

Abstract
Transgenic mice (Tg2576) overexpressing the Swedish mutation of the human amyloid precursor protein display biochemical, pathological, and behavioral markers consistent with many aspects of Alzheimer's disease, including impaired hippocampal function. Impaired, hippocampal-dependent, contextual fear conditioning (CFC) is observed in mice as young as 20 weeks of age. This impairment can be attenuated after treatment before training with the phosphodiesterase-4 inhibitor rolipram (0.1 mg/kg, i.p.). A rolipram-associated improvement is also observed in the littermate controls, suggesting that the effect of rolipram is independent of β-amyloid. Acute treatment before training (but not after training or before testing) with the γ-secretase inhibitor (GSI) N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine-t-butylester (DAPT), at a dose that reduces brain concentrations of β-amyloid (100 mg/kg), attenuates the impairment in 20- to 65-week-old Tg2576 mice. Importantly, DAPT had no effect on performance of control littermates. These data are supportive of a role of β-amyloid in the impairment of CFC in Tg2576 mice. Furthermore, they suggest that acute treatment with GSI may provide improved cognitive functioning as well as disease-modifying effects in Alzheimer's disease.
Keywords: Alzheimer's disease, amyloid, learning, memory, secretase, dementia
Introduction
Alzheimer's disease (AD) is characterized by a progressive decline in cognitive function and elevated levels of β-amyloid and amyloid plaques. β-Amyloid (Aβ) peptides are produced through proteolytic cleavages of the amyloid precursor protein (APP) by β- and γ-secretase (Selkoe, 2001). Mutations in APP or the cleavage enzymes have been identified in familial AD populations (Hardy, 1997). Mutations altering APP processing increase β-amyloid level and plaque deposition and produce cognitive dysfunction at younger ages than observed in sporadic AD. Transgenic mice (Tg2576) overexpressing a mutated form of the human APP (Hsiao et al., 1996) show increased Aβ40 and Aβ42/43 and plaque deposition at 9–12 months of age (Kawarabayashi et al., 2001; Terai et al., 2001). Early reports indicated these mice display behavioral deficits that parallel plaque deposition (Hsiao et al., 1996; Chapman et al., 1999). However, postmortem studies in AD patients show poor correlation between β-amyloid plaque deposition and memory function (Berg et al., 1998; Lue et al., 1999; McLean et al., 1999). Similarly, recent reports suggest that behavioral deficits observed in mutant APP mice are more tightly coupled to soluble β-amyloid and/or species rather than plaque deposition (Hsia et al., 1999; Dineley et al., 2002; Kotilinek et al., 2002; Westerman et al., 2002; Van Dam et al., 2003).
Both in vitro and in vivo studies support a relationship between elevated soluble β-amyloid and impaired synaptic physiology, including impaired long-term potentiation (LTP) (Vitolo et al., 2002; Walsh et al., 2002; Wang et al., 2002; Kamenetz et al., 2003). Furthermore, Vitolo and colleagues (2002) suggest that soluble β-amyloid, via an unidentified receptor/cell-surface protein, inhibits adenylate cyclase and cAMP response element-binding protein (CREB) phosphorylation, thus disrupting LTP. The altered signal transduction and synaptic physiology associated with soluble β-amyloid suggests that elevations in soluble β-amyloid may underlie some of the observed cognitive deficits in AD. Therefore, targeting the adenylate cyclase pathway to enhance cAMP, as occurs through the inhibition of phosphodiesterase-4 (PDE-4), may modulate the effects of β-amyloid and provide symptomatic treatment of AD.
Dovey and colleagues (2001) have described γ-secretase inhibitor (GSI) compounds that inhibit cleavage at the C terminal of β-amyloid and decrease levels of β-amyloid in vitro without altering levels of APP. Acute administration of N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine-t-butylester (DAPT) to mutant APP-overexpressing PDAPP mice produced a 40% reduction in cortical β-amyloid. DAPT provides an excellent tool to examine the impact of modulating β-amyloid levels on cognitive deficits in models of AD. If effective, a GSI could have dual therapeutic effects in AD; first, a disease-modifying effect via reduced β-amyloid levels and a reduction in plaque pathology and downstream neurodegenerative cascades; second, a concomitant symptomatic effect through inhibition of soluble β-amyloid production resulting in a restoration of cAMP levels, LTP, and synaptic function.
Dineley and colleagues (2002) reported that Tg2576 mice show hippocampal-dependent conditioning deficits beginning at 20 weeks of age. Because this deficit occurs before plaque deposition, elevated soluble β-amyloid observed in these younger animals may be responsible for the cognitive dysfunction. Additional behavioral training ameliorated the deficit, suggesting the impairment could be overcome. Here, we provide the first evidence that a small molecule targeting β-amyloid processing will improve cognition in the Tg2576 mouse model of AD.
Materials and Methods
Subjects. Male Tg2576 mice (Hsiao et al., 1996), expressing a double mutation (K670N/M671L) of human APP, were obtained from Taconic (Germantown, NY) and used for subsequent breeding. Transgenic mice heterozygous for the K670N/M671L transgene were maintained on a hybrid C57BL/6/SJL background through breeding hemizygous males with B6SJLF1 females. Genotypes were confirmed by PCR, and animals homozygous for the retinal degeneration mutation were excluded. Individually housed mice, maintained on a 12 h light/dark cycle, had ad libitum access to food and water. Studies were performed comparing heterozygous transgenic (Tg2576) males to age-matched transgene-negative littermates (controls).
β-Amyloid levels. Male Tg2576 mice (20 weeks of age) were given vehicle or 100 mg/kg DAPT orally. Brains from saline-perfused mice (DAPT-treated mice killed 0.5–24 h and vehicle-treated 4 h after dosing) were homogenized in 10:1 (v/w) cold 5 m guanidine, 50 mm Tris-HCl, shaken at room temperature 3 h, then stored at –80°. One milligram of total protein from each sample was mixed with casein buffer, centrifuged, and immunoprecipitated overnight at 4° with 6E10 (Signet, Dedham, MA). Samples were fractionated on Tris-Tricine gels (Bio-Rad, Hercules, CA), transferred to nitrocellulose (Amersham Biosciences, Arlington Heights, IL), and immunoblotted with 6E10. β-Amyloid was detected with biotinylated sheep anti-mouse IgG and 35S-labeled streptavidin (Amersham Biosciences) and visualized with a PhosphorImager (Molecular Dynamics, Sunnyvale, CA). For quantification, integrated values for β-amyloid in experimental samples were compared with a standard curve of synthetic amyloid peptide (AnaSpec, San Jose, CA).
Testing apparatus. Contextual fear conditioning (CFC) occurred in 30 × 24 × 21 cm operant chambers (Med Associates, Georgia, VT). Each chamber was equipped with a stainless-steel rod floor through which a footshock could be administered, two stimulus lights, one house light, and a solenoid, all controlled by Med-PC (Med Associates) computer software. The chambers were located in a sound-isolated room in the presence of red light.
Conditioning. Mice (n = 8–12 per genotype per treatment) were trained and tested on 2 consecutive days (see Fig. 1 A). Training consisted of placing a subject in a chamber, illuminating stimulus and house lights, and allowing exploration for 2 min. Afterward, an auditory cue [2 Hz clicking via the solenoid; conditioned stimulus (CS)] was presented for 15 s. The 2 s footshock [1.5 mAmp; unconditioned stimulus (US)] was administered for the final 2 s of the CS. This procedure was repeated, and mice were removed from the chamber 30 s later.
Figure 1.
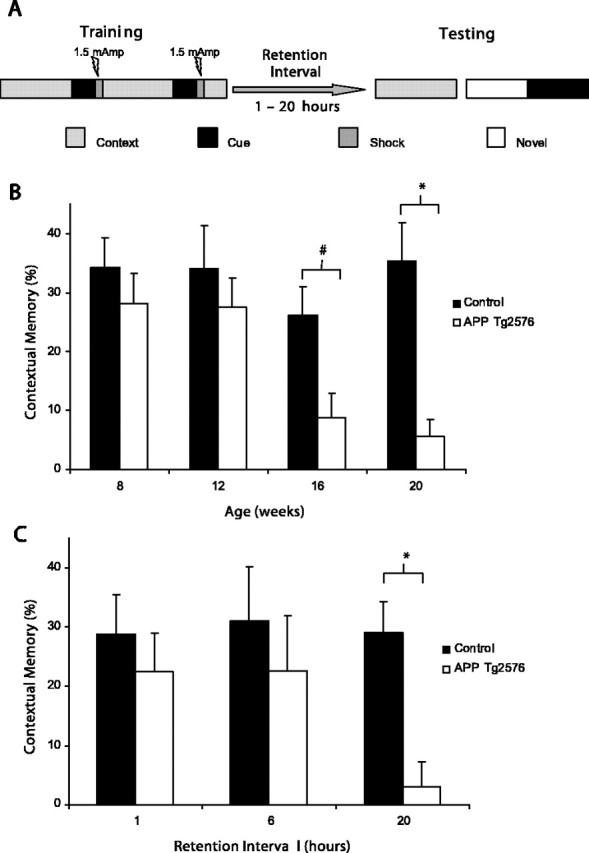
A, Schematic representation of CFC procedure. Training consisted of two cue: footshock pairings during an ∼5 min exposure to the context. Testing, unless otherwise indicated, began 18–20 h after training. B, Tg2576 mice display impaired memory for context after fear conditioning beginning as early as 20 weeks of age with a trend for an impairment observed at 16 weeks (*p < 0.05; #p = 0.054 vs control). C, Contextual memory was impaired at 20 h (*p < 0.05 vs control) but not at 1 or 6 h after training in 20-week-old Tg2576 mice. Error bars represent SEM.
Twenty hours after training, mice were returned to the same chambers in which training occurred (context), and freezing behavior was recorded by the experimenter using time sampling (10 s intervals). Freezing was defined as lack of movement except that required for respiration. At the end of the 5 min context test, mice were returned to their home cage. Approximately 1 h later, freezing was recorded in a novel environment and in response to the cue. The novel environment consisted of modifications including an opaque Plexiglas divider diagonally bisecting the chamber, a Plexiglas floor, and decreased illumination. Mice were placed in the novel environment, and time sampling was used to score freezing for 3 min. The auditory cue (CS) was then presented for 3 min, and freezing was again scored. Freezing scores for each subject were expressed as a percentage for each portion of the test. Memory for the context (contextual memory) for each subject was obtained by subtracting the percent freezing in the novel environment from that in the context.
Locomotor activity. Locomotor activity was collected using AccuScan Instruments (Columbus, OH) activity monitors. Mice (20 weeks of age) were placed in 20 × 20 cm fields, and infrared beam breaks were collected for 30 min. Activity was analyzed using a two-way repeated-measures ANOVA.
Drug treatment. Rolipram was suspended in 1% methylcellulose at a concentration of 0.01 mg/ml and administered intraperitoneally 30 min before training. DAPT was administered orally formulated in corn oil: ethanol (90:10). For CFC, DAPT was administered 3 h before training for all experiments, unless otherwise described.
Data analysis. Contextual memory was analyzed using a two-way ANOVA followed by Fisher's least significant difference (LSD) pairwise comparisons of group mean differences using SAS software (SAS Institute, Cary, NC) with an α level of p < 0.05. The effect of DAPT on β-amyloid was analyzed using a one-way ANOVA followed by Fisher's LSD comparison to the vehicle-treated group. For presentation purposes, β-amyloid level was converted to percent of the vehicle-treated group. All data are presented as mean ± SEM.
Results
Age-associated cognitive impairment
Consistent with previous reports, Tg2576 mice demonstrated a robust deficit in contextual fear conditioning by 20 weeks of age (Fig. 1B) (Corcoran et al., 2002; Dineley et al., 2002). Two-way ANOVA revealed a significant main effect of genotype (F(1,80) = 12.354; p < 0.001) and age (F(3,80) = 3.113; p < 0.05); however, the interaction was not significant (F(3,80) = 1.759). No significant difference in contextual memory was observed in 8- to 12-week-old mice. In contrast, Tg2576 mice tended to perform worse than controls beginning at 16 weeks of age (p = 0.054) with the difference becoming significant at 20 weeks (p < 0.05). No significant effect of genotype was observed in hippocampal-independent cue conditioning (data not shown). Consistent with results reported by Dineley et al. (2002), additional training (five US:CS pairs) produced significant contextual memory in 20-week-old, but not 36-week-old, Tg2576 mice (data not shown).
Tg2576 mice (20 weeks of age), tested at intervals shorter than the standard 20 h between training and testing, were able to display contextual conditioning that was not significantly different from controls (Fig. 1C). The two-way ANOVA revealed a significant main effect of genotype (F(1,47) = 5.452; p < 0.05) and no significant effect of training interval (F(2,47) = 1.391) or interaction (F(2,47) = 1.154). Planned comparisons revealed a significant difference between control and Tg2576 mice at the 20 h interval (p < 0.05), whereas neither the 1 nor 6 h interval resulted in a significant difference between the genotypes.
No difference between genotypes was observed in activity (control, 15,424 ± 741; Tg2576, 17,486 ± 1952 counts).
Cognitive enhancing effect of PDE-4 inhibition
The PDE-4 inhibitor rolipram, administered 30 min before training, improved conditioning in Tg2576 mice at both 24 and 36 weeks of age (Fig. 2). At 24 weeks, a significant main effect of genotype (F(1,28) = 25.906; p < 0.001) and treatment (F(1,28) = 12.57; p < 0.01) were observed; however, the interaction was not significant (F(1,28) = 0.835). Rolipram-treated Tg2576 mice had contextual memory scores that were not different from vehicle-treated controls, indicating memory for the context. However, the increase over vehicle-treated Tg2576 mice did not reach statistical significance (p < 0.1). At 36 weeks of age, significant main effects of genotype (F(1,28) = 14.678; p < 0.001) and treatment (F(1,28) = 6.787; p < 0.05) were observed; however, the interaction was not significant (F(1,28) = 2.423). At 36 weeks, rolipram produced a significant improvement in contextual memory in the Tg2576 mice compared with vehicle-treated Tg2576 mice (p < 0.05), resulting in contextual memory scores that were not different from vehicle-treated controls. Consistent with the effect of PDE-4 inhibition on cognitive tasks reported by others (Imanishi et al., 1997; Barad et al., 1998), rolipram significantly improved contextual conditioning in the 24-week-old controls (p < 0.05). No effect on β-amyloid level was observed (p = 0.465) in brains of 20-week-old Tg2576 mice 30 min after treatment with rolipram (0.1 mg/kg). These data suggest that the rolipram-induced improvements in contextual memory are independent of changes in β-amyloid levels.
Figure 2.

Rolipram significantly improved conditioning in 36-week-old Tg2576 mice with a trend observed in 24 weeks, such that conditioning not significantly different from vehicle-treated controls. Rolipram increased contextual memory scores in the 24-week-old controls. Brackets denote group mean comparisons (*p < 0.05; #p < 0.1). Error bars represent SEM.
GSI and β-amyloid lowering
An oral dose of 100 mg/kg of DAPT in Tg2576 mice resulted in reductions in brain levels of Aβ40 (F(6,48) = 10.912; p < 0.0001) and Aβ42 (F(6,48) = 3.706; p < 0.005) (Fig. 3A). Significant reductions were observed in Aβ40 1–8 h and Aβ42 4–8 h after dosing relative to vehicle-treated Tg2576 mice. Levels of Aβ were no longer reduced 24 h after drug administration. Figure 3B indicates the period of reduced levels of β-amyloid relative to the various training and testing procedures.
Figure 3.

A, Oral DAPT (100 mg/kg) in 20-week-old Tg2576 mice produced a significant reduction in Aβ40 and Aβ42 at 1–8 and 4–8 h, respectively (*p < 0.05 vs vehicle treatment). B, Schematic representation of the effect of drug treatment (triangle) on brain levels of β-amyloid levels relative to CFC training and testing. Drug treatment before training results in β-amyloid reductions during and after the training session but not during the testing session. Drug treatment immediately after training results in β-amyloid reduction during the retention interval. Treatment before testing decreased β-amyloid at the time of testing, with the usual elevated levels during training and the retention interval.C, Acute treatment with DAPT(100mg/kg, orally) 3 h before training improved conditioning in Tg2576 mice from 20 to 65 weeks of age. DAPT-treated Tg2576 mice at 20, 36, and 54 weeks of ages had contextual memory scores significantly greater than vehicle-treated Tg2576 and not significantly different from DAPT-treated controls. No effect of DAPT was observed on the performance of controls at any age. Vehicle-treated Tg2576 mice demonstrated a significant impairment at all ages tested. Brackets denote group mean comparisons (*p < 0.05). D, Twenty-week-old Tg2576 mice treated with vehicle immediately after training displayed significant impairments in contextual memory (*p < 0.05 vs controls). DAPT (100 mg/kg, orally) administered either after training (lowering β-amyloid during the retention interval) or 2 h before testing (loweringβ-amyloid during testing) failed to attenuate the contextual memory deficit in the Tg2576 mice (*p < 0.05 vs controls). Error bars represent SEM.
GSI and cognitive impairment
Acute administration of DAPT 3 h before training resulted in improved contextual memory in Tg2576 mice (Fig. 3C). At 20 weeks, a significant main effect of genotype (F(1,29) = 8.588; p < 0.05) was observed; however, neither the treatment main effect (F(1,29) = 1.213) nor the interaction (F(1,29) = 3.278) were significant. Vehicle-treated Tg2576 mice displayed no contextual conditioning and had memory scores significantly different from those observed in vehicle-treated controls (p < 0.05). In contrast, DAPT-treated (100 mg/kg) Tg2576 mice had memory scores that were not significantly different from those observed in the 20-week-old DAPT-treated controls (p = 0.4124) and displayed a significant increase relative to vehicle-treated Tg2576 mice (p < 0.05). In 36- and 54-week-old mice, significant group-by-treatment interactions were observed (36-week-old, F(1,28) = 5.751, p < 0.05; 54-week-old, F(1,26) = 4.465, p < 0.05). At both ages, DAPT-treated Tg2576 mice displayed significantly greater contextual memory than vehicle-treated Tg2576 mice (p < 0.05) and were not significantly different from DAPT-treated controls. In 65-week-old mice, as a result of animal availability, vehicle-treated Tg2576 were not tested. No significant difference was observed between control and Tg2576 mice treated with DAPT (F(1,18) = 1.212). No effect of DAPT was observed in controls at any age.
DAPT administered immediately after training or 2 h before testing had no effect on contextual memory (Fig. 3D). A significant main effect of genotype was observed (F(1,52) = 26.784; p < 0.0001) with no main effect of treatment (F(2,52) = 0.947) or interaction (F(2,52) = 0.002). Tg2576 mice treated with DAPT displayed significant impairments in contextual memory after both treatment regimens (p < 0.05) comparable with what is observed in Tg2576 mice dosed with vehicle immediately after training. No significant differences in contextual memory were observed across treatment conditions in controls.
Discussion
The data presented in this report demonstrate that acute treatment with the GSI DAPT is associated with improved contextual fear conditioning in a transgene-specific manner in the Tg2576 model of AD, but only when given before training. In addition, an acute pharmacological manipulation (PDE-4 inhibition) that does not alter brain levels of β-amyloid improves conditioning in both Tg2576 and control mice, similar to the effect of additional training. Finally, a previous finding of a cognitive deficit in Tg2576 mice detected using the contextual fear conditioning model, in the absence of locomotor activity changes, was replicated and extended by evaluating mice at more advanced ages.
Inhibition of PDE-4 has been demonstrated to increase cAMP levels, activate PKA, and increase the levels of activated CREB, all of which are associated with the late phase of LTP, synaptic plasticity, and long-term memory (for review, see Silva et al., 1998; Bozon et al., 2003). In vivo, rolipram has been demonstrated to ameliorate the cognitive deficits induced by scopolamine as well as improve retention in cognitively intact animals and other AD transgenic models using contextual fear conditioning (Barad et al., 1998; Zhang and O'Donnell, 2000; Gong et al., 2004). The cognitive improvements in the Tg2576 mice described here after rolipram treatment or after additional training clearly indicate that the learning and memory system, although impaired, is intact in these animals. The more robust improvement in Tg2576 performance, compared with controls, after rolipram treatment at 36 weeks, coupled with the data from Vitolo and colleagues (2001), suggesting a direct effect of β-amyloid on the cAMP signaling pathway, indicates that the cognitive dysfunction associated with AD may be particularly sensitive to the inhibition of PDE-4. However, the effect on control performance suggests that these improvements are, at least partially, independent of β-amyloid and therefore are more consistent with symptomatic rather than disease-modifying therapeutic approaches (e.g., GSI).
In contrast to the genotype-independent cognitive improvements described above, acute inhibition of APP cleavage with a GSI during learning and consolidation improved performance only in Tg2576 mice. This improvement is observed even at ages when plaques have been reported to be present in the hippocampus of these mice (Hsiao et al., 1996). The acute nature of this effect on cognition, the limited time frame in which inhibition of γ-secretase is required, and the transgene-specific nature of these effects suggests β-amyloid contributes to the cognitive dysfunction in Tg2576 mice. In addition to effects on β-amyloid, GSI can affect other proteins, such as Notch (Selkoe, 2001; Presente et al., 2004) and DCCα (Parent et al., 2005), implicated in learning and memory. Indeed, loss of presenilin function, a component of γ-secretase, has been shown to result in impaired memory and synaptic plasticity (Saura et al., 2004). However, because our current results indicate that DAPT only improves cognitive performance in Tg2576 animals and has no effect on controls, the impact of DAPT in this learning paradigm does not appear to be mediated by non-β-amyloid mechanisms (i.e., via changes in Notch or DCCα processing).
In vitro and in vivo studies provide evidence for a role of soluble β-amyloid in altering synaptic function. Wang et al. (2002) have demonstrated that soluble β-amyloid impairs LTP in a hippocampal slice. Furthermore, Walsh et al. (2002) suggest that the effects are a function of small oligomers of β-amyloid rather than monomers. Specifically, LTP disruption is not affected by the addition of insulin-degrading enzyme, which selectively degrades β-amyloid monomers, but is reversed by GSI at concentrations that selectively inhibit multimeric β-amyloid. In addition, soluble oligomeric β-amyloid is reported to be necessary and sufficient to rapidly and transiently impair learning (Cleary et al., 2005).
A number of recent reports have demonstrated reduced β-amyloid, plaque load, and improved cognitive function after long-term experimental manipulations, particularly in very old transgenic animals (Ray et al., 2000; Dodart et al., 2002; Kotilinek et al., 2002; Matsuoka et al., 2003). The current results suggest that short-term, relatively modest decrease in β-amyloid is sufficient to allow normal synaptic function and thus memory encoding in Tg2576 mice. Neutralizing antibodies, able to bind synaptotoxic oligomeric species of β-amyloid, are reported to rapidly reverse LTP deficits in vivo (Klyubin et al., 2005). In addition, acute administration of an antibody producing a rapid decrease in brain levels of β-amyloid is reported to improve performance on object recognition and hole-board cognition tasks in mouse AD models (DeMattos et al., 2001; Dodart et al., 2002). Thus, soluble β-amyloid within the brain may be playing a critical role in the cognitive impairment in AD, as modeled in these transgenic animals. Moreover, different approaches to lowering or sequestering β-amyloid may yield effective cognitive therapies.
In summary, the current results are consistent with the hypothesis that at least some of the cognitive impairments observed in AD are associated with soluble β-amyloid, occur before plaque deposition, and may be reversed by acute pharmacological treatment. These pharmacological approaches could target downstream signal transduction pathways, as occurs with the PDE-4 inhibitor, or directly modulate β-amyloid, as seen with the GSI. In addition, the acute effect of GSI on cognition in the Tg2576 animal model suggests that improved cognitive function in AD patients may be the earliest indication of therapeutic efficacy of disease modifying strategies in the clinic.
Footnotes
We thank Drs. Mark Day, Sheree Logue, Peter Reinhart, and Margaret Zaleska for their valuable comments and critical review of this manuscript.
Correspondence should be addressed to T. A. Comery, Discovery Neuroscience, CN8000, Princeton, NJ 08543-8000. E-mail: comeryt@wyeth.com.
DOI:10.1523/JNEUROSCI.2693-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258898-05$15.00/0
References
- Barad M, Bourtchouladze R, Winder DG, Golan H, Kandel E (1998) Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc Natl Acad Sci USA 95: 15020–15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg L, McKeell Jr DW, Miller JP, Storandt M, Rubin EH, Morris JC, Baty J, Coats M, Norton J, Goate AM, Price JL, Gearing M, Mirra SS, Saunders AM (1998) Clinicopathologic studies in cognitively healthy aging and Alzheimer disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol 55: 326–335. [DOI] [PubMed] [Google Scholar]
- Bozon B, Kelly A, Josselyn SA, Silva AJ, Davis S, Laroche S (2003) MAPK, CREB and zif268 are all required for the consolidation of recognition memory. Philos Trans R Soc Lond B Biol Sci 358: 805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TVP, Hyman BT, Younkin SG, Hsiao KK (1999) Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 2: 271–276. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH (2005) Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat Neurosci 8: 79–84. [DOI] [PubMed] [Google Scholar]
- Corcoran KA, Lu Y, Turner RS, Maren S (2002) Overexpression of hAPP-swe impairs rewarded alternation and contextual fear conditioning in a transgenic mouse model of Alzheimer's disease. Learn Mem 9: 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart J-C, Paul SM, Holtzman DM (2001) Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 98: 8850–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Xia X, Bui D, Sweatt D, Zheng H (2002) Accelerated plaque accumulations, associative learning deficits, and up-regulation of a7 nicotinic receptor protein in transgenic mice co-expressing mutant human Presenilin-1 and amyloid precursor proteins. J Biol Chem 277: 22768–22780. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM (2002) Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci 5: 452–457. [DOI] [PubMed] [Google Scholar]
- Dovey HF, John V, Anderson JP, Chen LZ, de Saint Andrieu P, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, Hu KL, Johnson-Wood KL, Kennedy SL, Kholodenko D, Knops JE, Latimer LH, Lee M, Liao Z, Liederburg IM, Motter RN, et al. (2001) Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem 76: 173–181. [DOI] [PubMed] [Google Scholar]
- Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O (2004) Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest 114: 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J (1997) Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci 20: 154–159. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu G-Q, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L (1999) Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA 96: 3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274: 99–102. [DOI] [PubMed] [Google Scholar]
- Imanishi T, Sawa A, Ichimaru Y, Miyashiro M, Kato S, Yamamoto T, Ueki S (1997) Ameliorating effects of rolipram on experimentally induced impairments of learning and memory in rodents. Eur J Pharmacol 321: 273–278. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R (2003) APP processing and synaptic function. Neuron 37: 925–937. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG (2001) Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci 21: 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ (2005) Amyloid β protein immunotherapy neutralizes Aβ oligomers that disrupt synaptic plasticity in vivo. Nat Med 11: 556–561. [DOI] [PubMed] [Google Scholar]
- Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH (2002) Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J Neurosci 22: 6331–6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue L-F, Kuo Y-M, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J (1999) Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 155: 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Saito M, LaFrancois J, Saito M, Gaynor K, Olm V, Wang L, Casey E, Lu Y, Shiratori C, Lemere C, Duff K (2003) Novel therapeutic approach for the treatment of Alzheimer's disease by peripheral administration of agents with affinity to β-amyloid. J Neurosci 23: 29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL (1999) Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46: 860–866. [DOI] [PubMed] [Google Scholar]
- Parent AT, Barnes NY, Taniguch Y, Thinakaran G, Sisodia SS (2005) Presenilin attenuates receptor-mediated signaling and synaptic function. J Neurosci 25: 1540–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presente A, Boyles RS, Serway CN, deBell JS, Andres AJ (2004) Notch is required for long-term memory in Drosophila Proc Natl Acad Sci USA 101: 1764–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray I, Chauhan A, Wegiel J, Chauhan VPS (2000) Gelsolin inhibits the fibrillization of amyloid beta-protein, and also defibrillizes its preformed fibrils. Brain Res 853: 344–351. [DOI] [PubMed] [Google Scholar]
- Saura CA, Choi S-Y, Beglopoulos V, Malkani S, Zhang D, Rao BSS, Chattarji S, Kelleher RJ, Kandel ER, Duff K, Kirkwood A, Shen J (2004) Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42: 23–36. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81: 741–766. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S (1998) CREB and memory. Annu Rev Neurosci 21: 127–148. [DOI] [PubMed] [Google Scholar]
- Terai K, Iwai A, Kawaba S, Tasaki Y, Watanabe T, Miyata K, Yamaguchi T (2001) β-Amyloid deposits in transgenic mice expressing human β-amyloid precursor protein have the same characteristics as those in Alzheimer's disease. Neuroscience 104: 299–310. [DOI] [PubMed] [Google Scholar]
- Van Dam D, D'Hooge R, Staufenbiel S, Van Ginneken C, Van Meir F, De Deyn PP (2003) Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci 17: 388–396. [DOI] [PubMed] [Google Scholar]
- Vitolo OV, Sant'Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M (2002) Amyloid β-peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci USA 99: 13217–13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416: 535–539. [DOI] [PubMed] [Google Scholar]
- Wang H-W, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL (2002) Soluble oligomers of β amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res 924: 133–140. [DOI] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin S, Ashe KH (2002) The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci 22: 1858–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HT, O'Donnell JM (2000) Effects of rolipram on scopolamine-induced impairment of working and reference memory in the radial-arm maze tests in rats. Psychopharmacology 150: 311–316. [DOI] [PubMed] [Google Scholar]