

Abstract
Cyclin-dependent kinase 5 (cdk5) is a member of the cyclin-dependent kinase family whose activity is localized mainly to postmitotic neurons attributable to the selective expression of its activating partners p35 and p39. Deregulation of cdk5, as a result of calpain cleavage of p35 to a smaller p25 form, has been suggested to be a central component of neuronal death underlying numerous neurodegenerative diseases. However, the relevance of cdk5 in apoptotic death that relies on the mitochondrial pathway is unknown. Furthermore, evidence that cdk5 can also promote neuronal survival has necessitated a more complex understanding of cdk5 in the control of neuronal fate. Here we explore each of these issues using apoptotic and excitotoxic death models. We find that apoptotic death induced by the DNA-damaging agent camptothecin is associated with early transcription-mediated loss of p35 and with late production of p25 that is dependent on Bax, Apaf1, and caspases. In contrast, during excitotoxic death induced by glutamate, neurons rapidly produce p25 independent of the mitochondrial pathway. Analysis of the localization of p35 and p25 revealed that p35 is mainly cytoplasmic, whereas p25 accumulates selectively in the nucleus. By targeting a dominant-negative cdk5 to either the cytoplasm or nucleus, we show that cdk5 has a death-promoting activity within the nucleus and that this activity is required in excitotoxic death but not apoptotic death. Moreover, we also find that cdk5 contributes to pro-survival signaling selectively within the cytoplasm, and manipulation of this signal can modify death induced by both excitotoxicity and DNA damage.
Keywords: apoptosis, cell cycle, p35, cdk5, excitotoxicity, DNA damage
Introduction
Cyclin-dependent kinases (CDKs) are a family of proline-directed serine/threonine kinases best known for their role in controlling proliferation. Once bound to an activating cyclin protein, CDKs control progression through the phases of the cell cycle by phosphorylation of specific substrates. For example, at the G1–S phase transition, cyclinD/cdk4/6 and cyclinA/cdk2 complexes phosphorylate and inactivate the retinoblastoma protein (pRb), a major break on the cell cycle (Johnson and Walker, 1999; Ekholm and Reed, 2000; Massague, 2004). Inactivation of pRb allows for E2F-mediated transcription of genes, which promote S phase (Dyson, 1998).
Some members of the CDK family, however, are involved in functions unrelated to the cell cycle. One such member, cdk5, is inactive in most tissues except for the CNS because of the neuronspecific expression of its non-cyclin-activating partners p35 and p39. Cdk5 is a multifunctional kinase required for a variety of normal neuronal functions, including cortical development, axon migration, cell adhesion, and synaptic activity (Dhavan and Tsai, 2001).
Evidence suggests that DNA damage may be a central event in neurodegeneration. For instance, DNA strand breaks occur as an early event after reperfusion of ischemic brain (Tobita et al., 1995; Chen et al., 1997; Cui et al., 2000), and DNA lesions have been reported in Parkinson's and Alzheimer's diseases (Robison and Bradley, 1984; Alam et al., 1997; Gabbita et al., 1998; Jenner, 1998; Lovell and Markesbery, 2001). Furthermore, mutations in DNA repair genes have been linked to the development of neurodegenerative conditions in humans (Date et al., 2001; Moreira et al., 2001; Takashima et al., 2002), and an increase in DNA damage within the promoters of genes involved with synaptic plasticity and neuronal survival may underlie the neuronal loss seen in the aged human brain (Lu et al., 2004). Neuronal death attributable to DNA damage can be modeled in vitro using camptothecin (CA), an inhibitor of topoisomerase 1 (Morris and Geller, 1996; Liu et al., 2000). Treatment of cultured cortical neurons with CA induces apoptotic death that is dependent on p53, Bax, and the mitochondrial death pathway (Xiang et al., 1998; Morris et al., 2001).
Interestingly, a wide range of evidence has accumulated implicating cell cycle CDKs as required signals in neuronal death induced by DNA damage. For example, CA induces cyclinD-associated kinase activity and leads to an increase in pRb phosphorylation (Park et al., 1998, 2000). Inhibition of cell cycle CDKs by pharmacological means or by expression of dominant-negative cdk4/6 prevents DNA damage-induced death (Park et al., 1997a, 1998). Cell cycle CDKs have also been implicated in other models of neuronal death, including trophic factor withdrawal and β-amyloid-induced death (Park et al., 1997b; Copani et al., 1999; Giovanni et al., 1999; Padmanabhan et al., 1999; Konishi et al., 2002).
More recently, cdk5 has also received considerable attention as a regulator of neuronal death. Evidence suggests that cdk5 becomes an inducer of death when its activator p35 is cleaved into a smaller p25 form by calpains (Patrick et al., 1999; Kusakawa et al., 2000; Lee et al., 2000). Accumulation of p25 and/or involvement of cdk5 has been observed in neurons treated with excitotoxins, β-amyloid, and oxidative stress (Alvarez et al., 1999; Lee et al., 2000; Gong et al., 2003), as well as in animal models of stroke (Wang et al., 2003), Parkinson's disease (Smith et al., 2003), and amyotrophic lateral sclerosis (Nguyen et al., 2001). Importantly, each of these models is thought to involve Ca+2 signaling and do not have a strict reliance on the mitochondrial death pathway. Production of p25 and/or involvement of p25/cdk5 complexes in death models that are dependent on the conserved apoptotic mitochondrial pathway, such as CA-induced death, have yet to be carefully examined.
Although the evidence implicating cdk5 in neuronal death is compelling, some reports have provided contradictory evidence that cdk5 acts instead as a pro-survival signal (Cheung and Ip, 2004). For example, under certain conditions, cdk5 has been shown to activate phosphatidylinositol 3-kinase/Akt signaling (Li et al., 2003). Also, cdk5 can prevent apoptosis induced by UV irradiation through negative regulation of c-Jun-N-terminal kinase 3 (JNK3) (Li et al., 2002). Furthermore, cdk5 null mice, in addition to showing extensive abnormalities in cortical layering and cerebellar foliation, display degeneration of brainstem and spinal cord neurons (Ohshima et al., 1996).
Together, the above observations highlight two important questions with regards to the role cdk5 plays in neuronal death: (1) is cdk5, like cell cycle CDKs, important in a delayed model of neuronal apoptosis that has a strict dependence on the mitochondrial pathway, and 2) what factors determine whether cdk5 acts as a pro-survival or pro-death signal? To address these questions, we examined p25 formation and the effect of cdk5 inhibition in apoptotic death induced by CA and in excitotoxic death induced by glutamate. We provide evidence that p35 can regulate a prosurvival activity of cdk5 within the cytoplasm, whereas p25 regulates cdk5 pro-death activity within the nucleus. Moreover, although nuclear p25/cdk5 complexes accumulate in both excitotoxic and apoptotic death, they are functionally relevant only in the former, in which they are produced early in the death process. In apoptotic death initiated by CA, p25 complexes are only produced as a late consequence of the mitochondrial death pathway.
Materials and Methods
Primary neuronal cultures. For cortical neuron isolation, brains from embryonic day 15 CD1 or transgenic embryos were dissected as described previously (Xiang et al., 1996; Fortin et al., 2001), and neurons were maintained in Neurobasal media supplemented with B-27, N2, 0.5 mm glutamine, and 0.05 mg/ml penicillin–streptomycin (all from Invitrogen, San Diego, CA). Cortical neurons were plated in Nunc (Naperville, IL) six-well dishes (2.5–3 × 106 cells per well) for Western blot, kinase assay, caspase assay, and reverse transcription (RT)-PCR, and in Costar (Cambridge, MA) four-well dishes (3–5 × 105 cells per well) for immunocytochemistry and survival assay. Cerebellar granule neurons (CGNs) were cultured from CD1 or transgenic mice at postnatal day 7 or 8 as described previously (Cregan et al., 1999). CGNs were maintained in Eagles' MEM (Sigma, St. Louis, MO) containing 10% dialyzed FBS, 25 mm KCl, 0.1 mg/ml gentamycin, 2 mm glutamine, and 25 mm glucose. CGNs were plated in Costar four-well dishes (3–5 × 105 cells per well in 750 μl of media). All dishes were coated with poly-d-lysine (Sigma). AraC (10 μm) was added to CGN cultures that were used for glutamate toxicity (see Figs. 9, 10, 11) but not to cultures used for camptothecin toxicity (see Fig. 12).
Figure 9.

p25 is produced early after glutamate exposure. A, Primary cultures of CGNs were treated with 20 μm glutamate (Glut.) with or without 5 μm MK801 for the indicated times. Total cellular protein was collected and analyzed by Western blot for p35, p25, and cdk5 levels. Densitometry analysis of p35, p25, and cdk5 levels over two experiments is shown below. B, In vitro kinase assay of cdk5 activity from 30 μg of whole-cell lysate reveals a peak in activity at 1.0 h. CB, Coomassie blue. C, Neuronal survival was assessed at the indicated times using the technique described in Materials and Methods. Error bars represent the mean ± SEM for data from three independent experiments. D, CGNs were fixed at the indicated times after glutamate treatment. Neurons were immunostained for p35/p25 as described in Materials and Methods, and nuclei were stained with Hoescht. The percentage of neurons displaying overlap of p35/p25 signal with Hoescht was quantified as described for Figure 6. Error bars represent the mean ± SEM for data from three independent experiments.
Figure 10.

The mitochondrial pathway is not required for p25 production during excitotoxicity. A, CGNs from Bax +/– and Bax –/– littermates were treated with glutamate (Glut.) for 1 and 3 h, and total cellular protein was analyzed for p35 and p25 levels. B, Protein lysate from glutamate-treated CGN cultures was analyzed for DEVD-AFC cleavage activity. Protein from neurons treated with low levels (5 mm) of K+ was included as a positive control for caspase activity. Each error bar represents the mean ± SEM of data from three independent experiments.
Figure 11.

Nuclear inhibition or cytoplasmic facilitation of cdk5 protects against excitotoxic death in CGNs. After 5 d in culture, CGNs were infected with the indicated adenoviral constructs at a multiplicity of infection of 20 and subjected to glutamate 2–3 d later. At 3 h after glutamate treatment, cells were fixed and immunostained for MAP2. Nuclei were stained with Hoescht. The total number of GFP-positive live neurons (MAP2-positive cells) were counted in a circumscribed area. Survival is represented relative to non-glutamate-treated cultures infected with the same construct. Adenoviral infections did not alter survival in nonglutamate-treated cultures. Each error bar represents mean ± SEM from three independent experiments. *p < 0.001, **p < 0.01 by ANOVA. Protection with MK801 verifies that death is NMDA dependent.
Figure 12.

DNA damage-induced death of CGNs is associated with caspase-dependent p25 production and sensitization by DN-NES. A, Western blot analysis of total cellular protein from CGNs treated 2 d after plating with camptothecin (campto) alone or camptothecin plus BAF (100 μm) for the indicated times. Quantification of relative p35 and p25 levels is shown on the right. Density values for each band were normalized to actin and then to the control time point. B, CGNs treated with camptothecin alone or camptothecin plus BAF (100 μm) were assayed for survival at the indicated times according to the protocol outlined in Materials and Methods. C, CGNs were infected at the time of plating with a multiplicity of infection of 20. At 48 h after plating/infection, neurons were treated with 10 μm camptothecin for the indicated times. Survival was assessed after 24 h of camptothecin treatment. GFP-positive neurons were classified as either dead or alive according to the appearance of Hoescht. The ratio of live to total cells counted was used as a measure of survival. Error bars represent the mean ± SEM for data from three independent experiments each done in triplicate. *p < 0.05 by ANOVA. A minimum of 100 cells were counted per well. Infected cultures not treated with camptothecin showed low toxicity (>85% survival for all constructs).
DNA damage and survival assays. All treatments with CA (Sigma) were at 10 μm. Cortical neurons were treated with CA 3.5–4 d after plating, and CGNs were treated 2 d after plating. For survival assays, in Figures 1C, 5, 9C, and 12 B, cell membranes were lysed and healthy nuclei were counted as described previously (Rukenstein et al., 1991; O'Hare et al., 2000). The percentage of surviving neurons is expressed relative to untreated control wells. For survival of adenoviral infected cortical and CGN cultures exposed to CA (see Figs. 8, 12C), neurons were fixed and immunostained for microtubule associated protein 2 (MAP2), followed by nuclear staining with Hoescht. Green fluorescent protein (GFP)-expressing MAP2-positive cells were counted as either alive or dead according to the appearance of Hoescht staining. Live neurons displayed large intact nuclei, whereas dead neurons displayed condensed and/or fragmented nuclei. Survival is expressed as the percentage of total cells that were classified as live.
Figure 1.

p35 decreases early and p25 increases late in response to DNA damage. A, Representative Western blot showing relative p35, p25, and cdk5 levels from cortical neuron whole-cell lysates after treatment with 10 μm camptothecin (campto) or 6 μm ionomycin or from HEK 293 cells transfected with pCMV-p25 (p25) or empty pCMV (C). Vehicle (DMSO) treatment did not alter p35, p25, or cdk5 levels. Probing for β-actin shows relative protein loading between lanes. B, Quantification of Western blots from three separate experiments is shown (error bars represent the mean ± SEM). Density values for each band were first normalized to actin and then to the zero time point. C, Survival assay showing that the majority of neuronal death occurs between 8 and 16 h. D, DEVD-AFC cleavage assay indicates that caspase3-like activity increases within 8 h of camptothecin treatment and peaks at 16 h. Each data point represent the mean ± SEM from three experiments. E, In vitro kinase assay of immunoprecipitated cdk5 from 30 μg of whole-cell lysate using histone H1 as substrate. CB, Coomassie blue.
Figure 5.

p35-deficient cortical neurons are not resistant to DNA damage. Cortical neurons were cultured from p35 null (–/–), heterozygous (+/–), or wild-type (+/+) littermates and treated with camptothecin for 12 h. Neurons from the indicated number of mice with each genotype were cultured and tested individually. Cell membranes were lysed, and intact healthy nuclei were counted according to the protocol outlined in Materials and Methods. Survival was expressed relative to untreated cells for each animal and then grouped according to genotype. Untreated neurons from p35 –/– embryos did not display any difference in survival compared with +/+ or +/– neurons. Each error bar represents the mean ± SEM.
Figure 8.

Nuclear inhibition of Cdk5 does not protect from DNA damage, but cytoplasmic inhibition of Cdk5 sensitizes to DNA damage. Cortical neuron cultures plated 24–36 h before were infected with adenoviral constructs expressing GFP, DN-NLS, or DN-NES at a multiplicity of infection of 75. Two days later, neurons were treated with camptothecin for 14 h. Cultures were immunostained for MAP2, and nuclei were labeled with Hoescht. GFP-expressing MAP2-positive cells were classified as either dead or alive according to the appearance of Hoescht. The ratio of live to total cells counted was used as a measure of survival. Error bars represent the mean ± SEM for data from three independent experiments each done in triplicate. *p < 0.05 by ANOVA. A minimum of 150 cells were counted per well (450 cells per experiment). Infected cultures not treated with camptothecin showed low toxicity (>85% survival for all constructs).
Western blot. Whole-cell protein lysate was collected from six-well plates (containing equal numbers of neurons) in solubilization buffer (0.0625 m Tris, 2.5 mm EDTA, 2.5 mm EGTA, 10% glycerol, 2% SDS, 0.001% bromophenol blue, and 5% β-mercaptoethanol). Equivalent volumes of lysate were separated on 10 or 8% SDS-polyacrylamide gels and transferred to nitrocellulose membranes according to standard procedures. After blocking, membranes were probed with anti-p35 (C-19, sc-820, 1:2000; Santa Cruz Biotechnology, Santa Cruz, CA), anti-cdk5 (C-8, sc-173, 1:2000; Santa Cruz Biotechnology), anti-β-actin (1:3000; Sigma), anti-pRb (554136, 1:500; BD PharMingen, San Diego, CA), anticathepsinB (1910-8004, 1:3000; Biogenesis, Poole, UK), or anticalpastatin (1:1000; kind gift from Edon Melloni, University of Genoa, Genoa, Italy). Before blocking, some membranes were stained with PonceauS to determine equivalent protein loading. Densitometry analysis was performed using NIH ImageJ software (http://rsb.info.nih.gov/ij). For subcellular protein isolation (see Fig. 6 K), cells were collected and washed in 1× PBS. Pelleted cells were resuspended in buffer 1 [0.1× PBS, pH 7.4, 0.4 mm Na2HPO4, 0.15 mm KH2PO4, 13.5 mm NaCl, 0.25 mm KCl, 0.5% Triton X-100, 2 mm MgCl2, and protease inhibitor cocktail from Roche (Indianapolis, IN), which dissolves cell membranes]. Nuclei were pelleted and the supernatant was harvested as the cytosolic fraction. Nuclei were washed three times with buffer 1 before lysis in solubilization buffer (see above).
Figure 6.
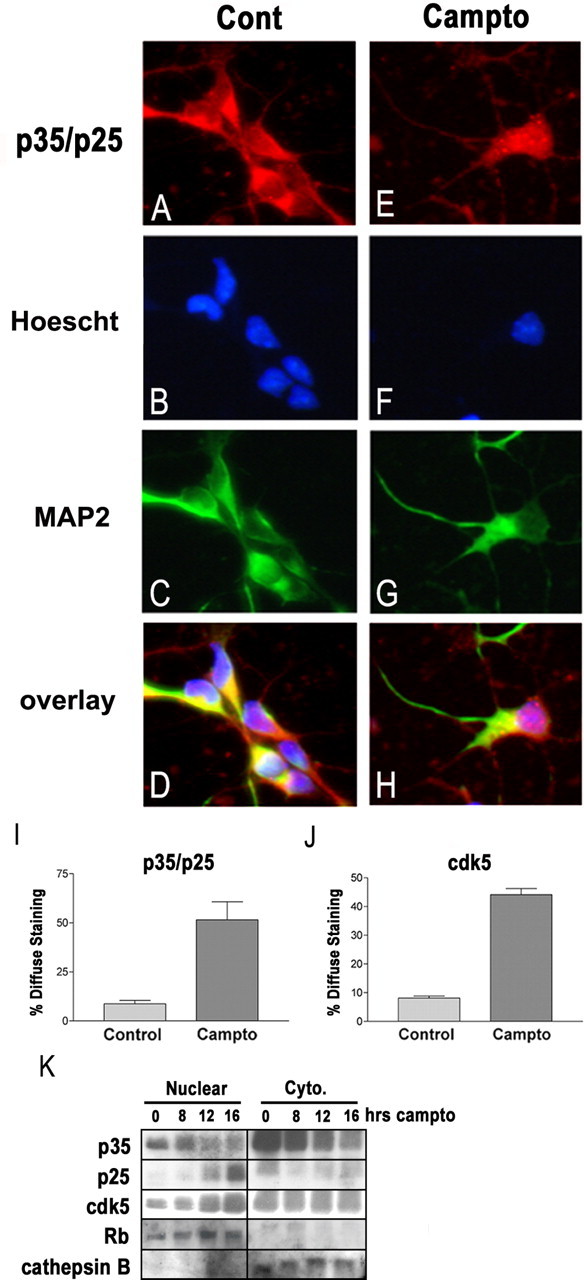
p25 and cdk5 accumulate selectively within the nucleus. A–D, Untreated neurons and neurons treated for 16 h with camptothecin (E–H; Campto) were fixed and immunostained for p35/p25 and MAP2. Nuclei were stained with Hoescht. Under control conditions (Cont), the p35/p25 signal predominates in the cytoplasm. After camptothecin treatment, many neurons display more diffuse p35/p25 staining that overlaps with Hoescht. I, The percentage of p35 C-19-stained neurons showing such a diffuse signal inclusive of the nucleus were quantified under control and camptothecin-treated conditions. J, Similar immunostaining for cdk5 also showed a predominantly cytoplasmic signal under control conditions and an increase in the percentage of neurons showing diffuse staining after camptothecin treatment. Each error bar represents the mean ± SEM of data from three independent experiments. K, Protein from cytoplasmic and nuclear fractions was isolated as described in Materials and Methods. Western blot shows that p25 levels rise selectively within the nuclear fraction. Cdk5 levels also rise only within the nuclear fraction. Purity of nuclear and cytoplasmic fractions was verified using pRb as a marker for nuclear protein and cathepsinB as a marker for cytoplasmic protein.
Kinase assay. Neurons were collected and pelleted (at 4°C) in 1× PBS and then resuspended in immunoprecipitation buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 1 mm EDTA, 2.5 mm EGTA, 1 mm DTT, 0.1% Tween 20, and protease inhibitor cocktail from Roche). Lysates were incubated on ice for 10 min, sonicated twice for 10 s at mid power, and then centrifuged at 10,000 × g for 6 min. Supernatant was collected, and protein concentrations were determined by Bradford assay. Equal amounts of protein were incubated with 1 μg of anti-cdk5 antibody (C-8; Santa Cruz Biotechnology) for 3 h and precipitated with protein A-Sepharose beads (Sigma). Immunoprecipitated complexes were incubated with 2 μg of histone H1 and 5 μCi of [32P]ATP, and, after 20 min at 30°C, the reaction was stopped with the addition of Western blot solubilization buffer. Protein was separated by SDS-PAGE. The gel was stained with Coommassie blue, dried, and then exposed to film.
Immunocytochemistry. At the indicated times, neurons were fixed in 4% paraformaldehyde (containing 0.2% picric acid in 0.1 m phosphate buffer, pH 6.9) for 15 min at room temperature. After washing, neurons were incubated with the primary antibodies anti-p35 (C-19, 1:500; Santa Cruz Biotechnology), anticdk5 (C-8, 1:500; Santa Cruz Biotechnology), or anti-MAP2 (HM2, 1:500; Sigma) overnight at 4°C. Secondary antibody to p35 and cdk5 was AlexaFluor 594 (Molecular Probes, Eugene, OR) and to MAP2 was AlexaFluor 488 (see Fig. 6) or AlexaFluor 594 (see Fig. 7), all for 1 h at room temperature. Nuclei were stained with Hoescht 33258 (0.25 μg/ml) for 20 min at room temperature. Neurons were viewed using either a Zeiss (Oberkochen, Germany) Axiovert 100 or Axioskop2 fluorescence microscopes. Images were captured using a Sony (Tokyo, Japan) 3CCD color digital video camera and Northern Eclipse version 6.0 software. Final images were assembled using Adobe Photoshop version 6.0 (Adobe Systems, San Jose, CA).
Figure 7.
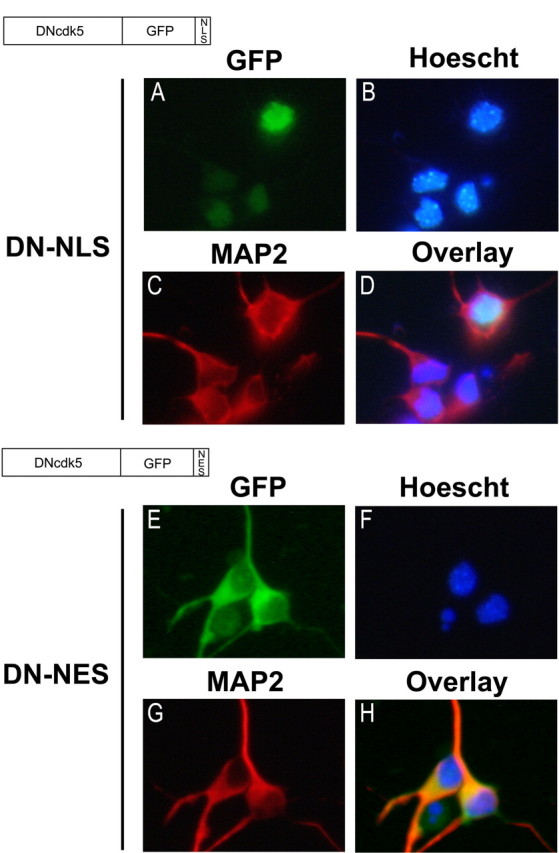
Characterization of DN-NLS and DN-NES adenoviral constructs. Recombinant adenovirus expressing the fusion constructs DNcdk5-GFP-NLS or DNcdk5-GFP-NES were used to infect cortical neurons 24 h after plating at a multiplicity of infection of 75. After 36 h, neurons were fixed and stained for MAP2 and Hoescht. DN-NLS overlaps strongly with Hoescht but not with MAP2 (A–D). DN-NES signal overlaps strongly with MAP2 but not with Hoescht (E–H).
Semiquantitative RT-PCR. Total RNA was isolated using TRIzol reagent (Invitrogen) according to the instructions of the manufacture. A total of 50 ng of RNA was used for RT-PCR using the SuperScript One-Step RT-PCR with Platinum Taq kit (Invitrogen). The following p35-specific primers were used at a final concentration of 500 nm: upper, 5′-CTGCTGCGCTGCCTGGGTGAGTTT-3′; and lower, 5′-CAGGAAGGGCTTGAGCGGGTAGGA-3′. After DNase treatment, cDNA synthesis was performed at 45°C for 45 min. This was followed by 25 cycles of 94°C for 30 s, 67°C for 30 s, and 72°C for 1 min. Preliminary experiments determined that 25 cycles fell within the linear range of amplification. These conditions generate a 297 bp product. Ribosomal S12 RT-PCR was performed as described previously (Cregan et al., 2004).
Caspase activity assay. Total cellular protein was harvested in caspase lysis buffer (1 mm KCl, 10 mm HEPES, pH 7.4, 1.5 mm MgCl2, 1 mm DTT, 1 mm PMSF, 5 μg/ml leupeptin, 2 μg/ml aprotinin, and 10% glycerol) as described previously (O'Hare et al., 2000). A total of 7.5 μgof protein was incubated with 15 μm DEVD-AFC (N-acetyl-Asp-Glu-Val-Asp-AFC) in buffer as described previously (Stefanis et al., 1996). Fluorescence at 505 nm was measured using an SLM 8000 fluorometer (PerkinElmer, Wellesley, MA) using an excitation wavelength of 400 nm and slit length of 10.
Adenoviral vectors. cDNA for dominant-negative (D144N) or wild-type (WT) versions of cdk5 were kind gifts from Dr. Li-Huei Tsai (Harvard University, Boston, MA). The 3′ end of D144N was ligated in-frame to GFP containing either a nuclear localization sequence [NLS from SV40 large T antigen, PKKKRKV (Kalderon et al., 1984; Groulx et al., 2000)] or nuclear exclusion sequence [NES from HIV Rev, LPPLERLTL (Fischer et al., 1995; Lee et al., 1999)] fused to the C terminus. Also, the 3′ end of wild-type cdk5 was ligated to GFP containing an NES. The resultant constructs (cdk5D144N-GFP-NES, cdk5D144N-GFP-NLS, and cdk5WT-GFP-NES, referred to as DN-NES, DN-NLS, and WT-NES, respectively) were inserted into pShuttle-cytomegalovirus (CMV). All constructs were verified by sequencing. pShuttle constructs were processed using the AdEasy system as described previously (Sedarous et al., 2003) to yield final adenoviral products. Adenovirus expressing GFP-calpastatin fusion protein was described previously (Sedarous et al., 2003). Virus titers were assessed by plaque assay. Multiplicity of infection equates to plaque forming units per cell.
Knock-out mice. All animal experimentation was approved by the University of Ottawa Animal Care Committee and conformed to guidelines set by the Canadian Council on Animal Care. Bax null mice were maintained on C57BL/6 background and genotyped as reported previously (Knudson et al., 1995). Apaf-1-deficient mice have been described previously (Cecconi et al., 1998) and were maintained on a C57BL/6 genetic background. Genotyping of Apaf-1 mice was performed as reported previously (Fortin et al., 2001). Generation of p35-deficient animals, their characterization, and genotyping have been reported previously (Hallows et al., 2003).
Glutamate toxicity. After 7–8 d in culture, glutamate was added to CGNs at a final concentration of 20 μm with or without 5 μm MK801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate]. Neurons were incubated at 37°C/5%CO2 for 20 min. Each well was then washed two times with 500 μl of fresh media. After the final wash, 600 μl of 1:1 conditioned media to fresh media was added. Neurons were then returned to 37°C/5%CO2 for the indicated times.
Materials. DEVD-AFC was obtained from Biomol (Plymouth Meeting, PA). DEVD-FMK (z-Asp-Glu-Val-Asp-FMK) and BAF (BOC-Asp-FMK) were obtained from Enzyme Systems Products (Livermore, CA). Carbobenzoxy-valinyl-phenylalaninal [MDL28170 (MDL)] was obtained from Calbiochem (San Diego, CA). Camptothecin was obtained from Sigma.
Results
The significance of cdk5 signaling as regulated by p35 and p25 in different forms of neuronal death is not completely clear. Some reports have indicated that certain death models rely on a p25/cdk5 pathway, whereas others have suggested that cdk5 is instead a protective factor (Cheung and Ip, 2004). We propose that both roles are possible and that the exact function of cdk5 depends on both whether it is regulated by p35 or p25 and the nature of the death stimulus. Although clear evidence has implicated cdk5 signaling in oxidative and excitotoxic death, we first asked whether cdk5 is involved in a classic form of apoptotic death. As a model for such death, we first used embryonic cortical neurons treated with the DNA-damaging agent CA. These neurons undergo apoptotic death dependent on Bax and the mitochondrial pathway of caspase activation (Xiang et al., 1998; Keramaris et al., 2000; Morris et al., 2001).
p25 is produced in response to DNA damage
We began by examining whether p25 is produced in primary cortical neurons after treatment with CA. Immunoblot on total cell lysate using an antibody directed against the C terminus of p35 (p35-C19, which recognizes both p35 and p25) reveals that p35 levels decrease and p25 is formed after DNA damage. Levels of p35 begin to decrease as early as 4 h, whereas p25 is detectable within ∼12 h and reaches maximal levels at 16 h (Fig. 1A,B). It is important to note that the level of basal p25 in untreated control neurons is somewhat variable between cultures. However, even in cultures with detectable basal levels of p25, there is a sharp increase in this protein between 12 and 16 h. To ensure that the p25 band formed after DNA damage is the same as that reported by other groups under different death conditions, we also examined protein from neurons treated with ionomycin, a Ca+2 ionophore that directly activates calpains, as well as protein from HEK293 cells transfected with pCMV-p25. The p25 band seen after DNA damage comigrates with that produced by ionomycin and with p25 exogenously expressed from pCMV-p25 (Fig. 1A). A second antibody directed against the N terminus of p25 also confirmed that DNA damage induces an increase in p25 (data not shown). Interestingly, levels of cdk5 also increase after DNA damage at approximately the same time as p25 (Fig. 1A,B).
To determine the effect of DNA damage on cdk5 activity, we performed in vitro kinase assay of immunoprecipitated cdk5 at various times after CA treatment. Cdk5 activity falls sharply within 4 h (Fig. 1E). This is consistent with and parallels the loss of p35. Cdk5 activity rises again at 16 h, coinciding with the increase in p25 levels. This suggests that the early fall in cdk5 activity is in part attributable to loss of p35, whereas the late gain in activity may be attributable to p25 formation. However, other mechanisms of control may also be important, as suggested by the sharp decrease in cdk5 activity even in the presence of relatively abundant p35 (e.g., at 4 and 8 h).
Importantly, production of p25 after DNA damage occurs much later than signaling events that are known to be critical for the death process in this model. For example, increased cyclinD-associated kinase activity (Park et al., 1998) and upregulation of p53 and c-Jun (Morris et al., 2001; Ghahremani et al., 2002) protein are observed within 4 h of CA treatment. Although a decrease in p35 levels occurs within this time frame, levels of p25 reach reliably detectable levels only at 12 h. At this time point, caspases are activated sevenfold over control and survival has dropped to 50% (Fig. 1C,D). These findings present two important questions with regards to the regulation of p35 and p25 after DNA damage: (1) are p35 levels regulated independent from cleavage to p25, and (2) is p25 production a consequence or cause of late apoptotic processes?
With regards to the first question, previous reports have shown that p35 levels can be regulated through proteosomal degradation (Patrick et al., 1998). However, proteosome inhibitors failed to block the early loss of p35 induced by DNA damage (data not shown). We therefore examined whether p35 was regulated at the transcriptional level. Semiquantitative RT-PCR revealed that p35 mRNA levels decrease significantly between 0 and 4 h of DNA damage induction (Fig. 2), suggesting that down-regulation of p35 transcription accounts for the early reduction in p35 protein, although this evidence is indirect. This result not only suggests that p35 protein levels are regulated independent from cleavage to p25 but also raises the question of whether early loss of p35 is relevant to death signaling. This possibility is more closely examined later.
Figure 2.

p35 mRNA decreases early in response to DNA damage. A, RNA was extracted from cortical neurons at the indicated times after camptothecin (campto) treatment and analyzed for p35 mRNA expression using semiquantitative RT-PCR. S12 levels are shown as a control for equal input. B, p35 and S12 bands from three independent experiments were analyzed by densitometry using NIH Image version 1.61. Data for p35 expression were normalized to S12 and expressed relative to the zero time point. Each point represents the mean ± SEM.
p25 formation is regulated downstream of caspase activation
To address the question of whether p25 production after DNA damage is a cause or consequence of late apoptotic events, we examined whether caspase activity was required for production of p25. Caspase activation in this model requires both Bax-mediated cytochrome c release from the mitochondria and formation of the apoptosome complex consisting of cytochrome c, Apaf-1, and pro-caspase 9 (Xiang et al., 1998; Keramaris et al., 2000; Fortin et al., 2001). Cotreatment of CA with either of the caspase inhibitors BAF or DEVD-FMK inhibited the production of p25 (Fig. 3A,B). Furthermore, neurons cultured from mice deficient for either Bax or Apaf-1 showed no production of p25 (Fig. 3C,D). These results show that p35 cleavage to p25 can be caspase dependent. Interestingly, Bax-deficient neurons displayed higher levels of p35 compared with heterozygous neurons at 16 h (Fig. 3C). This relative increase in p35 level correlates with robust long-lasting protection seen in Bax-deficient neurons (Morris et al., 2001), which is in contrast to the more transient protection seen in Apaf-1-deficient neurons and neurons treated with caspase inhibitors (Cregan et al., 2002).
Figure 3.

p25 formation requires activation of the mitochondrial death pathway. Left panels display representative Western blots, and center and right panels display quantitative analysis of p35 and p25, respectively, from three or more separate experiments (error bars represent the mean ± SEM). Density values for each band were first normalized to loading control and then to the zero time point. A, B, Cortical neurons were treated with camptothecin (campto) alone or with camptothecin plus 100 μm BAF (A) or campto plus 50 μm DEVD-FMK (B) for 8 and 16 h. Total cellular protein was subjected to Western blot and analyzed for p35 and p25 levels. Caspase inhibition with BAF or DEVD-FMK prevents production of p25 after DNA damage. C, D, Neurons from individual Bax (C) or Apaf-1 (D) -deficient embryos and from appropriate littermate controls were treated with camptothecin, and levels of p35 and p25 were analyzed by Western blot.
The dependency of p25 formation on caspases is not likely to be direct for two reasons: (1) p35 has no known caspase cleavage site within the region cleaved in the production of p25, and (2) purified p35 cannot be cleaved to p25 by caspase-3, the predominant caspase in neurons (Lee et al., 2000). Therefore, calpains are still the most likely candidate responsible for p35 cleavage to p25 after DNA damage. To test whether calpains are required for late production of p25, we cotreated neurons with CA and the calpain inhibitor MDL. We found that calpain inhibition with MDL impeded the production of p25 (Fig. 4A). Similar results were found using additional inhibitors calpeptin and PD150606 (data not shown).
Figure 4.

p25 formation requires calpain and caspase-mediated downregulation of calpastatin. A, Cortical neurons were treated with camptothecin (campto) alone or camptothecin plus the calpain inhibitor MDL28170 (75 μm) for 8 and 16 h. p35 and p25 levels were analyzed by Western blot. Quantification of relative p35 (B) and p25 (C) levels from three independent trials (each error bar represents the mean ± SEM). Density values for each band were first normalized to loading control and then to the control time point. D, At 36 h after plating, cortical neurons were infected with either GFP-expressing or GFP-calpastatin-expressing adenovirus using a multiplicity of infection of 75. At 48 h later, neurons were treated with camptothecin for 12 h and then fixed and immunostained using p35 C-19 antibody. Nuclei were stained with Hoescht, and overlap of p35 C-19 staining and Hoescht was quantified. Each error bar represents the mean ± SEM of data from three independent experiments. Significantly less GFP-calpastatin-expressing neurons treated with camptothecin show overlap of p35 C-19 and Hoescht compared with camptothecin-treated neurons expressing GFP alone (ANOVA, p < 0.001; n = 3). E, F, Cortical neurons were treated with camptothecin alone or camptothecin plus 100 μm BAF (E) or camptothecin plus 50 μm DEVD-FMK (F) for 8 and 16 h. Total cellular protein was analyzed for calpastatin levels (∼85 kDa). G, Quantification of calpastatin levels at 16 h from three independent experiments (mean ± SEM). Density values for each band were first normalized to loading control and then to the control time point.
As we will show later, when p25 is produced in response to DNA damage, it accumulates selectively within the nucleus (see Fig. 6K). Associated with this accumulation is a switch in the profile of p35/p25 immunostaining from predominantly cytoplasmic to a more diffuse staining in both the cytoplasm and nucleus (see Fig. 6A–I). We tested whether calpastatin, the endogenous inhibitor of calpains, could prevent this switch in localization of p35/p25 immunosignal. Cortical cultures infected with adenovirus expressing GFP-calpastatin showed significantly less neurons with diffuse p35/p25 staining after CA treatment compared with cultures expressing GFP alone (Fig. 4D). This ability of calpastatin to prevent the change in p35/p25 staining suggests that cleavage of p35 to p25 is mediated by calpains.
By what mechanism could both caspases and calpains be responsible for p35 cleavage to p25? Previous reports have shown that caspases can regulate calpain activity by cleaving and inactivating calpastatin (Porn-Ares et al., 1998; Wang et al., 1998; Kato et al., 2000). Similarly, we found that CA treatment induces a reduction in calpastatin protein levels and that this reduction is prevented by the caspase inhibitors BAF and DEVD-FMK (Fig. 4E–G). Together, these observations are consistent with a model in which p35 cleavage by calpains occurs as a consequence of caspase degradation of calpastatin.
Addressing the functional importance of cdk5 in DNA damage
We have shown that the accumulation of p25 in neurons after DNA damage occurs downstream of caspase activation. Although this demonstrates that p35 cleavage to p25 is not a proximal event in this apoptotic model, it is still possible that the formation of p25 is essential for completion of the apoptotic program. As we have also shown, however, transcriptional-mediated downregulation of p35 is a much earlier event, and this may represent a functionally important loss of cdk5 survival signaling.
To determine whether either of these possibilities is correct, we treated neurons from p35 null mice (–/–), along with wild-type (+/+) and heterozygous (+/–) littermate controls, with CA and examined either for protection or sensitization. Untreated neurons from p35 –/– animals appeared similar to +/– or +/+ neurons and did not display any difference in basal survival (data not shown). After CA treatment, p35 –/– neurons did not show any difference in survival compared with +/+ or +/– littermates (Fig. 5).
The above result suggests that p25 is not required for neuronal death induced by DNA damage and that p35 is not involved in pro-survival cdk5 signaling in this model. However, it is also possible that the homologous cdk5 activator p39 could be compensating in the absence of p35 (Tang et al., 1995). For this reason, we sought to inhibit cdk5 activity more directly through expression of a dominant-negative mutant of cdk5 (DN). However, this approach is also problematic because, if cdk5 regulates both pro-survival as well as pathogenic functions depending on whether it is regulated by p35 or p25, global inhibition would not allow the discrete effects of either to be observed. To avoid this problem, we sought to inhibit pathogenic p25/cdk5 activity independent of p35/cdk5.
Some reports have shown that the localization of p25 differs markedly from that of p35, likely attributable to the loss of a myristoylation sequence at the N terminus of p35 (Patrick et al., 1999; Kusakawa et al., 2000). This difference in localization could afford an opportunity to specifically inhibit p25/cdk5 through targeted expression of DN. Consequently, we investigated the subcellular localization of p35/p25, as well as cdk5, before and after DNA damage. Immunohistochemical staining using the p35 C-19 antibody, which does not distinguish between p35 and p25, revealed a change in signal localization after DNA damage (Fig. 6). Before CA treatment, when p35 is much more prevalent than p25, neuronal cell bodies show strong p35/p25 staining predominantly within the cytoplasm and relative absence of nuclear staining (Fig. 6A–D). After 16 h of CA treatment, when p25 levels are maximal, many neurons begin to show p35/p25 staining that is more diffuse throughout both the cytoplasm and nucleus (Fig. 6E–H). Only 10% of control neurons show this diffuse p35/p25 staining, whereas 51% of neurons treated with CA for 16 h show such staining (Fig. 6I). Immunostaining for cdk5 was remarkably similar to that seen for p35. Quantification of cdk5 immunostaining revealed that only 9% of control neurons, but 44% of CA-treated neurons, displayed diffuse staining (Fig. 6J and data not shown). Together with our observation that total cellular p25 and cdk5 reach maximal levels 16 h after CA treatment (Fig. 1), these results suggest the possibility that p25/cdk5 complexes accumulate within the nucleus.
Because immunostaining for p35/p25 does not distinguish between p35 and p25, we performed immunoblotting on subcellular protein fractions to more directly determine whether p25 is selectively accumulating in the nucleus. Our previous whole-cell Western blots had shown that, under control conditions, p35 levels are much higher relative to p25 and that, 16 h after CA treatment, p25 levels reach maximum. Subcellular fractionation reveals that the majority of p35 is cytoplasmic and that p25, once it accumulates, is present predominantly within the nucleus (Fig. 6K). Moreover, we found that cdk5 levels increase only within the nuclear compartment (Fig. 6K). Combined with the immunochemistry data above, these results reveal a dramatic down-regulation of p35/cdk5 in the cytoplasm and an increased formation of p25/cdk5 in the nucleus.
This unique localization of p35 and p25 allowed us to devise a strategy to selectively inhibit cdk5 activity regulated by each. We constructed adenoviral vectors expressing DN-cdk5 C-terminal fused to GFP carrying either a nuclear localization sequence (DN-NLS) or a nuclear exclusion sequence (DN-NES) (Fig. 7). Expression of DN-NLS was localized to the nucleus as judged by overlap with Hoescht and expression of DN-NES was localized to the cytoplasm as judged by nonoverlap with Hoescht and overlap with MAP2 (Fig. 7A–D,E–H). In vitro kinase assays of cdk5 immunoprecipitated from neurons infected with these constructs confirmed that they effectively suppress cdk5 activity (data not shown).
Cortical neurons were infected 24 h after plating with DN-NES, DN-NLS, or a control GFP expressing adenovirus and treated with CA 48 h later. After CA treatment, neurons were fixed and nuclei were stained with Hoescht. GFP-positive neurons were assayed for survival according to the appearance of Hoescht staining. Importantly, expression of DN-NLS did not provide any significant protection against DNA damage (Fig. 8). Together with the observations that nuclear p25 formation occurs late after DNA damage and that p35 null neurons are not protected, this suggests that the nuclear production of p25/cdk5 is not required for neuronal death induced by DNA damage.
Interestingly, expression of DN-NES caused a significant sensitization to DNA damage (Fig. 8). Sensitization attributable to cytoplasmic inhibition of cdk5 would be expected if p35/cdk5 were performing a pro-survival role within the cytoplasm. Our observations that p35 mRNA and protein levels decrease early after CA treatment are also consistent with a pro-survival role for cytoplasmic p35/cdk5. Together, these data suggest that, in this apoptotic model of death, p25/cdk5 complexes increase and are present in the nucleus. However, this increase occurs too late to be of functional significance. In contrast, early loss of p35/cdk5 complexes in the cytoplasm may lead to sensitization.
Nuclear and cytoplasmic roles for cdk5 in excitotoxicity
The relative contributions of p35-regulated versus p25-regulated cdk5 activity have not been addressed in nonapoptotic models of neuronal death in which cdk5 has been implicated, such as excitotoxic death. For this reason, we examined the involvement of nuclear p25/cdk5 complexes and cytoplasmic p35/cdk5 complexes in excitotoxic death of CGNs exposed to glutamate. We found that CGNs treated with 20 μm glutamate rapidly cleave p35 to p25 (Fig. 9A). Levels of p25 increase within 30 min and reach maximal within 2 h. Levels of cdk5 also increase within 30 min of glutamate treatment (Fig. 9A). In vitro kinase assay of cdk5 activity shows an early increase in activity 1 h after exposure (Fig. 9B). This increase is concurrent with the early rise in cdk5 and appearance of p25. However, the return of cdk5 activity to basal levels at 2 and 3 h, even in the presence of increased p25 and cdk5, indicates that mechanisms in addition to protein levels contribute to cdk5 activity.
In contrast to what is seen after DNA damage-induced death, cell survival after glutamate treatment begins to decline only after the appearance of p25. Reduced cell survival is detectable within 1 h of glutamate exposure, and death proceeds rapidly between 1 and 3 h (Fig. 9C). Potent protection with MK801 indicates that death occurs in an NMDA-dependant manner (Fig. 9C). Cotreatment with BAF did not inhibit death, suggesting that caspases are not involved in this death model (Fig. 9C). Although production of p25 after glutamate has been reported previously (Lee et al., 2000), the localization of this endogenously produced p25 has not been addressed. We found that >70% of CGNs treated with glutamate for 30 min display diffuse p35/p25 immunoreactivity in both cytoplasm and nucleus (Fig. 9D) compared with only ∼25% of nontreated control cells. This finding is consistent with nuclear accumulation of p25.
We next tested whether p25 production during excitotoxicity was dependent on the mitochondrial death pathway, as it is after DNA damage. We found that p25 production in response to glutamate is not dependent on the presence of Bax (Fig. 10A) and occurs in the absence of any increase in caspase activity (Fig. 10B). Together with the data presented in Figure 3, these results imply distinct differences in the mechanisms controlling p35 cleavage to p25 in rapid excitotoxic death versus delayed apoptotic death.
To determine whether the rapid caspase-independent increase in p25 is functionally required for glutamate-induced death, we tested the ability of DN-NLS to protect. Indeed, expression of DN-NLS is significantly protective after glutamate treatment (Fig. 11). This again contrasts with the DNA damage model, in which the identical construct was ineffective in providing protection. These results are the first to suggest that inhibition of cdk5 exclusively within the nucleus is sufficient to prevent neuronal death.
Interestingly, although DN-NES expression did not sensitize to excitotoxicity as it did to DNA damage, we did find that wild-type cdk5 expression selectively in the cytoplasm (WT-NES) provided protection (Fig. 11). Much like our finding that DN-NES sensitizes cortical neurons to DNA damage, this result again supports the idea that cdk5 performs a pro-survival role within the cytoplasm. However, because DN-NES did not sensitize to glutamate treatment within the time frame examined, it is unlikely that endogenous cytoplasmic cdk5 activity plays a functionally important role during glutamate-induced death. It is also possible that, during glutamate-induced death, strong pro-death signals initiated by nuclear p25/cdk5 (and other death signals) overwhelm pro-survival signaling by cytoplasmic p35/cdk5. Importantly, the localization of DN-NLS and DN-NES in CGNs was similar to that observed in cortical neurons, and the WT-NES construct also showed selective cytoplasmic localization (data not shown).
We next asked whether the differences we have observed in the contribution of cdk5 to apoptotic and excitotoxic death could be attributable simply to differences in the respective culture systems used. To address this question, we subjected CGNs to DNA damage induced by CA. Similar to cortical neurons treated with CA, CGNs also upregulate p25 late in the death process (Fig. 12A). The kinetics of CA-induced death in CGNs is slightly delayed compared with that in cortical neurons (Fig. 12B). However, the rise in p25 levels is also delayed, with the peak coming at 24 h. At this time, >50% of neurons have died (Fig. 12B). Caspase inhibition by BAF significantly delays CA-induced CGN death and also suppresses the production of p25 (Fig. 12A,B). The transient inhibition of death seen with BAF is similar to that described in cortical neurons (Cregan et al., 2002). Most importantly, the effect of expressing DN-NLS or DN-NES in CGNs exposed to DNA damage was analogous to that seen in cortical neurons. At 24 h after CA treatment, DN-NLS expression did not change survival with respect to GFP expression control, whereas DN-NES expression resulted in a small but significant reduction in survival (Fig. 12C). These results indicate that, even within the same cell type (CGNs), cdk5 plays very distinct roles in delayed apoptotic death and rapid excitotoxic death. Furthermore, these results also suggest that the differences observed between excitotoxic and apoptotic death are not an artifact of the different culture systems used.
In summary, we propose that, in apoptotic death induced by DNA damage, downregulation of p35/cdk5 activity in the cytoplasm plays a modulatory role in survival, whereas caspase-dependent nuclear p25 formation occurs temporally late and does not play a role. In contrast, in a glutamate model of death, rapid accumulation of nuclear p25 is sufficient and required to induce death, whereas driving cytoplasmic cdk5 can be protective.
Discussion
cdk5 in DNA damage signaling induced by CA
We had initially identified two questions of interest in regards to the role of cdk5 in neuronal death. The first was whether cdk5 was functionally important in a model of neuronal death that is dependent on the mitochondrial pathway. We raised this question because a number of previous reports have suggested that cdk5 is a common regulator of neuronal death in multiple types of neurodegenerative conditions, yet few reports have examined the role of cdk5 in core apoptotic signaling. We examined this question using a model in which neuronal death occurs as a result of DNA damage.
A number of signaling pathways have been found to be required for neuronal death induced by DNA damage. For example, neurons lacking p53, a major response protein to DNA damage in mammalian cells, are robustly protected against a variety of DNA damage inducers, including CA (Xiang et al., 1998; Morris et al., 2001). In response to DNA damage in neurons, p53 acts as the main upstream activator of the mitochondrial death pathway (Cregan et al., 1999; Fortin et al., 2001; Morris et al., 2001). Activation of cell cycle CDKs and phosphorylation of pRb are also known to be critical for induction of the mitochondrial pathway and death in this model (Park et al., 1997a, 2000; Morris et al., 2001). The mitochondrial pathway involves cytochrome c release from the mitochondria induced by BH3-only members of the Bcl-2 family such as Bax, followed by formation of the apoptosome complex (Apaf-1, cytochrome c, dATP, and procaspase 9) and activation of caspases (Green and Kroemer, 2004). We provide several lines of evidence that p25 production after DNA damage occurs downstream of this pathway. First, levels of p25 accumulate relatively late in the death process, only after caspase activity has begun to increase. Second, caspase inhibitors prevent p25 production. Third, Bax- or Apaf-1-deficient neurons do not display any increase in p25. Together, these observations indicate that p35 cleavage to p25 can occur as a late, caspase-dependent apoptotic event. This finding is consistent with results from others showing that staurosporine-induced apoptotic death of cortical neurons is associated with a late increase in p25 production (Kusakawa et al., 2000). Combined with our observations that neither p35 deficiency nor selective inhibition of p25/cdk5 activity (using DN-NLS) afforded protection against DNA damage, we conclude that the late appearance of p25 is a result of the death process rather than a required component. This result indicates that the presence of p25 in neurodegenerative or neurotoxic conditions, particularly those that involve delayed apoptotic signaling, does not necessarily point toward a direct requirement for cdk5 in the death process.
Despite the late caspase-dependent appearance of p25, we do not believe that caspases directly mediate p35 cleavage. Calpains are the only proteases shown to be capable of cleaving p35 in the region necessary to produce p25 (Kusakawa et al., 2000; Lee et al., 2000). However, multiple reports have shown that caspases can regulate calpain activity by cleaving and inactivating calpastatin, an endogenous calpain inhibitory protein (Porn-Ares et al., 1998; Wang et al., 1998; Kato et al., 2000). Our results show that, in response to DNA damage, calpastatin levels decrease in a caspase-dependent manner, and calpain inhibition (either with MDL or with overexpression of calpastatin) prevents both p25 production and appearance of nuclear p35/p25 immunosignal. These results are consistent with a model in which calpain-mediated cleavage of p35 to p25 occurs as a result of caspase degradation of calpastatin.
Our previous results had shown that calpains were activated early after DNA damage and that this activation was required for the upregulation of p53 (Sedarous et al., 2003). An important question then is why do we not also observe an early cleavage of p35 to p25? Calpains are a large family of distinct but homologous proteases, some with tissue-specific expression, some with ubiquitous expression, and each with a unique Ca2+ concentration requirement (Goll et al., 2003). Some reports have also identified nuclear-specific calpains and/or nuclear transport of select calpains (Mellgren et al., 1993; Mellgren and Lu, 1994; Ma et al., 2001; Gil-Parrado et al., 2003). This variety and specificity in localization may explain why calpain regulation of p53, a predominantly nuclear protein, is distinct from that of p35, a cytoplasmic/cell membrane protein. Additional studies are required to determine whether these two proteins are associated with distinct pools of calpain that are activated at different stages after DNA damage.
p35/cdk5 promotes survival within the cytoplasm, whereas p25/cdk5 promotes death within the nucleus
Our second objective was to resolve contradictory reports in the literature that suggest that cdk5 can act as both a pro-survival and a pro-death signal. Most findings have agreed that cdk5 prodeath signaling is regulated by p25 (Patrick et al., 1999; Kusakawa et al., 2000; Lee et al., 2000; Cheung and Ip, 2004). By default, therefore, pro-survival signaling seems likely to be regulated by p35. However, the relative contributions of p25/cdk5 and p35/cdk5 to death/survival in single models of neuronal death have not been addressed. One reason for this has been the inability to study cdk5 function regulated by p35 independent from that regulated by p25. Traditional means of cdk5 inhibition (pharmacological, molecular, and genetic) do not distinguish between p35/cdk5 and p25/cdk5. Our finding that p35 is localized predominantly to the cytoplasm whereas p25 is found mainly within the nucleus allowed us to target inhibition of either p35/cdk5 or p25/cdk5 by localizing a dominant-negative mutant of cdk5 to the cytoplasm or nucleus, respectively. Using this approach in both DNA damage and glutamate excitotoxicity, we find evidence supporting a model in which pro-survival signaling by cdk5 occurs mainly within the cytoplasm, whereas pro-death cdk5 signaling is contained to the nucleus.
Our evidence that p35 regulates a pro-survival cdk5 function within the cytoplasm is threefold. First, neurons actively down-regulate p35 mRNA and protein in the early phase after DNA damage. This downregulation may represent a damping of prosurvival cdk5 signaling. Consistent with this is our observation that cytoplasmic cdk5 inhibition sensitizes neurons to DNA damage-induced death. Third, cytoplasmic expression of wild-type cdk5 provides protection against glutamate toxicity. Furthermore, we have observed that CGNs undergoing death induced by low levels of K+ also downregulate p35 protein, without production of p25 (data not shown). These findings are consistent with the reported findings that p35 protein decreases without p25 production under a variety of neurotoxic conditions, including treatment with cyclosporine A, etoposide, okadaic acid, and 4-hydroxynonenal (Kerokoski et al., 2001).
What is the nature of this potential pro-survival cytoplasmic signal activated by p35/cdk5? Previous reports have shown that cdk5 can phosphorylate and inactivate the JNK3 and that this property of cdk5 can inhibit neuronal death induced by UV irradiation (Li et al., 2002). Also, our previous work has identified JNK phosphorylation of c-Jun as a required event in neuronal death induced by CA (Ghahremani et al., 2002). Together, these results suggest the possibility that p35/cdk5 promotes survival in neurons exposed to CA by negatively regulating the JNK/c-Jun pathway. Additional work will be needed to test this possibility.
We also shown that, in the context of glutamate excitotoxicity, p25 promotes a pro-death cdk5 activity selectively within the nucleus. In support of this, we show first that p25 is formed rapidly and localizes almost exclusively to the nuclear compartment, and second that inhibition of cdk5 only within the nucleus is sufficient to inhibit glutamate-induced death. Although we have not identified here the relevant nuclear cdk5 target(s), work from others has shown that, in response to glutamate and oxidative stress, cdk5 phosphorylation of the myocyte enhancer factor 2 (MEF2) transcription factor is essential for death (Gong et al., 2003). This phosphorylation of MEF2 results in inactivation of its pro-survival activity.
In conclusion, our evidence points to a model by which the functional relevance of cdk5 to different neuronal death models depends on at least two critical factors, localization and timing. Localization, which is controlled by binding to either p35 or p25, determines whether cdk5 suppresses or activates cell death mechanisms. Timing, in the production of p25, determines the importance of cdk5 in death. Late caspase-dependent production of p25 can occur as a consequence of apoptotic death, but early noncaspase-dependent p25 production plays a strong role in promoting death.
Footnotes
This work was supported by funds from the Canadian Institutes of Health Research (CIHR), the Center for Stroke Recovery, the Canadian Stroke Network, the Heart and Stroke Foundation of Ontario, and the Parkinson's Research Consortium (D.S.P). M.J.O. and N.K. were supported by CIHR Doctoral Research Awards. We thank Dr. Stephen Lee for sharing GFP-NLS/NES-containing constructs, Dr. Li-Huei Tsai for sharing cdk5 wild-type and mutant constructs, Dr. Edon Melloni for sharing the anti-calpastatin antibody, and Andre Fortin and Nicole Arbour for kind assistance with animal breeding.
Correspondence should be addressed to Dr. David S. Park, Ottawa Health Research Institute, Neuroscience Group, University of Ottawa, 451 Smyth Road, Ottawa, Ontario, Canada K1H 8M5. E-mail: dpark@uottawa.ca.
DOI:10.1523/JNEUROSCI.2899-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258954-13$15.00/0
References
- Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B (1997) Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem 69: 1196–1203. [DOI] [PubMed] [Google Scholar]
- Alvarez A, Toro R, Caceres A, Maccioni RB (1999) Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett 459: 421–426. [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P (1998) Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 94: 727–737. [DOI] [PubMed] [Google Scholar]
- Chen J, Jin K, Chen M, Pei W, Kawaguchi K, Greenberg DA, Simon RP (1997) Early detection of DNA strand breaks in the brain after transient focal ischemia: implications for the role of DNA damage in apoptosis and neuronal cell death. J Neurochem 69: 232–245. [DOI] [PubMed] [Google Scholar]
- Cheung ZH, Ip NY (2004) Cdk5: mediator of neuronal death and survival. Neurosci Lett 361: 47–51. [DOI] [PubMed] [Google Scholar]
- Copani A, Condorelli F, Caruso A, Vancheri C, Sala A, Giuffrida Stella AM, Canonico PL, Nicoletti F, Sortino MA (1999) Mitotic signaling by betaamyloid causes neuronal death. FASEB J 13: 2225–2234. [PubMed] [Google Scholar]
- Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS (1999) Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci 19: 7860–7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS (2002) Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol 158: 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Arbour NA, Maclaurin JG, Callaghan SM, Fortin A, Cheung EC, Guberman DS, Park DS, Slack RS (2004) p53 activation domain 1 is essential for PUMA upregulation and p53-mediated neuronal cell death. J Neurosci 24: 10003–10012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Holmes EH, Greene TG, Liu PK (2000) Oxidative DNA damage precedes DNA fragmentation after experimental stroke in rat brain. FASEB J 14: 955–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, Koike R, Hiroi T, Yuasa T, Awaya Y, Sakai T, Takahashi T, Nagatomo H, Sekijima Y, Kawachi I, Takiyama Y, Nishizawa M, Fukuhara N, Saito K, Sugano S, Tsuji S (2001) Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet 29: 184–188. [DOI] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH (2001) A decade of CDK5. Nat Rev Mol Cell Biol 2: 749–759. [DOI] [PubMed] [Google Scholar]
- Dyson N (1998) The regulation of E2F by pRB-family proteins. Genes Dev 12: 2245–2262. [DOI] [PubMed] [Google Scholar]
- Ekholm SV, Reed SI (2000) Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr Opin Cell Biol 12: 676–684. [DOI] [PubMed] [Google Scholar]
- Fischer U, Huber J, Boelens WC, Mattaj IW, Luhrmann R (1995) The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell 82: 475–483. [DOI] [PubMed] [Google Scholar]
- Fortin A, Cregan SP, MacLaurin JG, Kushwaha N, Hickman ES, Thompson CS, Hakim A, Albert PR, Cecconi F, Helin K, Park DS, Slack RS (2001) APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J Cell Biol 155: 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbita SP, Lovell MA, Markesbery WR (1998) Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem 71: 2034–2040. [DOI] [PubMed] [Google Scholar]
- Ghahremani MH, Keramaris E, Shree T, Xia Z, Davis RJ, Flavell R, Slack RS, Park DS (2002) Interaction of the c-Jun/JNK pathway and cyclin-dependent kinases in death of embryonic cortical neurons evoked by DNA damage. J Biol Chem 277: 35586–35596. [DOI] [PubMed] [Google Scholar]
- Gil-Parrado S, Popp O, Knoch TA, Zahler S, Bestvater F, Felgentrager M, Holloschi A, Fernandez-Montalvan A, Auerswald EA, Fritz H, Fuentes-Prior P, Machleidt W, Spiess E (2003) Subcellular localization and in vivo subunit interactions of ubiquitous mu-calpain. J Biol Chem 278: 16336–16346. [DOI] [PubMed] [Google Scholar]
- Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS (1999) Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F × DP, in B-amyloid-induced neuronal death. J Biol Chem 274: 19011–19016. [DOI] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The calpain system. Physiol Rev 83: 731–801. [DOI] [PubMed] [Google Scholar]
- Gong X, Tang X, Wiedmann M, Wang X, Peng J, Zheng D, Blair LA, Marshall J, Mao Z (2003) Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron 38: 33–46. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305: 626–629. [DOI] [PubMed] [Google Scholar]
- Groulx I, Bonicalzi ME, Lee S (2000) Ran-mediated nuclear export of the von Hippel-Lindau tumor suppressor protein occurs independently of its assembly with cullin-2. J Biol Chem 275: 8991–9000. [DOI] [PubMed] [Google Scholar]
- Hallows JL, Chen K, DePinho RA, Vincent I (2003) Decreased cyclin-dependent kinase 5 (cdk5) activity is accompanied by redistribution of cdk5 and cytoskeletal proteins and increased cytoskeletal protein phosphorylation in p35 null mice. J Neurosci 23: 10633–10644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P (1998) Oxidative mechanisms in nigral cell death in Parkinson's disease. Mov Disord 13 [Suppl 1]: 24–34. [PubMed] [Google Scholar]
- Johnson DG, Walker CL (1999) Cyclins and cell cycle checkpoints. Annu Rev Pharmacol Toxicol 39: 295–312. [DOI] [PubMed] [Google Scholar]
- Kalderon D, Roberts BL, Richardson WD, Smith AE (1984) A short amino acid sequence able to specify nuclear location. Cell 39: 499–509. [DOI] [PubMed] [Google Scholar]
- Kato M, Nonaka T, Maki M, Kikuchi H, Imajoh-Ohmi S (2000) Caspases cleave the amino-terminal calpain inhibitory unit of calpastatin during apoptosis in human Jurkat T cells. J Biochem (Tokyo) 127: 297–305. [DOI] [PubMed] [Google Scholar]
- Keramaris E, Stefanis L, MacLaurin J, Harada N, Takaku K, Ishikawa T, Taketo MM, Robertson GS, Nicholson DW, Slack RS, Park DS (2000) Involvement of caspase 3 in apoptotic death of cortical neurons evoked by DNA damage. Mol Cell Neurosci 15: 368–379. [DOI] [PubMed] [Google Scholar]
- Kerokoski P, Suuronen T, Salminen A, Soininen H, Pirttila T (2001) The levels of cdk5 and p35 proteins and tau phosphorylation are reduced during neuronal apoptosis. Biochem Biophys Res Commun 280: 998–1002. [DOI] [PubMed] [Google Scholar]
- Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ (1995) Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 270: 96–99. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A (2002) Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell 9: 1005–1016. [DOI] [PubMed] [Google Scholar]
- Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S (2000) Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem 275: 17166–17172. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405: 360–364. [DOI] [PubMed] [Google Scholar]
- Lee S, Neumann M, Stearman R, Stauber R, Pause A, Pavlakis GN, Klausner RD (1999) Transcription-dependent nuclear-cytoplasmic trafficking is required for the function of the von Hippel-Lindau tumor suppressor protein. Mol Cell Biol 19: 1486–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BS, Zhang L, Takahashi S, Ma W, Jaffe H, Kulkarni AB, Pant HC (2002) Cyclin-dependent kinase 5 prevents neuronal apoptosis by negative regulation of c-Jun N-terminal kinase 3. EMBO J 21: 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BS, Ma W, Jaffe H, Zheng Y, Takahashi S, Zhang L, Kulkarni AB, Pant HC (2003) Cyclin-dependent kinase-5 is involved in neuregulin-dependent activation of phosphatidylinositol 3-kinase and Akt activity mediating neuronal survival. J Biol Chem 278: 35702–35709. [DOI] [PubMed] [Google Scholar]
- Liu LF, Desai SD, Li TK, Mao Y, Sun M, Sim SP (2000) Mechanism of action of camptothecin. Ann NY Acad Sci 922: 1–10. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Markesbery WR (2001) Ratio of 8-hydroxyguanine in intact DNA to free 8-hydroxyguanine is increased in Alzheimer disease ventricular cerebrospinal fluid. Arch Neurol 58: 392–396. [DOI] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429: 883–891. [DOI] [PubMed] [Google Scholar]
- Ma H, Fukiage C, Kim YH, Duncan MK, Reed NA, Shih M, Azuma M, Shearer TR (2001) Characterization and expression of calpain 10. A novel ubiquitous calpain with nuclear localization. J Biol Chem 276: 28525–28531. [DOI] [PubMed] [Google Scholar]
- Massague J (2004) G1 cell-cycle control and cancer. Nature 432: 298–306. [DOI] [PubMed] [Google Scholar]
- Mellgren RL, Lu Q (1994) Selective nuclear transport of mu-calpain. Biochem Biophys Res Commun 204: 544–550. [DOI] [PubMed] [Google Scholar]
- Mellgren RL, Song K, Mericle MT (1993) m-Calpain requires DNA for activity on nuclear proteins at low calcium concentrations. J Biol Chem 268: 653–657. [PubMed] [Google Scholar]
- Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, Mendonca P, Costa M, Barros J, Yanagisawa T, Watanabe M, Ikeda Y, Aoki M, Nagata T, Coutinho P, Sequeiros J, Koenig M (2001) The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet 29: 189–193. [DOI] [PubMed] [Google Scholar]
- Morris EJ, Geller HM (1996) Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell cycle-independent toxicity. J Cell Biol 134: 757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris EJ, Keramaris E, Rideout HJ, Slack RS, Dyson NJ, Stefanis L, Park DS (2001) Cyclin-dependent kinases and P53 pathways are activated independently and mediate Bax activation in neurons after DNA damage. J Neurosci 21: 5017–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MD, Lariviere RC, Julien JP (2001) Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron 30: 135–147. [DOI] [PubMed] [Google Scholar]
- O'Hare MJ, Hou ST, Morris EJ, Cregan SP, Xu Q, Slack RS, Park DS (2000) Induction and modulation of cerebellar granule neuron death by E2F-1. J Biol Chem 275: 25358–25364. [DOI] [PubMed] [Google Scholar]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB (1996) Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA 93: 11173–11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan J, Park DS, Greene LA, Shelanski ML (1999) Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J Neurosci 19: 8747–8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Greene LA, Geller HM (1997a) G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J Neurosci 17: 1256–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Levine B, Ferrari G, Greene LA (1997b) Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci 17: 8975–8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA (1998) Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol 143: 457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Bremner R, Keramaris E, Padmanabhan J, Rosenbaum M, Shelanski ML, Geller HM, Greene LA (2000) Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. J Neurosci 20: 3104–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zhou P, Kwon YT, Howley PM, Tsai LH (1998) p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5) is degraded by the ubiquitin-proteasome pathway. J Biol Chem 273: 24057–24064. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402: 615–622. [DOI] [PubMed] [Google Scholar]
- Porn-Ares MI, Samali A, Orrenius S (1998) Cleavage of the calpain inhibitor, calpastatin, during apoptosis. Cell Death Differ 5: 1028–1033. [DOI] [PubMed] [Google Scholar]
- Robison SH, Bradley WG (1984) DNA damage and chronic neuronal degenerations. J Neurol Sci 64: 11–20. [DOI] [PubMed] [Google Scholar]
- Rukenstein A, Rydel RE, Greene LA (1991) Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J Neurosci 11: 2552–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedarous M, Keramaris E, O'Hare M, Melloni E, Slack RS, Elce JS, Greer PA, Park DS (2003) Calpains mediate p53 activation and neuronal death evoked by DNA damage. J Biol Chem 278: 26031–26038. [DOI] [PubMed] [Google Scholar]
- Smith PD, Crocker SJ, Jackson-Lewis V, Jordan-Sciutto KL, Hayley S, Mount MP, O'Hare MJ, Callaghan S, Slack RS, Przedborski S, Anisman H, Park DS (2003) Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson's disease. Proc Natl Acad Sci USA 100: 13650–13655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanis L, Park DS, Yan CY, Farinelli SE, Troy CM, Shelanski ML, Greene LA (1996) Induction of CPP32-like activity in PC12 cells by withdrawal of trophic support. Dissociation from apoptosis. J Biol Chem 271: 30663–30671. [DOI] [PubMed] [Google Scholar]
- Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR (2002) Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet 32: 267–272. [DOI] [PubMed] [Google Scholar]
- Tang D, Yeung J, Lee KY, Matsushita M, Matsui H, Tomizawa K, Hatase O, Wang JH (1995) An isoform of the neuronal cyclin-dependent kinase 5 (Cdk5) activator. J Biol Chem 270: 26897–26903. [DOI] [PubMed] [Google Scholar]
- Tobita M, Nagano I, Nakamura S, Itoyama Y, Kogure K (1995) DNA single-strand breaks in postischemic gerbil brain detected by in situ nick translation procedure. Neurosci Lett 200: 129–132. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu S, Fu Y, Wang JH, Lu Y (2003) Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat Neurosci 6: 1039–1047. [DOI] [PubMed] [Google Scholar]
- Wang KK, Posmantur R, Nadimpalli R, Nath R, Mohan P, Nixon RA, Talanian RV, Keegan M, Herzog L, Allen H (1998) Caspase-mediated fragmentation of calpain inhibitor protein calpastatin during apoptosis. Arch Biochem Biophys 356: 187–196. [DOI] [PubMed] [Google Scholar]
- Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS (1996) Evidence for p53-mediated modulation of neuronal viability. J Neurosci 16: 6753–6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS (1998) Bax involvement in p53-mediated neuronal cell death. J Neurosci 18: 1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]