.31 | 81.1 ± 6.39 | 0.568 |
| HR (bpm) |
397 ± 27 |
414 ± 17 |
0.667 |
MBP, Mean blood pressure; HR, heart rate.
t test.
Hypoxia–reoxygenation induces an increase in PrPC immunoreactivity in primary cortical cultures
To examine the effect of hypoxia–reoxygenation on PrPC immunoreactivity in primary cortical cultures, cells were placed in a hypoxic chamber (95% N2 and 5% CO2, 37°C) for 12 h and then returned to a normoxic incubator (95% air and 5% CO2, 37°C) for varying lengths of time (1, 3, 10, and 24 h), after which they were immunostained for PrPC. The results revealed an increase in the immunoreactivity of PrPC after hypoxia-reoxygenation treatment (Fig. 6A) in a time-dependent manner compared with the control (Fig. 6B). Compared with control cells, hypoxia-reoxygenation treatment significantly increased PrPC-immunoreactive cell density 10 h after treatment (Fig. 6C).
Figure 6.

Hypoxia–reoxygenation induces an increase in PrPC immunoreactivity in primary cortical cultures. A, B, Representative microscopic features of primary cortical cultures were immunoreacted for PrPC in conditions of hypoxia–reoxygenation or control, respectively. C, Quantitative analysis of PrPC-immunoreactive cells. Increased immunoreactivity of PrPC was time dependent and reached its peak level 24 h after hypoxia–reoxygenation compared with non-hypoxia-treated cells, indicated as “C.” The mean ± SEM is shown. *p < 0.05 versus control. Scale bar, 40 μm.
Hypoxia–reoxygenation induces translation of PrPC and ERK1/2 in primary cortical cultures
To examine the induction of increased protein expression in PrPC, ERK1/2, p38, and JNK, primary cortical cultures were placed in a hypoxic chamber (95% N2 and 5% CO2, 37°C) for 12 h and returned to a normoxic incubator (95% air and 5% CO2, 37°C) for differing lengths of time (1, 3, 10, and 24 h), after which total cell protein was extracted. It was found that there was a time-dependent increase in the expression of PrPC after hypoxia-reoxygenation treatment (Fig. 7A). The ratio of PrPC/actin protein reached a peak level of approximately a twofold increase over control cells 24 h after hypoxia-reoxygenation treatment (Fig. 7B).
Figure 7.

Hypoxia–reoxygenation induces translation of PrPC and ERK1/2 in primary cortical cultures. A, primary cortical cultures were placed in a hypoxic chamber (95% N2 and 5% CO2, 37°C) for 12 h and returned to a normoxic incubator (95% air and 5% CO2, 37°C) for different lengths of time (1, 3, 10, and 24 h), after which, total protein was extracted for Western blot analysis of PrPC. B, Densitometric analysis of the Western blot, the ratio of PrPC/actin was taken as 1 for nonhypoxic cells, which represents the control (C). The mean ± SEM is shown. *p < 0.05 versus control. The time-dependent changes in PrPC expression are depicted. C, Primary cortical cultures were treated as above, and total protein was then extracted for Western blot analysis of ERK1/2, p38, and JNK expression. D, Densitometric analysis of the Western blot, the ratio of ERK1/2, p38, and JNK/actin were taken as 1 for nonhypoxic cells, which represents the control. E, Primary cortical cultures were treated as above and total protein was then extracted for Western blot analysis of PrPC expression with or without addition of inhibitor of PD98059. F, Densitometric analysis of the Western blot, the ratio of PrPC/Actin were taken as 1 for nonhypoxic cells, which represents the control. G, The results show that N18 cells infected with rAd-PGK-PrPC-Flag subjected to hypoxia for 12 h exhibited significantly reduced caspase-3 activity compared with that of rAd-PGK-eGFP. H, The rAd-PGK-PrPC-Flag-infected N18 cells showed much reduced LDH activity after 24 h of hypoxic treatment compared with that of rAd-PGK-eGFP. The mean ± SEM is shown. *p < 0.05 versus control. C, Control.
To examine the expression of activated ERK1/2, p38, and JNK, primary cortical cultures were treated under the aforementioned conditions, after which total protein was extracted. We found an increase in the expression of activated ERK1/2 3 h after hypoxia-reoxygenation treatment (Fig. 7C). At this time, the ratio of ERK1/2/actin protein reached a peak level of approximately a twofold increase in treated cells compared with the control cells (Fig. 7D). The expression levels of p38 and JNK showed no statistically significant differences between treated and control cells (Fig. 7D). However, the increased expression of PrPC returned to the control level after the addition of the specific inhibitor of activated ERK1/2 (PD98059) to treated cells (Fig. 7E, F).
Overexpression of PrPC transduces antiapoptotic signals in vitro
To examine whether overexpression of PrPC would be neuroprotective under hypoxic conditions, we examined caspase-3 activity and LDH activity in the neuroblastoma cells line (N18) by adenovirus-mediated overexpression of PrPC. The results, obtained using a fluorometric method, showed that N18 cells infected with rAd-PGK-PrPC-Flag subjected to 12 h of hypoxic treatment had increased expression of PrPC and significantly reduced caspase-3 activity compared with the cells infected with rAd-PGK-eGFP (Fig. 7G). In addition, rAd-PGK-PrPC-Flag-infected cells showed remarkably reduced LDH activity after 24 h of hypoxia treatment compared with those injected with rAd-PGK-eGFP (Fig. 7H).
Hypoxia–reoxygenation induced activation of PrP-luciferase reporter gene constructs containing HSE
To determine whether the molecular regulation of PrPC expression took place through activated transcriptional factor binding to HSE in the promoter region of rat PrPC gene, N18 cells were transiently transfected with 6 μg of various PrPC reporter constructs and pCMV β-gal using lipofectamine, and luciferase activity was then analyzed. After transfection, cells were grown overnight and were either subjected or not subjected to hypoxia for 12 h, after which they were reoxygenated for up to 24 h. Luciferase activities of each reporter plasmid were measured in triplicate and expressed as fold-inductions after normalization by β-gal assay. We found a significant increase in luciferase activities between hypoxia-reoxygenation-treated and untreated cells. A 10- to 15-fold increase in the luciferase activity of praPrP(-2831)-luc and praPrP(-1026)-luc was observed compared with praPrP(-514)-luc and the pGL2 control under conditions of hypoxia (Fig. 8). The luciferase activity of praPrP(-2831M)-luc showed a threefold increase compared with the praPrP(-514)-luc and pGL2-control (Fig. 8).
Figure 8.

Hypoxia–reoxygenation induces activation of a PrP-luciferase reporter gene containing HSE. N18 cells were transiently transfected using lipofectamine with 6 μg of reporter plasmids [praPrP(-2831)-luc, praPrP(-1026)-luc, praPrP(-514)-luc, and pGL2-control] and pCMVβ-gal, and the luciferase activity of these constructs were analyzed. Luciferase activities of each reporter plasmid were measured in triplicate and expressed as fold-inductions after normalization byβ-gal assay. The results show a 10- to 15-fold increase in luciferase activity of praPrP(-2831)-luc and praPrP(-1026)-luc compared with praPrP(-514)-luc and pGL2-control under conditions of maximal induction. The luciferase activity of praPrP(-2831M)-luc shows a threefold increase compared with praPrP(-514)-luc and the pGL2-control.
Discussion
Although the biological function of PrPC remains unknown, several lines of research have suggested a protective role for PrPC in the cellular response to oxidative damage (Brown and Sassoon, 2002; McLennan et al., 2004). In this study, ischemic conditions resulted in a time-dependent increase in PrPC expression over the penumbric area in cerebral ischemic animals. Immunohistochemical analysis of brain tissue also demonstrated that PrPC-positive cells significantly increased after cerebral ischemia compared with the nonischemic control. In the cerebral ischemic animal model, PrPC expression was present in neuronal, glial, and vascular endothelial cells. The functional importance of PrPC having a neuroprotective role after neuronal damage has been further emphasized in studies using ischemic rats. Intracerebral administration of rAd-PGK-PrPC-Flag to cerebral ischemic rats improved neurological dysfunction and diminished the infarct volume, further reinforcing the impression of a neuroprotective role for PrPC. We also found increased expression of PrPC and an increased number of PrPC-immunoreactive cells under hypoxia-reoxygenation conditions in primary cortical cultures. In addition, expression of activated ERK increased after hypoxiareoxygenation treatment, and the upregulation of PrPC was blocked by the ERK-specific inhibitor (PD98059). After hypoxiareoxygenation treatment, N18 cells transfected with luciferase reporter constructs of the rat PrPC promoter, which contained HSE, expressed higher luciferase activities (3- to 10-fold) than those cells transfected with the constructs containing no HSE. In conclusion, from these findings, we can propose that HSTF-1 (Shyu et al., 2002), phosphorylated by ERK1/2, could in turn interact with HSE in the promoter of PrPC resulting in PrPC gene expression.
In previous studies, it was found that PrPC could cause neuronal cells to respond to many types of environmental stress, including heat shock (Shyu et al., 2000, 2002) and oxidative stress (Shyu et al., 2004). Some investigators found that, under ischemic conditions, PrPC expression is upregulated (McLennan et al., 2004; Weise et al., 2004). These previous in vivo results, in either the human or rodent brain, indicate that PrPC has a protective role against neurotoxicity. In our study, we have again demonstrated that expression of PrPC dose protects the neural tissue from ischemic and hypoxia injury in the rat brain and primary cortical neuron. Furthermore, we also found increased expression of PrPC in the penumbric region after cerebral ischemia peaking 3 d after 90 min cerebral ischemia was induced in rats. In contrast, a recent report demonstrated that transient cerebral ischemia (60 min) does not induce any significant increase in PrPC expression (Weise et al., 2004). Therefore, we can speculate that more severe neuronal injuries would bring about a greater change in the extent and temporal profile of PrPC upregulation.
Many growth factors and neuroprotective substances, such as transforming growth factor-β1 and glial cell-derived neurotrophic factor, show increased expression after brain insult (Humpel et al., 1994; Knuckey et al., 1996). This indicates that neuroprotective signals are transduced immediately after environmentally stressful conditions. In our investigation, expression of PrPC increased time dependently under ischemia/hypoxia conditions. We also demonstrate that increased expression of PrPC in the local ischemic brain region could exert a neuroprotective effect that improves neurological dysfunction and reduces the volume of cerebral infarction. As shown in this study, the signaling pathway of increased expression of PrPC definitely occurs through a mitogen-activated protein kinase of ERK1/2, because enhanced expression of PrPC was inhibited by adding ERK1/2 inhibitor (PD98059) to the primary cortical culture medium. In addition, according to previous reports, PrPC may transduce neuroprotective signals through a cell-surface ligand (Chiarini et al., 2002; Zanata et al., 2002) and is capable of protecting neuronal cell lines from apoptosis (Kuwahara et al., 1999). Therefore, we suggest that PrPC may play a neuroprotective role in vivo. Understanding the physiological role of PrPC may help elucidate the pathological processes associated with prion diseases. Furthermore, according to this study, overexpression of PrPC by adenovirus-mediated gene targeting could be adopted in the future as a therapeutic strategy for the treatment of ischemic stroke patients.
Proliferative glial cells responding to mitogenic factors released from microglia increase PrPC expression under certain stresses (Brown et al., 1998b). In addition, the neuroprotective effect of PrPC in glial cells in response to stress has been demonstrated in vivo by transplantation experiments between Prnp0/0 mice and mice overexpressing PrPC (Brandner et al., 1996). These experiments indicated that PrPC exerts a strong neuroprotective effect on brain tissue and might be expressed mainly from glial cells. In this study, we also demonstrate increased expression of PrPC in the penumbric region of ischemic glial cells (GFAP-immunoreactive cells). In a previous report, PrPC expression in astroglial cells was suggested to be related to a regulation in cellular resistance to free-radical damage by oxidative stress (Brown et al., 1998b). Similar findings also suggested that PrPC expression in neurons can aid resistance to oxidative stress (Brown et al., 1997). Thus, we speculate that increased expression of PrPC in glial cells and neurons of the ischemic brain might protect neural tissue from neurotoxic substances after ischemic injury.
In a previous study, a green fluorescent protein reporter gene under the control of bovine PrPC gene regulatory sequences was expressed in the mucosal capillary endothelial cells of the intestine of transgenic mice (Lemaire-Vieille et al., 2000). Reporter gene expression in endothelial and epithelial cells strongly suggests that these cell types normally express the PrPC gene. Subsequently, some reports have demonstrated that endothelial cells of both macrovascular and microvascular origin express high levels of PrPC (Simak et al., 2002; Starke et al., 2002). Our study has shown increased expression of PrPC in the penumbric region of ischemic brain cells and that this expression is colocalized with markers for vascular endothelial cells, as observed by double immunohistochemical confocal microscopy. Compared with previous studies (Simak et al., 2002; Starke et al., 2002), which found only endothelial cell expressing PrPC outside the CNS, we found that brain vascular endothelial cells (vWF-immunoreactive cells) produce PrPC in the ischemic region. This indicates that increased expression of PrPC in endothelial cells in the ischemic brain region may be neuroprotective compared with normal brains, in which there is no increase in PrPC expression. According to a previous report, which investigated caveolin-1 (cav-1) in endothelial cells, PrPC is bound to cav-1 and, by recruiting Fyn kinase, can participate in signal transduction events related to cell survival, differentiation, and even angiogenesis (Massimino et al., 2002). In summary, taking our results in combination with these investigations (Massimino et al., 2002; Simak et al., 2002; Starke et al., 2002), we speculate that increased expression of PrPC in neuron, glial, and endothelial cells of the ischemic brain might be involved in aiding neuroplasticity and angiogenesis to attenuate ischemic injury in the stroke rat.
Footnotes
This work was supported in part by research grants from the Chen-Han Foundation for Education, the Academia Sinica (AS92IMB3), and the National Science Council (NSC93-2314-B-303-009). We thank Dr. K. Deen for his critical reading of this manuscript and Dr. Onodera for providing us the templates of PrP-promoter luciferase contructs.
Correspondence should be addressed to Dr. Hung Li, Institute of Molecular Biology, Academia Sinica, Taipei, Taiwan 115. E-mail: hungli@ccvax.sinica.edu.tw.
DOI:10.1523/JNEUROSCI.1115-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258967-11$15.00/0
W.-C.S. and S.-Z.Y. contributed equally to this work.
References
- Bolton DC, McKinley MP, Prusiner SB (1982) Identification of a protein that purifies with the scrapie prion. Science 218: 1309–1311. [DOI] [PubMed] [Google Scholar]
- Borlongan CV, Hida H, Nishino H (1998) Early assessment of motor dysfunctions aids in successful occlusion of the middle cerebral artery. NeuroReport 9: 3615–3621. [DOI] [PubMed] [Google Scholar]
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A (2001) Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem 276: 39145–39149. [DOI] [PubMed] [Google Scholar]
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A (1996) Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 379: 339–343. [DOI] [PubMed] [Google Scholar]
- Brown DR, Sassoon J (2002) Copper-dependent functions for the prion protein. Mol Biotechnol 22: 165–178. [DOI] [PubMed] [Google Scholar]
- Brown DR, Schulz-Schaeffer WJ, Schmidt B, Kretzschmar HA (1997) Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp Neurol 146: 104–112. [DOI] [PubMed] [Google Scholar]
- Brown DR, Schmidt B, Groschup MH, Kretzschmar HA (1998a) Prion protein expression in muscle cells and toxicity of a prion protein fragment. Eur J Cell Biol 75: 29–37. [DOI] [PubMed] [Google Scholar]
- Brown DR, Schmidt B, Kretzschmar HA (1998b) A prion protein fragment primes type 1 astrocytes to proliferation signals from microglia. Neurobiol Dis 4: 410–422. [DOI] [PubMed] [Google Scholar]
- Brown DR, Besinger A, Herms JW, Kretzschmar HA (1998c) Microglial expression of the prion protein. NeuroReport 9: 1425–1429. [DOI] [PubMed] [Google Scholar]
- Brown DR, Wong BS, Hafiz F, Clive C, Haswell SJ, Jones IM (1999) Normal prion protein has an activity like that of superoxide dismutase. Biochem J 344: 1–5. [PMC free article] [PubMed] [Google Scholar]
- Brown DR, Nicholas RS, Canevari L (2002) Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J Neurosci Res 67: 211–224. [DOI] [PubMed] [Google Scholar]
- Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C (1993) Mice devoid of PrP are resistant to scrapie. Cell 73: 1339–1347. [DOI] [PubMed] [Google Scholar]
- Caughey B, Chesebro B (2001) Transmissible spongiform encephalopathies and prion protein interconversions. Adv Virus Res 56: 277–311. [DOI] [PubMed] [Google Scholar]
- Chen J, Graham SH, Zhu RL, Simon RP (1996) Stress proteins and tolerance to focal cerebral ischemia. J Cereb Blood Flow Metab 16: 566–577. [DOI] [PubMed] [Google Scholar]
- Chen ST, Hsu CY, Hogan EL, Maricq H, Balentine JD (1986) A model of focal ischemic stroke in the rat: reproducible extensive cortical infarction. Stroke 17: 738–743. [DOI] [PubMed] [Google Scholar]
- Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R (2002) Cellular prion protein transduces neuroprotective signals. EMBO J 21: 3317–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu JJ, Ng ML (2003) The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. J Gen Virol 84: 3305–3314. [DOI] [PubMed] [Google Scholar]
- Humpel C, Hoffer B, Stromberg I, Bektesh S, Collins F, Olson L (1994) Neurons of the hippocampal formation express glial cell line-derived neurotrophic factor messenger RNA in response to kainate-induced excitation. Neuroscience 59: 791–795. [DOI] [PubMed] [Google Scholar]
- Hung TH, Skepper JN, Burton GJ (2001) In vitro ischemia-reperfusion injury in term human placenta as a model for oxidative stress in pathological pregnancies. Am J Pathol 159: 1031–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G, Jones G, Wegrzyn RD, Masison DC (2000) A role for cytosolic hsp70 in yeast [PSI(+)] prion propagation and [PSI(+)] as a cellular stress. Genetics 156: 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G, Jones G, Masison DC (2002) Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion-curing by guanidine, prion propagation, and thermotolerance. Proc Natl Acad Sci USA 99: 9936–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckey NW, Finch P, Palm DE, Primiano MJ, Johanson CE, Flanders KC, Thompson NL (1996) Differential neuronal and astrocytic expression of transforming growth factor beta isoforms in rat hippocampus following transient forebrain ischemia. Brain Res Mol Brain Res 40: 1–14. [DOI] [PubMed] [Google Scholar]
- Kretzschmar HA, Prusiner SB, Stowring LE, DeArmond SJ (1986) Scrapie prion proteins are synthesized in neurons. Am J Pathol 122: 1–5. [PMC free article] [PubMed] [Google Scholar]
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, Saeki K, Matsumoto Y, Yokoyama T, Itohara S, Onodera T (1999) Prions prevent neuronal cell-line death. Nature 400: 225–226. [DOI] [PubMed] [Google Scholar]
- Lee KS, Magalhaes AC, Zanata SM, Brentani RR, Martins VR, Prado MA (2001) Internalization of mammalian fluorescent cellular prion protein and N-terminal deletion mutants in living cells. J Neurochem 79: 79–87. [DOI] [PubMed] [Google Scholar]
- Lemaire-Vieille C, Schulze T, Podevin-Dimster V, Follet J, Bailly Y, Blanquet-Grossard F, Decavel JP, Heinen E, Cesbron JY (2000) Epithelial and endothelial expression of the green fluorescent protein reporter gene under the control of bovine prion protein (PrP) gene regulatory sequences in transgenic mice. Proc Natl Acad Sci USA 97: 5422–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SZ, Hoffer BJ, Kaplan P, Wang Y (1999) Osteogenic protein-1 protects against cerebral infarction induced by MCA ligation in adult rats. Stroke 30: 126–133. [DOI] [PubMed] [Google Scholar]
- Lin TN, He YY, Wu G, Khan M, Hsu CY (1993) Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke 24: 117–121. [DOI] [PubMed] [Google Scholar]
- Luckenbill-Edds L (1997) Laminin and the mechanism of neuronal out-growth. Brain Res Brain Res Rev 23: 1–27. [DOI] [PubMed] [Google Scholar]
- Martins VR, Graner E, Garcia-Abreu J, de Souza SJ, Mercadante AF, Veiga SS, Zanata SM, Neto VM, Brentani RR (1997) Complementary hydropathy identifies a cellular prion protein receptor. Nat Med 3: 1376–1382. [DOI] [PubMed] [Google Scholar]
- Massimino ML, Griffoni C, Spisni E, Toni M, Tomasi V (2002) Involvement of caveolae and caveolae-like domains in signalling, cell survival and angiogenesis. Cell Signal 14: 93–98. [DOI] [PubMed] [Google Scholar]
- McLennan NF, Brennan PM, McNeill A, Davies I, Fotheringham A, Rennison KA, Ritchie D, Brannan F, Head MW, Ironside JW, Williams A, Bell JE (2004) Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol 165: 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra OP, Zubrow AB, Ashraf QM (2004) Nitric oxide-mediated activation of extracellular signal-regulated kinase (ERK) and c-jun N-terminal kinase (JNK) during hypoxia in cerebral cortical nuclei of newborn piglets. Neuroscience 123: 179–186. [DOI] [PubMed] [Google Scholar]
- Moser M, Colello RJ, Pott U, Oesch B (1995) Developmental expression of the prion protein gene in glial cells. Neuron 14: 509–517. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, Coyle JT (1990) Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J 4: 1624–1633. [PubMed] [Google Scholar]
- Negro A, Ballarin C, Bertoli A, Massimino ML, Sorgato MC (2001) The metabolism and imaging in live cells of the bovine prion protein in its native form or carrying single amino acid substitutions. Mol Cell Neurosci 17: 521–538. [DOI] [PubMed] [Google Scholar]
- Perera WS, Hooper NM (2001) Ablation of the metal ion-induced endocytosis of the prion protein by disease-associated mutation of the octarepeat region. Curr Biol 11: 519–523. [DOI] [PubMed] [Google Scholar]
- Prusiner SB (1991) Molecular biology of prion diseases. Science 252: 1515–1522. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE (1998) Prion protein biology. Cell 93: 337–348. [DOI] [PubMed] [Google Scholar]
- Rafiee P, Shi Y, Pritchard Jr KA, Ogawa H, Eis AL, Komorowski RA, Fitzpatrick CM, Tweddell JS, Litwin SB, Mussatto K, Jaquiss RD, Baker JE (2003) Cellular redistribution of inducible Hsp70 protein in the human and rabbit heart in response to the stress of chronic hypoxia: role of protein kinases. J Biol Chem 278: 43636–43644. [DOI] [PubMed] [Google Scholar]
- Saeki K, Matsumoto Y, Hirota Y, Matsumoto Y, Onodera T (1996) Threeexon structure of the gene encoding the rat prion protein and its expression in tissues. Virus Genes 12: 15–20. [DOI] [PubMed] [Google Scholar]
- Shyu WC, Hsu YD, Kao MC, Tsao WL (1996) Panencephalitic Creutzfeldt-Jakob disease in a Chinese family. Unusual presentation with PrP codon 210 mutation and identification by PCR-SSCP. J Neurol Sci 143: 176–180. [DOI] [PubMed] [Google Scholar]
- Shyu WC, Kao MC, Chou WY, Hsu YD, Soong BW (2000) Heat shock modulates prion protein expression in human NT-2 cells. NeuroReport 11: 771–774. [DOI] [PubMed] [Google Scholar]
- Shyu WC, Harn HJ, Saeki K, Kubosaki A, Matsumoto Y, Onodera T, Chen CJ, Hsu YD, Chiang YH (2002) Molecular modulation of expression of prion protein by heat shock. Mol Neurobiol 26: 1–12. [DOI] [PubMed] [Google Scholar]
- Shyu WC, Lin SZ, Saeki K, Kubosaki A, Matsumoto Y, Onodera T, Chiang MF, Thajeb P, Li H (2004) Hyperbaric oxygen enhances the expression of prion protein and heat shock protein 70 in a mouse neuroblastoma cell line. Cell Mol Neurobiol 24: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyue SK, Tsai MJ, Liou JY, Willerson JT, Wu KK (2001) Selective augmentation of prostacyclin production by combined prostacyclin synthase and cyclooxygenase-1 gene transfer. Circulation 103: 2090–2095. [DOI] [PubMed] [Google Scholar]
- Simak J, Holada K, D'Agnillo F, Janota J, Vostal JG (2002) Cellular prion protein is expressed on endothelial cells and is released during apoptosis on membrane microparticles found in human plasma. Transfusion 42: 334–342. [DOI] [PubMed] [Google Scholar]
- Starke R, Drummond O, MacGregor I, Biggerstaff J, Gale R, Camilleri R, Mackie I, Machin S, Harrison P (2002) The expression of prion protein by endothelial cells: a source of the plasma form of prion protein? Br J Haematol 119: 863–873. [DOI] [PubMed] [Google Scholar]
- Wang X, Grammatikakis N, Siganou A, Calderwood SK (2003) Regulation of molecular chaperone gene transcription involves the serine phosphorylation, 14-3-3 epsilon binding, and cytoplasmic sequestration of heat shock factor 1. Mol Cell Biol 23: 6013–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lin SZ, Chiou AL, Williams LR, Hoffer BJ (1997) Glial cell line-derived neurotrophic factor protects against ischemia-induced injury in the cerebral cortex. J Neurosci 17: 4341–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weise J, Crome O, Sandau R, Schulz-Schaeffer W, Bahr M, Zerr I (2004) Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci Lett 372: 146–150. [DOI] [PubMed] [Google Scholar]
- White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S, Anstee D, Collinge J, Hawke S (2003) Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 422: 80–83. [DOI] [PubMed] [Google Scholar]
- Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, Freitas AR, Cabral AL, Lee KS, Juliano MA, de Oliveira E, Jachieri SG, Burlingame A, Huang L, Linden R, Brentani RR, Martins VR (2002) Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J 21: 3307–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoldhelyi P, McNatt J, Xu XM, Loose-Mitchell D, Meidell RS, Clubb Jr FJ, Buja LM, Willerson JT, Wu KK (1996) Prevention of arterial thrombosis by adenovirus-mediated transfer of cyclooxygenase gene. Circulation 93: 10–17. [DOI] [PubMed] [Google Scholar]