

Abstract
Inflammation is a critical component of the pathogenesis of Alzheimer's disease (AD). Although not an initiator of this disorder, inflammation nonetheless plays a pivotal role as a driving force that can modulate the neuropathology. Here, we characterized the time course of microglia activation in the brains of a transgenic model of AD (3xTg-AD) and discerned its relationship to the plaque and tangle pathology. We find that microglia became activated in a progressive and age-dependent manner, and this activation correlated with the onset of fibrillar amyloidβ-peptide plaque accumulation and tau hyperphosphorylation. To determine whether microglial activation can exacerbate the pathology, we exposed young 3xTg-AD mice to lipopolysaccharide (LPS), a known inducer of CNS inflammation. Although amyloid precursor protein processing appeared unaffected, we find that LPS significantly induced tau hyperphosphorylation at specific sites that were mediated by the activation of cyclin-dependent kinase 5 (cdk5) through increased formation of the p25 fragment. We further show that administration of roscovitine, a selective and potent inhibitor of cdk5, markedly blocked the LPS-induced tau phosphorylation in the hippocampus. Therefore, this study clearly demonstrates that microglial activation exacerbates key neuropathological features such as tangle formation.
Keywords: microglia, amyloid, transgenic, cdk5, tau, interleukin, APP
Introduction
The Alzheimer's disease (AD) brain is marked by the buildup of two aberrant protein aggregates, amyloid plaques, comprising the amyloid β peptide (Aβ), and neurofibrillary tangles, which consist of hyperphosphorylated tau protein (Selkoe, 2001). Other degenerative changes occur as well, including progressive neuronal and synaptic loss and a reduction in selective neurotransmitter systems (DeKosky and Scheff, 1990; Terry et al., 1991; Dringenberg, 2000). The activation of inflammatory cells is another prominent feature that is performed by microglia and astrocytes, and there is ample evidence showing that these cells are upregulated in the AD brain as well as in transgenic animals (McGeer et al., 1988; Frautschy et al., 1998; Akiyama et al., 2000; Sasaki et al., 2002).
Microglia play a pivotal role in the brain under normal and pathological conditions in an effort to maintain neuronal function. The precise trigger of inflammation in the AD brain remains to be established, although Aβ itself has been shown to act as a pro-inflammatory agent (Matsuoka et al., 2001; Giovannini et al., 2002). The activation of microglia may be duplicitous, because they secrete cytokines including interleukins (ILs), tumor necrosis factor α (TNFα), and chemokines and release reactive oxygen species and nitrogenous compounds, all of which may exacerbate the pathology (Akiyama et al., 2000; Grammas and Ovase, 2002). For example, high levels of IL-1 have been reported in AD or Down syndrome brains or after post-traumatic head injury (Griffin et al., 1989, 1994), a purported risk factor for AD. IL-1 is a pluripotent and pro-inflammatory molecule that affects expression of other inflammatory cytokines and inflammation-associated proteins to amplify immune and inflammatory responses (Mrak and Griffin, 2001). Acute levels of IL-1 in the brain cause a marked elevation of amyloid precursor protein (APP) expression in neurons (Griffin et al., 1994). Furthermore, IL-1 has been reported to accelerate tangle formation in cultured cortical neurons, indicating that it may play a pivotal role in the progression of AD-related pathology (Li et al., 2003).
To elucidate the role of inflammation in the pathogenesis of AD, and in particular its effect on tau pathology, we used the 3xTg-AD mice harboring three mutant human genes (APPK670N;M671L, PS1M146V, and tauP301L) that develop Aβ and tau pathology in an age-dependent manner in disease-relevant brain regions (Oddo et al., 2003). Here, we define the temporal profile of inflammation of these mice and discern its relationship to the Aβ and tau-laden lesions. To determine whether inflammation plays a pathological role, we administered lipopolysaccharide (LPS) to young 3xTg-AD mice and assessed the consequences on the plaque and tangle pathology. We find that the LPS-induced inflammation did not affect Aβ; however, it caused a marked increase in tau hyperphosphorylation. The mechanism underlying this effect is specifically attributable to an increase in p25 and the subsequent activation of cyclin-dependent kinase 5 (cdk5), because this effect can be blocked by roscovitine, a selective inhibitor of this kinase. Based on these findings, we conclude that activation of microglia, either by amyloid plaques or by LPS, exacerbates the tau neuropathology.
Materials and Methods
LPS treatment. LPS (from Escherichia coli 055:B5; Sigma, St. Louis, MO) was dissolved in PBS at a concentration of 0.1 mg/ml and administered intraperitoneally to 4-month-old 3xTg-AD or nontransgenic (NonTg) mice (n = 12; 6 males and 6 females) twice per week for 6 weeks at a dose of 0.5 mg/kg body weight. A control group of 3xTg-AD mice (n = 11; 6 males and 5 females) received injections in the same manner with PBS only. The body weight of each mouse was recorded weekly. Mice were killed 24 h after the last injection, the brain was isolated, and one-half of the brain was fixed in 4% paraformaldehyde, whereas the other half was snap frozen in dry ice and stored at –80°C until analysis.
Intraventricular infusion. Roscovitine (Calbiochem, La Jolla, CA), a potent and selective cdk5 inhibitor, was dissolved in DMSO and further diluted to the desired concentration. Because roscovitine does not cross the blood–brain barrier, it was delivered intraventricularly by using micro-osmotic mini-pumps (model 1002; Alzet, Cupertino, CA). Roscovitine (300 nmol per day) was infused during the last 2 weeks of the LPS treatment. This concentration of roscovitine was used because it was shown previously to significantly inhibit cdk5 activity (Zhang et al., 2004). The brain infusion apparatus (brain infusion kit III; Alzet) was assembled as described in the manufacturer's guide. After the infusion of roscovitine or vehicle only, mice were killed, and hippocampi were removed for additional analyses.
Isolation of mRNA and quantification of ILs by real-time PCR. Total RNA was isolated from half brain (cortex and hippocampus) using TRI reagent (Molecular Research Center, Cincinnati, OH). Traces of DNA were removed by RNase-free DNase I treatment (New England Biolabs, Beverly, MA). One microgram of RNA was subsequently used for one-cycle reverse transcriptase reaction to make cDNA using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) and was subjected to real-time PCR (RT-PCR) to quantify expressions of IL-1β, IL-6, and TNFα using iQ SYBR Green supermix (Bio-Rad). The mouse IL-1β primers were 5′-AAATGCCTCGTGCTGTCTGACC-3′ and 5′-CTGCTTGAGAGGTGCTGATGTACC-3′ (Tanda et al., 1998). The mouse IL-6 primers were 5′-AGTTGCCTTCTTGGGACTGA-3′ and 5′-TCCACGATTTCCCAGAGAAC-3′. The mouse TNFα primers were 5′-CTGGGACAGTGACCTGGACT-3′ and 5′-GCACCTCAGGGAAGAGTCTG-3′. The mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers were 5′-AACTTTGGCATTGTGGAAGG-3′ and 5′-ACACATTGGGGGTAGGAACA-3′. Threshold cycle (Ct) values were calculated with MyiQ software (Bio-Rad), and the quantitative fold changes in mRNA were determined as relative to GAPDH mRNA levels in each treatment group.
Immunohistochemistry. The primary antibodies used in this study are summarized in Table 1. Secondary biotinylated antibodies (anti-mouse, anti-rat, and anti-rabbit), normal sera (Vector Laboratories, Burlingame, CA), and secondary antibodies for immunofluorescent staining (AlexaFluor 488, 546, or 568 for anti-mouse and anti-rabbit; Molecular Probes, Eugene, OR) were also used in this study.
Table 1.
Primary antibodies used in this study
|
Antibody |
Immunogen |
Host |
Specificity |
Application |
Source |
|---|---|---|---|---|---|
| 6E10 | aa 1-17 of Aβ | Mouse | H, M | IHC, IF, WB | Signet (Dedham, MA) |
| CT20 | aa 751-770 of human APP | Rabbit | H, M | WB | Calbiochem |
| GFAP | Bovine GFAP | Rabbit | H, M, R | IHC, IF | DAKO (Carpinteria, CA) |
| CD11b | Mouse B-cells | Rat | M | IHC | Serotec (Raleigh, NC) |
| CD45 | Mouse B-cells | Rat | M | IHC | Serotec |
| Iba1 | C terminus of Iba1 | Rabbit | H, M, R | IF | WAKO (Richmond, VA) |
| HT7 | aa 159-163 of tau | Mouse | H | IHC, WB | Innogenetics (Alpharetta, GA) |
| AT8 | Peptides containing phospho-S202/T205 | Mouse | H, M, R | IHC, IF, WB | Innogenetics |
| AT180 | Peptides containing phospho-T231/S235 | Mouse | H, M, R | IHC, WB | Innogenetics |
| PHF1 | Peptides containing phospho-S396/S404 | Mouse | H, M, R | IHC, WB | a |
| β-Actin | C terminus of actin | Rabbit | Wide range | WB | Sigma |
| Aβ40 | aa 35-40 of Aβ | Mouse | H, M, R, D | ELISA | Mayo Clinic (Jacksonville, FL) |
| Aβ42 | aa 35-42 of Aβ | Mouse | H, M, R, D | WB | Mayo Clinic |
| p35/p25 | C terminus of human p35 | Rabbit | H, M, R | WB | Santa Cruz Biotechnology (Santa Cruz, CA) |
| cdk5 | aa 16 of human cdk5 | Rabbit | H, M, R | WB | Calbiochem |
| JNK | Human JNK2 | Rabbit | H, M, R | WB | Cell Signaling (Beverly, MA) |
| Phospho-JNK | Phosphopeptides around The183/Tyr185 | Mouse | H, M, R | WB | Cell Signaling |
| GSK-3β | N terminus of rat GSK-3β | Mouse | H, M, R, D, C | WB | BD Transduction (San Diego, CA) |
| Phospho-GSK-3β | Phosphopeptides around Tyr216 | Mouse | H, M, R, D | WB | BD Transduction |
| Phospho-GSK-3β | Phosphopeptides around Ser9 | Rabbit | H, M, R | WB | Cell Signaling |
| p38 | Human p38 | Rabbit | H, M, R | WB | Cell Signaling |
| Phospho-p38 |
Phosphopeptides around Thr180/Tyr182 |
Rabbit |
H, M, R |
WB |
Cell Signaling |
H, Human; M, mouse; R, rat; D, dog; C, chicken; IHC, immunohistochemistry; IF, immunofluorescent staining; WB, Western blotting.
Generous gift from P. Davis (Albert Einstein College of Medicine, Bronx, NY).
Immunohistochemical staining for Aβ and tau were essentially as described previously (Oddo et al., 2003). For activated microglia staining, free-floating sections (50 μm thickness) were first pretreated with Tris-buffered saline (TBS) containing 3% hydrogen peroxide and 10% methanol for 30 min to block endogenous peroxidase activity. After a TBS wash, sections were incubated once in TBS with 0.1% Triton X-100 (TBST) for 15 min and once with TBST with 2% BSA for 30 min. The sections were incubated overnight with primary antibody [anti-mouse CD45 (1:500) or anti-mouse CD11b (1:500) in TBS plus 5% normal rabbit serum] at 4°C; this was followed by treatment with a secondary antibody [biotinylated anti-rat (1:200) in TBS plus 5% normal rabbit serum] for 1 h at room temperature. The sections were visualized with the Vector ABC kit and stained with DAB (Vector Laboratories).
For double immunofluorescent staining with thioflavin S (Sigma) and microglia, free-floating sections were incubated with 0.5% thioflavin S in 50% ethanol for 10 min. Sections were washed twice with 50% ethanol for 5 min each, once with water for 5 min, once with TBST for 15 min, and once with TBST with 2% BSA for 30 min. The sections were then incubated with primary antibody [Iba1 (1:200) in TBS plus 5% normal goat serum] overnight at 4°C, followed by secondary AlexaFluor 546 anti-rabbit antibody (1:200 in TBS plus 5% normal goat serum) for 1 h at room temperature. Staining was visualized under a confocal microscope.
Immunoblotting. The brains were homogenized in T-PER (tissue protein extraction reagent) (Pierce, Rockford, IL) in the presence of a protease inhibitor mixture (Roche Applied Science, Indianapolis, IN) and phosphatase inhibitors (5 mm sodium fluoride and 50 μm sodium orthovanadate) and centrifuged at 100,000 × g for 1 h at 4°C. Supernatants were collected as the detergent-soluble fraction. Pellets were resuspended in 70% formic acid (FA), homogenized, and centrifuged at 100,000 × g for 1 h at 4°C. The resultant supernatants were collected as the detergent-insoluble or FA fraction. These fractions were immunoblotted with antibodies that recognize APP, C99, total tau, phosphorylated tau, cdk5, p35/p25, glycogen synthase kinase-3β (GSK-3β), phospho-GSK-3β, p38–mitogen-activated protein kinase (MAPK), phospho-p38-MAPK, c-Jun NH2-terminal kinase (JNK), or phospho-JNK. Membranes were reprobed with antibody against β-actin to control for protein loading. Band intensity was measured using Quantity One software (Bio-Rad).
ELISA for Aβ40 and Aβ42. Aβ40 and Aβ42 were detected in both the detergent-soluble and -insoluble fractions by ELISA. Soluble fractions were loaded directly onto ELISA plates, and FA fractions were diluted 1:20 in neutralization buffer (1 m Tris base and 0.5 m Na2HPO) before loading. MaxiSorp immunoplates (Nunc, Naperville, IL) were coated with mAβ20.1 antibody at a concentration of 25 μg/ml in coating buffer (0.1 m NaCO3 buffer, pH 9.6) and blocked with 3% BSA. Synthetic Aβ standards were defibrillated by dissolving in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) at 1 mg/ml, and the HFIP evaporated with a stream of nitrogen. The defibrillated Aβ was then dissolved in DMSO at 1 mg/ml. Standards of both Aβ40 and Aβ42 were made in antigen capture buffer (20 mm NaH2PO4, 2 mm EDTA, 0.4 m NaCl, 0.5 g of 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, and 1% BSA, pH 7.0) and loaded onto ELISA plates in duplicate. Samples were loaded in duplicate and incubated overnight at 4°C. The plates were washed and probed with either HRP-conjugated anti-Aβ35–40 (MM32–13.1.1 for Aβ1–40) or anti-Aβ35–42 (MM40–21.3.4 for Aβ1–42) overnight at 4°C. 3,3′,5,5′-Tetramethylbenzidine was used as the chromagen, and the reaction was stopped by 30% O-phosphoric acid and read at 450 nm on a Molecular Dynamics (Sunnyvale, CA) plate reader.
Immunoprecipitation and cdk5 and GSK-3β kinase assays. Immunoprecipitation and kinase assays were performed as described previously with a slight modification (Cruz et al., 2003; Noble et al., 2003). Briefly, 100 μg of brain samples were immunoprecipitated with protein A-agarose and cdk5 antibody or protein G-agarose and GSK-3β antibody. The kinase assays were then performed in a total volume of a 50 μl reaction mixture containing 20 mm 4-morpholinepropanesulfonic acid, pH 7.2, 5 mm MgCl2, 1 mm sodium orthovanadate, 5 mm NaF, 100 μm ATP, 2.5 μCi [γ-32P]ATP, and 0.2 mm cdk5 substrate (Calbiochem) for cdk5 kinase assay or 0.2 mm GSK-3β substrate (Calbiochem) for GSK-3β assay. The reaction took place for 1 h at 30°C, and the 35 μl supernatant was placed on P81 phosphocellulose squares (Upstate, Waltham, MA). The squares were washed in 0.3% phosphoric acid and counted in a scintillation counter to determine the kinase activity.
Statistical analysis. All data were analyzed using one-way ANOVA or an unpaired t test, and p < 0.05 or lower was considered to be significant.
Results
Age-related inflammatory response
The first pathological manifestation in the 3xTg-AD mice is the buildup of intraneuronal Aβ within the neocortex, which occurs by 4 months of age and is followed shortly thereafter by a similar buildup within pyramidal cells of the hippocampus (Oddo et al., 2003). At this time point, no thioflavin S-positive plaques are apparent (data not shown). Likewise, we find no evidence of microglial activation, as determined by CD45 immunostaining (data not shown). Therefore, far more extensive pathology is likely required to trigger inflammation in these murine brains. Diffuse Aβ plaques as well as thioflavin S-positive fibrillar Aβ plaques start to develop at ∼9 months of age within the subiculum (Fig. 1A,D). Yet, activated microglia are not found in or around these plaques (Fig. 1H). Although still relatively sparse and small, 12 months of age corresponds to the development of more fibrillar Aβ deposits, particularly within the CA1 and subicular subfields of the hippocampus and the septum, as detected by 6E10 immunostaining and thioflavin S staining (Fig. 1B,E). Likewise, the tau pathology also develops in a progressive manner and by 12 months of age includes significant numbers of MC1-, AT180-, and AT8-immunopositive CA1 pyramidal neurons (Oddo et al., 2003). It was within this region that we first detected CD45-immunoreactive activated microglia in the 3xTg-AD brains (Fig. 1I). Many of the activated microglia are in or around the thioflavin S-positive plaques. By 24 months of age, CD45 immunoreactivity was more widespread and could be observed in the neocortex, subiculum, CA1 subfield, and amygdala, brain regions that harbored extensive numbers of thioflavin S-positive plaques (Fig. 1C,F,J). These CD45-immunopositive microglia showed increased cell body and process size (Fig. 1M), whereas age-matched NonTg mice did not express CD45 immunoreactivity in the brain, even at very advanced ages (Fig. 1L). Another activated microglia marker, CD11b, also showed a similar pattern to CD45 at all time points (data not shown). Quantitative analysis of the amyloid burden in the 3xTg-AD brain showed an age-related increase, such that by 24 months of age it was increased nearly five times compared with 12-month-old mice (Fig. 1G). Notably, the activation of microglia closely paralleled the age-related increase in the amyloid burden (Fig. 1K). These data clearly show that microglia become activated after the formation of fibrillar plaques in the transgenic brain.
Figure 1.

Temporal profile of brain inflammation in the 3xTg-AD mice. The time course of intraneuronal and extraneuronal Aβ buildup and its relationship to activated microglia were described in the 3xTg-AD mice. Both intraneuronal and extraneuronal Aβ deposits were determined by 6E10 immunostaining of 3xTg-AD brains at 9 (A), 12 (B), and 24 (C) months of age. Fibrillar amyloid plaques were detected by thioflavin S staining at 9 (D), 12 (E), and 24 (F) months of age. Serial sections were stained with anti-CD45 to detect activated microglia at 9 (H), 12 (I), and 24 (J) months of age. Amyloid burden (G) and CD45-immunopositive-activated microglia (K) were quantitatively analyzed by measuring the percentage of occupied area by thioflavin S (amyloid burden) or CD45-immunopositive staining (microglia burden). Data are presented as mean±SEM (n=5 in each age). Higher-magnification images of CD45-immunopositive staining in the subicular subfield of the hippocampus from a 24-month-old NonTg mouse (L) and 3xTg-AD mouse (M) are shown. GFAP immunostaining revealed no gliosis in layers III–IV of the neocortex of a 24-month-old NonTg mouse (N), whereas there was a marked increase in GFAP-immunoreactive astrocytes detected in 24-month-old 3xTg-AD mice (O).
In addition to the age-dependent activation of microglia, we also found increased numbers of GFAP-immunoreactive astrocytes in the neocortex of 3xTg-AD mice (Fig. 1O) relative to age-matched NonTg mice (Fig. 1N).
In the AD brain, microglia are often found in close proximity to amyloid plaques and frequently surround these proteinaceous aggregates (Akiyama et al., 2000). To further define the relationship between the microglial activation and plaque buildup in the 3xTg-AD mice (Fig. 2), we double immunolabeled brain sections for Aβ (blue) and microglia (brown). Activated microglia were colocalized around extraneuronal Aβ plaques in the neocortex, amygdala, and hippocampus (Fig. 2A–C). Most notably, most of the amyloid plaques surrounded by activated microglia were predominantly fibrillar in nature, because analysis of serial sections indicated that these structures were also thioflavin S positive (Fig. 2D). Confocal microscopic analysis further established that the microglia (red) surrounding fibrillar Aβ plaques (green) showed physical signs of activation because they exhibited an amoeboid structure and increased cytosolic volume (Fig. 2E) compared with those observed in age-matched NonTg mice (data not shown). We also observed a close physical relationship between microglia and fibrillar amyloid plaques in the hippocampus of human AD brains (Fig. 2F). Likewise, we found that a high proportion of the fibrillar deposits were also surrounded by astrocytes in the 3xTg-AD brain (Fig. 2G). Therefore, the build up of diffuse plaques appears insufficient to trigger a robust microglial or astrocytic response in the 3xTg-AD brains; rather, we found that the inflammatory response coincides with the accumulation of fibrillar deposits and appears qualitatively similar to the response observed in the human AD brain.
Figure 2.
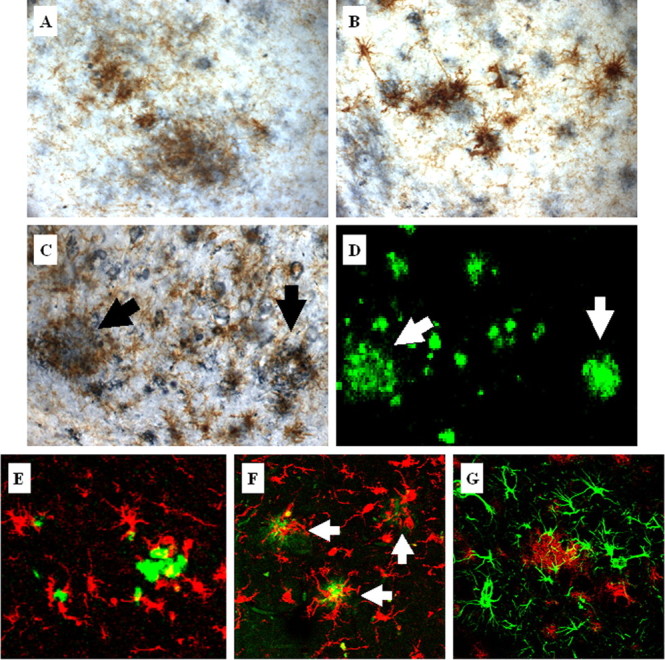
Activated microglia colocalize with plaques in aged 3xTg-AD mice. Activated microglia detected by CD45 immunoreactivity (brown) colocalize with extraneuronal Aβ-immunopositive deposits (blue) in the cortex (A), amygdala (B), and hippocampus (C) in 24-month-old 3xTg-AD mice. Serial brain sections of the hippocampus were stained with thioflavin S (D). Arrows point to activated microglia around thioflavin S-positive plaques. Additional double-staining fluorescent analysis using thioflavin S (green) and Iba1 (red) to detect microglia demonstrates that 3xTg-AD mice (E) show similar microglia and plaque interactions as observed in the hippocampus of the human AD brain (F). Arrows point to microglia associated with the cores of amyloid plaques in the human AD brain. G, Double fluorescent labeling of GFAP-immunopositive astrocytes (green) and amyloid-containing plaques (red) detected by 6E10 in 24-month-old 3xTg-AD mice shows colocalization of astrocytes around plaques.
LPS induces microglial activation in the 3xTg-AD mice
To determine whether inflammation can modulate the onset and severity of the Aβ and tau pathology, LPS (0.5 mg/kg body weight, twice per week) was administered intraperitoneally for a 6 week period to 4-month-old 3xTg-AD mice. After this time, all mice were killed, and their brains were harvested. We found that chronic LPS treatment markedly increased the number of activated microglia, as determined by CD45 immunostaining, throughout the neocortex and hippocampus compared with sham-treated mice (Fig. 3A,B). The microglia burden in PBS- and LPS-treated mice was 0.6 ± 0.4 and 7.8 ± 0.7, respectively, and the levels of activated microglia in the LPS-treated group were significantly greater than those in the PBS-treated group (p < 0.01). The number of activated microglia observed in these LPS-treated, 6-month-old 3xTg-AD mice was slightly higher than that observed in untreated 12-month-old 3xTg-AD mice (Fig. 1K). Because activated microglia secrete various cytokines, we monitored the expression of selected cytokines, including IL-1β, IL-6, and TNFα, which have been suggested to play important roles in the progression of AD (Griffin et al., 1989; Dickson et al., 1993; Huell et al., 1995). We found that IL-1β levels were markedly increased in the LPS-treated brain, being nearly five times higher than in the PBS-treated brains (Fig. 3C). In contrast, IL-6 and TNFα expression levels were not significantly altered by LPS treatment (Fig. 3C). The mRNA levels of IL-1β, IL-6, and TNFα in PBS-treated brains were relatively the same, with Ct values of 28.58 ± 0.21, 27.81 ± 0.19, and 28.76 ± 0.83, respectively.
Figure 3.

LPS activates microglia in the 3xTg-AD mice. LPS (0.5 mg/kg body weight) or PBS was injected intraperitoneally 4-month-old 3xTg-AD mice twice per week for 6 weeks, and the brains were analyzed by immunohistochemistry. CD45 immunoreactivity was compared in the hippocampus of PBS- and LPS-treated 3xTg-AD mice. The brains from PBS-treated mice show only basal levels of microglial activation (A), whereas LPS-treated mice show significantly increased activated microglia (B). At least five mice per group were analyzed. C, Quantitative RT-PCR of IL-1β and IL-6 was performed in total RNA extracts from PBS- or LPS-treated 3xTg-AD mice brains. In each mouse, the expression levels of mRNA were normalized relative to the levels of GAPDH mRNA (Ct values for the PBS- and LPS-treated brain are 14.54 ± 0.22 and 14.55 ± 0.18, respectively), and fold differences were calculated relative to the PBS-treated group for each IL. **p < 0.01 compared with the PBS-treated mice group (n = 3 in group).
We next assessed the consequences of the LPS-induced activation of microglia on APP metabolism. We monitored brain levels of APP and its derivatives, C99, C83, Aβ40, and Aβ42, by immunoblotting or ELISA. We found that the LPS-induced CNS inflammation did not significantly alter the steady-state levels of full-length APP (Fig. 4A,B). We also found that C99 or C83 levels were not significantly different between the treatments as determined by immunoblotting with the CT20 (APP C-terminal) antibody (Fig. 4A,B). Likewise, measurement of detergent-soluble and FA-soluble Aβ levels by ELISA revealed that 6 week exposure to LPS did not cause a significant change in either Aβ40 or Aβ42 (Fig. 4C). The levels of detergent-soluble and FA-soluble Aβ fragments in LPS-treated mice were found to range from 0.056 to 0.093 pg/g and from 0.21 to 0.97 pg/g, respectively, whereas those in PBS-treated mice were slightly lower, ranging from 0.046 to 0.049 pg/g and from 0.18 to 0.97 pg/g, respectively. Moreover, the pattern and extent of Aβ immunoreactivity with 6E10 was not significantly different between the LPS-versus PBS-treated groups (Fig. 4D,E). Therefore, based on our analysis, although LPS treatment caused a marked activation of microglial cells in the brains of the 3xTg-AD mice, it did not produce any discernible effect on APP processing, nor did it affect Aβ deposition.
Figure 4.

Effect of LPS-induced inflammation on APP processing in the 3xTg-AD mice. A, Immunoblotting of APP, C99, and C83 in PBS- or LPS-treated 3xTg-AD mice, withβ-actin used as a protein loading control. B, Densitometric analysis of steady-state levels of full-length APP, C99, or C83 between PBS- and LPS-treated 3xTg-AD mice. Data are presented as mean ± SEM (n = 4 each group). C, Total Aβ40 and Aβ42 from detergent-soluble and -insoluble fractions of PBS- and LPS-treated 3xTg-AD mice were measured by ELISA. □, PBS-treated 3xTg-AD mice; ▪, LPS-treated 3xTg-AD mice. Data are presented as mean ± SEM (n = 8 each group). D, Fluorescent intensity of 6E10-immunopositive neurons in the cortex and hippocampus was quantitatively analyzed using Quantity One software. Data are presented as mean intensity ± SEM (n = 5 each group). E, Representative immunofluorescent staining of APP and Aβ fragments by 6E10 in the neocortex and hippocampus of 3xTg-AD mice treated with PBS or LPS. a.u., Arbitrary units; HC, hippocampus; CX, cortex.
Our results did not match the previous report that 12 week LPS treatment in old APPswe transgenic mice increased full-length APP, C99, Aβ40, and Aβ42 (Sheng et al., 2003). The disparity may be attributable to the age of the animals used for the experiments, the duration of the treatment, or the background strain. We used young mice (4 months old) that were treated for a shorter duration and observed only a slight increase in APP levels in LPS-treated 3xTg-AD mice, indicating that longer LPS exposure may cause a significant increase in APP as described by Sheng et al. (2003).
LPS-induced inflammation modulates selective tau phosphorylation
Unlike the minimal effect on APP, we found that the LPS treatment significantly exacerbated the tau pathology in the 3xTg-AD mice. Although the steady-state levels of total human tau protein as recognized by the human-specific HT7 antibody on immunoblots were comparable between the LPS- and PBS-treated groups as measured by densitometric analysis, we found that the steady-state levels of AT8 immunoreactivity, which recognizes tau phosphorylated at Ser202 and Thr205, were more than twofold higher in the LPS-treated mice (Fig. 5A), and the change was significant (p < 0.05). The LPS-induced effect on AT8 immunoreactivity was further evident by immunohistochemistry (Fig. 5B). In contrast, we found no evidence of AT8 immunoreactivity in PBS-treated 3xTg-AD mice (Fig. 5B). Typically, we first observe AT8 immunoreactivity when the mice are ∼1 year of age. Nevertheless, although the LPS-treated mice were only 6 months of age at the time they were killed, AT8-positive pyramidal neurons were clearly evident in the CA1 subfield of the hippocampus (Fig. 5B). The effect was mainly limited to the hippocampus because cortical AT8 immunoreactivity was not detected. The reason underlying this region-specific effect is unknown, although the hippocampus is the first region to develop tau pathology in the 3xTg-AD mice.
Figure 5.

Inflammation-induced enhancement of tau phosphorylation. A, Immunoblotting analysis of total tau (HT7) and phosphorylated tau (AT8) in PBS- and LPS-treated 3xTg-AD mice, with β-actin used as a protein loading control. Data represent at least five mice per group. B, Immunohistochemical staining of total human tau and phosphorylated tau in hippocampal neurons of 3xTg-AD mice treated with PBS and LPS. The inset in the bottom right panel is a magnified view of AT8-immunopositive neurons in the hippocampus.
We next tested other phospho-specific tau antibodies in PBS- and LPS-treated 3xTg-AD mice brains. LPS treatment did not alter tau phosphorylation at Ser396/Ser404, which is recognized by antibody PHF1 (Fig. 6 A, B). However, a nearly twofold increase in the steady-state level of phosphorylated tau recognized by AT180 was found in LPS-treated 3xTg-AD mice (Fig. 6 A, B). Additional immunohistochemical staining with AT180 supported a marked accumulation of AT180-positive neurons including cell bodies and neurites in the CA1 region of the hippocampus (Fig. 6C). Because AT180 antibody recognizes tau phosphorylated at residues Thr231/Ser235, LPS-induced inflammation modulated selective phosphorylation sites of tau in the brain, indicative of the activation of a certain kinase(s).
Figure 6.

Site-specific phosphorylation of tau after LPS exposure. A, Immunoblotting of AT180- and PHF1-positive phospho-tau in LPS- or PBS-treated 3xTg-AD mice brain, with β-actin used as a protein loading control. B, Densitometric analysis of steady-state levels of PHF1 or AT180 between PBS- and LPS-treated 3xTg-AD mice. Data are presented as mean ± SEM (n = 5). **p < 0.01. C, Representative immunohistochemical staining of AT180-positive tau in hippocampal neurons of 3xTg-AD mice treated with PBS and LPS. D, Six-month-old NonTg mice were treated with 0.5 mg/kg LPS or PBS intraperitoneally twice per week for 6 weeks. After the treatment period, the hippocampus and cortex were isolated, and proteins were analyzed by immunoblotting for AT8-positive phosphorylated tau. a.u., Arbitrary units.
To further validate that LPS-induced tau phosphorylation is not mediated by APP/Aβ-related pathological changes, 6-month-old NonTg mice were treated with LPS in the same manner for 6 weeks. The hippocampus and cortex were isolated after the completion of the treatment, and tau phosphorylation was determined by immunoblotting. AT8-immunoreactive tau was markedly elevated in these brains (Fig. 6D). Densitometric analysis of the steady-state levels of AT8 between PBS- and LPS-treated NonTg mice showed a significant (p < 0.01) increase in AT8-positive tau in LPS-treated mice (n = 4; data not shown). Because NonTg mice do not normally develop any tau pathology, our study shows that LPS-induced inflammation has a profound effect on tau phosphorylation even in NonTg mice; in contrast, Aβ levels were unaffected.
LPS-induced inflammation exacerbates tau pathology via cdk5
To elucidate the molecular basis underlying the LPS-induced selective phosphorylation of tau in the 3xTg-AD mice, we assayed several putative tau kinases. Specially, we measured the activated forms of cdk5, GSK-3β, p38–MAPK, and JNK because these kinases are known to phosphorylate tau in vitro and in vivo (Atzori et al., 2001; Savage et al., 2002; Sun et al., 2002; Liu et al., 2003). Among them, the activities of GSK-3β, p38–MAPK, and JNK are all regulated via phosphorylation at specific amino acids. For example, GSK-3β is activated through the phosphorylation at Tyr216 or is inhibited when Ser9 is phosphorylated by Akt activity (Cohen and Frame, 2001). We found that LPS-induced inflammation did not alter the activation status of GSK-3β, p38–MAPK, or JNK, as determined by immunoblotting with selective phospho-specific epitope antibodies (Fig. 7A).
Figure 7.

LPS-induced inflammation causes increased p25 formation. A, Immunoblotting for p35/p25, cdk5, phosphorylated/total GSK-3β, phosphorylated/total p38–MAPK, and phosphorylated/total JNK of PBS- and LPS-treated 3xTg-AD mice, withβ-actin used as a protein loading control. Densitometric analysis revealed a significant (p < 0.05) increase in p25 levels in LPS-treated 3xTg-AD mice (n = 5; data not shown). B, Immunoblotting for p35/p25 and cdk5 of PBS- and LPS-treated NonTg mice, with β-actin used as a protein loading control. Densitometric analysis revealed a significant (p < 0.05) increase in p25 levels in LPS-treated NonTg mice (n = 4; data not shown).
We did find that LPS treatment affected cdk5. cdk5 is considered to be a major tau kinase, whereby its activation is accomplished by binding of activator proteins p35, p39, or p25, a cleaved fragment of p35 (Humbert et al., 2000; Lee and Tsai, 2003). p35, a molecule with a short half-life, is localized to cell membranes, whereas its proteolytic product, the p25 fragment, is diffused throughout the cytosol and nucleus and has a longer half-life (Lee and Tsai, 2003). Potent neurotoxicity of p25 has been documented, and significantly increased p25 levels have been reported in postmortem AD brain tissues (Patrick et al., 1999; Lee et al., 2000). We detected no difference in the steady-state levels of cdk5 between the LPS- and saline-treated mice, but we did detect a significant elevation in the steady-state levels of p25 in the LPS-treated 3xTg-AD mice (Fig. 7A), and densitometric analysis revealed that p25 levels were increased nearly twofold (p < 0.05; data not shown). Likewise, enhanced p25 levels were also found in LPS-treated NonTg mice (Fig. 7B), further supporting the fact that cdk5/p25 activation is mediated by inflammation.
To further address the role of cdk5 in the LPS-induced increase in tau phosphorylation, we directly measured cdk5 enzymatic activity. At least eight mouse brains per treatment were analyzed for cdk5 activity. The cdk5 activity was nearly twofold higher in the LPS-treated 3xTg-AD brain compared with sham-treated 3xTg-AD mice, and the difference was statistically significant (p < 0.01; data not shown). In contrast, the kinase activity of GSK-3β did not differ between the two groups (data not shown), in agreement with the immunoblot data, which showed no difference in steady-state levels of phosphorylated GSK-3β. In contrast, the higher steady-state levels of p25 is consistent with it leading to a prolonged activation of cdk5, thereby resulting in increased phosphorylation of tau at residues Ser202/Thr205 (recognized by AT8) and Ser235 (recognized by AT180) but not at non-cdk5 sites such as Ser404 that (recognized by PHF1) (Ishiguro et al., 1991; Baumann et al., 1993; Patrick et al., 1998, 1999). Overall, these findings are consistent with our immunohistochemical observations in which we found marked increases in AT8 and AT180 immunoreactivity in the brain of LPS-treated 3xTg-AD mice, whereas PHF1 immunoreactivity was unaffected by the LPS treatment.
To confirm that the LPS-induced phosphorylation of tau was mediated by cdk5, we treated mice with roscovitine, a cdk5-specific inhibitor (Zhang et al., 2004). Roscovitine was chronically delivered during the final 2 weeks of LPS exposure. We first determined whether roscovitine modulated APP and tau steady-state levels in the LPS-treated mice by immunoblotting. As shown in Figure 8, total human APP (6E10) and tau (HT7) did not differ between the treatments, consistent with the data shown above. Notably, AT8- and AT180-positive tau phosphorylation, however, were markedly decreased in the roscovitine-treated mice (Fig. 8). As expected, PHF1-positive tau remained unchanged. Therefore, based on these findings, we conclude that the mechanism underlying the LPS-induced inflammation and subsequent phosphorylation of tau is specifically mediated by the activation of the cdk5/p25 pathway.
Figure 8.

Roscovitine inhibits the cdk5-dependent phosphorylation of tau in LPS-treated 3xTg-AD mice. 3xTg-AD mice were treated with roscovitine (300 nmol per day) or vehicle (50% DMSO) through an intraventricular infusion during the last 2 weeks of the 6 week LPS treatment. The hippocampus was isolated and tested for phosphorylation status of tau by immunoblotting. The steady-state levels of human APP and tau were not altered by roscovitine treatment. Densitometric analysis revealed the steady-state levels of AT8- and AT180-positive tau resulted in nearly a twofold and threefold reduction in roscovitine treatment, respectively, whereas PHF1-positive tau levels were not affected by LPS (data not shown). Membranes were reprobed with β-actin to control the equal protein loading (n = 3 each group).
Discussion
In this study, we characterized the inflammatory response that develops in the CNS of the 3xTg-AD mice and discerned its relationship to the plaque and tangle pathology. We found that microglia become activated in an age-dependent manner and are highly localized to Aβ-affected brain regions including the neocortex, amygdala, and the subiculum and CA1 subfields of the hippocampus. Marked activation of microglia was first evident at ∼11–12 months of age, which corresponds to the time when Aβ plaque numbers start to increase in these brain regions and to the onset of fibrillar plaques. Many activated microglia could be found adjacent to or surrounding Aβ plaques, and similar temporal and spatial patterns have been documented previously in other APP transgenic mouse models (Frautschy et al., 1998; Stalder et al., 1999; Gordon et al., 2002) as well as in human AD (Perlmutter et al., 1990). Collectively, these data support a role for extraneuronal Aβ serving as a major trigger for the onset of inflammation.
Inflammation, and in particular microglia, appears to play a dichotomous role in the pathogenesis of AD. On one hand, these reactive cells clearly play a beneficial role by surrounding amyloid plaques, presumably isolating it from the neighboring neuropil (Akiyama et al., 2000). Activated microglia can phagocytose amyloid, thereby facilitating its clearance from the brain (Frautschy et al., 1992; Funato et al., 1998; Weldon et al., 1998). Immunotherapeutic-based strategies also seem to depend on microglial activation to clear amyloid deposits (Schenk et al., 1999; Bard et al., 2000, 2003; Morgan et al., 2000). On the other hand, epidemiological evidence suggests that use of a subset of nonsteroidal anti-inflammatory drugs can reduce the risk for AD, presumably by lessening the inflammatory component (Stewart et al., 1997; Anthony et al., 2000; Broe et al., 2000; Lim et al., 2000; Weggen et al., 2001; Zhou et al., 2003). Furthermore, as our studies clearly demonstrate, inflammation may also have a detrimental effect on the onset and development of the tau pathology.
After administering LPS to the 3xTg-AD mice for 6 weeks, the expression of IL-1β was significantly increased in these brains, whereas the expression levels of IL-6 or TNFα were not altered. This finding is consistent with a previous in vitro study showing that LPS caused a marked elevation of IL-1β mRNA in microglial cells isolated from mouse brain (Hetier et al., 1988). Although the levels of all of these cytokines are found to be elevated in postmortem AD brains (Griffin et al., 1989; Dickson et al., 1993; Huell et al., 1995), IL-1β may be an early component during disease development and progression and potentially modulate tau pathology, as our data and others have suggested (Griffin et al., 1995; Apelt and Schliebs, 2001; Bellucci et al., 2004). Work in other transgenic models further supports this hypothesis. For example, in Tg2576 mice, astrocytes immunoreactive for IL-1β are found in and around early-stage plaques, whereas increased levels of IL-6 are not found until later ages (Apelt and Schliebs, 2001). Elevation of IL-6 expression was followed by the treatment of IL-1β and Aβ peptides in a cell-culture model (Holmlund et al., 2002). Finally, high levels of IL-1β in both the spinal cord and brain were reported in a tau transgenic model, with the elevated IL-1β levels correlating with the tau pathology (Bellucci et al., 2004).
These inflammatory responses may lead to a marked increase in AT8- and AT180-immunoreactive neurons in the CA1 subfield of the hippocampus but not in the steady-state levels of PHF1-positive tau. This selective phosphorylation of tau was mediated by an activation of cdk5. Based on our results, three critical findings support a central role for cdk5 in the inflammation-mediated phosphorylation of tau. First, the steady-state levels of p25 were markedly higher in the LPS-treated mice, which is significant because elevated levels of p25 cause a prolonged activation of cdk5 (Tsai et al., 1994; Alvarez et al., 1999; Ahlijanian et al., 2000; Lee et al., 2000). Second, the enhancement of p25 levels in the LPS-treated group resulted in the activation of cdk5 as evidenced by directly monitoring its kinase activity. Finally, roscovitine abolished the LPS-mediated effects on tau phosphorylation at the cdk5-specific sites (i.e., Ser202 and Ser235) in the 3xTg-AD mice. Notably, cdk5 activation appears to be one of the earliest tau-associated pathological changes documented in postmortem studies, because activation of cdk5 and the presence of AT8 immunoreactivity occurs in neurons with early stages of AD neurofibrillary degeneration, but the colocalization of cdk5 and AT8 immunoreactivity in neurons is not apparent in late stages (Pei et al., 1998; Augustinack et al., 2002).
In our model, the levels of PHF1-positive tau did not significantly change in LPS-treated mice, although several in vivo and in vitro studies reported that cdk5 activation increased PHF1-positive tau phosphorylation (Evans et al., 2000; Lund et al., 2001; Cruz et al., 2003; Shelton et al., 2004). The inability of LPS to increase PHF1 immunoreactivity may be explained by the age of mice used in these experiments. In the 3xTg-AD mice, PHF1-positive tau is evident at older ages (at least 12 months or older) (Oddo et al., 2003). Because PHF1-tau is considered to be a late component of tau pathology, it is possible that higher levels or a longer duration of cdk5/p25 activation may be required to alter PHF1 immunoreactivity.
Previously, Li et al. (2003) reported that IL-1-induced tau hyperphosphorylation is mediated by the activation of p38–MAPK and the application of an IL-1 receptor antagonist significantly reduced phosphorylated tau, activated p38–MAPK, and neuronal loss in rat primary cortical neurons. Furthermore, IL-1-treated rats exhibited significantly increased numbers of phosphorylated tau-containing neurons, and these neurons were also immunoreactive with phosphorylated p38–MAPK (Sheng et al., 2001). It was also reported that JNK was markedly activated within 3 h of LPS injection in rats, and the activation of JNK was mediated by IL-1β (Lynch et al., 2004). These data implicate other kinases in the inflammation-mediated tau phosphorylation, yet interestingly, we did not find any significant activation of these kinases after 6 week LPS exposure in young 3xTg-AD mice, although we cannot exclude that they played an early transient role.
In conclusion, we report the temporal profile of microglia and astrocyte activation in the brains of the 3xTg-AD mice and show that it appears to be best associated with the development of fibrillar Aβ deposits in the brain. The effect of LPS-induced inflammation on tau pathology is particularly intriguing because it provides strong experimental evidence showing that brain inflammation can induce a pathological outcome in the brain. Our studies suggest that the mechanism underlying the inflammation-induced effect on tau is mediated by the cdk5 kinase, because administering roscovitine to these mice specifically blocked this effect. To our knowledge, this study represents one of the first reports to show that inflammation specifically targets this kinase and leads to an event that many consider to be an adverse effect (i.e., tau hyperphosphorylation) in tauopathies such as AD. Finally, because the inflammatory response that naturally develops in the 3xTg-AD brains best correlates with the buildup of fibrillar Aβ plaques, it is plausible that Aβ-induced inflammation likewise leads to a similar effect, targeting cdk5 kinase and resulting in tau hyperphosphorylation. Therefore, these data suggest a novel pathway by which the Aβ and tau pathology may be linked further.
Footnotes
This work was supported by a grant from the Alzheimer's Association and by National Institutes of Health Grants AG17968 and AG0212982.
Correspondence should be addressed to Dr. Frank M. LaFerla, Department of Neurobiology and Behavior, 1109 Gillespie Neuroscience Facility, University of California–Irvine, Irvine, CA 92697-4545. E-mail: laferla@uci.edu.
DOI:10.1523/JNEUROSCI.2868-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258843-11$15.00/0
References
- Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD (2000) Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA 97: 2910–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez A, Toro R, Caceres A, Maccioni RB (1999) Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett 459: 421–426. [DOI] [PubMed] [Google Scholar]
- Anthony JC, Breitner JC, Zandi PP, Meyer MR, Jurasova I, Norton MC, Stone SV (2000) Reduced prevalence of AD in users of NSAIDs and H2 receptor antagonists: the Cache County study. Neurology 54: 2066–2071. [DOI] [PubMed] [Google Scholar]
- Apelt J, Schliebs R (2001) Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res 894: 21–30. [DOI] [PubMed] [Google Scholar]
- Atzori C, Ghetti B, Piva R, Srinivasan AN, Zolo P, Delisle MB, Mirra SS, Migheli A (2001) Activation of the JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J Neuropathol Exp Neurol 60: 1190–1197. [DOI] [PubMed] [Google Scholar]
- Augustinack JC, Sanders JL, Tsai LH, Hyman BT (2002) Colocalization and fluorescence resonance energy transfer between cdk5 and AT8 suggests a close association in pre-neurofibrillary tangles and neurofibrillary tangles. J Neuropathol Exp Neurol 61: 557–564. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. (2000) Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6: 916–919. [DOI] [PubMed] [Google Scholar]
- Bard F, Barbour R, Cannon C, Carretto R, Fox M, Games D, Guido T, Hoenow K, Hu K, Johnson-Wood K, Khan K, Kholodenko D, Lee C, Lee M, Motter R, Nguyen M, Reed A, Schenk D, Tang P, Vasquez N, et al. (2003) Epitope and isotype specificities of antibodies to beta-amyloid peptide for protection against Alzheimer's disease-like neuropathology. Proc Natl Acad Sci USA 100: 2023–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E (1993) Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett 336: 417–424. [DOI] [PubMed] [Google Scholar]
- Bellucci A, Westwood AJ, Ingram E, Casamenti F, Goedert M, Spillantini MG (2004) Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. Am J Pathol 165: 1643–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broe GA, Grayson DA, Creasey HM, Waite LM, Casey BJ, Bennett HP, Brooks WS, Halliday GM (2000) Anti-inflammatory drugs protect against Alzheimer disease at low doses. Arch Neurol 57: 1586–1591. [DOI] [PubMed] [Google Scholar]
- Cohen P, Frame S (2001) The renaissance of GSK3. Nat Rev Mol Cell Biol 2: 769–776. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH (2003) Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40: 471–483. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol 27: 457–464. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C (1993) Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer's disease. Glia 7: 75–83. [DOI] [PubMed] [Google Scholar]
- Dringenberg HC (2000) Alzheimer's disease: more than a “cholinergic disorder”—evidence that cholinergic-monoaminergic interactions contribute to EEG slowing and dementia. Behav Brain Res 115: 235–249. [DOI] [PubMed] [Google Scholar]
- Evans DB, Rank KB, Bhattacharya K, Thomsen DR, Gurney ME, Sharma SK (2000) Tau phosphorylation at serine 396 and serine 404 by human recombinant tau protein kinase II inhibits tau's ability to promote microtubule assembly. J Biol Chem 275: 24977–24983. [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Cole GM, Baird A (1992) Phagocytosis and deposition of vascular beta-amyloid in rat brains injected with Alzheimer beta-amyloid. Am J Pathol 140: 1389–1399. [PMC free article] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM (1998) Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 152: 307–317. [PMC free article] [PubMed] [Google Scholar]
- Funato H, Yoshimura M, Yamazaki T, Saido TC, Ito Y, Yokofujita J, Okeda R, Ihara Y (1998) Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. Am J Pathol 152: 983–992. [PMC free article] [PubMed] [Google Scholar]
- Giovannini MG, Scali C, Prosperi C, Bellucci A, Vannucchi MG, Rosi S, Pepeu G, Casamenti F (2002) Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38MAPK pathway. Neurobiol Dis 11: 257–274. [DOI] [PubMed] [Google Scholar]
- Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KW, Connor K, Melachrino J, O'Callaghan JP, Morgan D (2002) Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp Neurol 173: 183–195. [DOI] [PubMed] [Google Scholar]
- Grammas P, Ovase R (2002) Cerebrovascular transforming growth factor-beta contributes to inflammation in the Alzheimer's disease brain. Am J Pathol 160: 1583–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White III CL, Araoz C (1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 86: 7611–7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Gentleman SM, Graham DI, Mrak RE, Roberts GW (1994) Microglial interleukin-1 alpha expression in human head injury: correlations with neuronal and neuritic beta-amyloid precursor protein expression. Neurosci Lett 176: 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Roberts GW, Mrak RE (1995) Interleukin-1 expression in different plaque types in Alzheimer's disease: significance in plaque evolution. J Neuropathol Exp Neurol 54: 276–281. [DOI] [PubMed] [Google Scholar]
- Hetier E, Ayala J, Denefle P, Bousseau A, Rouget P, Mallat M, Prochiantz A (1988) Brain macrophages synthesize interleukin-1 and interleukin-1 mRNAs in vitro. J Neurosci Res 21: 391–397. [DOI] [PubMed] [Google Scholar]
- Holmlund L, Cortes Toro V, Iverfeldt K (2002) Additive effects of amyloid beta fragment and interleukin-1beta on interleukin-6 secretion in rat primary glial cultures. Int J Mol Med 10: 245–250. [PubMed] [Google Scholar]
- Huell M, Strauss S, Volk B, Berger M, Bauer J (1995) Interleukin-6 is present in early stages of plaque formation and is restricted to the brains of Alzheimer's disease patients. Acta Neuropathol (Berl) 89: 544–551. [DOI] [PubMed] [Google Scholar]
- Humbert S, Dhavan R, Tsai L (2000) p39 activates cdk5 in neurons, and is associated with the actin cytoskeleton. J Cell Sci 113: 975–983. [DOI] [PubMed] [Google Scholar]
- Ishiguro K, Omori A, Sato K, Tomizawa K, Imahori K, Uchida T (1991) A serine/threonine proline kinase activity is included in the tau protein kinase fraction forming a paired helical filament epitope. Neurosci Lett 128: 195–198. [DOI] [PubMed] [Google Scholar]
- Lee MS, Tsai LH (2003) Cdk5: one of the links between senile plaques and neurofibrillary tangles? J Alzheimers Dis 5: 127–137. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405: 360–364. [DOI] [PubMed] [Google Scholar]
- Li Y, Liu L, Barger SW, Griffin WS (2003) Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38–MAPK pathway. J Neurosci 23: 1605–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM (2000) Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci 20: 5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Zhang AH, Li HL, Wang Q, Deng HM, Netzer WJ, Xu H, Wang JZ (2003) Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. J Neurochem 87: 1333–1344. [DOI] [PubMed] [Google Scholar]
- Lund ET, McKenna R, Evans DB, Sharma SK, Mathews WR (2001) Characterization of the in vitro phosphorylation of human tau by tau protein kinase II (cdk5/p20) using mass spectrometry. J Neurochem 76: 1221–1232. [DOI] [PubMed] [Google Scholar]
- Lynch AM, Walsh C, Delaney A, Nolan Y, Campbell VA, Lynch MA (2004) Lipopolysaccharide-induced increase in signalling in hippocampus is abrogated by IL-10—a role for IL-1 beta? J Neurochem 88: 635–646. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, Olschowka JA, Fonseca MI, O'Banion MK, Tenner AJ, Lemere CA, Duff K (2001) Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer's disease. Am J Pathol 158: 1345–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, McGeer EG (1988) Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol (Berl) 76: 550–557. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW (2000) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408: 982–985. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS (2001) Interleukin-1, neuroinflammation, and Alzheimer's disease. Neurobiol Aging 22: 903–908. [DOI] [PubMed] [Google Scholar]
- Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna, Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K (2003) Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38: 555–565. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39: 409–421. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zhou P, Kwon YT, Howley PM, Tsai LH (1998) p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5) is degraded by the ubiquitin-proteasome pathway. J Biol Chem 273: 24057–24064. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402: 615–622. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Grundke-Iqbal I, Iqbal K, Bogdanovic N, Winblad B, Cowburn RF (1998) Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer's disease neurofibrillary degeneration. Brain Res 797: 267–277. [DOI] [PubMed] [Google Scholar]
- Perlmutter LS, Barron E, Chui HC (1990) Morphologic association between microglia and senile plaque amyloid in Alzheimer's disease. Neurosci Lett 119: 32–36. [DOI] [PubMed] [Google Scholar]
- Sasaki A, Shoji M, Harigaya Y, Kawarabayashi T, Ikeda M, Naito M, Matsubara E, Abe K, Nakazato Y (2002) Amyloid cored plaques in Tg2576 transgenic mice are characterized by giant plaques, slightly activated microglia, and the lack of paired helical filament-typed, dystrophic neurites. Virchows Arch 441: 358–367. [DOI] [PubMed] [Google Scholar]
- Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW (2002) Activation of c-Jun N-terminal kinase and p38 in an Alzheimer's disease model is associated with amyloid deposition. J Neurosci 22: 3376–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, et al. (1999) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400: 173–177. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81: 741–766. [DOI] [PubMed] [Google Scholar]
- Shelton SB, Krishnamurthy P, Johnson GV (2004) Effects of cyclin-dependent kinase-5 activity on apoptosis and tau phosphorylation in immortalized mouse brain cortical cells. J Neurosci Res 76: 110–120. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Jones RA, Zhou XQ, McGinness JM, Van Eldik LJ, Mrak RE, Griffin WS (2001) Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer's disease: potential significance for tau protein phosphorylation. Neurochem Int 39: 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE (2003) Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis 14: 133–145. [DOI] [PubMed] [Google Scholar]
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M (1999) Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol 154: 1673–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ (1997) Risk of Alzheimer's disease and duration of NSAID use. Neurology 48: 626–632. [DOI] [PubMed] [Google Scholar]
- Sun W, Qureshi HY, Cafferty PW, Sobue K, Agarwal-Mawal A, Neufield KD, Paudel HK (2002) Glycogen synthase kinase-3beta is complexed with tau protein in brain microtubules. J Biol Chem 277: 11933–11940. [DOI] [PubMed] [Google Scholar]
- Tanda N, Ohyama H, Yamakawa M, Ericsson M, Tsuji T, McBride J, Elovic A, Wong DT, Login GR (1998) IL-1 beta and IL-6 in mouse parotid acinar cells: characterization of synthesis, storage, and release. Am J Physiol 274: G147–G156. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: 572–580. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness Jr VS, Chae T, Harlow E (1994) p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 371: 419–423. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH (2001) A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414: 212–216. [DOI] [PubMed] [Google Scholar]
- Weldon DT, Rogers SD, Ghilardi JR, Finke MP, Cleary JP, O'Hare E, Esler WP, Maggio JE, Mantyh PW (1998) Fibrillar β-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo J Neurosci 18: 2161–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Li J, Chakrabarty P, Bu B, Vincent I (2004) Cyclin-dependent kinase inhibitors attenuate protein hyperphosphorylation, cytoskeletal lesion formation, and motor defects in Niemann-Pick type C mice. Am J Pathol 165: 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Su Y, Li B, Liu F, Ryder JW, Wu X, Gonzalez-DeWhitt PA, Gelfanova V, Hale JE, May PC, Paul SM, Ni B (2003) Nonsteroidal antiinflammatory drugs can lower amyloidogenic Abeta42 by inhibiting Rho. Science 302: 1215–1217. [DOI] [PubMed] [Google Scholar]