

Abstract
In layers 2/3 in the rat visual cortex, glutamatergic synapses, between pyramidal neurons and GABAergic interneurons, show target-specific depression or facilitation. To study the mechanisms regulating these short-term synaptic modifications, we recorded from synaptically connected pyramidal neurons (presynaptic) and multipolar or bitufted interneurons (postsynaptic). Evoked AMPA receptor (AMPAR)- or NMDA receptor (NMDAR)-mediated EPSCs were pharmacologically isolated at these pyramidal-to-interneuron synapses while altering release probability (Pr) by changing the extracellular Ca2+ concentration ([Ca2+]o). At the pyramidal-to-multipolar synapse, which shows paired-pulse depression, elevation of [Ca2+]o from physiological concentrations (2 mm) to 3 mm increased the amplitude of the initial AMPAR-mediated EPSC and enhanced paired-pulse depression. In contrast, the initial NMDAR-mediated EPSC did not change in amplitude with raised Pr nor was paired-pulse depression altered. This lack of an increase of NMDAR-mediated currents is not a result of Ca2+-dependent effects on the NMDAR. Rather, at the pyramidal-to-multipolar synapse, raised Pr increases the transient glutamate concentration at individual release sites, possibly reflecting multivesicular release. In contrast, at the pyramidal-to-bitufted synapse, which shows facilitation, AMPAR- and NMDAR-meditated EPSCs showed parallel increases in response to raised Pr. Thus, our results reveal differential recruitment of AMPA and NMDARs at depressing and facilitating synapses in layers 2/3 of the cortex and suggest that the mechanisms regulating dynamic aspects of synaptic transmission are target specific.
Keywords: glutamatergic synapses, interneurons, short-term synaptic plasticity, glutamate receptors, multivesicular release, NMDA
Introduction
Short-term changes in synaptic efficacy, lasting on the time scale of a millisecond to seconds, are key mechanisms regulating information processing in local circuits (Abbott et al., 1997; Tsodyks and Markram, 1997; Cook et al., 2003). Numerous presynaptic and postsynaptic factors modify synaptic amplitudes and their frequency dependence (Zucker and Regehr, 2002). Although release probability (Pr) defines the efficacy of synaptic activity, other presynaptic factors such as the degree of vesicle emptying (Gandhi and Stevens, 2003), and whether single or multiple vesicles are released (Tong and Jahr, 1994; Wadiche and Jahr, 2001), can alter the amount of transmitter released with each release event. In contrast, postsynaptic factors such as the distribution of postsynaptic receptors relative to the release site (Uteshev and Pennefather, 1996; Franks et al., 2003), the degree of their saturation (Mainen et al., 1999; McAllister and Stevens, 2000; Harrison and Jahr, 2003), and biophysical processes like receptor desensitization (Rozov et al., 2001b) further modify synaptic amplitudes.
In layers 2/3 of the neocortex, GABAergic interneurons can be distinguished into several classes based on well-characterized morphological, biochemical, and electrophysiological properties (Cauli et al., 1997, 2000; Gonchar and Burkhalter, 1997). Two of the most studied types are parvalbumin-positive multipolar (or fast-spiking) and somatostatin-positive bitufted (or regularspiking) interneurons (Thomson and Bannister, 2003). Both types can be identified according to their firing pattern in response to current injection (Reyes et al., 1998; Rozov et al., 2001a). In addition, the excitatory glutamatergic input they receive from local pyramidal neurons shows target-specific short-term plasticity. Pyramidal-to-multipolar synapses exhibit paired-pulse depression (PPD), whereas pyramidal-to-bitufted synapses show paired-pulse facilitation (PPF) (Markram et al., 1998; Reyes et al., 1998; Rozov et al., 2001a). Despite arising from the same pyramidal neuron, both PPD and PPF are presynaptic in origin reflecting strong differences in the initial Pr. At the pyramidal-to-multipolar synapse, the initial Pr is high, with few or no failures, whereas at the pyramidal-to-bitufted synapse, it is extremely low.
In the present study, we recorded from synaptically connected pairs (pyramidal-to-interneurons) in layers 2/3 in the visual cortex of juvenile rats. To study mechanisms of synaptic release dynamics and variability, we pharmacologically isolated postsynaptic currents mediated by different glutamate receptor (GluR) subtypes on the interneuron and altered Pr by changing extracellular Ca2+ concentration ([Ca2+]o). We find that at the pyramidal-to-multipolar synapse, the amplitude of the first AMPA receptor (AMPAR)-mediated EPSCs increased with raised Pr, whereas that of subsequent EPSCs did not, leading to stronger PPD. Surprisingly, the amplitudes of all NMDA receptor (NMDAR)-mediated responses and consequently PPD did not change with elevated [Ca2+]o. We conclude that this differential dependence of postsynaptic AMPAR- and NMDAR-mediated responses to elevated [Ca2+]o arises because increases in Pr at the pyramidal-to-multipolar connections alter the glutamate transient at individual release sites, possibly because of multivesicular release (MVR). In contrast, at the pyramidal-to-bitufted synapse, the dynamics of facilitation involves recruitment of additional release sites with raised Pr. These results suggest that mechanisms regulating the dynamics of vesicle release are fundamentally different between the pyramidal-to-multipolar and pyramidal-to-bitufted synapses.
Materials and Methods
Slice preparation. Sprague Dawley rats [postnatal day 12 (P12) to P16] were deeply anesthetized using halothane (inhalation) and then decapitated in accordance with the institutional animal ethics committee. The brain was dissected out in ice-cold Ringer's solution (composition below). Neocortical slices (transverse; 250-300 μm thick) were cut on a vibratome (Sigma Elektronik, Hüffenhardt, Germany). A hemisphere was glued at the surface of the sagittal plane onto a block, which was mounted at a 30° angle such that the blade cuts from the upper border of the neocortex toward the caudal border and down toward the midline. Slices were incubated for 30 min at 34°C and then at room temperature (22-25°C) until transfer to the recording chamber (22-25°C).
Solutions. The Ringer's solution consisted of the following (in mm): 125 NaCl, 2.5 KCl, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, and 1 MgCl2, pH 7.2 (NaOH), which was saturated with 95% O2/5% CO2 under all conditions. In some instances, where noted, we used the same solution but without added MgCl2 (see Figs. 1, 2). All pharmacological agents were added to the external solutions without substitution. Our standard internal solution consisted of the following (in mm): 105 K-gluconate, 30 KCl, 10 HEPES, 10 phosphocreatine, 4 Mg-ATP, and 0.3 GTP, pH 7.3 (KOH). In some instances, we replaced K-gluconate (10 mm) with equimolar K-glutamate. When recording GluR currents from nucleated patches, our external solution consisted of the following (in mm): 140 NaCl and 10 HEPES, pH 7.2 (NaOH), to which various concentrations of CaCl2 (1-4 mm) were added.
Figure 1.

AMPAR- and NMDAR-mediated EPSCs at the pyramidal-to-multipolar synapse. Simultaneous recordings of presynaptic pyramidal APs (bottom traces) and EPSCs from multipolar interneurons (top traces). EPSCs are an average of ∼100 sweeps. The far left trace in each panel was recorded in the absence of GluR antagonists. In the right two traces, the bathing solution contained either APV (A) (100 μm), an NMDAR competitive antagonist yielding the AMPAR-mediated component, or CNQX (B) (10 μm), an AMPAR competitive antagonist yielding the NMDAR-mediated component. These synapses do not have a significant kainate receptor component (Rozov et al., 2001a). The experimental paradigm was as follows: initial recordings were made in the 2 mm [Ca2+]o Ringer's solution with no GluR antagonists (left traces; control recording) (none of these solutions contained added MgCl2). Subsequently, recordings were made in the same solution with added APV or CNQX (middle traces; baseline recording) and then in 3 mm [Ca2+]o, APV, or CNQX (right traces; test recording) and finally back to 2 mm [Ca2+]o, APV, or CNQX (data not shown) (washout recording).
Figure 2.

Changes in [Ca2+]o differentially altered PPR and EPSC amplitudes as monitored by AMPA and NMDARs. Comparison of the PPR (A, B) and the underlying EPSC amplitudes (C, D) (EPSC1, E1; EPSC2, E2) in a test [Ca2+]o (1, 3, or 4 mm) normalized to that recorded in the same pair in 2 mm [Ca2+]o. All experiments were performed as outlined in Figure 1, isolating either the AMPAR-mediated (A, C) or NMDAR-mediated (B, D) components. Number of pairs, from left to right, 6, 18, 7, 5 (A), and 7, 27, 10, 10 (B). In C and D, recordings are from the same set of pairs as shown in A and B. Solid symbols or bars in this and all subsequent figures indicate values significantly different from control or baseline responses.
Electrophysiology. Membrane potential or currents were recorded at room temperature (22-25°C) using EPC-7 amplifiers with PULSE software (HEKA Elektronik, Lambrecht, Germany), low-pass filtered at 3 kHz, and digitized at 10 kHz. Pipettes had resistances of 5-7 MΩ when filled with the pipette solution and measured in the Ringer's solution. External solutions were bath applied at a perfusion rate of ∼1 ml/min. When recording currents from interneurons, they were clamped at -70 mV. For nucleated patches, external solutions were applied using a piezodriven double-barrel application system. One barrel contained the external solution plus 10 μm glycine, whereas the other barrel contained the same solution with added 200 μm glutamate.
Infrared differential contrast video microscopy-guided selection of pairs of neurons. We initially identified layers 2/3 pyramidal neurons and interneurons morphologically using infrared differential contrast video microscopy with an upright microscope (Olympus BX50WI, fitted with a 40× W/0.80 numerical aperture objective; Olympus Optical, Tokyo, Japan) (Stuart et al., 1993) and subsequently based on their firing patterns (Reyes et al., 1998). Pyramidal neurons had triangular somata with distinct basal and apical dendrites and, when injected with a suprathreshold current step, exhibited a burst of action potentials (APs) that have a slow afterhyperpolarization, peak amplitude adaptation, and burst-frequency accommodation. Multipolar interneurons had round somata with dendrites extending radially and, when injected with a suprathreshold current step, exhibited high-frequency action potentials, which had a fast afterhyperpolarization, no peak amplitude adaptation, and no spike frequency accommodation. Bitufted interneurons had ovoid somata with dendrites extending only in the apical and distal directions and, when injected with a suprathreshold current step, displayed action potentials that also had a fast afterhyperpolarization but did exhibit peak amplitude adaptation and spike frequency accommodation.
Slices were placed in the recording chamber such that the pial surface (dorsal) was right and anterior up. We initially patched an interneuron. Synaptically connected (presynaptic) pyramidal neurons were then searched for around this interneuron. A pyramidal neuron was typically selected such that its main apical dendrite was nearly parallel to the surface and that its apical dendrite and axon were approximately parallel to those of the interneuron. To test for connectivity, we evoked a train of APs (3-5 at 10 Hz) in the presynaptic pyramidal neuron with an injection of suprathreshold current steps and recorded EPSPs. In most cases, the interneuron was reciprocally connected to the pyramidal neuron. We also tested for electrical coupling (i.e., gap junctions) by injecting a long hyperpolarizing current step into the pyramidal cell. In the infrequent instances where the interneuron showed correlated hyperpolarization, we assumed electrical coupling and discarded the pair.
Pharmacological compounds. To examine currents mediated by GluR subtypes in isolation, we blocked NMDARs using either 50-100 μm d(-)-2-amino-5-phosphonopentanoic acid (APV) or 1 mm MgCl2 and AMPARs using 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) added to the Ringer's (bath) solution. In some instances, we added cyclathiozide (CTZ; 30-50 μm) to the bath solution to reduce AMPAR desensitization. All reagents, unless otherwise noted, were obtained from either Sigma Aldrich (St. Louis, MO) or Tocris Cookson (Ellisville, MO).
Data collection and analysis. The stimulus intensity in presynaptic pyramidal neurons was adjusted until the current necessary to produce an all-or-none response was minimized. During a single sweep, pyramidal neurons were injected with two to five suprathreshold current pulses (width, 10 ms) at 10 Hz. We recorded either EPSPs or EPSCs. Typically, 50-120 sweeps were recorded per each recording condition. Within an individual sweep, postsynaptic responses were aligned based on the timing of the initial presynaptic action potential in the pyramidal neuron. Sweeps that showed epileptic activity were discarded. Unless otherwise noted, displayed recordings are averages of recorded sweeps. Our typical experimental protocol included a baseline recording, a test recording, and then a washout recording in which we returned the bath solution to that used during the baseline recording. We did not include in the final analysis pairs in which the average current amplitude in the washout was not at least 70% of that in the baseline recording.
Analysis of individual sweeps. We used the average of the initial 100 ms of an individual sweep as the baseline. To estimate the noise, 10 500-μs windows were randomly selected in the baseline, and local maxima within each window were determined and averaged as the noise. We only sampled noise in the positive direction from the baseline to prevent spontaneous EPSCs from affecting our noise estimate and assumed that noise is of equal magnitude in the positive and negative directions.
Using the averaged record as a template, we defined a time window to determine the EPSC amplitude of an individual sweep. The local minima within the window was identified and then a 1-3 ms sample, encompassing the local minima, was averaged to generate peak EPSC amplitude. An EPSC amplitude less than that of noise was classified as a failure.
Statistical analysis. All curve fitting was done using Igor Pro (WaveMetrics, Lake Oswego, OR). Results are reported as mean ± SEM and shown graphically as mean ± 2 SEM. The Student's t test and an ANOVA were used to test for statistical differences with the Tukey test used for multiple comparisons. Significance was assumed if p < 0.05.
Results
Short-term synaptic depression at the pyramidal-to-multipolar synapse in physiological Ca2+ concentrations
Multipolar interneurons and pyramidal neurons were identified by the infrared video microscopy image based on their appearance, location, and shape of the cell body and subsequently on firing patterns. In all cases where they were synaptically connected, EPSCs recorded in the multipolar interneurons (Fig. 1A,B, left traces), elicited by trains of APs in the presynaptic pyramidal neuron, were always depressing as found previously (Reyes et al., 1998; Rozov et al., 2001a) (Table 1, control recordings). Initially, we wanted to test whether AMPAR- and NMDAR-mediated components of the EPSCs undergo the same level of PPD. We therefore recorded evoked EPSCs from multipolar interneurons either in the presence of APV or CNQX isolating AMPAR- and NMDAR-mediated synaptic currents, respectively.
Table 1.
EPSC amplitudes and PPRs at the pyramidal-to-multipolar synapse
|
|
AMPAR |
NMDAR |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
Amplitude (pA) |
PPR |
n
|
Amplitude (pA) |
PPR |
n
|
||||
| Control recording | −47.9 ± 8.6 | 0.57 ± 0.04 | 19 | −53.0 ± 7.9 | 0.60 ± 0.03 | 36 | ||||
| GluR component (Baseline recording) |
−49.0 ± 14.2 |
0.58 ± 0.04 |
|
−22.3 ± 3.9 |
0.60 ± 0.04 |
|
||||
Values shown are mean ± SEM. Initial recordings were made in the 2 mm [Ca2+]o Ringer's solution with no GluR antagonists (Control recording) (none of these solutions contained added MgCl2). Subsequently, recordings were made in the same solution with added APV or CNQX (Baseline recording) and then in various [Ca2+]o, APV, or CNQX (test recording). Test recordings for most of the same pairs are summarized in Figures 1 and 2.
Both AMPAR- and NMDAR-mediated EPSCs strongly depressed at 10 Hz. For the AMPAR-mediated component in 2 mm [Ca2+]o (Fig. 1A, middle trace), the amplitude of EPSC1 (approximately -49 pA) (Table 1) was comparable with that recorded from the same pairs before the application of APV (approximately -48 pA), as was the paired-pulse ratio (PPR) (EPSC2/EPSC1) (0.58 ± 0.04 and 0.57 ± 0.04, respectively) (mean ± SEM). For the NMDAR-mediated component (Fig. 1B, middle trace), the amplitude of EPSC1 (approximately -22 pA) was less than half of that recorded in the absence of CNQX (approximately -53 pA). Still, the PPR for this NMDAR-mediated component (0.60 ± 0.04) was identical to that recorded in the absence of CNQX (0.60 ± 0.03). The similarity of the PPR suggests that neither APV nor CNQX interfere with synaptic release and that the AMPA and NMDARs are seeing the same pool of presynaptically released glutamate.
Changes in [Ca2+]o differentially alter PPRs as monitored by AMPA or NMDARs
Assuming AMPA and NMDARs have a homogeneous subsynaptic distribution and are equally sensitive to changes in the transient glutamate concentration, one would expect changing Pr should equally affect the AMPAR- and NMDAR-mediated EPSCs. To alter Pr, we changed [Ca2+]o from an approximate physiological concentration (2 mm) to 1, 3, or 4 mm. The far right traces in Figure 1, A and B, show example recordings of the AMPAR- and NMDAR-mediated EPSCs in 3 mm [Ca2+]o. Consistent with an increase in Pr, the amplitude of the AMPAR-mediated EPSC1 (Fig. 1A) increased with elevated [Ca2+]o (to -98 pA compared with -68 pA in 2 mm [Ca2+]o in the example shown). Correspondingly, PPR was smaller in 3 mm (≈0.38) than in 2 mm (≈0.51) [Ca2+]o, yielding a PPR3Ca/PPR2Ca of ∼0.75. In contrast and quite surprisingly, the same manipulation did not increase the amplitude of the NMDAR-mediated EPSC1 (-48 pA in 3 mm [Ca2+]o and -49 pA in 2 mm [Ca2+]o) nor did it alter the PPR (0.52 and 0.55, respectively), yielding a PPR3Ca/PPR2Ca close to unity.
Figure 2 summarizes average PPRs (Fig. 2A,B) and relative EPSC amplitudes (Fig. 2C,D) for the AMPAR-mediated (Fig. 2A,C) and NMDAR-mediated (Fig. 2B,D) EPSCs measured in various test [Ca2+]o (1, 3, or 4 mm). For all plots, because of the variability in the magnitude of PPRs and EPSC amplitudes between pairs, we normalized the test [Ca2+]o values to those recorded in 2 mm [Ca2+]o within the same pair.
For the AMPAR- and NMDAR-mediated EPSCs, PPD was weaker in 1 than in 2 mm [Ca2+]o, yielding a PPRtest/PPR2Ca greater than unity. This weaker PPD in 1 mm [Ca2+]o arises because, whereas the amplitude for the first and second AMPAR- and NMDAR-mediated EPSCs are reduced, EPSC2 is less affected. For AMPARs, PPD was stronger in 3 or 4 mm [Ca2+]o than in 2 mm [Ca2+]o, yielding a PPRtest/PPR2Ca less than unity. The stronger PPD in the higher [Ca2+]o for AMPARs arises because EPSC1 was significantly greater in amplitude in 3 and 4 mm [Ca2+]o than in 2 mm, whereas EPSC2 was unchanged. In contrast to the AMPAR-mediated component, PPRs and EPSC amplitudes for NMDARs were comparable in 3 or 4 mm relative to those in 2 mm [Ca2+]o.
In summary, the results shown in Figure 2 suggest that the AMPAR- and NMDAR-mediated postsynaptic responses, presumably to the same pool of presynaptically released glutamate, are different. For the AMPAR-mediated EPSCs, the raised Pr increases the amplitude of EPSC1 and, correspondingly, leads to a stronger PPD (Zucker et al., 2002). One possible explanation for this observation is that with elevated Pr, additional release sites are recruited during the initial release event. However, such an interpretation is inconsistent with the surprising observation that the amplitude of NMDAR-mediated EPSC1 was unaffected under the same conditions. It is possible that the elevated glutamate transient produced by the raised Pr can be sensed by AMPARs but not by NMDARs, the latter resulting from receptor saturation. Nevertheless, other alternatives have to be considered.
Analysis of individual AMPAR- and NMDAR-mediated EPSC amplitudes
The results in Figures 1 and 2 are derived from averages of at least 100 sweeps per condition. Correspondingly, unanticipated changes in the amplitudes or frequency of failures of individual events with the different [Ca2+]o might underlie the difference between the AMPAR- and NMDAR-mediated EPSC1. However, as shown in Figure 3 and summarized in Table 2, the pattern of amplitude histograms is consistent with the average responses. The amplitude distribution histograms for AMPAR-mediated EPSC1 (Fig. 3A) showed a clear rightward shift when [Ca2+]o was elevated from 2 mm (top left) to 3 mm (bottom left). Similarly, both amplitude distribution histogram and the mean value for EPSC2 were comparable at 2 mm (top right) and 3 mm (bottom right) [Ca2+]o, although shifted leftward relative to EPSC1. In contrast, raising [Ca2+]o did not significantly change the amplitude distributions of NMDAR-mediated EPSC1 or EPSC2 compared with those obtained in 2 mm [Ca2+]o (Fig. 3B). In addition, synaptic failure rates were very low and comparable for both AMPAR- and NMDAR-mediated EPSC1 (Table 2).
Figure 3.
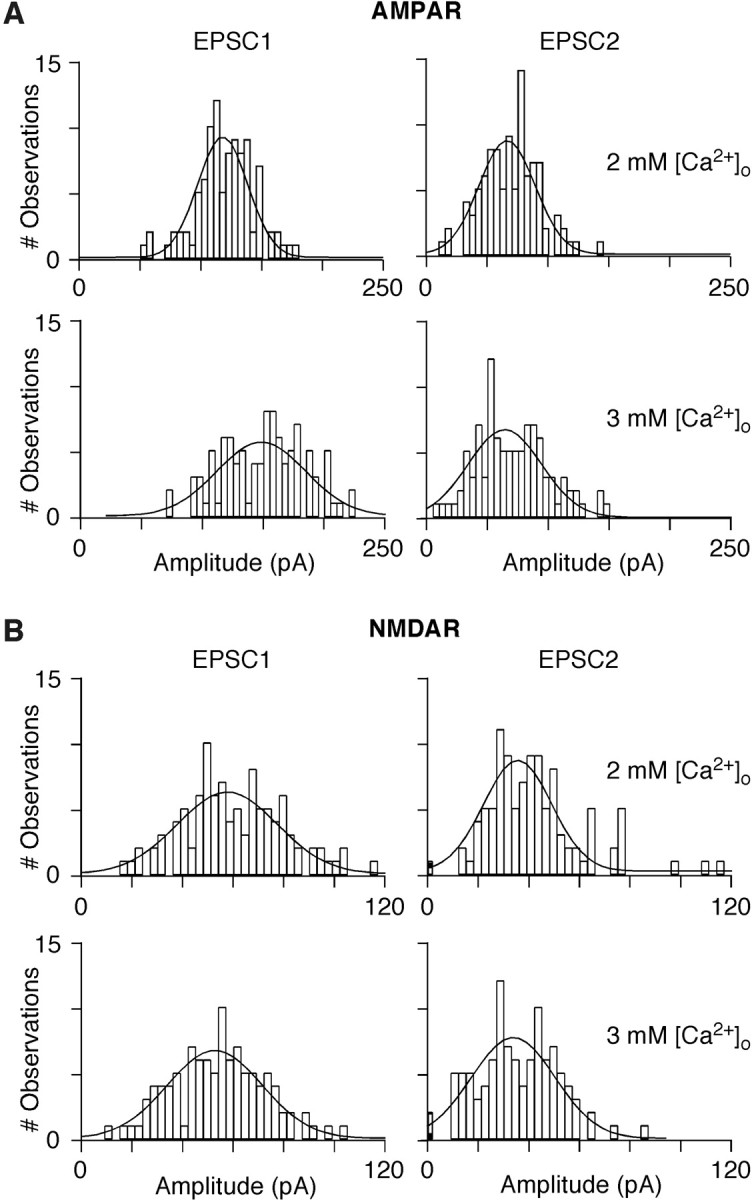
Amplitude histograms of individual EPSCs mediated by AMPA and NMDARs. A comparison of individual EPSC amplitudes for pyramidal-to-multipolar pairs recorded in APV (A) or CNQX (B). Within a panel, the amplitudes for EPSC1 (left column) and EPSC2 (right column) are shown in 2 mm [Ca2+]o (top row) or in 3 mm [Ca2+]o (bottom row). Bin width is 5 pA for the AMPAR and 3 pA for the NMDAR component. Failures are shown in shaded boxes. Number of failures (amplitudes were within noise)/trials: 2 mm [Ca2+]o: EPSC1, 0/100; EPSC2, 0/100; 3 mm [Ca2+]o: EPSC1, 0/100; EPSC2, 0/100 (A); 2 mm [Ca2+]o, EPSC1, 0/104; EPSC2, 1/104; 3 mm [Ca2+]o: EPSC1, 0/104; EPSC2, 2/104 (B). Continuous lines represent Gaussian fits.
Table 2.
Summary of individual AMPAR-and NMDAR-mediated EPSCs at the pyramidal-to-multipolar synapse
|
|
[Ca2+]o |
Normalized |
||
|---|---|---|---|---|
|
|
2 mm
|
3 mm
|
(3 mm [Ca2+]o/2 mm [Ca2+]o) |
|
| AMPAR |
|
|
|
|
| EPSC1 | ||||
| Amplitude (pA) | −66.3 ± 21.1 | −88.9 ± 30.7 | 1.26 ± 0.06 | |
| Failures (%) | 10 ± 8 | 7 ± 6 | ||
| CV | 0.35 ± 0.08 | 0.30 ± 0.05 | ||
| EPSC2 | ||||
| Amplitude (pA) | −39.2 ± 13.4 | −45.8 ± 19.3 | 1.05 ± 0.08 | |
| Failures (%) | 20 ± 9 | 20 ± 10 | ||
| CV |
0.46 ± 0.05 |
0.43 ± 0.03 |
|
|
| NMDAR |
|
|
|
|---|---|---|---|
| EPSC1 | |||
| Amplitude (pA) | −40.4 ± 5.8 | −39.7 ± 5.9 | 0.98 ± 0.07 |
| Failures (%) | 6 ± 4 | 3 ± 1 | |
| CV | 0.36 ± 0.03 | 0.34 ± 0.01 | |
| EPSC2 | |||
| Amplitude (pA) | −25.5 ± 3.9 | −23.9 ± 3.4 | 0.96 ± 0.07 |
| Failures (%) | 19 ± 8 | 21 ± 8 | |
| CV |
0.44 ± 0.03 |
0.43 ± 0.01 |
|
Values shown are mean ± SEM. Individual AMPAR-and NMDAR-mediated EPSCs were recorded in 2 or 3 mm [Ca2+]o (n = 8) (see Materials and Methods for details). Amplitudes exclude failures. Failures are shown as a percentage (typically 200-400 sweeps per pair).
Another alternative to account for the differential effect of raised Pr on AMPAR- and NMDAR-mediated currents is that AMPA and NMDARs are located at different synapses and therefore are activated by different pools of presynaptically released glutamate. However, the coefficient of variance (CV = SD/mean) values of individual EPSC1 as well as EPSC2 were almost identical for AMPAR- and NMDAR-mediated currents (Table 2) and did not change significantly when [Ca2+]o was increased from 2 to 3 mm [Ca2+]o. Thus, based on these results, we can conclude that, first, no new release sites with a new pool of vesicles are being recruited in response to raised Pr, and second, both AMPARs and NMDARs are colocalized at the same synapses and are exposed to the same concentration of glutamate during synaptic transmission.
The lack of an increase in the NMDAR-mediated EPSC1 is not attributable to Ca2+-dependent effects on NMDARs
Ca2+ can directly or indirectly decrease NMDAR-mediated currents via channel block by extracellular Ca2+ (Jahr and Stevens, 1993) or inactivation by intracellular Ca2+ (Westbrook et al., 1997). Hence, an increase in the NMDAR-mediated EPSC1 caused by raised Pr (induced by an elevated [Ca2+]o) could be counteracted by channel block and/or Ca2+-dependent inactivation. We therefore tested for the effect of changes in extracellular and intracellular Ca2+ on NMDAR-mediated currents in multipolar interneurons (Fig. 4).
Figure 4.

Effect of extracellular and intracellular Ca2+ on NMDAR in multipolar interneurons. A, Recordings of NMDAR-mediated currents in nucleated patches taken from presumed multipolar interneurons. Patches were bathed either in 2 mm [Ca2+]o (thick traces) before and after a recording in 1 mm (left) or 3 mm (right) (lighter traces) [Ca2+]o. Currents were elicited by glutamate (200 μm; horizontal bar) at a holding potential of -60 mV. In all instances, the external solution contained added glycine (10 μm) and CNQX (10 μm). B, Average relative peak current amplitudes in a test [Ca2+]o (1, 3, or 4 mm) (Ipeak(test)) normalized to that in 2 mm [Ca2+]o. Number of recordings, from left to right: 5, 5, 4. C, Simultaneous recordings of presynaptic pyramidal APs (bottom traces) and average EPSCs from multipolar interneurons (top traces) in the presence of CNQX (10 μm). The far left trace was recorded in the 2 mm [Ca2+]o Ringer's solution (without added MgCl2) using our standard (no Ca2+ buffer) pipette solution. Subsequently, the patch-pipette recording of the postsynaptic multipolar interneuron was replaced with one containing the same internal solution plus 2 mm BAPTA. After an ∼5 min wait period, EPSCs were initially recorded in 2 mm (center trace) and then in 3 mm (right trace) [Ca2+]o as in Figure 1 A. D, Amplitude of EPSC1 normalized to that in 2 mm [Ca2+]o. Left bar, EPSC1test was recorded in 2 mm [Ca2+]o with 2 mm BAPTA in the pipette and was normalized to records with 0 BAPTA in the pipette (n = 6). Right bar, EPSC1test was recorded in 3 mm [Ca2+]o and 2 mm BAPTA and normalized to records with 2 mm BAPTA in the pipette (n = 6). E, Paired-pulse ratio under the different recording conditions shown in C.
Figure 4A shows NMDAR-mediated currents recorded from nucleated patches taken from multipolar interneurons in various [Ca2+]o (1-4 mm) in the application solution. Reducing [Ca2+]o from 2 to 1 mm caused a 19% increase in current amplitudes (Fig. 4B). In contrast, current amplitudes were not significantly different in 2 or 3 (or 4) mm [Ca2+]o. Therefore, assuming somatic and synaptic NMDARs in multipolar interneurons are identical, channel block by Ca2+ already reaches saturation at 2 mm. Hence, this process would not further affect current amplitudes at higher [Ca2+]o concentrations.
To test for Ca2+-dependent inactivation at the pyramidal-to-multipolar synapse, we initially recorded NMDAR-mediated EPSCs in 2 mm [Ca2+]o using our standard internal solution (Fig. 4C, left trace). We then repatched the postsynaptic multipolar interneuron with a pipette in which the internal solution contained 2 mm BAPTA, a fast Ca2+ buffer (middle trace). In five of six pairs, the NMDAR-mediated EPSC1 amplitude was potentiated (61 ± 29%; n = 5), suggesting that postsynaptic Ca2+ might tonically inhibit NMDARs possibly via Ca2+-dependent inactivation. However, when [Ca2+]o was increased from 2 to 3 mm with BAPTA in the pipette, the amplitude of EPSC1 remained unaffected (Fig. 4C, right trace, D). Similarly, as in the absence of internal BAPTA, the PPR was indistinguishable in 2 and 3 mm [Ca2+]o (Fig. 4E). Hence, although Ca2+-dependent inactivation may modulate NMDAR-mediated synaptic amplitudes, it does not contribute to the lack of an increase in the NMDAR-mediated EPSC1 amplitude with elevated [Ca2+]o.
In summary, channel block and Ca2+-dependent inactivation may affect the amplitude of NMDAR-mediated EPSCs at the pyramidal-to-multipolar synapse. Still, these processes are at saturation in 2 mm [Ca2+]o and, therefore, do not limit current amplitudes at higher [Ca2+]o.
Elevated [Ca2+]o does not alter release latency
At the pyramidal-to-multipolar synapse, the distribution of the latency of the postsynaptic response, following the presynaptic AP, is quite narrow (Rozov et al., 2001a). Still, elevated [Ca2+]o might further synchronize vesicle release from different release sites, a process that would mainly influence the amplitude of the fast-rising AMPAR but not the slow-rising NMDAR-mediated responses. However, the latency of the AMPAR-mediated EPSC1, measured as the time difference between the peak of the presynaptic AP and the peak of the postsynaptic response, was the same in 2 mm (2.38 ± 0.33 ms; n = 7) and 3 mm (2.34 ± 0.33 ms) [Ca2+]o, nor was there any difference in the CV (0.14 ± 0.03 and 0.13 ± 0.03, respectively). Thus, the significant increase of the amplitude of AMPAR-mediated EPSC1 at elevated [Ca2+]o is not attributable to enhanced synchronicity of vesicle release.
Changes in the glutamate transient at pyramidal-to-multipolar release sites
At the pyramidal-to-multipolar synapse, elevation of [Ca2+]o might lead to an increase in the amount of glutamate released at an individual release site. This hypothesis would be consistent with the selective enhancement of AMPAR-mediated EPSCs, assuming that at 2 mm [Ca2+]o, the glutamate concentration in the cleft is already high enough to saturate NMDARs. To test how the transient glutamate concentration in the synaptic cleft depends on [Ca2+]o and whether it can be dynamically regulated during a train of APs, we used the low-affinity AMPAR competitive antagonist γ-d-glutamylglycine (γ-DGG) (Liu et al., 1999; Wadiche and Jahr, 2001). Because of its low affinity, γ-DGG rapidly unbinds from AMPARs permitting ambient glutamate to rebind. Hence, AMPAR-mediated EPSCs arising from a higher relative glutamate concentration would be inhibited less effectively by γ-DGG. To exclude the possible contribution of channel desensitization to EPSC amplitudes, we performed these experiments in the presence of CTZ, an allosteric modulator that reduces AMPAR desensitization (Partin et al., 1994). Although CTZ alters release probability at certain synapses (Diamond and Jahr, 1995; Ishikawa and Takahashi, 2001), it apparently does not do so at the pyramidal-to-multipolar synapse (Rozov et al., 2001b). Indeed, the PPR was not significantly different in the absence or presence of CTZ either in 2 mm [Ca2+]o (0.55 ± 0.04, n = 10, and 0.55 ± 0.04, respectively) or in 3 mm [Ca2+]o (0.40 ± 0.04, n = 15, and 0.44 ± 0.05, respectively).
Figure 5A shows AMPAR-mediated EPSCs at the pyramidal-to-multipolar synapse in 3 mm [Ca2+]o either in the absence (left trace) or presence (right two traces) of CTZ (50 μm). CTZ (middle trace) both increased the amplitudes and slowed the decay of AMPAR-mediated EPSCs. Application of γ-DGG (0.5 mm; far right trace) reduced the amplitudes of both EPSC1 and EPSC2. Figure 5B summarizes the percentage block by γ-DGG of EPSC1 and EPSC2 under various conditions. The percentage block of EPSC1 was significantly less than that of EPSC2 both in 2 mm [Ca2+]o (27 ± 2 vs 32 ± 2%; n = 9; p < 0.02) and in 3 mm [Ca2+]o (21 ± 2 vs 32 ± 1%; n = 8; p < 0.0009), although this difference was more notable in 3 mm [Ca2+]o. These results are consistent with the idea that the cleft concentration of glutamate can be greater during the first AP in the train and declines with subsequent ones. In addition, the stronger differential effect of γ-DGG in 3 mm [Ca2+]o, relative to that in 2 mm [Ca2+]o, arises solely because of a reduced block for EPSC1. Indeed, the percentage block of EPSC1 in 3 mm [Ca2+]o was significantly less than that in 2 mm [Ca2+]o (p < 0.04). The preferential effect of γ-DGG on EPSC1 supports the idea of an increased glutamate transient with elevated [Ca2+]o.
Figure 5.

Differential effect of γ-DGG on AMPAR-mediated EPSCs. A, Average AMPAR-mediated EPSCs from multipolar interneurons (top traces) recorded in the 3 mm [Ca2+]o Ringer's solution (with added 1 mm MgCl2) either in the absence (left trace) or presence (right two traces) of CTZ (50 μm). γ-DGG (0.5 mm; far right trace) was applied in the presence of CTZ. B, Percentage block of EPSC1 and EPSC2 by γ-DGG recorded either in 2 or 3 mm [Ca2+]o and CTZ. Number of pairs, 2 mm [Ca2+]o (n = 9) and 3 mm [Ca2+]o (n = 8). The various symbols indicate significant differences (t test) in the percentage block between EPSC1 and EPSC2 in 2 mm [Ca2+]o (*p < 0.02), EPSC1 and EPSC2 in 3 mm [Ca2+]o (**p < 0.0009), or EPSC1 in 2 mm [Ca2+]o and EPSC1 in 3 mm [Ca2+]o (#p < 0.04).
To verify that the differential effect of γ-DGG on EPSC1 and EPSC2 was not a result of CTZ, we performed the same experiments in 3 mm [Ca2+]o, although in the absence of CTZ. The percentage block by γ-DGG of EPSC1 (43 ± 5%; n = 6) and EPSC2 (54 ± 3%) was again significantly different (p < 0.02).
Short-term synaptic facilitation at the pyramidal-to-bitufted synapse
Pyramidal neurons in layers 2/3 also innervate somatostatin-positive bitufted interneurons. This connection is characterized by a low initial Pr and strong short-term synaptic facilitation (Reyes et al., 1998; Rozov et al., 2001a). With high-frequency stimulation, Pr increases with every subsequent AP. Therefore, to contrast pyramidal-to-bitufted and pyramidal-to-multipolar synapses (the latter showing a high initial Pr), we used a 10 Hz train of five APs for the pyramidal-to-bitufted connection ensuring that Pr would be high during later release events.
Figure 6 shows AMPAR-mediated (Fig. 6A) and NMDAR-mediated (Fig. 6B) EPSCs recorded either in 2 mm (left traces) or 3 mm (right traces) [Ca2+]o from a bitufted interneuron in response to five pyramidal APs. Both AMPAR- and NMDAR-mediated components showed strong facilitation. In 2 mm [Ca2+]o, for example, the amplitude of EPSC2 relative to EPSC1 was increased by 45 ± 10% (n = 10) and 28 ± 7% (n = 8) for the AMPAR- and NMDAR-mediated components, respectively. Although the degree of relative enhancement, measured as the EPSCn/EPSCn-1 ratio, was greatest for EPSC2, facilitation also occurred for all subsequent EPSCs (Fig. 6C). Consistent with facilitation being presynaptic in origin (Reyes et al., 1998; Rozov et al., 2001a), it was accompanied by a strong reduction in the failure rate (Table 3, 2 mm [Ca2+]o).
Figure 6.
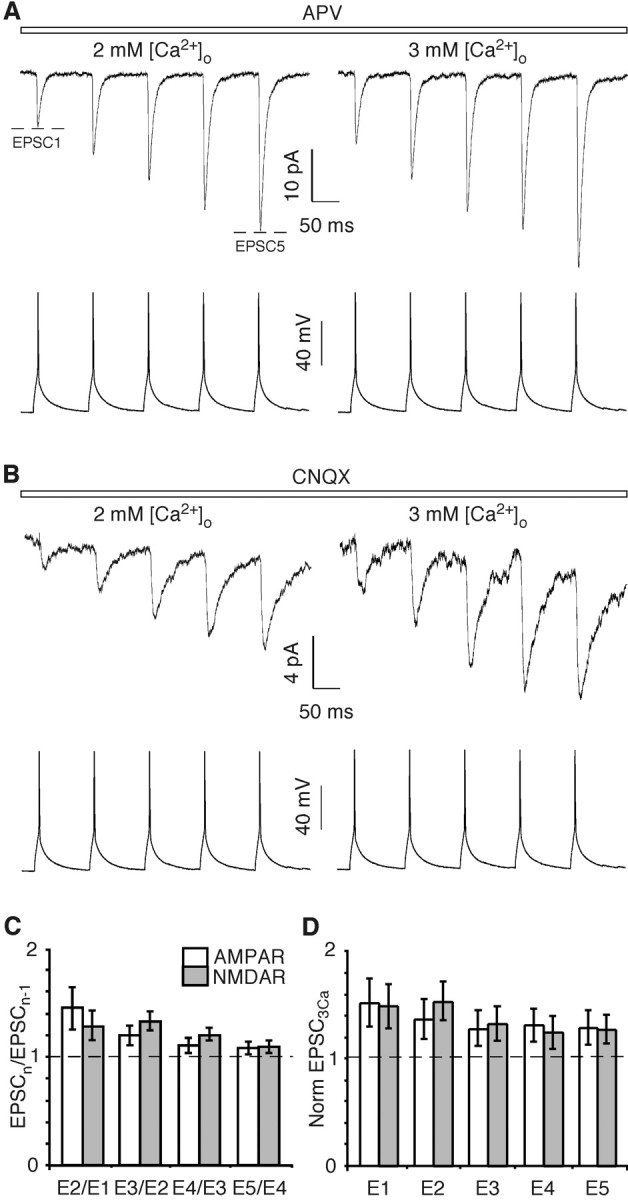
Effect of increases in [Ca2+]o on AMPAR- and NMDAR-mediated EPSCs at the pyramidal-to-bitufted synapse. A, B, Simultaneous recordings of presynaptic pyramidal APs (bottom traces) and average AMPAR-mediated (A) and NMDAR-mediated (B) EPSCs from bitufted interneurons (top traces) in either 2 mm (left) or 3 mm (right) [Ca2+]o. Experimental protocol, solutions, and analysis are as in Figure 1, except that five presynaptic APs were stimulated (at 10 Hz). C, Ratio of the amplitude of successive AMPAR-mediated (n = 10) or NMDAR-mediated (n = 8) EPSCs. Recordings were made in 2 mm [Ca2+]o. All ratios were significantly greater than unity, with the AMPAR-mediated E2/E1 ratio significantly different from the others. D, AMPAR- and NMDAR-mediated EPSC amplitudes in 3 mm [Ca2+]o normalized to that recorded in the same pair in 2 mm [Ca2+]o. All ratios were significantly greater than unity but were not significantly different from each other.
Table 3.
Summary of individual AMPAR-mediated EPSCs at the pyramidal-to-bitufted synapse
|
|
[Ca2+]o |
Normalized |
||
|---|---|---|---|---|
|
|
2 mm
|
3 mm
|
(3 mm [Ca2+]o/2 mm [Ca2+]o) |
|
| EPSC1 | ||||
| Amplitude (pA) | −17.1 ± 3.2 | −19.7 ± 4.1 | 1.15 ± 0.04 | |
| Failures (%) | 39 ± 6 | 26 ± 8 | 0.55 ± 0.11 | |
| Failures (norm) | 1.00 | |||
| EPSC2 | ||||
| Amplitude (pA) | −20.0 ± 3.8 | −21.9 ± 3.4 | 1.15 ± 0.06 | |
| Failures (%) | 30 ± 6 | 18 ± 6 | 0.49 ± 0.07 | |
| Failures (norm) | 0.70 ± 0.06 | |||
| EPSC3 | ||||
| Amplitude (pA) | −21.3 ± 3.7 | −23.2 ± 3.4 | 1.13 ± 0.06 | |
| Failures (%) | 27 ± 6 | 17 ± 6 | 0.47 ± 0.10 | |
| Failures (norm) | 0.61 ± 0.08 | |||
| EPSC4 | ||||
| Amplitude (pA) | −22.5 ± 3.8 | 25.3 ± 3.9 | 1.15 ± 0.06 | |
| Failures (%) | 24 ± 6 | 15 ± 6 | 0.51 ± 0.09 | |
| Failures (norm) | 0.54 ± 0.08 | |||
| EPSC5 | ||||
| Amplitude (pA) | −23.7 ± 4.1 | −26.3 ± 3.8 | 1.16 ± 0.05 | |
| Failures (%) | 20 ± 5 | 12 ± 5 | 0.47 ± 0.12 | |
| Failures (norm) |
0.44 ± 0.07 |
|
|
|
Values shown are mean ± SEM. Individual AMPAR-mediated EPSCs were recorded in 2 or 3 mm [Ca2+]o (n = 10). Amplitudes exclude failures. The number of failures is shown as a percentage of the total number of sweeps (typically 200-300 per pair) or normalized either to the failures in EPSC1 (2 mm [Ca2+]o) or to that for an EPSC in 2 mm [Ca2+]o (far right column). For each EPSC, the CV was ∼0.60 in 2 mm [Ca2+]o and 0.55 in 3 mm [Ca2+]o but were not significantly different.
At the pyramidal-to-multipolar synapse, which has a high initial Pr and is depressing, elevation of [Ca2+]o increased the amplitude of the initial EPSC, specifically that mediated by AMPARs but not subsequent AMPAR-mediated EPSCs nor those mediated by NMDARs. In contrast, at low initial Pr facilitating synapses (pyramidal-to-bitufted), raising [Ca2+]o from 2 to 3 mm substantially increased relative amplitudes of all EPSCs recorded and enhanced both the AMPAR- and NMDAR-mediated EPSCs equally well, the relative change of which depends only on the number of the response in the train (Fig. 6D). For instance, AMPAR- and NMDAR-mediated EPSCs are enhanced by 53 ± 11 and 49 ± 10%, respectively, for EPSC13Ca/EPSC12Ca and similarly indistinguishable for EPSC53Ca/EPSC52Ca, although not so strongly enhanced (29 ± 8 and 28 ± 7%, respectively).
The most likely explanation for the parallel increase in the AMPAR- and NMDAR-mediated EPSCs amplitudes in response to a raised Pr is the recruitment of additional release sites. Consistent with this idea was the strong and uniform reduction in the failure rate in 3 mm [Ca2+]o compared with that in 2 mm [Ca2+]o for each EPSC (Table 3).
γ-DGG has no differential effect on AMPAR-meditated EPSCs at the pyramidal-to-bitufted synapse
Based on the above results, it seems likely that at the pyramidal-to-bitufted synapse, raised Pr resulting from either elevated [Ca2+]o or repetitive stimulation, enhances current amplitudes by recruiting additional release sites. In addition, however, raised Pr might also increase the glutamate transient at individual release sites like at the pyramidal-to-multipolar synapse. To test this possibility, we again took advantage of γ-DGG. Figure 7A shows AMPAR-mediated EPSCs at the pyramidal-to-bitufted synapse either in the absence or presence of γ-DGG (0.5 mm) recorded in 3 mm [Ca2+]o and CTZ, conditions identical to those used at the pyramidal-to-multipolar synapse (compare Fig. 5). At the pyramidal-to-bitufted synapse, application of CTZ increased the amplitude of EPSC1 more dramatically (120 ± 19%; n = 7) than that at the pyramidal-to-multipolar (25 ± 8%; n = 15). However, like at the pyramidal-to-multipolar synapse, CTZ had a comparable potentiating effect on all EPSCs such that facilitation of each EPSCn/EPSCn-1 ratio remained the same (data not shown).
Figure 7.

Effect of γ-DGG on AMPAR-mediated EPSC amplitudes. A, Average EPSCs from bitufted interneurons (top traces) recorded in the 3 mm [Ca2+]o Ringer's solution (with added 1 mm MgCl2). All shown traces were recorded in the presence of CTZ (50 μm). γ-DGG (0.5 mm; right trace) was applied in the presence of CTZ. B, Percentage block of EPSCs by γ-DGG recorded in 3 mm [Ca2+]o and CTZ (n = 7). None of the values were significantly different.
As illustrated in Figure 7A and summarized in Fig. 7B, γ-DGG reduced the amplitude of all EPSCs by approximately the same amount, even for EPSC5 in which Pr is relatively high. This result suggests that the relative glutamate concentration is comparable at all active release sites throughout the facilitation process in contrast to what is observed at the pyramidal-to-multipolar synapse. It also is consistent with the idea that potentiation of EPSCs by raising Pr is mainly attributable to increasing the number of active sites.
Discussion
To study dynamic mechanisms at pyramidal-to-interneuron synapses, we altered [Ca2+]o, starting with approximate physiological concentrations (2 mm), and assumed that the major action of this manipulation was to change efficacy of release. However, Ca2+ could also have some postsynaptic effects because, for instance, NMDARs undergo numerous Ca2+-dependent modifications (Jahr and Stevens, 1993; Westbrook et al., 1997). In the case of the pyramidal-to-multipolar synapse, we found that changes in [Ca2+]o did alter NMDAR channel block by Ca2+, but that this effect was maximal at 2 mm [Ca2+]o (Fig. 4B). Additionally, intracellular Ca2+ may produce some tonic inhibition of NMDARs, but elevated [Ca2+]o does not produce any greater inhibition (Fig. 4D). Hence, the main effect of elevating [Ca2+]o, at least on going from 2 mm to higher concentrations, is a change in Pr.
At the pyramidal-to-multipolar synapse, the amplitude of the AMPAR-mediated EPSC1 was very sensitive to raised Pr, whereas AMPAR-mediated EPSC2 as well as all those mediated by NMDARs were not. Correspondingly, factors that would increase Pr at the pyramidal-to-multipolar synapse would selectively augment the contribution of AMPARs, but not NMDARs, to synaptic transmission specifically during the initial release event in a train of APs. In contrast, at the pyramidal-to-bitufted synapse, the amplitudes of both AMPAR- and NMDAR-mediated EPSC1 as well as all subsequent EPSCs increased in parallel with raised Pr (Fig. 6), primarily because of decreases in failure rates (Table 3). Both of these lines of evidence along with the γ-DGG experiments (Fig. 7), where differences in glutamate transient were not observed, indicate that raised Pr at the pyramidal-to-bitufted synapse increases the number of active release sites with both AMPARs and NMDARs recruited equally.
Mechanism underlying the differential increase in the AMPAR- and NMDAR-mediated EPSC1 at the pyramidal-to-multipolar synapse
Various alternatives might account for the differential effect of raised Pr on the AMPAR- and NMDAR-mediated EPSC1 at the pyramidal-to-multipolar synapse. One possibility is that, like at the pyramidal-to-bitufted synapse, raised Pr recruits additional (homogenous) release sites (which it presumably does, although to a very limited extent) with Ca2+-dependent effects on NMDARs limiting the increase in the NMDAR-mediated EPSC1. However, based on results shown in Figure 4, this latter alternative seems unlikely. In addition, if Ca2+-dependent effects are limiting NMDAR-mediated EPSC1, they should also limit EPSC2, yet this did not occur (relative AMPAR- and NMDAR-mediated EPSC2 amplitudes were unchanged in higher [Ca2+]o) (Fig. 2D).
Alternatively, the release sites at the pyramidal-to-multipolar synapses, rather than being homogeneous, might be heterogeneous (Murthy et al., 1997; Craig and Boudin, 2001; Franks et al., 2002). Heterogeneity could arise because of purely postsynaptic factors such as differential distribution of synaptic GluR subtypes relative to release sites (Franks et al., 2002). Although this alternative cannot be completely ruled out, we think it is unlikely because PPD in 2 mm [Ca2+]o was quantitatively the same for both the AMPAR- and NMDAR-mediated EPSCs, despite large differences in their amplitudes relative to that in the absence of APV or CNQX (Table 1). In addition, the CVs for the AMPAR- and NMDAR-mediated EPSCs were indistinguishable (Table 2), again arguing against the idea that they are seeing different pools of glutamate.
Other mechanisms could also give rise to synaptic heterogeneity. One such possibility is that there are two separate populations of release sites, one with a normal Pr and synaptic AMPA and NMDA receptors and another population with a lower Pr and only synaptic AMPARs. This latter population would be recruited only at high Pr. However, if the increase in Pr is a result of the recruitment of an additional pool of release sites, one would expect a significant increase in the amplitude of AMPAR-mediated EPSC2, because a new set of release sites are recruited, but this does not occur (Fig. 2C). In addition, such a model cannot account for the differential effect of γ-DGG on EPSC1 versus EPSC2 in 2 and 3 mm [Ca2+]o (Fig. 5).
A final alternative is that raised Pr increases the glutamate transient at individual release sites. Such a mechanism would explain the differential effect of raised Pr on AMPAR- and NMDAR-mediated EPSCs assuming NMDAR saturation (see below). In addition, this alternative is strongly supported by experiments with γ-DGG, in which we found that the AMPAR-mediated EPSC1 was less effectively blocked by γ-DGG than EPSC2 in 2 or 3 mm [Ca2+]o with the more prominent effect in 3 mm [Ca2+]o arising solely because of changes in EPSC1 (Fig. 5B). Previous studies (Liu et al., 1999; Wadiche and Jahr, 2001) have proposed that such a differential effect of γ-DGG arises because of differences in the glutamate concentration. Hence, results from these experiments are consistent with the idea that the concentration of glutamate at an individual release site increases during EPSC1, but not during EPSC2, with raised Pr.
An elevation in the transient glutamate concentration, a process that can be detected by AMPARs but not NMDARs, suggests that at the pyramidal-to-multipolar synapse, NMDARs but not AMPARs are saturated during a unitary release event. Given their low steady-state affinity for glutamate, the possibility that AMPARs are not saturated is reasonable (Liu et al., 1999; McAllister and Stevens, 2000). In contrast, NMDARs have more than a 100- to 1000-fold greater steady-state affinity for glutamate than the presumed glutamate concentration during a unitary release event (Clements et al., 1992), yet do not appear to be saturated (Mainen et al., 1999; McAllister and Stevens, 2000). In part, this disparity may reflect kinetic features of NMDARs in that they can exist in a long-lived glutamate-bound closed state that enters the open state only 50% of the time (Popescu et al., 2004). In addition, other features of synaptic release, such as the duration of the glutamate transient or the anatomical distribution of NMDARs relative to the release site, may influence receptor saturation.
Possible presynaptic mechanisms underlying the increase in glutamate transient
Our results suggest that raised Pr at the pyramidal-to-multipolar synapse increases the glutamate transient at individual release sites. Nevertheless, the mechanism underlying this effect remains unknown. One possibility is that a raised Pr might result in a more efficient emptying of fused vesicles (Gandhi et al., 2003). Alternatively, the increase in the glutamate transient might be a result of MVR where more than one vesicle is released during a unitary event. MVR occurs at other central synapses (Tong and Jahr, 1994; Auger et al., 1998; Prange and Murphy, 1999; Wadiche and Jahr, 2001; Oertner et al., 2002) and likely underlies the increase in the glutamate transient at the pyramidal-to-multipolar synapse. Nevertheless, additional experiments are needed to fully define the mechanism underlying this phenomenon.
Physiological significance
In our study, we found that presynaptic terminals of pyramidal neurons exhibit different target-specific dynamics of vesicle release. Although the glutamate transient at the pyramidal-to-bitufted synapse is consistent with the classic one vesicle per release site model, leading to a fairly uniform glutamate transient, the pyramidal-to-multipolar synapse shows properties consistent with a variable glutamate transient, possibly reflecting MVR. Hence, facilitation at the pyramidal-to-bitufted synapses appears to occur mainly by recruiting additional release sites. In contrast, various mechanisms contribute to PPD at the pyramidal-to-multipolar synapse. In part, PPD arises because the enhanced glutamate transient at individual release sites occurs predominantly and perhaps exclusively during EPSC1 (Fig. 5B). Indeed, an enhanced glutamate transient during EPSC1 is most pronounced with raised Pr, and this presumably underlies the greater PPD under these conditions. Hence, PPD at the pyramidal-to-multipolar synapse arises not only because of a reduction in the number of active release sites resulting from vesicle depletion but also because of a reduced frequency of MVR with each release event. The relative contribution of these two processes to PPD is unknown.
Although increasing [Ca2+]o is highly unphysiological, there are endogenous modulators of Pr such as endocannabinoids (Alger, 2002; Trettel and Levine, 2002), presynaptic ionotropic and metabotropic glutamate receptors (Llano et al., 1991; Pitler and Alger, 1992; Wu and Saggau, 1997), and adenosine (Song et al., 2000; Arrigoni et al., 2001) among many others. How these various modulations of Pr converge onto the pyramidal-to-multipolar as well as pyramidal-to-bitufted synapse is unknown. Similarly, they might modulate synaptic amplitudes either via altering the number of active release sites or changing specific properties of individual release site.
Footnotes
This work was supported by National Institutes of Health-National Institute of Neurological Disorders and Stroke RO1 Grant N539102. We thank Drs. G. Matthews, A. Sobolevsky, and C. Hewlett for comments on the experiments and this manuscript.
Correspondence should be addressed to Dr. Lonnie P. Wollmuth, Department of Neurobiology and Behavior, State University of New York at Stony Brook, Stony Brook, NY 11794-5230. E-mail: lwollmuth@notes1.cc.sunysb.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/251024-10$15.00/0
References
- Abbott LF, Varela JA, Sen K, Nelson SB (1997) Synaptic depression and cortical gain control. Science 275: 220-224. [DOI] [PubMed] [Google Scholar]
- Alger BE (2002) Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol 68: 247-286. [DOI] [PubMed] [Google Scholar]
- Arrigoni E, Rainnie DG, McCarley RW, Greene RW (2001) Adenosine-mediated presynaptic modulation of glutamatergic transmission in the laterodorsal tegmentum. J Neurosci 21: 1076-1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger C, Kondo S, Marty A (1998) Multivesicular release at single functional synaptic sites in cerebellar stellate and basket cells. J Neurosci 18: 4532-4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Audinat E, Lambolez B, Angulo MC, Ropert N, Tsuzuki K, Hestrin S, Rossier J (1997) Molecular and physiological diversity of cortical nonpyramidal cells. J Neurosci 17: 3894-3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Porter JT, Tsuzuki K, Lambolez B, Rossier J, Quenet B, Audinat E (2000) Classification of fusiform neocortical interneurons based on unsupervised clustering. Proc Natl Acad Sci USA 97: 6144-6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL (1992) The time course of glutamate in the synaptic cleft. Science 258: 1498-1501. [DOI] [PubMed] [Google Scholar]
- Cook DL, Schwindt PC, Grande LA, Spain WJ (2003) Synaptic depression in the localization of sound. Nature 421: 66-70. [DOI] [PubMed] [Google Scholar]
- Craig AM, Boudin H (2001) Molecular heterogeneity of central synapses: afferent and target regulation. Nat Neurosci 4: 569-578. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE (1995) Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron 15: 1097-1107. [DOI] [PubMed] [Google Scholar]
- Franks KM, Bartol Jr TM, Sejnowski TJ (2002) A Monte Carlo model reveals independent signaling at central glutamatergic synapses. Biophys J 83: 2333-2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks KM, Stevens CF, Sejnowski TJ (2003) Independent sources of quantal variability at single glutamatergic synapses. J Neurosci 23: 3186-3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi SP, Stevens CF (2003) Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature 423: 607-613. [DOI] [PubMed] [Google Scholar]
- Gonchar Y, Burkhalter A (1997) Three distinct families of GABAergic neurons in rat visual cortex. Cereb Cortex 7: 347-358. [DOI] [PubMed] [Google Scholar]
- Harrison J, Jahr CE (2003) Receptor occupancy limits synaptic depression at climbing fiber synapses. J Neurosci 23: 377-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Takahashi T (2001) Mechanisms underlying presynaptic facilitatory effect of cyclothiazide at the calyx of Held of juvenile rats. J Physiol (Lond) 533: 423-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahr CE, Stevens CF (1993) Calcium permeability of the N-methyl-d-aspartate receptor channel in hippocampal neurons in culture. Proc Natl Acad Sci USA 90: 11573-11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Choi S, Tsien RW (1999) Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron 22: 395-409. [DOI] [PubMed] [Google Scholar]
- Llano I, Leresche N, Marty A (1991) Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron 6: 565-574. [DOI] [PubMed] [Google Scholar]
- Mainen ZF, Malinow R, Svoboda K (1999) Synaptic calcium transients in single spines indicate that NMDA receptors are not saturated. Nature 399: 151-155. [DOI] [PubMed] [Google Scholar]
- Markram H, Wang Y, Tsodyks M (1998) Differential signaling via the same axon of neocortical pyramidal neurons. Proc Natl Acad Sci USA 95: 5323-5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Stevens CF (2000) Nonsaturation of AMPA and NMDA receptors at hippocampal synapses. Proc Natl Acad Sci USA 97: 6173-6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Sejnowski TJ, Stevens CF (1997) Heterogeneous release properties of visualized individual hippocampal synapses. Neuron 18: 599-612. [DOI] [PubMed] [Google Scholar]
- Oertner TG, Sabatini BL, Nimchinsky EA, Svoboda K (2002) Facilitation at single synapses probed with optical quantal analysis. Nat Neurosci 5: 657-664. [DOI] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Mayer ML (1994) Cyclothiazide differentially modulates desensitization of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor splice variants. Mol Pharmacol 46: 129-138. [PubMed] [Google Scholar]
- Pitler TA, Alger BE (1992) Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci 12: 4122-4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu G, Robert A, Howe JR, Auerbach A (2004) Reaction mechanism determines NMDA receptor response to repetitive stimulation. Nature 430: 790-793. [DOI] [PubMed] [Google Scholar]
- Prange O, Murphy TH (1999) Analysis of multiquantal transmitter release from single cultured cortical neuron terminals. J Neurophysiol 81: 1810-1817. [DOI] [PubMed] [Google Scholar]
- Reyes A, Lujan R, Rozov A, Burnashev N, Somogyi P, Sakmann B (1998) Target cell specific facilitation and depression in neocortical circuits. Nat Neurosci 1: 279-285. [DOI] [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B, Neher E (2001a) Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol (Lond) 531: 807-826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Jerecic J, Sakmann B, Burnashev N (2001b) AMPA receptor channels with long-lasting desensitization in bipolar interneurons contribute to synaptic depression in a novel feedback circuit in layer 2/3 of rat neocortex. J Neurosci 21: 8062-8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WJ, Tkatch T, Surmeier DJ (2000) Adenosine receptor expression and modulation of Ca2+ channels in rat striatal cholinergic interneurons. J Neurophysiol 83: 322-332. [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Dodt HU, Sakmann B (1993) Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflügers Arch 423: 511-518. [DOI] [PubMed] [Google Scholar]
- Thomson AM, Bannister AP (2003) Interlaminar connections in the neocortex. Cereb Cortex 13: 5-14. [DOI] [PubMed] [Google Scholar]
- Tong G, Jahr CE (1994) Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron 12: 51-59. [DOI] [PubMed] [Google Scholar]
- Trettel J, Levine ES (2002) Cannabinoids depress inhibitory synaptic inputs received by layer 2/3 pyramidal neurons of the neocortex. J Neurophysiol 88: 534-539. [DOI] [PubMed] [Google Scholar]
- Tsodyks MV, Markram H (1997) The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc Natl Acad Sci USA 94: 719-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uteshev VV, Pennefather PS (1996) A mathematical description of miniature postsynaptic current generation at central nervous system synapses. Biophys J 71: 1256-1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE (2001) Multivesicular release at climbing fiberPurkinje cell synapses. Neuron 32: 301-313. [DOI] [PubMed] [Google Scholar]
- Westbrook GL, Krupp JJ, Vissel B (1997) Cytoskeletal interactions with glutamate receptors at central synapses. Soc Gen Physiol Ser 52: 163-175. [PubMed] [Google Scholar]
- Wu LG, Saggau P (1997) Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci 20: 204-212. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355-405. [DOI] [PubMed] [Google Scholar]