

Abstract
The roles of oxidative stress and structural alterations in the cerebrovascular dysfunctions associated with Alzheimer's disease (AD) were investigated in transgenic mice overexpressing amyloid precusor protein (APP+) or transforming growth factor-β1 (TGF+). Age-related impairments and their in vitro reversibility were evaluated, and underlying pathogenic mechanisms were assessed and compared with those seen in AD brains. Vasoconstrictions to 5-HT and endothelin-1 were preserved, except in the oldest (18-21 months of age) TGF+ mice. Despite unaltered relaxations to sodium nitroprusside, acetylcholine (ACh) and calcitonin gene-related peptide-mediated dilatations were impaired, and there was an age-related deficit in the basal availability of nitric oxide (NO) that progressed more gradually in TGF+ mice. The expression and progression of these deficits were unrelated to the onset or extent of thioflavin-S-positive vessels. Manganese superoxide dismutase (SOD2) was upregulated in pial vessels and around brain microvessels of APP+ mice, pointing to a role of superoxide in the dysfunctions elicited by amyloidosis. In contrast, vascular wall remodeling associated with decreased levels of endothelial NO synthase and cyclooxygenase-2 and increased contents of vascular endothelial growth factor and collagen-I and -IV characterized TGF+ mice. Exogenous SOD or catalase normalized ACh dilatations and NO availability in vessels from aged APP+ mice but had no effect in those of TGF+ mice. Increased perivascular oxidative stress was not evidenced in AD brains, but vascular wall alterations compared well with those seen in TGF+ mice. We conclude that brain vessel remodeling and associated alterations in levels of vasoactive signaling molecules are key contributors to AD cerebrovascular dysfunctions.
Keywords: amyloid-β, transforming growth factor-β1, transgenic mouse models, cerebral blood vessels, superoxide dismutase, vascular endothelial growth factor
Introduction
Alzheimer's disease (AD) is characterized by neurofibrillary tangles, oxidative stress, progressive neuronal deficits, increased levels of amyloid β (Aβ) peptides, and their deposition in neuritic plaques and cerebral blood vessels [cerebral amyloid angiopathy (CAA)] (Hardy and Selkoe, 2002; McGeer and McGeer, 2003). Recent evidence indicates that cerebrovascular pathologies, ranging from structural alterations (Shi et al., 2000), atherosclerotic lesions (Roher et al., 2003), and impaired hemodynamic responses at rest and during functional activation (Smith et al., 1999; Bookheimer et al., 2000; Johnson and Albert, 2000), are an early if not a primary feature of AD (for review, see Iadecola, 2004). Moreover, increased risk factors for AD include stroke, hypertension, diabetes, atherosclerosis, and hypercholesterolemia (Kalaria, 2000; Iadecola, 2003; de la Torre, 2004; Casserly and Topol, 2004), and their aggregation further increases the risk of AD (Luchsinger et al., 2005). These pathologies, akin to AD (Akiyama et al., 2000; McGeer and McGeer, 2003), are associated with chronic inflammation and altered blood vessel responsiveness (Cai and Harrison, 2000; Iadecola, 2003).
In vitro studies on the effects of Aβ on cerebral blood vessels (Niwa et al., 2001; Price et al., 2001; Paris et al., 2003) and in vivo investigations with transgenic mice that overproduce Aβ (Niwa et al., 2000; Mueggler et al., 2002; Beckman et al., 2003; Paris et al., 2004) have provided convincing evidence that inflammatory mediators and, particularly, reactive oxygen species (ROS) drive the cerebrovascular pathology in these mice (Paris et al., 2000, 2003; Park et al., 2004). However, microvessels from AD brains not only display Aβ pathology and high levels of inflammatory molecules (Grammas and Ovase, 2001), but they also exhibit profound structural alterations (Perlmutter and Chui, 1990; Vinters et al., 1994; Zarow et al., 1997). Therefore, increased levels of the angiogenic cytokine transforming growth factor-β1 (TGF-β1) are found in microvessels from AD brains (Chao et al., 1994; Wyss-Coray et al., 1997; Grammas and Ovase, 2002). In CSF of AD patients, increased levels of TGF-β1 and vascular endothelial growth factor (VEGF) have been ascribed to vascular remodeling (Tarkowski et al., 2002), consistent with elevated TGF-β1 levels as a marker for vascular diseases (Das, 2004). Moreover, genetic polymorphisms of the TGF-β1 gene represent an increased risk factor for hypertension (Luedecking et al., 2000) or AD (Yamada et al., 2002). Accordingly, transgenic mice that overproduce TGF-β1 in the brain display several features of AD cerebral angiopathy, particularly those related to the vascular basement membrane (Wyss-Coray et al., 1997, 2000).
These observations point to the importance of better understanding the respective role of amyloidosis and angiogenic mediators such as TGF-β1 in the cerebrovascular pathology associated with AD. Hence, we studied the age-dependent changes in responsiveness of cerebral arteries from transgenic mice that overproduce Aβ or TGF-β1. Furthermore, we investigated in vitro their reversibility at different stages of the pathology and assessed alterations in protein expression for specific vascular or inflammatory markers. Additionally, we sought to establish the relevance of these changes for the cortical microcirculation in both transgenic mouse models and neuropathologically confirmed cases of AD.
Materials and Methods
Transgenic mouse models. All experiments were approved by the Animal Ethics Committee of the Montreal Neurological Institute and abided by the guidelines of the Canadian Council on Animal Care. Two transgenic mouse models, characterized for amyloidosis [amyloid precusor protein (APP+) mice, line J20] (Mucke et al., 2000) or cerebrovascular pathology (TGF+ mice) (Wyss-Coray et al., 2000), were used at 4, 12, or 18-21 months of age (body weight, 40 ± 10 g). Heterozygous transgenic founders were backcrossed with wild-type C57BL/6 mice, and all mice were screened for transgene expression by touchdown PCR using tail-extracted DNA (Wyss-Coray et al., 1997). Controls were wild-type littermates, with males and females in approximately equal number in each group.
Vascular and brain tissues. The first segment of the middle cerebral artery (MCA) (average intraluminal diameter, ∼ 40-70 μm) was isolated from wild-type control and transgenic mice killed by cervical dislocation and collected in cold Krebs' solution (4°C, pH 7.4 ± 0.1) containing the following (in mm): 118 NaCl, 4.5 KCl, 2.5 CaCl2, 1 MgSO4, 1 KH2PO4, 25 NaHCO3, and 11 glucose. All remaining vessels of the circle of Willis and their cortical branches, referred to as pial vessels, were removed under a dissecting microscope, immediately frozen on dry ice, and stored at -80°C. Another series of mice of the same ages (n = 4 of 6 per age group for both wild-type and transgenic mice) were perfusion fixed through the heart under deep anesthesia (65 mg/kg sodium pentobarbital, i.p.) with a 4% paraformaldehyde phosphate-buffered solution, pH 7.4, and the brains were removed and postfixed (overnight at 4°C) before sucrose cryoprotection, freezing, and sectioning (20 μm thick) on a freezing microtome (Aucoin et al., 2005).
Sections from human cerebral cortex. Freezing microtome sections (50 μm thick) from immersion-fixed human temporal cortex were obtained and prepared as described previously (Tong and Hamel, 1999) from neuropathologically confirmed cases of AD (clinically diagnosed for AD for 2-11 years; n = 5 females) and non-demented elderly controls (n = 7; two females and five males). An additional group of paraffin-embedded sections (5 μm thick) from AD (n = 5; three females and two males) and non-demented elderly controls (n = 5; two females and three males, four from the same donors as for thick sections) was used for Sirius red staining of total collagen (see below). Donors (10 AD and 8 controls) were matched as much as possible for age (79.1 ± 3.2 vs 74.0 ± 4.3 years old, AD vs controls; not significant), postmortem delay (27.5 ± 7.1 vs 24.1 ± 5.8 h; not significant), and documented history of AD-related risk factors that may alter vessel wall structure such as cardiovascular diseases (hypertension, two AD and one control; coronary heart disease/infarcts, one AD and two controls; hypercholesterolemia, one AD and 0 control) and diabetes (one AD and one control). Atherosclerosis (light to severe) of brain vessels was present in virtually all cases. Human tissues were obtained through the Department of Neuropathology at McGill University and the Douglas Hospital Research Centre Brain Bank (Verdun, Québec, Canada) with the patients' and/or family's approval.
Cerebrovascular reactivity studies. MCA segments (∼ 2 mm in length) were mounted in a superfusion chamber system for on-line videomicroscopy (Living Systems Instrumentation, Burlington, VT) (Elhusseiny and Hamel, 2000). They were cannulated on a glass micropipette (∼ 40 μm diameter) at one end, sealed to another glass micropipette on the other end, and filled with oxygenated (5% CO2 in 95% O2) Krebs' solution (37 ± 1°C, pH 7.4 ± 0.1). A pressure-servo micropump (Living Systems) was used to maintain intraluminal pressure at 60 mmHg. Vessels were superfused (6 ml/min) with Krebs' solution and allowed to stabilize and acquire basal tone (30-45 min). On-line measurements of intraluminal diameter were performed using a closed-circuit video system (National Electronics, Taiwan) coupled with a video caliper (Image Instrumentation, Trenton, NJ). All compounds were applied extraluminally to the superfusion solution for a 3 min period.
Contractile responses were assessed with serotonin (5-HT) (10-9 to 10-5 m) and endothelin-1 (ET-1) (10-10 to 3 × 10-7 m) on vessels at basal tone, whereas dilatory responses were tested with acetylcholine (ACh) (10-10 to 10-5 m), calcitonin gene-related peptide (CGRP) (10-10 to 10-6 m), or sodium nitroprusside (SNP) (10-10 to 10-4 m) on vessels preconstricted submaximally with 5-HT (10-8 to 2 × 10-7 m). The tonic production of nitric oxide (NO) was evaluated in vessels at basal tone by superfusion (35 min) of the nitric oxide synthase (NOS) inhibitor Nω-nitro-l-arginine (l-NNA) 10-5 m) (Erdos et al., 2002). In some vessels, reversibility of the vascular deficit in response to ACh or l-NNA application was tested before and/or after preincubation (30-60 min) of the arterial segments with the free radical scavenging enzyme superoxide dismutase (SOD) or catalase (120 or 1000 U/ml, respectively; Sigma, St. Louis, MO).
Calculations and statistical analysis of cerebrovascular responses. Responses to 5-HT, ET-1, ACh, CGRP, and SNP were compared on the basis of their dose-dependent and maximal (EAmax) responses (expressed as percentage of change in vessel diameter from basal or pharmacologically induced tone) and/or potency (pD2 values or -log of EC50) expressed as mean ± SEM. Statistical differences between transgenic mice of different ages and their respective wild-type controls were determined by one-way ANOVA, followed by a post hoc Newman-Keuls multiple comparison test. For l-NNA, the extent of the contractile response as a function of time was similarly compared by ANOVA. All statistics and analyses were performed with the software Prism 4 (Graph Pad, San Diego, CA). A p value of < 0.05 was considered significant.
Western blot. Frozen pial vessels from each of the oldest APP+ and TGF+ mice were powdered with a mortar on dry ice, dissolved in 20 μl of Laemmli buffer (62.5 mm Tris, pH 6.8, 6 m urea, 160 mm 1,4-DTT, 2% SDS, and 0.001% bromophenol blue), boiled (5 min), protein assayed (assay kit; Bio-Rad, Hercules, CA), loaded (∼ 10 μg) onto a 10% acrylamide SDS gel, and transferred to nitrocellulose membrane (Schleicher & Schuell, Keene, NH) (Csiszar et al., 2002). The membrane was blocked (1 h at room temperature) with 5% skim milk in Tris-buffered saline-Tween 20 (20 mm Tris, 137 mm NaCl, and 0.1% Tween 20) and incubated overnight (4°C) with primary antibodies against endothelial NOS (eNOS) (1:500; Transduction Laboratories, Lexington, KY), inducible NOS (iNOS) (1:100; BD Bioscience, Mississauga, Ontario, Canada), SOD1 (1:3000; Streegen, Victoria, British Columbia, Canada), SOD2 (1:3000; Streegen), cyclooxygenase-1 (COX-1) (1:250; Cayman Chemical, Ann Arbor, MI), COX-2 (1:2000; Cayman Chemical), VEGF (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), or β-tubulin (1:500; Santa Cruz Biotechnology). The blots were then incubated (1 h) with secondary antibody (1:2000; horseradish peroxidase-conjugated anti-mouse and anti-rabbit; Jackson ImmunoResearch, West Grove, PA), and the membranes were developed with ECL (Amersham Biosciences, Baie d'Urfé, Québec, Canada) and analyzed with densitometry. Tubulin was used to normalize for loading variation. Readings from individual mice from both the wild-type and transgenic groups were averaged and compared by Student's t tests. A p value of < 0.05 was considered significant.
Detection of amyloid deposits. Free-floating sections were stained for amyloid deposits with 1% thioflavin-S solution (Wyss-Coray et al., 2000). Thioflavin-S-positive pial and cortical penetrating vessels in APP+ and TGF+ mice (n = 4-6 per group) were counted under a Leitz Aristoplan light microscope equipped with epifluorescence using an FITC filter (Leica, Montréal, Québec, Canada) and expressed as number of vessels per hemisphere. In some sections, blood-vessel walls were immunostained with a mouse anti-laminin antibody (1:6000; Chemicon, Temecula, CA) detected with a donkey cyanine 3 (Cy3)-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch) before thioflavin-S staining. Sections were examined under a laser scanning confocal microscope (LSM 510; Zeiss, Jena, Germany) using simultaneous double-channel visualization with emission intensities of 488 nm (thioflavin-S) and 543 nm (Cy3), respectively.
Immumohistochemistry. Changes in pial and/or intracortical microvessels in transgenic mice (perfusion-fixed brains) and AD patients (50-μm-thick sections) were assessed by immunostaining for the following markers: SOD2 (rabbit anti-MnSOD; 1:400; Stressgen), nitrosative stress (mouse anti-nitrotyrosine; 1:1000; Upstate Biotechnology, Lake Placid, NY), basement membrane proteins collagen-I (goat anti-collagen type I; 1:400; Southern Biotechnology, Birmingham, AL) and collagen-IV (goat anti-collagen type IV; 1: 400; Chemicon), and astroglial (GFAP, rabbit anti-GFAP; 1:300; Dako, Glostrup, Denmark) or microglial (rat anti-CD11b; 1:100; Serotec, Raleigh, NC) activation. Sections were incubated (overnight at room temperature) with primary antibody, rinsed (phosphate buffer), and incubated (1 h, 30 min) with species-specific biotinylated IgG (Vector Laboratories, Burlingame, CA) and the ABComplex (1 h, 15 min), and the reaction was visualized with a 0.05% solution of 3,3′-diaminobenzidine (DAB) (brown precipitate; Sigma) activated with 0.005% H2O2 or, in the case of SOD2, with the slate-gray reagent (SG kit; Vector Laboratories). Double immunofluorescence for nitrotyrosine (detected with a Cy3-labeled secondary antibody) and CD11b or GFAP (Cy2-labeled secondary antibody) was performed on a few sections by simultaneous overnight incubation of the two primary antibodies and observed under confocal microscopy as described above. For finer inspection of collagen-I staining, 18-month-old wild-type controls and TGF+ mice (n = 3 per group) were perfusion fixed as above, their brains were removed and processed for paraffin embedding (Leica ASP300 paraffin tissue processor), and 5 μm sections were obtained with a Leica rotary microtome. Dewaxed paraffin sections were then incubated (overnight at room temperature) with goat anti-collagen-I (1:300), and the reaction was visualized with DAB as above. To control for nonspecific staining, preimmune serum was substituted for primary antibodies.
Sirius red staining for collagen content. Sirius red staining was used to detect collagen accumulation (Orlandi et al., 2004) in brain vessels. Fivemicrometerthick paraffin sections from 18- to 21-month-old TGF+ mice and wild-type littermate controls (n = 3 per group) or neuropathologically confirmed cases of AD and non-demented elderly controls (n = 5 per group) were dewaxed, rinsed in H2O, and incubated (50 min) with 1% Sirius red saturated with picric acid. After rinsing, sections were dehydrated, defatted, and observed under a Leitz Aristoplan light microscope (Leica).
Quantitative analysis of immunochemical and histocytochemical stainings. In 18- to 21-month-old TGF+ mice and their wild-type controls, the thickness and staining intensity (optical density) of the pial membrane, taken at three different locations in total collagen-stained (Sirius red staining), collagen-I-stained, and collagen-IV-stained sections (n = 6-8 per group), were measured with the MetaMorph 6.1r3 program (Universal Imaging, Downington, PA) on digital pictures taken under the same conditions of illumination and magnification. Total collagen accumulation around vessels was evaluated by (1) counting the number of vessels with thickened walls directly under the microscope and (2) measuring staining intensity (optical density) of the material around laden vessels. For this purpose, triplicate readings were made (from six to eight vessels per mouse; n = 3 per group), averaged, and expressed as a ratio over background intensity. All values obtained from TGF+ mice and wild-type littermates were averaged and compared by Student's t test. In human tissues, a semi-quantitative analysis was performed in the cortical gray matter by three independent observers, two of them blind to the identity of the sections. Arbitrary scores (0-3, with increasing scores reflecting increased pathology) (Yermakova and O'Banion, 2001) were assigned based on (1) the relative amount of stained vessels and their staining intensity (light to dark) for collagen-I, (2) the appearance of the vessel walls (smooth to damaged or ragged and/or vacuolized) for collagen-IV, and (3) the relative amount of darkly stained vessels and vessels with increased wall thickness for total collagen (sirius red). The scores of the three observers were averaged for AD samples and controls and compared by Student's t test. In all cases, p < 0.05 was considered significant.
Results
Vasomotor dysfunctions in APP+ and TGF+ mice
In APP+ mice of both ages, the contractile responses to 5-HT and ET-1 were comparable to those of control littermates (Fig. 1 and supplemental Table 1, available at www.jneurosci.org as supplemental material). In contrast, despite preserved relaxation to SNP (supplemental Table 1, available at www.jneurosci.org as supplemental material), the dilatations to CGRP and ACh were significantly reduced in APP+ mice compared with controls at both 4 months of age (↓ 48 and 38%, respectively; p < 0.01) and 12 months of age (↓ 56 and 47%, respectively; p < 0.01 and 0.001), without any change in affinity at the cerebrovascular receptors (supplemental Table 1 available at www.jneurosci.org as supplemental material). The tonic production of NO, as evaluated by the contraction elicited by inhibition of NOS with l-NNA, was slightly reduced at 4 months of age (not significant) and almost completely abolished (↓ ≥70%) in 12-month-old APP+ mice (Fig. 1 and supplemental Table 1, available at www.jneurosci.org as supplemental material). In TGF+ mice, except for a reduced contractile response to ET-1 at 18-21 months of age (↓ 35%; p < 0.01) (Fig. 2), the efficacy and potency of 5-HT and ET-1 in inducing contraction were comparable between wild-type and transgenic mice (supplemental Table 2, available at www.jneurosci.org as supplemental material). Akin to APP+ mice, the relaxation to SNP was unaltered, but the dilatations to CGRP and ACh decreased with increasing age with no (ACh) or small (CGRP) changes in potency, and the overall response to CGRP was even reversed to a contraction in 12-month-old TGF+ mice (Fig. 2 and supplemental Table 2, available at www.jneurosci.org as supplemental material). There was also an age-dependent decrease in the tonic production of NO that became significant at 21 months of age (↓ ≥54%; p < 0.01) (Fig. 2 and supplemental Table 2, available at www.jneurosci.org as supplemental material).
Figure 1.

Cerebrovascular responses of the MCA to ET-1, CGRP, ACh, and NOS inhibition in APP+ (▴) and wild-type (•) mice at 4 and 12 months of age. Although the contractile response to ET-1 is preserved, the ability of CGRP and ACh to induce dilatation is significantly impaired at both ages. Also, decreased responsiveness of the vessels after NOS inhibition (10-5 m l-NNA) is apparent at both ages but significant only in the aged APP+ mice (n = 4-11 per group). ★p <0.05; ★★p <0.01; ★★★p <0.001. Error bars represent SEM.
Figure 2.

Cerebrovascular responses of the MCA to ET-1, CGRP, ACh, and NOS inhibition in TGF+ (▪) and wild-type (•) mice at 4, 12, or 18-21 months of age. In young mice, only the ACh-induced dilatation is significantly decreased. At 12 months, impairments in the dilatory responses to ACh and CGRP are highly significant, and impairment in the dilatory response to CGRP was reversed to a constriction. Inhibition of NOS with l-NNA reveals a small, nonsignificant decrease in basal NO production. At 18-21 months, impairments in contractile and dilatory responses are highly significant. Note the small but significant (∝p <0.05) decrease in the maximal ACh-mediated dilatation in elderly wild-type controls compared with that in 4- and 12-month-old controls (n = 4-9 per group). ★p < 0.05; ★★p < 0.01; ★★★p < 0.001. Error bars represent SEM.
Effect of in vitro treatment with SOD and catalase
Superfusion of vessels with SOD or catalase resulted in a complete normalization of ACh-mediated dilatations and l-NNA-induced contractile responses in 12-month-old APP+ mice (Fig. 3 and supplemental Table 3, available at www.jneurosci.org as supplemental material), indicating that basal NO production was normal in these vessels but that NO was unavailable because of its trapping by superoxide (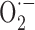 ) anions. In contrast, SOD or catalase had no beneficial effect in TGF+ mice of any age, and vessels still exhibited an impaired ability to dilate to ACh or constrict in response to l-NNA application (Fig. 3 and supplemental Table 4, available at www.jneurosci.org as supplemental material). SOD or catalase had no effect on vessels from wild-type controls, except for the normalizing effect of SOD on the small decrease in the ACh-mediated dilatation in 18- to 21-month-old wild-type controls from TGF+ mice (Fig. 3 and supplemental Table 4, available at www.jneurosci.org as supplemental material), suggesting the presence of age-related vascular oxidative stress in these elderly mice.
) anions. In contrast, SOD or catalase had no beneficial effect in TGF+ mice of any age, and vessels still exhibited an impaired ability to dilate to ACh or constrict in response to l-NNA application (Fig. 3 and supplemental Table 4, available at www.jneurosci.org as supplemental material). SOD or catalase had no effect on vessels from wild-type controls, except for the normalizing effect of SOD on the small decrease in the ACh-mediated dilatation in 18- to 21-month-old wild-type controls from TGF+ mice (Fig. 3 and supplemental Table 4, available at www.jneurosci.org as supplemental material), suggesting the presence of age-related vascular oxidative stress in these elderly mice.
Figure 3.
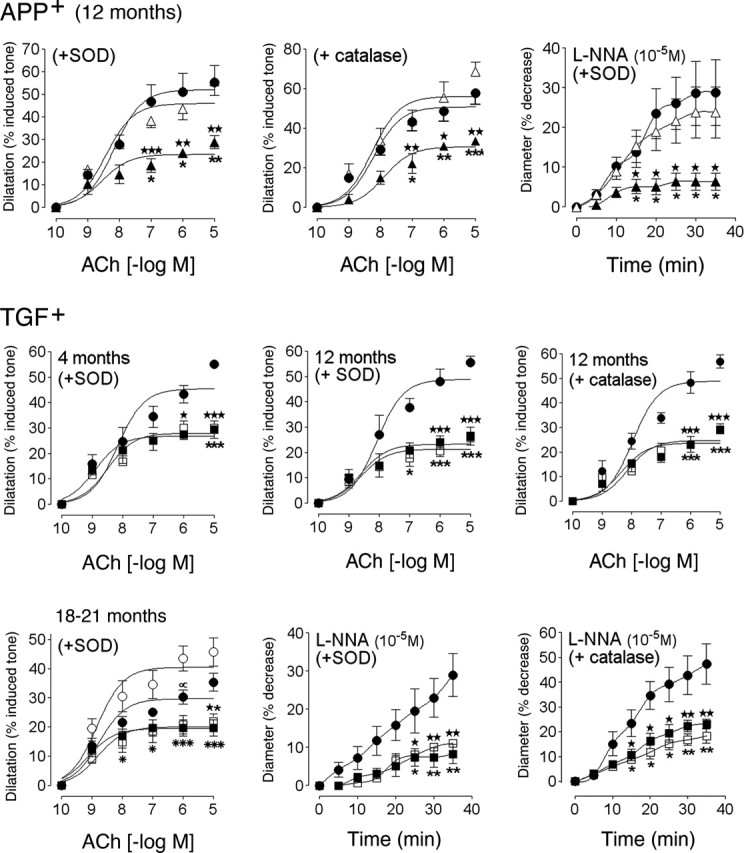
Effects of SOD or catalase on the cerebrovascular deficits in APP+ and TGF+ mice. Untreated wild-type (•), APP+ (▴), or TGF+ (▪) mice and treated wild-type (○), APP+ (▵), or TGF+ (□) mice are shown. For clarity, the curves of SOD- or catalase-treated vessels from wild-type controls were omitted because they were superimposed on those of untreated vessels, except in 18- to 21-month-old wild-type controls (bottom left). In these elderly controls, the ACh-mediated dilatation was reduced in nontreated compared with SOD-treated vessels and was statistically significant at 10-6 m ACh (∝p < 0.05). In APP+ mice, SOD and catalase normalized the responses to ACh and NOS inhibition, but they had no beneficial effect in TGF+ mice at any age. ★ or ★p < 0.05, ★★ or ★★p < 0.01, and ★★★ or ★★★p < 0.001, in APP+ mice compared with untreated wild-type controls or with SOD- or catalase-treated APP+ mice. ★p < 0.05, ★★p < 0.01, and ★★★p < 0.001, in TGF+ mice compared with untreated TGF+ mice and wild-type controls. ★p < 0.05, ★★p < 0.01, and ★★★p < 0.001, in treated TGF+ mice compared with untreated or treated (data not shown, except in bottom left panel) wild-type mice. *p < 0.05, **p < 0.01, and ***p < 0.001, in TGF+ mice compared with wild-type control mice treated with SOD, for ACh only (n = 3-7 per group). Error bars represent SEM.
CAA and changes in vascular protein expression
No vascular amyloid deposits were detected with thioflavin-S staining in wild-type controls or in APP+ mice at 4 months of age. In 12-month-old APP+ mice, some pial vessels and penetrating arteries amounting to approximately three vessels per hemisphere exhibited thioflavin-S-positive plaques outside their basement membrane immunodetected with laminin (Fig. 4A). In TGF+ mice, a comparable number of vessels were thioflavin-S positive at all ages (13.4 ± 1.2 vessels per hemisphere for all age groups), in agreement with vascular thioflavin-S material reaching steady-state levels by 4 months in these mice (Lacombe et al., 2004). The stained material covered the overall surface of the vessel wall (Fig. 4B), and well defined perivascular plaques, as seen in APP+ mice, were virtually never detected in TGF+ mice (see Fig. 9). Western blot analysis of cerebral blood vessels from 12-month-old APP+ mice showed a significant increase in levels of SOD2 (↑ 12.5 ± 3.8%; p < 0.05) compared with wild-type controls (Fig. 4A). In contrast, in vessels from 18- to 21-month-old TGF+ mice, eNOS (↓ 14.8 ± 5.8%; p < 0.05) and COX-2 (↓ 15.6 ± 2.9%; p < 0.05) proteins were significantly decreased compared with controls, and a modest albeit significant increase in VEGF (↑ 4.5 ± 0.9%, p < 0.05) protein content was observed (Fig. 4B). None of the other proteins tested (iNOS, COX-1, SOD1) was significantly altered.
Figure 4.

Cerebrovascular amyloid pathology and changes in protein levels in pial vessels from APP+ (12 months of age) and TGF+ (18-21 months of age) mice. A, APP+ mice. Left, Perivascular amyloid plaques in APP+ mice detected with thioflavin-S (green) are located outside the blood vessel wall immunodetected with laminin (red). Middle, Western blot analysis in pial vessels revealed significant increases in SOD2 protein levels, as quantified in the right panel as percentage of change from wild-type controls. B, TGF+ mice. Left, Thioflavin-S staining (green) in vessels from TGF+ mice was regular along the vessel walls of intracortical (top) and penetrating (bottom) arteries. Middle, Western blot analysis indicated significant upregulation in the VEGF, whereas eNOS and COX-2 protein levels were decreased, as quantified in the right panel. WT, Wild type. Error bars represent SEM. ★p < 0.05.
Figure 9.

Collagen-IV immunoreactivity in TGF+ mice and AD. A, In wild-type (WT) controls, virtually all vessels were immunopositive for collagen-IV and exhibited smooth and regular walls. In TGF+ mice, collagen-IV was upregulated in the pia (open arrows) and in intracortical microvessels in the form of irregularities, vacuolization, and debris within and around the vessel walls. Colocalization of collagen-IV (red) and thioflavin-positive (green) amyloid deposits was present in some large penetrating vessels (yellow, merged image). Note the presence of a thioflavin-positive perivascular plaque (curved arrow). B, Similarly, irregularities and vacuolization (arrow) of vessels were readily detected by collagen-IV immunostaining in AD brains, whereas vessels in elderly controls displayed smooth and regular walls. A string vessel connecting two capillaries is shown in AD tissue (curved arrow). Scale bars: A, 40 μm; B, 20 μm.
Changes in intracortical microvessels in transgenic mice and in AD brains
Oxidative/nitrosative stress
To compare the changes in pial vessels with those of intracortical microvessels, we looked at SOD2 immunoreactivity in the cerebral cortex of APP+ and TGF+ mice, neuropathologically confirmed cases of AD, and their respective controls. In wild-type mice, SOD2 immunostaining was concentrated in small punctate structures in the cytoplasm of neuronal cell bodies (Fig. 5A, left inset), consistent with a mitochondrial localization of the enzyme (Lindenau et al., 2000). Perivascular expression of SOD2 was upregulated in APP+ mice at all ages and was detected in the form of small immunoreactive material distributed in cuffs and rims around some penetrating arteries and small intracortical microvessels. In contrast, in TGF+ mice, perivascular SOD2-immunoreactive material was not or only rarely observed, even in 18- to 21-month-old mice (Fig. 5A, right inset). In human cerebral cortex from elderly controls, SOD2 immunolabeling had a punctate cytoplasmic localization in neuronal cell bodies with occasional weak perivascular labeling and virtually no glial staining (Fig. 5B). In contrast, in AD brains, a few to many glial cells were intensely SOD2 immunoreactive and distributed either diffusely or in islands across the cerebral cortex with neuronal labeling that was barely detectable in most cases. As in TGF+ mice, there was no evidence of enhanced SOD2 perivascular labeling (Fig. 5B). Nitrosative stress was seen in small cortical microvessels, likely capillaries, of young but not 12-month-old APP+ mice compared with wild-type controls. The 12-month-old APP+ mice nonetheless exhibited nitrotyrosine-immunoreactive material throughout the cortical parenchyma, localized primarily in CD11b-immunoreactive microglia but also in some neurons (Fig. 6). In contrast, in TGF+ mice, glial labeling was of comparable intensity between transgenic and age-matched littermate controls, as shown in aged mice (Fig. 6).
Figure 5.

Cerebrovascular oxidative stress in APP+ and TGF+ mice, or AD. A, SOD2 immunoreactivity in wild-type (WT) mice is found in neurons and distributes in small puncta (arrows in left inset) within the cytoplasm, consistent with a mitochondrial localization of the enzyme. SOD2 is upregulated in perivascular cuffs and rims (open arrows) in both 4- and 12-month-old APP+ mice but not or only slightly (open arrows in right inset) in 18- to 21-month-old TGF+ mice. mo, Month. B, SOD2 immunoreactivity in elderly, nondemented controls distributes primarily in neurons (also located in small punctate structures; arrow in top inset) with a slight perivascular location (open arrows; bottom insets). In contrast, in AD tissues, SOD2 is upregulated in glial cells without any apparent increase in perivascular staining (open arrows; bottom inset). Scale bars: A, 75 μm; B, 40 μm.
Figure 6.

Nitrosative stress in the cerebral cortex of APP+ and TGF+ mice. Nitrotyrosine immunoreactivity is upregulated in the vessel walls of young APP+ mice, whereas it is distributed in the neuropil and around intraparenchymal blood vessels in 12-month-old APP+ mice. As shown in the adjacent panels, nitrotyrosine immunoreactivity (red) is mainly located in CD11b-immunopositive microglial processes (green; the merge with nitrotyrosine is yellow) and cells (open arrows), but some neurons are also nitrotyrosine immunopositive (small arrows). In TGF+ mice, nitrotyrosine immunoreactivity is primarily distributed to glial elements in both wild-type (WT) and transgenic mice. Scale bar, 75 μm. mo, Month.
Collagen accumulation
After dissection and manipulation, the MCAs and pia-arachnoid membrane from TGF+ mice were more rigid, at all ages, than those of wild-type littermates and APP+ mice, as reported previously in these mice (Buckwalter et al., 2002). We thus investigated collagen deposition in pial and intracortical microvessels because collagen and, particularly collagen-I, is the major extracellular matrix protein associated with vascular stiffness, remodeling, and dysfunctions in cardiovascular pathologies (Bishop et al., 1990). Similarly, these matrix proteins were examined in AD patients and elderly controls with similar history of pathologies known to affect blood-vessel structure (see Materials and Methods). Sirius red staining of total collagen in 18-month-old TGF+ mice revealed increased vascular collagen content compared with wild-type littermates. Wild-type mice showed no darkly stained or thickened vessels, but an average of 27 ± 3 vessels per hemisphere exhibited thickened walls, albeit to different extents, in TGF+ mice, with an overall twofold increase in Sirius red staining intensity compared with controls (16.3 ± 1.8 vs 7.1 ± 1.7; p < 0.01) (Fig. 7). A similar phenomenon was observed in AD cases with a larger proportion of brain vessels exhibiting increased collagen content compared with controls (scores of 2.2 ± 0.1 vs 0.9 ± 0.2, respectively; p < 0.01); several vessels also displayed a ragged and irregular surface as shown here in a penetrating cortical artery (Fig. 7).
Figure 7.

Sirius red staining of collagen in the cerebral cortex from TGF+ mice and AD. Perivascular accumulation of collagen is increased in cerebral blood vessels of various sizes in 18-month-old TGF+ mice and AD brains compared with wild-type (WT) and elderly controls, respectively. Note the irregular wall (curved arrow) of a cortical penetrating artery in AD. Insets, Small intracortical microvessels with thickened walls in AD compared with regular thin walls in controls. Scale bar, 100 μm.
Analysis of collagen-I-immunostained sections in TGF+ mice also indicated a thicker and/or darker pial membrane, with no obvious change in APP+ mice (data not shown). At 18 months, TGF+ mice exhibited both a significantly thicker (↑ 28%; 4.78 ± 0.33 vs 3.74 ± 0. 26 μm; p < 0.05) and more intensely stained (2.25-fold; p < 0.001) pial membrane compared with wild-type controls (Fig. 8A). Additional inspection in thin paraffin-embedded sections revealed collagen-I upregulation not only in the pia but also in a subset of small cortical microvessels (Fig. 8A). Similarly, in AD cerebral cortex, more cortical vessels showed moderate to dark collagen-I immunostaining compared with controls (scores of 2.2 ± 0.2 vs 0.7 ± 0.2; p < 0.01), and some immunolabeled vessels were decorated with thioflavin-S-positive plaques (Fig. 8 B).
Figure 8.

Collagen-I immunoreactivity in TGF+ mice and AD. A, Virtually no collagen-I immunostaining could be detected in wild-type (WT) controls or APP+ mice (data not shown), but collagen-I was upregulated in the pia (open arrows), pial vessels, and intracortical microvessels (black arrows) in TGF+ mice, as shown in 18-month-old mice. Thin paraffin sections revealed upregulation in collagen-I in the walls of some vessels (black arrows in middle and right panels). B, In AD brains, collagen-I immunostaining was upregulated in cortical microvessels, and some were associated with thioflavin-positive plaques, as shown in double-immunofluorescence staining for collagen-I (red) and thioflavin-S (green) (right). Scale bars: A, 40 μm; B, 50 μm.
A thickened pial membrane was also apparent, albeit not significant, in collagen-IV-immunolabeled brain sections of TGF+ mice (Fig. 9A) (7.62 ± 1.15 vs 4.95 ± 0.66 μm at 18 months; ↑ 54%; not significant). Most, if not all, vessels were positive for collagen-IV in both controls and transgenic mice at all ages. However, vessels in TGF+ mice were intensely stained, and the vast majority exhibited an irregular surface with perivascular accumulation of stained material, in contrast to wild-type controls that exhibited smooth vascular walls, as shown here in 18-month-old mice (Fig. 9A). Double immunostaining of collagen-IV and thioflavin-S revealed partial colocalization of the two markers in some vessels (Fig. 9A). A similar pattern of collagen-IV immunoreactivity was seen in AD brains, which displayed an increased ragged appearance and vacuolization of the blood-vessel walls compared with elderly controls (scores of 2.1 ± 0.4 vs 0.6 ± 0.1; p < 0.05) (Fig. 9B). These findings in a limited sampling of patients confirm previous, more exhaustive reports of vascular basement membrane alterations in AD (Perlmutter and Chui, 1990; Vinters et al., 1994; Kalaria and Pax, 1995; Zarow et al., 1997; Shi et al., 2000) but also suggest that cardiovascular diseases or diabetes are not the only contributing factors to this pathology.
Discussion
Our prominent finding is that APP+ and TGF+ mice, even at ages when CAA is absent or very limited, exhibited relatively similar cerebrovascular dysfunctions that can be summarized as a (1) decreased response to vasodilators such as ACh and CGRP, (2) reduced NO basal tone, and (3) preserved contractile capacity, except for a slightly reduced ET-1 response in old TGF+ mice. We confirmed that 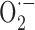 anions drive the readily reversible Aβ-induced dysfunctions in APP+ mice and found that vascular remodeling in TGF+ mice was associated with progressive cerebrovascular deficits that resisted antioxidant therapy. Increased perivascular oxidative stress was not evidenced in AD brains, which exhibited structural vascular alterations comparable to those of TGF+ mice. We conclude that vascular remodeling, and its associated alterations in key vasoactive signaling molecules, is an important contributor to AD cerebrovascular deficits.
anions drive the readily reversible Aβ-induced dysfunctions in APP+ mice and found that vascular remodeling in TGF+ mice was associated with progressive cerebrovascular deficits that resisted antioxidant therapy. Increased perivascular oxidative stress was not evidenced in AD brains, which exhibited structural vascular alterations comparable to those of TGF+ mice. We conclude that vascular remodeling, and its associated alterations in key vasoactive signaling molecules, is an important contributor to AD cerebrovascular deficits.
Vascular dysfunctions in APP+ mice
Our findings of altered cerebrovascular reactivity priming vasoconstriction support recent data in other APP+ mice of decreased responses to dilators but not constrictors (Iadecola et al., 1999; Niwa et al., 2001), reduction in functional hyperemia (Niwa et al., 2000), and compromised hemodynamic responses to pharmacological stimuli (Mueggler et al., 2002). Furthermore, these deficits are independent from CAA because ACh- and CGRP-mediated dilatations were altered at 4 months when there were no vascular Aβ deposits, in line with early perfusion deficits in other APP+ mice (Niwa et al., 2002; Beckmann et al., 2003; Park et al., 2004).
The complete recovery in the responses to ACh and NOS inhibition (and this in 12-month-old APP+ mice with fully developed dysfunctions) by in vitro SOD or catalase treatment unambiguously demonstrated that free radicals, and primarily 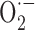 -mediated oxidative stress, account for the Aβ-induced cerebrovascular deficits. Such a statement agrees with previous reports, albeit in young APP+ mice, that endothelial impairments can be abrogated by coexpression of APP+ and SOD1, or topical SOD application (Iadecola et al., 1999). A remarkable finding from our study is that SOD2 was increased in meningeal vessels and around some intraparenchymal microvessels of APP+ mice at all ages, hence stressing that oxidative stress precedes Aβ deposition (Drake et al., 2003). Such perivascular localization also argues that
-mediated oxidative stress, account for the Aβ-induced cerebrovascular deficits. Such a statement agrees with previous reports, albeit in young APP+ mice, that endothelial impairments can be abrogated by coexpression of APP+ and SOD1, or topical SOD application (Iadecola et al., 1999). A remarkable finding from our study is that SOD2 was increased in meningeal vessels and around some intraparenchymal microvessels of APP+ mice at all ages, hence stressing that oxidative stress precedes Aβ deposition (Drake et al., 2003). Such perivascular localization also argues that 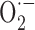 production is intimately associated with the vessel wall (Park et al., 2004). Because we found no upregulation in iNOS, eNOS, COX-1, and COX-2 proteins in pial vessels, increased enzyme activity rather than levels or, alternatively, other pathways may be involved in
production is intimately associated with the vessel wall (Park et al., 2004). Because we found no upregulation in iNOS, eNOS, COX-1, and COX-2 proteins in pial vessels, increased enzyme activity rather than levels or, alternatively, other pathways may be involved in 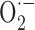 production. In this respect, xanthine and/or NADPH oxidase, the main sources of
production. In this respect, xanthine and/or NADPH oxidase, the main sources of 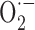 in vascular tissues (Cai and Harrison, 2000), may be implicated, as shown recently for NADPH oxidase in another APP+ mouse model (Park et al., 2005).
in vascular tissues (Cai and Harrison, 2000), may be implicated, as shown recently for NADPH oxidase in another APP+ mouse model (Park et al., 2005).
Signs of peroxynitrite (ONOO-) upregulation were detected in microvessels from young APP+ mice and in microglia and neurons in aged APP+ mice (Park et al., 2004). This suggests that NO trapping by increased 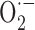 results in local nitrosative stress early in the pathology, but when
results in local nitrosative stress early in the pathology, but when 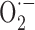 production exceeds its stabilization by NO, SOD2 is upregulated, likely as an ultimate effort from vascular and perivascular cells to maintain NO availability and endothelium-dependent relaxation by converting
production exceeds its stabilization by NO, SOD2 is upregulated, likely as an ultimate effort from vascular and perivascular cells to maintain NO availability and endothelium-dependent relaxation by converting 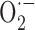 into hydrogen peroxide (Lacza et al., 2002). However, the prompt reversibility of the deficits by exogenous SOD indicates that SOD2 upregulation is insufficient or not in the right cellular compartment to fully counteract
into hydrogen peroxide (Lacza et al., 2002). However, the prompt reversibility of the deficits by exogenous SOD indicates that SOD2 upregulation is insufficient or not in the right cellular compartment to fully counteract 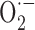 production and, furthermore, that structural changes are not yet manifest, as documented in other APP+ mice (Christie et al., 2001).
production and, furthermore, that structural changes are not yet manifest, as documented in other APP+ mice (Christie et al., 2001).
Increased ROS levels thus seem sufficient to alter cerebrovascular responsiveness. Both 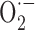 and ONOO- favor a contractile status (Elliott et al., 1998; Didion and Faraci, 2002):
and ONOO- favor a contractile status (Elliott et al., 1998; Didion and Faraci, 2002): 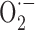 by trapping NO in the vessel wall and ONOO- because it induces alterations in ATP-sensitive K+ (KATP) channels.
by trapping NO in the vessel wall and ONOO- because it induces alterations in ATP-sensitive K+ (KATP) channels. 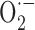 and ONOO- have also been shown to eliminate CGRP-mediated dilatations, but not the contractile responses to 5-HT, a selectivity attributed to their ability to impair KATP channels, important mediators of the CGRP-induced dilatation (DeWitt et al., 2001). Aβ-generated ROS reportedly also alter Ca2+ homeostasis and affect synthesis and release of vasoactive substances triggered by Ca2+ mobilization (Price et al., 2001).
and ONOO- have also been shown to eliminate CGRP-mediated dilatations, but not the contractile responses to 5-HT, a selectivity attributed to their ability to impair KATP channels, important mediators of the CGRP-induced dilatation (DeWitt et al., 2001). Aβ-generated ROS reportedly also alter Ca2+ homeostasis and affect synthesis and release of vasoactive substances triggered by Ca2+ mobilization (Price et al., 2001).
Vascular dysfunctions in TGF+ mice
As in APP+ mice, we observed a progressive loss of function in TGF+ mice that affected selective vasomotor responses. In contrast to APP+ mice, however, antioxidants had no beneficial effects, and there was no upregulation in levels of free radical-generating or antioxidant enzymes in TGF+ mice, excluding ROS as mediators of the dysfunctions. The deficits in TGF+ mice, when apparent, were seen in all arteries tested, whereas only a minority of vessels were thioflavin-S positive at all ages (Lacombe et al., 2004; present study), strongly arguing that, as in APP+ mice, vascular amyloid peptide accumulation cannot explain the vasomotor dysfunctions. Similarly, the unaltered dilatory and contractile capacity of the vessels to SNP and 5-HT, respectively, despite their increased rigidity, further suggests that selective functional deficits occur independently from fibrosis. Correspondingly, VEGF and collagen (∼1000-fold stiffer than elastin) (Mujumdar et al., 2001), both increased in AD cerebral blood vessels (Vinters et al., 1994; Kalaria and Pax, 1995; Kalaria et al., 1998; Tarkowski et al., 2002; present study), were upregulated in TGF+ mice in the face of decreased levels of eNOS and COX-2.
Decreased eNOS protein levels and SOD inefficacy in vessels of TGF+ mice indicated that NO unavailability was attributable, at least in part, to its diminished synthesis. Long-term NOS inhibition in animals induces collagen deposition and progressive structural changes in coronary resistance blood vessels (Takemoto et al., 1997), and homocysteinuria is associated with decreased vascular NO production and gradual replacement of elastin by collagen in vessel walls (Mujumdar et al., 2001). Furthermore, in brain vessels from Binswanger's disease patients (Lin et al., 2000), alterations like those reported here in TGF+ mice and AD patients result in profound microcirculatory disturbances, in conjunction with cerebral blood flow deficits. Similarly, downregulation of COX-2 [an enzyme constitutively expressed in brain neurons and endothelial cells, in which it catalyzes the synthesis of the endothelial-derived vasodilator prostacyclin (Parfenova et al., 2000; Ospina et al., 2004)] occurs in AD brains (Yermakova and O'Banion, 2001) and piglet cerebral microvessels after inhibition of NO production (Hauck et al., 1999) and is associated with increased collagen production and fibrosis of the lung (Hodges et al., 2004) and heart (Francois et al., 2005). These observations, together with our findings, imply that vascular remodeling and alterations in key vasoactive mediators are two independent facets of the same pathological process. Moreover, they indicate that vascular wall alterations independent from CAA, as in AD (Perry et al., 1998), drive the functional impairments in TGF+ mice. Because these changes also occur in microvessels (Figs. 6, 7) (Wyss-Coray et al., 2000), they likely explain the deficit in local brain perfusion recently reported in TGF+ mice (Gaertner et al., 2005).
Modifications in intrinsic properties of the vessel walls such as levels of NO, prostacyclin, and TGF-β1 could alter specific cerebrovascular responses or their signaling pathways. Decreased eNOS protein levels could explain the impaired basal NO production detected in aged TGF+ mice and the more severely lessened dilatations to receptor-mediated dilators such as CGRP and ACh. Both responses depend on endothelial NO, but to a different extent and via distinct transduction pathways (Elhusseiny and Hamel, 2000; Akerman et al., 2002; Xu et al., 2002) that could be differentially affected by TGF-β1. For instance, the altered ET-1 contraction in older TGF+ mice could be attributable to the ability of TGF-β1 to affect protein kinase C (Johnson and Waller, 2002) and p38 MAPK (mitogen-activated protein kinase) phosphorylation (Tong and Hamel, 2005), the activation of which is essential to the ET-1 response in cerebral arteries (Gorlach et al., 1998; Tong and Hamel, 2005).
Relevance to AD
In AD brains, upregulation in SOD2 immunoreactivity was restricted to glial cells, in accordance with previous studies (Pappolla et al., 1992; Furuta et al., 1995). Perivascular SOD labeling compared with that of elderly controls, likely reflecting normal oxidative stress of the aging vasculature (Taddei et al., 2001). However, the vascular wall alterations reported here in AD confirm and extend previous reports (Perlmutter and Chui, 1990; Vinters et al., 1994; Kalaria and Pax, 1995; Zarow et al., 1997) that together point to their similarity with those of TGF+ mice (Wyss-Coray et al., 1997, 2000; present study) and strongly support a role for vascular wall pathology in AD cerebrovascular deficits (Shi et al., 2000; Vagnucci and Li, 2003; de la Torre, 2004; Pogue and Lukiw, 2004). The exact cause of the structural alterations in AD brain vessels remains unknown, but our results suggest that factors other than or in addition to diabetes and vascular risk factors, such as chronic changes in levels of vasoactive mediators, VEGF, TGF-β1, soluble Aβ, and altered signaling or density of cerebrovascular receptors, may need to be considered. In view of the limited success of antioxidants in hypertension, atherosclerosis, and diabetes (Schwartz, 2001), we conclude that reducing oxidative stress alone is unlikely to alleviate the cerebrovascular deficits in AD. Rather, promising therapies may be those that target the onset and development of abnormal cerebrovascular remodeling or, maybe more relevant clinically, attempt to reverse it.
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) (MOP-53334 and MOP-64194 to E.H.) and the Alzheimer Society of Canada (E.H.) and by a CIHR studentship (N.N.). We are grateful to Dr. L. Mucke (University of California, San Francisco, CA) and Dr. T. Wyss-Coray (Stanford University Medical Center, Stanford, CA) for the hAPPSw, Ind and TGF-β1 transgenic mouse breeders, respectively. We are particularly in debt to the Douglas Hospital Brain Bank (McGill University, Montréal, Québec, Canada) and to Dr. J. Richardson (McGill University) for human material and paraffin-cut sections of human cerebral cortex. We also thank A. Niro-Tamburro and D. Cécyre (both from McGill University) for help in retrieving the clinical history of patients and elderly controls, N. Serluca for mouse genotyping, J.-P. Acco for help with the figures, and L. Michel for preparing this final manuscript.
Correspondence should be addressed to Dr. Edith Hamel, Professor, Department of Neurology and Neurosurgery, Montreal Neurological Institute, McGill University, Montréal, Québec, Canada H3A 2B4. E-mail: edith.hamel@mcgill.ca.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511165-10$15.00/0
References
- Akerman S, Williamson DJ, Kaube H, Goadsby PJ (2002) Nitric oxide synthase inhibitors can antagonize neurogenic and calcitonin gene-related peptide induced dilation of dural meningeal vessels. Br J Pharmacol 137: 62-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Barger SBS, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, et al. (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aucoin JS, Jiang P, Aznavour N, Tong XK, Buttini M, Descarries L, Hamel E (2005) Selective cholinergic denervation, independent from oxidative stress, in a mouse model of Alzheimer's disease. Neuroscience 132: 73-86. [DOI] [PubMed] [Google Scholar]
- Beckmann N, Schuler A, Mueggler T, Meyer EP, Wiederhold KH, Staufenbiel M, Krucker T (2003) Age-dependent cerebrovascular abnormalities and blood flow disturbances in APP23 mice modeling Alzheimer's disease. J Neurosci 23: 8453-8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JE, Greenbaum R, Gibson DG, Yacoub M, Laurent GJ (1990) Enhanced deposition of predominantly type I collagen in myocardial disease. J Mol Cell Cardiol 22: 1157-1165. [DOI] [PubMed] [Google Scholar]
- Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, Small GW (2000) Patterns of brain activation in people at risk for Alzheimer's disease. N Engl J Med 343: 450-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckwalter M, Pepper J-P, Gaertner RF, Von Euw D, Lacombe P, Wyss-Coray (2002) Molecular and functional dissection of TGF-β1-induced cerebrovascular abnormalities in transgenic mice. Ann NY Acad Sci 977: 87-95. [DOI] [PubMed] [Google Scholar]
- Cai H, Harrison DG (2000) Endothelial dysfunction in cardiovascular diseases. The role of oxidant stress. Circ Res 87: 840-844. [DOI] [PubMed] [Google Scholar]
- Casserly I, Topol E (2004) Convergence of atherosclerosis and Alzheimer's disease: inflammation, cholesterol, and misfolded proteins. Lancet 363: 1139-1146. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Frey WH, Ala TA, Tourtellotte WW, Peterson PK (1994) Transforming growth factor beta in Alzheimer's disease. Clin Diagn Lab Immunol 1: 109-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie R, Yamada M, Moskowitz M, Hyman B (2001) Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am J Pathol 158: 1065-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller AK (2002) Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res 90: 1159-1166. [DOI] [PubMed] [Google Scholar]
- Das UN (2004) Long-chain polyunsaturated fatty acids interact with nitric oxide, superoxide anion, and transforming growth factor-β to prevent human essential hypertension. Eur J Clin Nutr 58: 195-203. [DOI] [PubMed] [Google Scholar]
- de la Torre JC (2004) Is Alzheimer's disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol 3: 184-190. [DOI] [PubMed] [Google Scholar]
- DeWitt DS, Mathew BP, Chaisson JM, Prough DS (2001) Peroxynitrite reduces vasodilatory responses to reduced intravascular pressure, calcitonin gene-related peptide, and cromakalim in isolated middle cerebral arteries. J Cereb Blood Flow Metab 21: 253-261. [DOI] [PubMed] [Google Scholar]
- Didion SP, Faraci FM (2002) Effects of NADH and NADPH on superoxide levels and cerebral vascular tone. Am J Physiol Heart Circ Physiol 282: H688-H695. [DOI] [PubMed] [Google Scholar]
- Drake J, Link CD, Butterfield DA (2003) Oxidative stress precedes fibrillar deposition of Alzheimer's disease amyloid β-peptide (1-42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging 24: 415-420. [DOI] [PubMed] [Google Scholar]
- Elhusseiny A, Hamel E (2000) Muscarinic—but not nicotinic—acetylcholine receptors mediate a nitric oxide-dependent dilation in brain cortical arterioles: a possible role for the M5 receptor subtype. J Cereb Blood Flow Metab 20: 298-305. [DOI] [PubMed] [Google Scholar]
- Elliott SJ, Lacey DJ, Chilian WM, Brzezinska AK (1998) Peroxynitrite is a contractile agonist of cerebral artery smooth muscle cells. Am J Physiol 275: H1585-H1591. [DOI] [PubMed] [Google Scholar]
- Erdos B, Miller AW, Busija DW (2002) Impaired endothelium-mediated relaxation in isolated cerebral arteries from insulin-resistant rats. Am J Physiol Heart Cir Physiol 282: H2060-H2065. [DOI] [PubMed] [Google Scholar]
- Francois H, Athirakul K, Howell D, Dash R, Mao L, Kim H-S, Rockman HA, FitzGerald GA, Koller BH, Coffman TM (2005) Prostacyclin protects against elevated blood pressure and cardiac fibrosis. Cell Metab 2: 201-207. [DOI] [PubMed] [Google Scholar]
- Furuta A, Price DL, Pardo CA, Troncoso JC, Xu ZS, Taniguchi N, Martin LJ (1995) Localization of superoxide dismutases in Alzheimer's disease and Down's syndrome neocortex and hippocampus. Am J Pathol 146: 357-367. [PMC free article] [PubMed] [Google Scholar]
- Gaertner RF, Wyss-Coray T, Von Euw D, Lesne S, Vivien D, Lacombe P (2005) Reduced brain tissue perfusion in TGF-β1 transgenic mice showing Alzheimer's disease-like cerebrovascular abnormalities. Neurobiol Dis 19: 38-46. [DOI] [PubMed] [Google Scholar]
- Gorlach C, Benyo Z, Wahl M (1998) Endothelin-1-induced contraction in cerebral vessels mediated by phospholipase C/protein kinase C cascade. Kidney Int Suppl 67: S224-S225. [DOI] [PubMed] [Google Scholar]
- Grammas P, Ovase R (2001) Inflammatory factors are elevated in brain microvessels from Alzheimer's disease. Neurobiol Aging 22: 837-842. [DOI] [PubMed] [Google Scholar]
- Grammas P, Ovase R (2002) Cerebrovascular transforming growth factor-β contributes to inflammation in the Alzheimer's disease brain. Am J Pathol 160: 1583-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353-356. [DOI] [PubMed] [Google Scholar]
- Hauck W, Samlalsingh-Parker J, Glibetic M, Ricard G, Beaun MC, Noya FJ, Aranda JV (1999) Deregulation of cyclooxygenase and nitric oxide synthase gene expression in the inflammatory cascade triggered by experimental group B streptococcal meningitis in the newborn brain and cerebral microvessels. Semin Perinatol 23: 250-260. [DOI] [PubMed] [Google Scholar]
- Hodges RJ, Jenkins RG, Wheeler-Jones CPD, Copeman DM, Bottoms SE, Bellingan GJ, Nanthakumar CB, Laurent GJ, Hart SL, Foster ML, McAnulty RJ (2004) Severity of lung injury in cyclooxygenase-2-deficient mice is dependent on reduced prostaglandin E2 production. Am J Pathol 165: 1663-1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C (2003) Atherosclerosis and neurodegeneration. Unexpected conspirators in Alzheimer's dementia. Arterioscler Thromb Vasc Biol 23: 1951-1953. [DOI] [PubMed] [Google Scholar]
- Iadecola C (2004) Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Neurosci Rev 5: 347-360. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA (1999) SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci 2: 157-161. [DOI] [PubMed] [Google Scholar]
- Johnson JA, Waller J (2002) Transforming growth factor beta-1 attenuates endothelin-1-induced functions in neonatal cardiac myocytes. Life Sci 71: 99-113. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Albert MS (2000) Perfusion abnormalities in prodromal AD. Neurobiol Aging 21: 289-292. [DOI] [PubMed] [Google Scholar]
- Kalaria RN (2000) The role of cerebral ischemia in Alzheimer's disease. Neurobiol Aging 21: 321-330. [DOI] [PubMed] [Google Scholar]
- Kalaria RN, Pax AB (1995) Increased collagen content of cerebral microvessels in Alzheimer's disease. Brain Res 705: 349-352. [DOI] [PubMed] [Google Scholar]
- Kalaria RN, Cohen DL, Premkumar DRD, Nag S, LaManna JC, Lust WD (1998) Vascular endothelial growth factor in Alzheimer's disease and experimental cerebral ischemia. Mol Brain Res 62: 101-105. [DOI] [PubMed] [Google Scholar]
- Lacombe P, Mathews PM, Schmidt SD, Breidert T, Heneka MT, Landreth GE, Feinstein D, Galea E (2004) Effect of anti-inflammatory agents on transforming growth factor beta over-expressing mouse brains: a model revised. J Neuroinflammation 1: 1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacza Z, Puskar M, Kis B, Perciaccante JV, Miller AW, Busija DW (2002) Hydrogen peroxide acts as an EDHF in the piglet pial vasculature in response to bradykinin. Am J Physiol Heart Circ Physiol 283: H406-H411. [DOI] [PubMed] [Google Scholar]
- Lin JX, Tomimoto H, Akiguchi I, Matsuo A, Wakita H, Shibasaki H, Budka H (2000) Vascular cell components of the medullary arteries in Binswanger's disease brains. Stroke 31: 1838-1842. [DOI] [PubMed] [Google Scholar]
- Lindenau J, Noack H, Possel H, Asayama K, Wolf G (2000) Cellular distribution of superoxide dismutases in the rat CNS. Glia 29: 25-34. [DOI] [PubMed] [Google Scholar]
- Luchsinger JA, Reitz C, Honig LS, Tang M-X, Shea S, Mayeux R (2005) Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 65: 545-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedecking EK, DeKosky ST, Mehdi H, Ganguli M, Kamboh MI (2000) Analysis of genetic polymorphisms in the transforming growth factorbeta1 gene and the risk of Alzheimer's disease. Hum Genet 106: 565-569. [DOI] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL (2003) Inflammatory processes in Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry 27: 741-749. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L (2000) High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20: 4050-4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueggler T, Sturchler-Pierrat C, Baumann D, Rausch M, Staufenbiel M, Rudin M (2002) Compromised hemodynamic response in amyloid precursor protein transgenic mice. J Neurosci 22: 7218-7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujumdar VS, Aru GM, Tyagi SC (2001) Induction of oxidative stress by homocyst(e)ine impairs endothelial function. J Cell Biochem 82: 491-500. [DOI] [PubMed] [Google Scholar]
- Niwa K, Younkin L, Ebeling C, Turner SK, Westaway D, Younkin S, Ashe KH, Carlson GA, Iadecola C (2000) Aβ-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci USA 97: 9735-9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C (2001) Aβ-peptides enhance vasoconstriction in cerebral circulation. Am J Physiol Heart Circ Physiol 281: H2417-H2424. [DOI] [PubMed] [Google Scholar]
- Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C (2002) Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J PhysiolHeart Circ Physiol 283: H315-H323. [DOI] [PubMed] [Google Scholar]
- Orlandi A, Francesconi A, Marcellini M, Ferlosio A, Spagnoli LG (2004) Role of ageing and coronary atherosclerosis in the development of cardiac fibrosis in the rabbit. Cardiovasc Res 64: 544-552. [DOI] [PubMed] [Google Scholar]
- Ospina JA, Brevig HN, Krause DN, Duckles SP (2004) Estrogen suppresses IL-1β-mediated induction of COX-2 pathway in rat cerebral blood vessels. Am J Physiol Heart Circ Physiol 286: H2010-H2019. [DOI] [PubMed] [Google Scholar]
- Pappolla MA, Omar RA, Kim KS, Robakis NK (1992) Immunohistochemmical evidence of oxidative [corrected] stress in Alzheimer's disease. Am J Pathol 140: 621-628. [PMC free article] [PubMed] [Google Scholar]; [Erratum (1996) 149: 1770] [Google Scholar]
- Parfenova H, Massie V, Leffler CW (2000) Developmental changes in endothelium-derived vasorelaxant factors in cerebral circulation. Am J Physiol Heart Circ Physiol 278: H780-H788. [DOI] [PubMed] [Google Scholar]
- Paris D, Town T, Mori T, Parker TA, Humphrey J, Mullan M (2000) Soluble β-amyloid peptides mediate vasoactivity via activation of a pro-inflammatory pathway. Neurobiol Aging 21: 183-197. [DOI] [PubMed] [Google Scholar]
- Paris D, Humphrey J, Quadros A, Patel N, Crescentini R, Crawford F, Mullan M (2003) Vasoactive effects of Aβ in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer's disease: role of inflammation. Neurol Res 25: 642-651. [DOI] [PubMed] [Google Scholar]
- Paris D, Quadros A, Humphrey J, Patel N, Cresentini R, Crawford F, Mullan M (2004) Nilvadipine antagonizes both Aβ vasoactivity in isolated arteries, and the reduced cerebral blood flow in APPsw transgenic mice. Brain Res 999: 53-61. [DOI] [PubMed] [Google Scholar]
- Park L, Anrather J, Forster C, Kazama K, Carlson GA, Iadecola C (2004) Aβ-induced vascular oxidative stress and attenuation of functional hyperemia in mouse somatosensory cortex. J Cereb Blood Flow Metab 24: 334-342. [DOI] [PubMed] [Google Scholar]
- Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, Carlson GA, Iadecola C (2005) NADPH oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid β peptide. J Neurosci 25: 1769-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter LS, Chui HC (1990) Microangiopathy, the vascular basement membrane and Alzheimer's disease: a review. Brain Res Bull 24: 677-686. [DOI] [PubMed] [Google Scholar]
- Perry G, Smith AM, McCann CE, Siedlak SL, Jones PK, Friedland RP (1998) Cerebrovascular muscle atropy is a feature of Alzheimer's disease. Brain Res 791: 63-66. [DOI] [PubMed] [Google Scholar]
- Pogue AI, Lukiw AJ (2004) Angiogenic signaling in Alzheimer's disease. Mol Neurosci 15: 1507-1510. [DOI] [PubMed] [Google Scholar]
- Price JM, Chi X, Hellermann G, Sutton ET (2001) Physiological levels of β-amyloid induce cerebral vessel dysfunction and reduce endothelial nitric oxide production. Neurol Res 23: 506-512. [DOI] [PubMed] [Google Scholar]
- Roher AE, Esh C, Kokjohn TA, Kalback W, Luehrs DC, Seward JD, Sue LI, Beach TG (2003) Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer's disease. Arterioscler Thromb Vasc Biol 23: 2055-2062. [DOI] [PubMed] [Google Scholar]
- Schwartz SM (2001) A protective player in the vascular response to injury. Nat Med 7: 656-657. [DOI] [PubMed] [Google Scholar]
- Shi J, Perry G, Smith MA, Friedland RP (2000) Vascular abnormalities: the insidious pathogenesis of Alzheimer's disease. Neurobiol Aging 21: 357-361. [DOI] [PubMed] [Google Scholar]
- Smith CD, Andersen AH, Kryscio RJ, Schmitt FA, Kindy MS, Blonder LX, Avison MJ (1999) Altered brain activation in cognitively intact individuals at high risk for Alzheimer's disease. Neurology 53: 1391-1396. [DOI] [PubMed] [Google Scholar]
- Taddei S, Virdis A, Ghiadoni L, Salvetti G, Bernini G, Magagna A, Salvetti A (2001) Age-related reduction of NO availability and oxidative stress in humans. Hypertension 38: 274-279. [DOI] [PubMed] [Google Scholar]
- Takemoto M, Egashira K, Usui M, Numaguchi K, Tomita H, Tsutsui H, Shimokawa H, Sueishi K, Takeshita A (1997) Important role of tissue angiotensin-converting enzyme activity in the pathogenesis of coronary vascular and myocardial structural changes induced by long-term blockade of nitric oxide synthesis in rats. J Clin Invest 99: 278-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkowski E, Issa R, Sjogren M, Wallin A, Blennow K, Tarkowski A, Kumar P (2002) Increased intrathecal levels of the angiogenic factors VEGF and TGF-β in Alzheimer's disease and vascular dementia. Neurobiol Aging 23: 237-243. [DOI] [PubMed] [Google Scholar]
- Tong XK, Hamel E (1999) Regional cholinergic denervation of cortical microvessels and nitric oxide synthase-containing neurons in Alzheimer's disease. Neuroscience 92: 163-175. [DOI] [PubMed] [Google Scholar]
- Tong XK, Hamel E (2005) Chronic high levels of transforming growth factor-β (TGF-β1) alter endothelin-1 (ET-1)-induced contraction in brain vessels via upregulation of mitogen-activated protein kinase phosphatase 1 (MKP-1) and inactivation of p38 mitogen-activated protein kinase (p38-MAPK). Abstract 184. http://www.kenes.com/brain05/program/session1.asp.
- Vagnucci AH, Li WW (2003) Alzheimer's disease and angiogenesis. Lancet 361: 605-608. [DOI] [PubMed] [Google Scholar]
- Vinters HV, Secor DL, Read SL, Frazee JG, Tomiyasu U, Stanley TM, Ferreiro JA, Akers MA (1994) Microvasculature in brain biopsy specimens from patients with Alzheimer's disease: an immunohistochemical and ultrastructural study. Ultrastructural Pathol 18: 333-348. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L (1997) Amyloidogenic role of cytokine TGF-β1 in transgenic mice and in Alzheimer's disease. Nature 389: 603-606. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Sanan DA, Mucke L, Masliah E (2000) Chronic over-production of transforming growth factor-β1 by astrocytes promotes Alzheimer's disease-like microvascular degeneration in transgenic mice. Am J Pathol 156: 139-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H-L, Feinsten D, Santizo RA, Koenig HM, Pelligrino DA (2002) Agonist-specific differences in mechanisms mediating eNOS-dependent pial arteriolar dilation in rats. Am J Physiol Heart Circ Physiol 282: H237-H243. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Fujisawa M, Ando F, Niino NTM, Shimokata H (2002) Association of a polymorphism of the transforming growth factor-β1 gene with blood pressure in japanese individuals. J Hum Genet 47: 243-248. [DOI] [PubMed] [Google Scholar]
- Yermakova AV, O'Banion MK (2001) Downregulation of neuronal cyclooxygenase-2 expression in end stage Alzheimer's disease. Neurobiol Aging 22: 823-836. [DOI] [PubMed] [Google Scholar]
- Zarow C, Barron E, Chui HC, Perlmutter LS (1997) Vascular basement membrane pathology and Alzheimer's disease. Ann NY Acad Sci 826: 147-160. [DOI] [PubMed] [Google Scholar]