

Abstract
Spinal NMDA receptor (NMDAR), protein kinase C (PKC), and glucocorticoid receptor (GR) have all been implicated in the mechanisms of morphine tolerance; however, how these cellular elements interact after chronic morphine exposure remains unclear. Here we show that the expression of spinal NMDAR and PKCγ after chronic morphine is regulated by spinal GR through a cAMP response element-binding protein (CREB)-dependent pathway. Chronic morphine (10 μg, i.t.; twice daily for 6 d) induced a time-dependent upregulation of GR, the NR1 subunit of NMDAR, and PKCγ within the rat's spinal cord dorsal horn. This NR1 and PKCγ upregulation was significantly diminished by intrathecal coadministration of morphine with the GR antagonist RU38486 or a GR antisense oligodeoxynucleotide. Intrathecal coadministration of morphine with an adenylyl cyclase inhibitor (2′,5′-dideoxyadenosine) or a protein kinase A inhibitor (H89) also significantly attenuated morphine-induced NR1 and PKCγ expression, whereas intrathecal treatment with an adenylyl cyclase activator (forskolin) alone mimicked morphine-induced expression of GR, NR1, and PKCγ. Moreover, the expression of phosphorylated CREB was upregulated within the spinal cord dorsal horn after chronic morphine, and a CREB antisense oligodeoxynucleotide coadministered intrathecally with morphine prevented the upregulation of GR, NR1, and PKCγ. These results indicate that spinal GR through the cAMP-CREB pathway played a significant role in NMDAR and PKCγ expression after chronic morphine exposure. The data suggest that genomic interaction among spinal GR, NMDAR, and PKCγ may be an important mechanism that contributes to the development of morphine tolerance.
Keywords: glucocorticoid receptor, NMDA receptor, morphine tolerance, RU38486, protein kinase C, protein kinase A, adenylyl cyclase, cAMP response element-binding protein, CREB
Introduction
Opioid analgesic tolerance is a pharmacological phenomenon that hampers the clinical use of opioids. Recent research has shed light on the neurobiology of opioid tolerance, including studies on intracellular adenylyl cyclase (AC)-cAMP and cAMP-dependent protein kinase A (PKA) (Nestler and Aghajanian, 1997), G-protein signaling (Gintzler and Chakrabarti, 2001), β-arrestin (Bohn et al., 2000), μ-opioid receptor oligomerization (He et al., 2002; Bailey et al., 2003), NMDA receptor (NMDAR) and protein kinase C (PKC) (Trujillo and Akil, 1991; Mao et al., 1995; Zeitz et al., 2002; Xu et al., 2003), and glutamate transporter (Mao et al., 2002). Moreover, chronic morphine has been shown to upregulate and activate spinal glucocorticoid receptors (GRs) initiated by cAMP and PKA activity, which also contributed to the mechanisms of morphine tolerance in rats (Lim et al., 2005b,c). Although these studies have implicated multiple cellular elements in the mechanisms of morphine tolerance, how some of these cellular elements interact in this process remains unknown.
Activation of central GR has been shown to modulate morphine-induced antinociception (Pieretti et al., 1991; Capasso et al., 1992) and dopamine-dependent responses (Schoffelmeer et al., 1996; Marinelli et al., 1998). GR is expressed in spinal cord dorsal horn neurons, which contributes to spinal nociceptive processing (DeNicola et al., 1989; Cintra et al., 1993; Wang et al., 2004, 2005). Activation of central GR also has been shown to regulate neuronal plastic changes after neuronal injury (Cameron and Dutia, 1999) as well as the process of learning and memory (Quirarte et al., 1997; Oitzl et al., 1998; Roosendaal et al., 1999, 2003).
An increasing body of evidence indicates that activation of central GR modulates NMDAR through both genomic and nongenomic mechanisms. For example, the NMDAR-mediated response to excitatory amino acids of dopamine-sensitive neurons within the ventral tegmental area can be potentiated by GR activation (Cho and Little, 1999). Central GR also plays a role in NMDAR-mediated long-term depression (Coussens et al., 1997) and the regulation of NMDAR- and GR-mediated neuronal degeneration (Abraham et al., 2000; Lu et al., 2003). Indeed, peripheral nerve injury has been shown to upregulate spinal GR followed by a downstream upregulation and functional modulation of NMDAR (Wang et al., 2004, 2005). Moreover, the phosphorylated form of a cAMP response element-binding protein (p-CREB) was increased after nerve damage (Ma and Quirion, 2001; Ma et al., 2003; Miletic et al., 2004; Miyabe and Miletic, 2005), spinal cord injury (Crown et al., 2005), and chronic morphine (Guitart et al., 1992; Li and Clark, 1999; Ma et al., 2001), and there are interactions between GR and p-CREB (Imai et al., 1993; Sheppard et al., 1998; Focking et al., 2003; Bachmann et al., 2005). Thus, it is possible that GR may play an important role in the regulation of the cellular elements implicated in the mechanisms of morphine tolerance.
With a rat model of chronic intrathecal morphine administration, we examined the hypothesis that the expression of spinal NMDAR and PKCγ would be upregulated after chronic morphine exposure, which would be regulated by spinal GR expression and activation initiated by the intracellular cAMP-PKA-CREB pathway.
Materials and Methods
Experimental animals. Adult male Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) weighing 300-350 g were used. Animals were housed in cages with water and food pellets available ad libitum. The animal room was artificially illuminated from 7 A.M. until 7 P.M.. The general experimental protocol was approved through our Institutional Animal Care and Use Committee.
Intrathecal catheter implantation and drug delivery. An intrathecal catheter (PE-10) was implanted in each rat under intraperitoneal pentobarbital anesthesia (50 mg/kg) according to a previously published method (Yaksh and Rudy, 1976). Animals that exhibited neurological deficits (e.g., paralysis) after the catheter implantation were excluded from the experiments (Mao et al., 2002). Drugs were delivered intrathecally in a total volume of 10 μl followed by a saline flush. The following drugs were purchased from Sigma (St. Louis, MO): RU38486, morphine, MK-801, spironolactone, 2′,5′-dideoxyadenosine (ddA), H89, and forskolin. Morphine and MK-801 were dissolved in normal saline; the other drugs were dissolved in 10% ethanol solution (vehicle).
For the experiment with GR oligodeoxynucleotides (ONDs), sequences for antisense, sense, and mixed-base ONDs were chosen on the basis of previous studies (Engelmann et al., 1998; Wang et al., 2004; Lim et al., 2005b,c). The sequences overlapping the respective initiation codon (GenBank accession number M14053) used to target the rat's GR mRNA were as follows: antisense (TGGAGTCCATTGGCAAAT), sense (ATTTGCCAATGGACTCCA), and mixed base (TGAAGTTCAGTGTCAACT). For the experiment with CREB ONDs, the sequences for antisense, sense, and mixed-base ONDs were chosen on the basis of a previous study (Ma et al., 2003). The sequences overlapping the respective initiation codon used to target the rat's CREB mRNA (Ma et al., 2003) were as follows: antisense (TGGTCATCTAGTCACCGGTG), sense (CACCGGTGACTAGATGACCA), and mixed base (GACCTCAGGTAGTCGTCGTT). Sequencing was performed by MWG-Biotech (High Point, NC), and product stability was ensured at the time of delivery. ONDs were dissolved in 0.9% saline and injected intrathecally twice daily (5 μg for each GR OND; 10 μg for each CREB OND) for 6 consecutive days.
Induction of morphine tolerance and statistical analysis of behavioral data. Tolerance to the antinociceptive effect of morphine was induced by an intrathecal treatment regimen: 10 μg of morphine was given twice daily for 6 consecutive days. This same morphine regimen has been shown to induce reliable antinociceptive tolerance in rats (Lim et al., 2005b,c). The tail-flick test was used with baseline latencies of 3-4 s and a cutoff time of 10 s. At least two trials were made for each rat, with an intertrial interval of 1 min and with a change of the tail position receiving radiant heat stimulation at each trial. We used a probe dose of morphine (10 μg, i.t.) at different time points, as well as the generation of dose-response curves at the end of the morphine regimen, to compare differences in nociceptive responses between the morphine-treated and control groups. The percentage maximal possible effect (% MPE) was determined by comparing the tail-flick latency before baseline (BL) and after a drug injection (TL) using the following equation: % MPE = [(TL - BL)/(10 - BL)] × 100% (the constant 10 refers to the cutoff time). The data were analyzed by using two-way ANOVA with post hoc Newman-Keuls tests.
Immunocytochemistry. Rats were anesthetized with pentobarbital and transcardially perfused with 200 ml of normal saline followed by 200-300 ml of 4% paraformaldehyde in 0.1 m phosphate buffer. The lumbar spinal cords were dissected, postfixed for 1.5 h, transferred to 30% sucrose in 0.1 m phosphate buffer, and kept overnight. Tissues from both the experimental and control groups were then mounted together on the same block with the OCT compound and frozen on dry ice. The spinal cords were cut (20 μm sections) on a cryostat, mounted serially onto microscope slides, and stored at -80°C for later immunostaining. Immunocytochemical staining was used to detect GR (1:1000, rabbit polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA), NR1 (1:500, mouse monoclonal; Novus Biologicals, Littleton, CO), and PKCγ (1:500, mouse monoclonal; Santa Cruz Biotechnology). Lumbar spinal cord sections were blocked with 1% goat serum in 0.3% Triton for 1 h at room temperature and incubated overnight at 4°C with a primary antibody. Controls were made in the absence of a primary antibody, and antigen absorption (e.g., GR) was used whenever possible. The sections were then incubated for 1 h at room temperature with a corresponding fluorescein isothiocyanate- or cyanine 3-conjugated secondary antibody (1:300; Chemicon, Temecula, CA). For double staining, a second primary antibody was added after the incubation with the first primary antibody following the same procedure described above. Four to six nonadjacent spinal sections were randomly selected, analyzed with an Olympus fluorescence microscope, photographed with a digital camera, and processed with Adobe Photoshop.
Western blot. Rats were rapidly (<1 min) killed through decapitation after being anesthetized with pentobarbital. Lumbar spinal cord segments, divided into dorsal and ventral horns, were removed and homogenized in SDS sample buffer containing a mixture of proteinase inhibitors (Sigma). The lumbar segments were harvested because the drug delivery was aimed at this site. Protein samples were separated on SDS-PAGE gel (4-15% gradient gel; Bio-Rad, Hercules, CA) and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA). The membranes were blocked with 3% milk and incubated overnight at 4°C with a primary antibody [GR, 1:1000, rabbit polyclonal (Santa Cruz Biotechnology); PKCγ, 1:400, mouse monoclonal (Zymed, San Francisco, CA); NR1, 1:2000, mouse monoclonal (Novus Biologicals); p-CREB (Ser133), 1:1000, rabbit polyclonal (Cell Signaling Technology, Beverly, MA)] and for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibody (1:700; Amersham Biosciences, Arlington Heights, IL). The blots were visualized in enhanced chemiluminescence solution (DuPont NEN, Boston, MA) for 1 min and exposed to Hyperfilm (Amersham Biosciences) for 1-10 min. The blots were then incubated in a stripping buffer (67.5 mm Tris, pH 6.8, 2% SDS, and 0.7% β-mercaptoethanol) for 30 min at 50°C and reprobed with a polyclonal rabbit anti-β-actin antibody (1:20,000; Alpha Diagnostic International, San Antonio, TX) as a loading control. The Western analysis was made in triplicates. The band density was measured with a computer-assisted imaging analysis program and normalized against the corresponding loading control. Differences were compared with ANOVA followed by post hoc Newman-Keuls tests.
Reverse transcriptase-PCR. Total RNA was isolated from lumbar spinal cord segments taken according to the method described above, divided into the dorsal and ventral horns, and processed with Trizol reagent (Tel-Test, Friendswood, TX). After incubation for 15 min at 4°C, chloroform was added for the phase separation. The upper aqueous phase was collected, and RNA was precipitated after the sample was mixed with isopropyl alcohol. The RNA pellet was washed once with 75% ethanol, air dried, and finally redissolved in RNase-free water. The A260/A280 ratios (absorbance wave-length ratios) were between 1.6 and 1.8. RT-PCR was performed with the Titan One Tube RT-PCR System (Roche, Indianapolis, IN). Each PCR amplification with a volume of 25 μl contained 100 ng of total RNA, 0.4 μm each primer, 0.2 mm each deoxynucleotide (dNTP), 5 mm dithiothreitol, 5 U of RNase inhibitor, 1.5 mm MgCl2, and 0.5 μl of enzyme mix. The reactions were performed in an MJ Research thermocycler with the following programs: GR: forward primer (AAA TCT TTG TTG GGC TCA GGC TTC CTT GT), reverse primer (GAA GAG AAA CGA GCA AGC ATA G), program (95°C, 30 s; 43°C, 1 min; 72°C, 1 min); mineralocorticoid receptor (MR): forward primer (TAT CTG CAC TGT CGC TCT ATT ACT CTA CC), reverse primer (AAA TCT TTG TTG GGC TCA GGC TTC CTT GT), program (95°C, 30 s; 50°C, 1 min; 72°C, 1 min); NR1: forward primer (CTC ATC TCT AGC CAG GTC TA), reverse primer (TCG CAT CAT CTC AAA CCA GAC), program (94°C, 1 min; 55°C, 1 min; 72°C, 1 min); NR2: forward primer (TCC ATT CTT CTG TCA TCC TGC), reverse primer (AAG ACC GTC TCT CAC TCT TGC), program (95°C, 50 s; 55°C, 1 min; 72°C, 1 min); PKCγ: forward primer (GCT CCC ACA TCA GAT GAG ATC CA), reverse primer (CGT CCT GGG CTG GCA CCG AAG AA), program (95°C, 1 min; 55°C, 2 min; 72°C, 3 min); PKCα: forward primer (ACC CTC AGT GGA ATG AGT CCT TCA CGT), reverse primer (TTA GAT GGC TGC TTC CTG TCT TCT GAA), program (95°C, 1 min; 55°C, 2 min; 72°C, 3 min).
There were 35-37 cycles for each program (NR1 and NR2: 35 cycles; GR, MR, PKCγ, PKCα: 37 cycles). Each program ended after 7 min at 68°C, and products were stored at 4°C. Every PCR was accompanied by one negative control reaction without template RNA. PCR products were analyzed by gel electrophoresis on an ethidium bromide-stained 1% agarose gel (Sigma) in Tris borate-EDTA buffer. The amount of RNA per RT-PCR sample was normalized by using PCR with primers for β-actin (30 cycles each): forward primer (TAC AAC CTC CTT GCA CC); reverse primer (ACA ATG CCG TGT TCA ATG G); program (95°C, 1 min; 55°C, 1 min; 72°C, 1 min). Each band was then measured with a computer-assisted imaging analysis program and normalized against the corresponding loading control. Differences were compared with a one-way ANOVA followed by post hoc Newman-Keuls tests.
Results
Morphine-induced NR1 and PKCγ expression
Chronic morphine exposure (10 μg, i.t.; twice daily for 6 d) in rats induced antinociceptive tolerance on days 5 and 7 of the experimental period compared with the saline control group (Fig. 1A,B) (p < 0.05; n = 5-7). In association with the development of morphine tolerance, there was a time-dependent upregulation of the NR1 subunit of NMDAR and PKCγ within the spinal cord dorsal horn compared with the saline control group (Fig. 2) (p < 0.05; n = 5-7), as revealed by both RT-PCR and Western blot. Spinal NR1 expression began to increase on day 3 and continued to rise on days 5 and 7 of the experimental period, whereas PKCγ expression showed a significant difference from that of the saline control on days 5 and 7 (Fig. 2), indicating that increased PKCγ expression was preceded by NR1 expression, which occurred before the onset of morphine tolerance.
Figure 1.

Tolerance to the antinociceptive effect of morphine (MS) as assessed by the time course analysis (A) and cumulative dose-response curves on day 7 (B). % MPE, Percentage maximal possible effect. The ED50 values and 95% confidence interval (in parentheses) for the saline and morphine groups were 1.62 μg (0.17-2.25 μg) and 17.48 μg (12.46-41.70 μg), respectively. *p < 0.05 compared with the vehicle (saline) group. For all figures, the number of rats used in each group is shown in Results.
Figure 2.
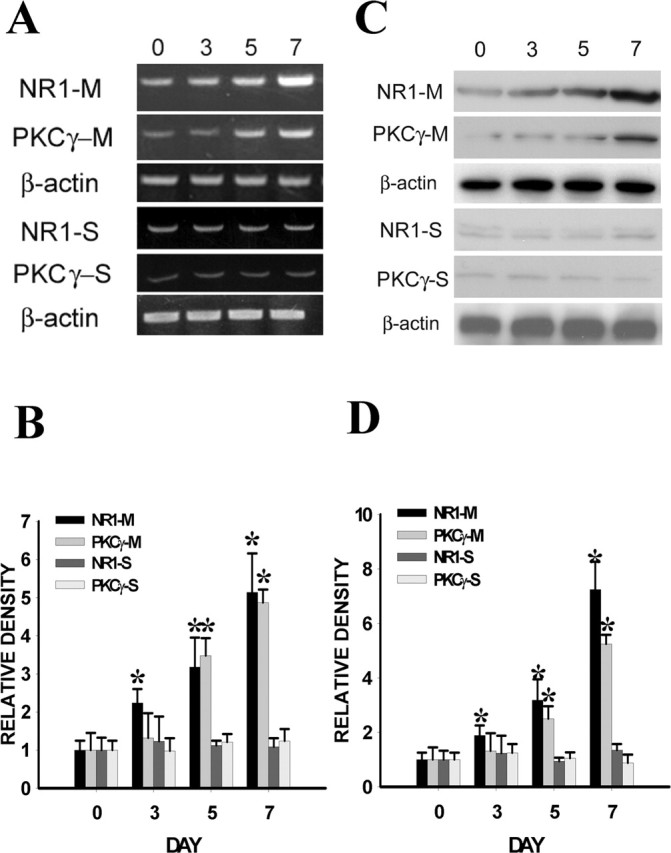
A, C, Time course (day 0 to day 7) of changes in mRNA level (NR1, 225 bp; PKCγ, 536 bp) (A) and protein content (NR1, 100 kDa; PKCγ, 80 kDa) (C) after morphine (M) (10 μg) or saline (S) treatment given intrathecally twice daily for 6 d. β-Actin is a loading control, and those bands measured by using the same blot were represented by a single β-actin loading band (also in other figures). B, D, Statistical analysis for the RT-PCR and Western data from A and C, respectively. *p < 0.05 compared with baseline (day 0).
There was no significant difference in the expression of the NR2 subunit of NMDAR and PKCα within the spinal cord dorsal horn between the morphine and saline treatment groups, nor was there a change in the expression of spinal MR on day 7 (Fig. 3) (p > 0.05; n = 5-7), indicating a selective change in the expression of NR1 and PKCγ within the spinal cord dorsal horn after chronic morphine exposure. Consistent with previous studies that showed a time-dependent increase in spinal GR expression after morphine exposure (Lim et al., 2005b,c), chronic morphine treatment induced the upregulation of GR within the spinal cord dorsal horn compared with the saline control on day 7 (Fig. 3) (p < 0.05, n = 5). This morphine-induced GR upregulation was attenuated in those rats receiving the combined treatment with morphine (10 μg) and RU38486 (2 μg) for 6 consecutive days (Fig. 3) (p < 0.05; n = 5), whereas RU38486 (2 μg) alone did not change baseline GR expression within the spinal cord dorsal horn (Fig. 3) (p > 0.05; n = 5). Because the expression of both NR1 and PKCγ was reliably upregulated on day 7 of the experimental period, we selected day 7 as the time point in the following experiments to examine the role of spinal GR in the regulation of this morphine-induced spinal NR1 and PKCγ expression.
Figure 3.
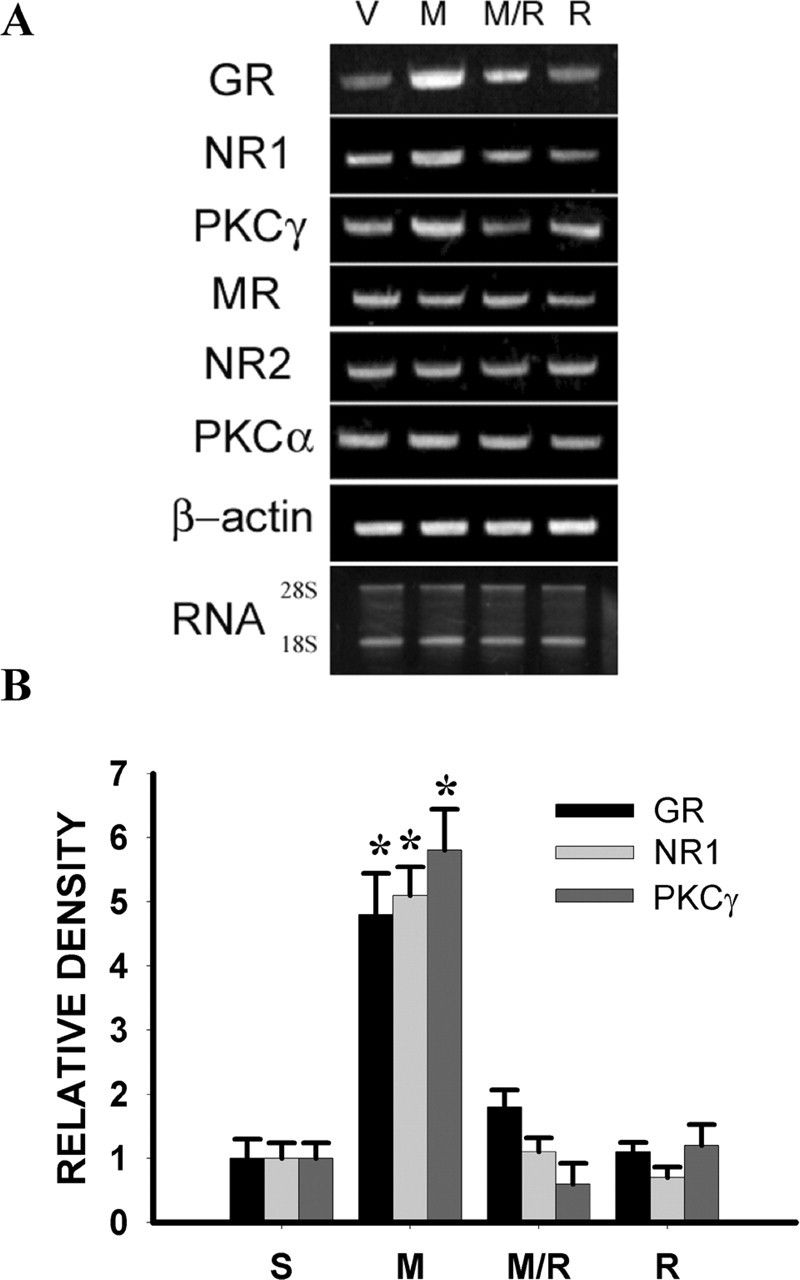
A, Upregulation of NR1, PKCγ, and GR (1157 bp) expression after morphine (M) (10 μg, i.t.; twice daily for 6 d), as assayed by RT-PCR, was blocked by coadministration of morphine and 2 μg of RU38486 (M/R). RU38486 (R) (2 μg) alone did not change baseline NR1 or PKCγ expression. There were no significant changes in the mRNA level of MR (583 bp), NR2 (224 bp), or PKCα (328 bp) on day 7 after chronic morphine. Total RNA loading (28 and 18S) for each group is shown at the bottom of A. B, Statistical data analysis for those groups with statistically significant changes shown in A. *p < 0.05 compared with the vehicle (V) group. S, Saline.
Effect of the GR antagonist RU38486 on NR1 and PKCγ expression
To examine whether spinal GR regulated the expression of NR1 and PKCγ after chronic morphine exposure, spinal cord dorsal horn samples were taken on day 7 from several groups of rats treated (intrathecally twice daily for 6 d) with 10 μg of morphine plus vehicle, 10 μg of morphine plus the GR antagonist RU38486 (0.25 or 2 μg), 10 μg of morphine plus the MR antagonist spironolactone (3 μg), 2 μg of RU38486 alone, or a vehicle. The spironolactone dose was chosen because it has been shown to be effective in blocking MR function (Marinelli et al., 1998), and RU38486 doses were chosen on the basis of the literature (Mao et al., 1992) and our previous dose-response experiments showing the preventive effect of 2 μg (but not 0.25 μg) of RU38486 on morphine tolerance (Lim et al., 2005b,c).
The results from this experiment showed that upregulation of NR1 and PKCγ expression within the spinal cord dorsal horn after chronic morphine was blocked by the coadministration of morphine with RU38486 (2 μg) on day 7 by both RT-PCR (Fig. 3) (p < 0.05; n = 4-5) and Western blot (Fig. 4A,B) (p < 0.05; n = 4-5). RU38486 (2 μg) alone did not change baseline NR1 or PKCγ expression (Figs. 3, 4) (p > 0.05; n = 4-5), nor did a low RU38486 dose (0.25 μg) attenuate the upregulation of NR1 and PKCγ expression after chronic morphine (Fig. 4C,D) (p > 0.05; n = 5). In contrast, coadministration of morphine with the MR antagonist spironolactone did not prevent the morphine-induced upregulation of NR1 and PKCγ (Fig. 4A,B) (p > 0.05; n = 4-5). RU38486 (2 μg) also did not change the expression of NR2, PKCα, and MR (Fig. 3) (p > 0.05; n = 4-5), indicating a selective effect of RU38486 on the expression of spinal NR1 and PKCγ after chronic morphine exposure. In addition, there was colocalization between GR and NR1 as well as GR and PKCγ within the superficial laminas of spinal cord dorsal horn as revealed by immunohistochemistry (Fig. 5).
Figure 4.

A, Upregulation of NR1 and PKCγ after morphine (10 μg, i.t.; twice daily for 6 d), as assayed by the Western blot, was blocked by coadministration of morphine and 2 μg of RU38486 (M/R), but not by morphine and 3 μg of spironolactone (M/SRL). RU38486 (R) (2 μg) alone did not change baseline NR1 or PKCγ expression. C, The dose-response effect of RU38486 (M/R) (0.25 or 2 μg) on NR1 and PKCγ expression. B, D, Statistical analysis for the groups shown in A and C, respectively. *p < 0.05 compared with the vehicle (V) group. S, Saline.
Figure 5.
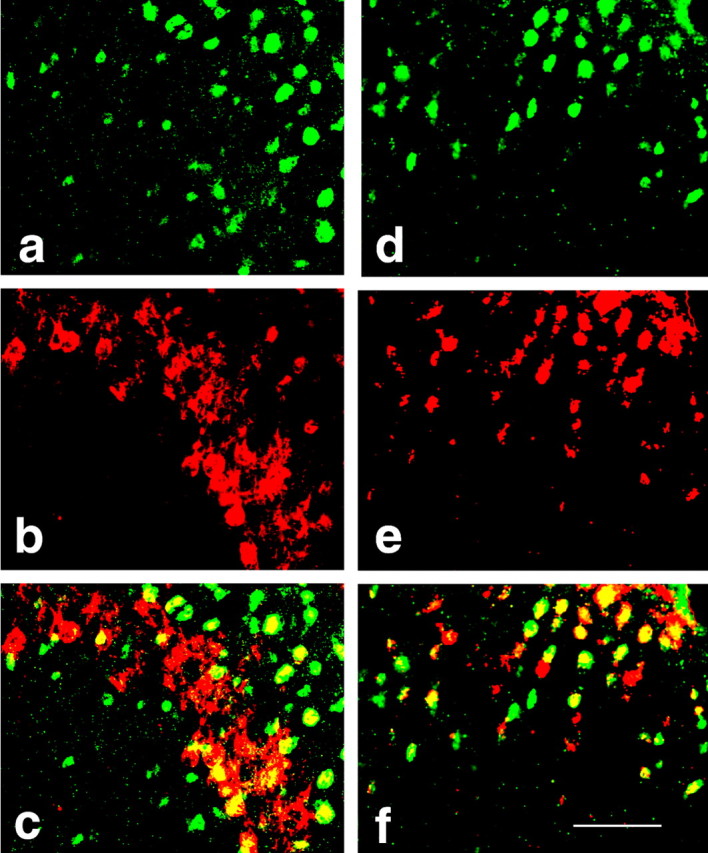
Photomicrographs of colocalization between GR (a, d) and NR1 (e) or PKCγ (b) within the spinal cord dorsal horn as shown in the merged (yellow) images (c, GR/PKCγ; f, GR/NR1). Spinal cord samples were taken from rats receiving morphine treatment on day 7. Scale bar, 100 μm.
Effect of GR antisense OND on NR1 and PKCγ expression
The role of spinal GR in the expression of both NR1 and PKCγ was examined further by downregulating spinal GR with a GR antisense OND. Although mouse models of genetically altered GR expression have been reported, these mouse models are either lethal at birth or unavailable (Tronche et al., 1999; Gartner et al., 2002; St-Hilaire et al., 2003). As an alternative, we used a GR antisense OND that was delivered regionally to the spinal site of GR action. In this set of experiments, each group of rats received 10 μg of morphine in combination with 5 μg each of GR antisense OND, sense OND, mixed-base OND, or vehicle given twice daily for 6 consecutive days, and the spinal expression of NR1, PKCγ, and GR was examined on day 7. This GR antisense treatment regimen significantly attenuated spinal GR expression (Fig. 6B) (p < 0.05; n = 5), which is consistent with previous findings that the same GR antisense treatment downregulated spinal GR expression and attenuated the development of morphine tolerance and neuropathic pain behaviors in rats (Wang et al., 2004, 2005; Lim et al., 2005b,c).
Figure 6.

A, A GR antisense OND (a/m), but not a sense (s/m) or mixed base (m/m) OND, significantly attenuated the upregulation of NR1 and PKCγ, as assayed by RT-PCR, within the spinal cord dorsal horn when examined on day 7. Each OND (5 μg, i.t.) was given in combination with morphine (10 μg, i.t.) for 6 d. B, A GR antisense OND (A), but not a sense (S) or mixed base (M) OND, substantially downregulated GR expression within the spinal cord dorsal horn. C, Statistical analysis for the groups shown in A. *p < 0.05 compared with the vehicle (V) group.
On day 7, the GR antisense OND treatment significantly attenuated the upregulation of spinal NR1 and PKCγ expression induced by chronic morphine exposure (Fig. 6A,C) (p < 0.05; n = 5-7). In contrast, the expression of NR1 and PKCγ remained elevated in the morphine plus mixed-base OND or sense OND group compared with the vehicle group on day 7 (Fig. 6A,C) (p < 0.05; n = 5-7).
Effect of the GR agonist dexamethasone on NR1 and PKCγ expression
To examine whether repeated treatment with an exogenous GR agonist would mimic the morphine-induced upregulation of NR1 and PKCγ expression within the spinal cord dorsal horn, dexamethasone (1 or 4 μg) alone was given intrathecally twice daily for 6 d. The dexamethasone treatment alone dose dependently (4 μg >1 μg = vehicle) induced upregulation of spinal NR1 and PKCγ expression compared with the vehicle group (Fig. 7) (p < 0.05; n = 5) on day 7. Coadministration (intrathecally twice daily for 6 d) of dexamethasone (4 μg) and the GR antagonist RU38486 (2 μg) prevented the dexamethasone-induced upregulation of spinal NR1 and PKCγ in naive rats (Fig. 7) (p < 0.05; n = 5), whereas RU38486 (2 μg) alone did not change the baseline NR1 and PKCγ expression. These results indicate that activation of GR was sufficient to induce NR1 and PKCγ expression within the spinal cord dorsal horn.
Figure 7.
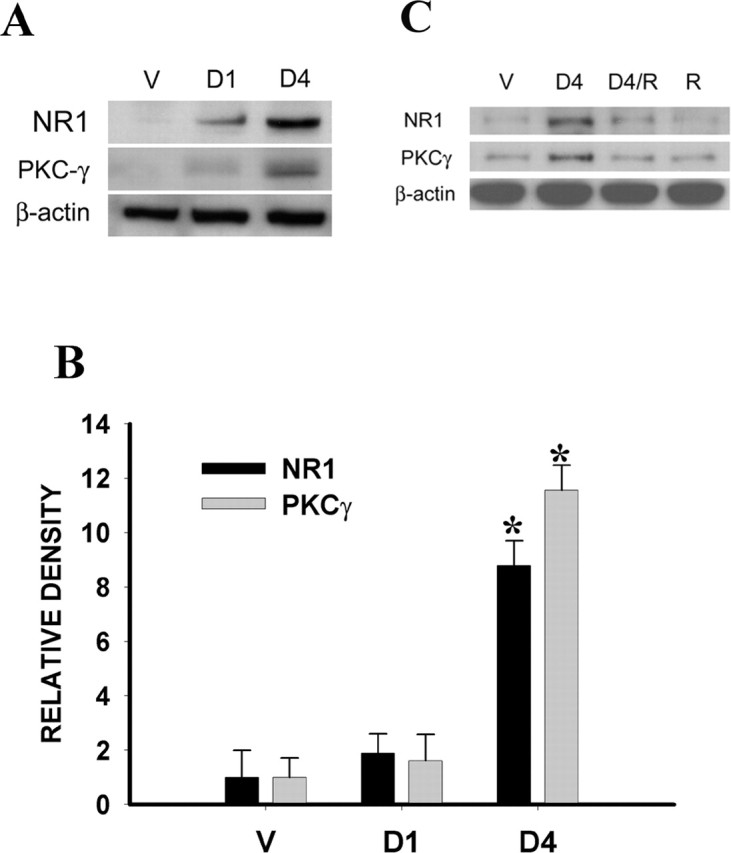
A, The dexamethasone treatment alone given intrathecally twice daily for 6 d dose-dependently (4 μg >1 μg = vehicle) induced the upregulation of NR1 and PKCγ expression within the spinal cord dorsal horn on day 7. B, Statistical data analysis for A. *p < 0.05 compared with the vehicle group. D1, 1 μg of dexamethasone; D4, 4 μg of dexamethasone. C, Coadministration of dexamethasone (4 μg, i.t.; twice daily for 6 d) and RU38486 (2 μg) prevented the dexamethasone-induced upregulation of spinal NR1 and PKCγ in naive rats, whereas RU38486 (2 μg) alone did not change baseline NR1 and PKCγ expression. V, Vehicle; R, RU38486.
Because NMDAR expression was downstream to spinal GR upregulation and activation and NMDAR activation has been shown to prevent the development of morphine tolerance (Trujillo and Akil, 1991), we examined whether inhibition of NMDAR would have a feedback effect on the expression of GR after chronic morphine exposure. The results showed that coadministration of morphine (10 μg, i.t.; twice daily) with the noncompetitive NMDAR antagonist MK-801 (10 nmol) for 6 d significantly diminished morphine-induced GR upregulation on day 7 compared with the vehicle or MK-801-alone group (Fig. 8) (p < 0.05; n = 5), indicating a feedback effect of NMDAR activation on GR expression after chronic morphine exposure.
Figure 8.
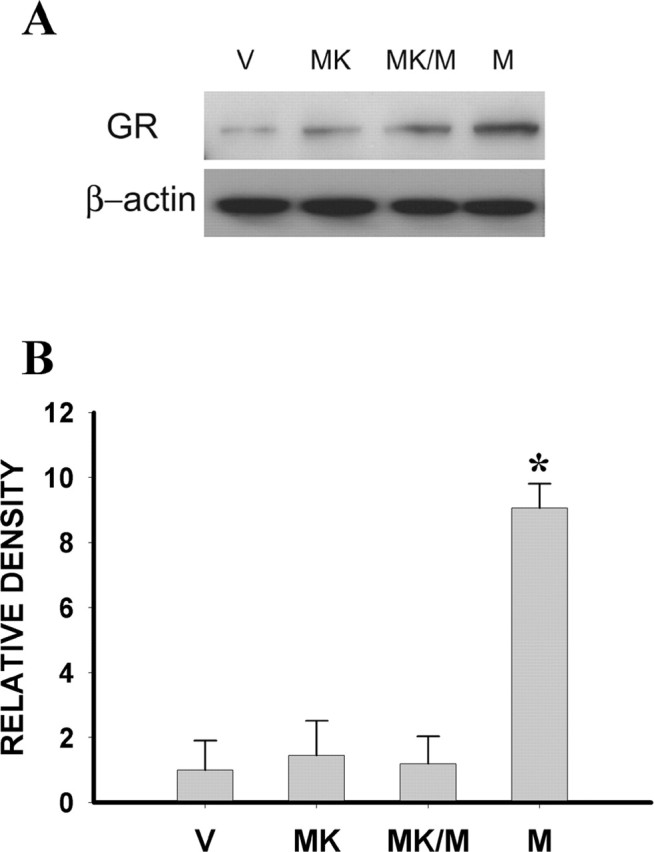
A, Upregulation of spinal GR after 10 μg of morphine, as assayed by Western blot, was attenuated by coadministration of morphine and 10 nmol of MK-801 (MK/M) given intrathecally twice daily for 6 d. MK-801 (MK) (10 nmol) alone did not change baseline GR expression. B, Statistical analysis for the groups shown in A examined on day 7. *p < 0.05 compared with the vehicle (V) group.
Role of AC and PKA activity in NR1 and PKCγ expression
Because the cAMP-PKA pathway regulated GR expression after chronic morphine exposure in our previous study (Lim et al., 2005b), we examined whether the cAMP-PKA pathway would mediate morphine-induced expression of NR1 and PKCγ. The results showed that the AC inhibitor ddA (1 μg, not 0.25 μg) or the PKA inhibitor H89 (10 μg, not 2.5 μg), given intrathecally twice daily for 6 d, nearly blocked the upregulation of spinal NR1 and PKCγ expression on day 7, compared with the morphine plus vehicle group (Fig. 9A-D) (p < 0.05; n = 4-5). In contrast, ddA (1 μg) or H89 (10 μg) treatment alone did not change baseline NR1 or PKCγ expression (Fig. 9A-D) (p > 0.05; n = 4-5). The selected dose range for ddA and H89 was based on previous studies that showed an effective inhibition of cAMP production and PKA activation, respectively (Aley and Levine, 1997; Jolas et al., 2000; Lim et al., 2005b).
Figure 9.
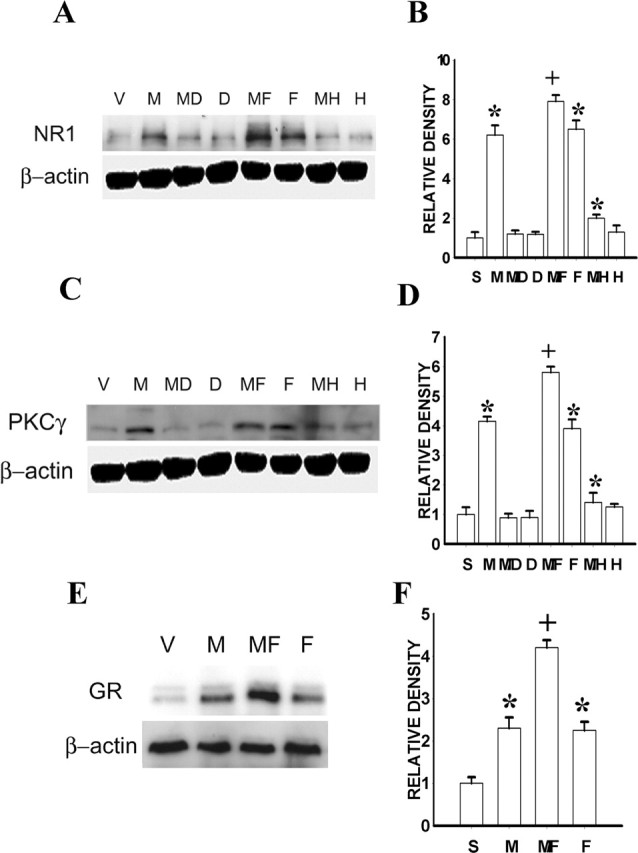
A, C, Upregulation of spinal NR1 (A) and PKCγ (C) expression after chronic morphine (10 μg) was blocked by coadministration of morphine with the AC inhibitor ddA (1 μg) or PKA inhibitor H89 (10 μg), given intrathecally twice daily for 6 d. The ddA or H89 treatment alone did not affect baseline NR1 or PKCγ expression. The AC activator forskolin (10 μg, i.t.; twice daily for 6 d) alone induced the NR1 and PKCγ expression within the spinal cord dorsal horn, and the combined treatment with morphine (10 μg) and forskolin (10 μg) further enhanced NR1 and PKCγ expression. B, D, Statistical data for A and C. E, The same forskolin (10 μg) treatment alone also induced spinal GR expression, and the combined treatment with morphine (10 μg) and forskolin (10 μg) further enhanced GR expression. F, Statistical data for E. S, Saline; V, vehicle; M, morphine; MD, morphine plus ddA; D, ddA alone; MF, morphine plus forskolin; F, forskolin alone; MH, morphine plus H89; H, H89 alone. *p < 0.05 compared with the vehicle group; +p < 0.05 compared with both vehicle and morphine-alone groups.
To further examine the role of cAMP in the expression of NR1 and PKCγ, either forskolin (an AC activator) (Jolas et al., 2000) or vehicle alone was administered intrathecally twice daily for 6 consecutive days. Forskolin (10 μg, not 2.5 μg) alone induced the expression of both NR1 and PKCγ within the spinal cord dorsal horn compared with the vehicle control on day 7 (Fig. 9A-D) (p < 0.05; n = 4-5), which mimicked increased NR1 and PKCγ expression after chronic morphine exposure at this same time point. When forskolin (10 μg) was coadministered with morphine (10 μg, i.t.; twice daily for 6 d), the expression of spinal NR1 and PKCγ was further increased compared with either the morphine-alone or the forskolin-alone group (Fig. 9A-D) (p < 0.05; n = 4-5). Forskolin treatment (10 μg, not 2.5 μg or vehicle, i.t.; twice daily for 6 d) alone also induced the upregulation of spinal GR expression on day 7 (Fig. 9E,F) (p < 0.05; n = 4-5). Moreover, spinal GR expression was further enhanced, compared with morphine (10 μg) alone, after forskolin (10 μg) was coadministered intrathecally with morphine (10 μg) twice daily for 6 d (Fig. 9E,F) (p < 0.05; n = 4-5).
Effect of CREB antisense OND on GR, NR1, and PKCγ expression
Chronic morphine exposure (10 μg, i.t.; twice daily for 6 d) induced the upregulation of p-CREB expression within the spinal cord dorsal horn on day 7 compared with the saline group (Fig. 10A,B) (p < 0.05; n = 5-6). CREB antisense OND treatment given twice daily for 6 d prevented the upregulation of spinal p-CREB expression on day 7 (Fig. 10A,B) (p < 0.05; n = 5), whereas CREB sense or mixed OND (10 μg each) treatment did not alter spinal p-CREB expression (Fig. 10C) (n = 5-6). CREB antisense OND (10 μg, i.t.) treatment also effectively attenuated the upregulation of spinal GR, NR1, and PKCγ expression induced by chronic morphine exposure on day 7 (Fig. 10A,B) (p < 0.05; n = 5-7), whereas CREB antisense treatment alone did not affect the baseline expression of GR, NR1, and PKCγ within the spinal cord dorsal horn (Fig. 10A,B) (p > 0.05; n = 5-6). These results indicate that the p-CREB upregulation was upstream to the expression of GR, NR1, and PKCγ after chronic morphine exposure.
Figure 10.
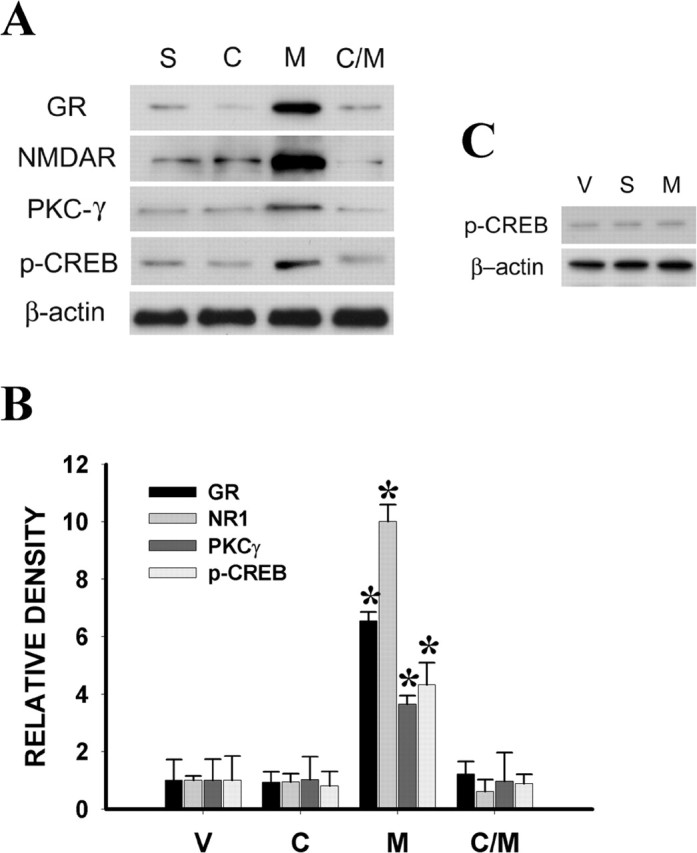
A, Chronic morphine (M) (10 μg, i.t.; twice daily for 6 d) induced the upregulation of p-CREB (43 kDa) expression with in the spinal cord dorsal horn on day 7. Coadministration of morphine and a CREB antisense OND (C/M) (10 μg, given intrathecally twice daily for 6 d, attenuated the morphine-induced upregulation of spinal GR, NR1, and PKCγ expression on day 7. The CREB antisense (C) treatment alone did not affect baseline expression of GR, NR1, and PKCγ. The CREB antisense OND treatment also prevented the upregulation of spinal p-CREB expression. B, Statistical data for A. C, The CREB sense (S) or mixed (M) OND (10 μg each) treatment given intrathecally twice daily for 6 d did not alter spinal p-CREB expression. *p < 0.05 compared with the vehicle (V) group.
Discussion
The present study shows that (1) chronic morphine exposure that resulted in antinociceptive tolerance induced a time-dependent upregulation of the NR1 subunit of NMDAR and PKCγ within the rat's spinal cord dorsal horn; (2) this upregulation of NR1 and PKCγ expression was diminished by coadministration of morphine with either the GR antagonist RU38486 or a GR antisense OND; (3) GR was colocalized with NR1 as well as PKCγ within the spinal cord dorsal horn; (4) coadministration of morphine with the AC inhibitor ddA or the PKA inhibitor H89 also significantly attenuated the upregulation of NR1 and PKCγ expression, whereas forskolin (an AC activator) alone mimicked the expression of spinal NR1, PKCγ, and GR induced by chronic morphine exposure; and (5) chronic morphine exposure induced the upregulation of p-CREB expression within the spinal cord dorsal horn, and CREB antisense OND treatment attenuated the upregulation of spinal GR, NR1, and PKCγ expression induced by chronic morphine exposure. Given that chronic morphine induced the upregulation of spinal GR that was preventable by blocking AC and PKA activity (Lim et al., 2005b), these results indicate that spinal GR through the cAMP-CREB pathway played a significant role in NMDAR and PKCγ expression after chronic morphine exposure.
Several methodological issues should be considered regarding interpretation of the data. Although RU38486 has been widely used as a GR antagonist, it also acts as an antiprogesterone (Mao et al., 1992). The antiprogesterone effect of RU38486 is unlikely to be contributory in this experimental paradigm because only male rats were used in the study. In addition, the selective effect of RU38486 on spinal GR after chronic morphine exposure was supported by the following: (1) the GR knock-down experiment using a GR antisense OND produced a result similar to that after RU38486; (2) RU38486 at 2 μg blocked NR1 and PKCγ expression induced by an exogenous GR agonist (dexamethasone) in naive rats; and (3) there was a dose-response effect of RU38486 on NR1 and PKCγ expression. Spinal MR also did not play a significant role in this process, because the baseline MR expression was not altered after chronic morphine exposure, and the MR antagonist spironolactone at a dose known to block MR function (Marinelli et al., 1998) did not prevent the morphine-induced upregulation of NR1 and PKCγ expression. It should be pointed out that the present experimental paradigm focused on the spinal cord dorsal horn because morphine was delivered through an intrathecal treatment regimen at this spinal site. This approach, however, does not rule out the possibility that a similar GR-mediated regulation of NR1 and PKCγ expression could be involved in brain regions known to be involved in the mechanisms of morphine tolerance.
For several reasons, we examined the relationship among the spinal cAMP-CREB pathway, GR expression and activation, and the expression of NMDAR and PKCγ after chronic morphine. First, a considerable number of previous studies have indicated that NMDAR and PKCγ play a significant role in the mechanisms of morphine tolerance (Trujillo and Akil, 1991; Mao et al., 1995; Zeitz et al., 2002; Xu et al., 2003). Second, our recent studies have shown that spinal GR contributed to the cellular mechanisms of morphine tolerance (Lim et al., 2005b,c) as well as neuropathic pain (Wang et al., 2004), and a number of studies have indicated that morphine tolerance and neuropathic pain may share a similar cellular mechanism involving both NMDAR and PKCγ (Mao et al., 1995). Third, chronic morphine treatment has been shown to increase the GR expression that was mediated through the cAMP and PKA pathway and prevented by the opioid receptor antagonist naloxone (Lim et al., 2005b,c), and the cAMP-PKA-CREB pathway can be activated after chronic morphine exposure (Guitart et al., 1992; Nestler and Aghajanian, 1997; Li and Clark, 1999; Ma et al., 2001). Thus, spinal GR is likely to play a critical role in the cellular mechanisms of morphine tolerance involving both NMDAR and PKCγ.
Our data indicate that at least part of the GR-mediated downstream response after chronic morphine exposure was an enhanced expression of NMDAR and PKCγ, two known contributors to the cellular mechanisms of neural plasticity related to learning, memory, and opioid tolerance (Olds et al., 1989; Collingridge and Singer, 1990; Madison et al., 1991; Trujillo and Akil, 1991; Mao et al., 1995, 2002; Mao, 2002; Zeitz et al., 2002). Moreover, the cAMP-PKA-CREB pathway played an active role in this process because inhibition of AC and PKA activity or a CREB antisense OND substantially diminished the upregulation of GR as well as NR1 and PKCγ expression after chronic morphine exposure. These findings are consistent with known GR actions within the CNS. For example, activation of central GR has been associated with (1) NMDAR-dependent long-term depression (Coussens et al., 1997) and an elevated intracellular Ca2+ concentration in hippocampal neurons (Takahashi et al., 2002), (2) modulation of the NMDAR function (Nair et al., 1998), (3) the potentiated response to NMDA of dopamine-sensitive neurons in the ventral tegmental area (Cho and Little, 1999), (4) neuronal apoptosis mediated through intracellular mitogen-activated protein kinases (Diem et al., 2003), and (5) inhibition of bradykinin-induced Ca2+ influx via PKC activation (Qiu et al., 2003). Thus, the role of GR in spinal NR1 and PKCγ expression is indicative of a broad cellular mechanism of GR regulation under various conditions, including chronic morphine exposure.
The present data using both RT-PCR and Western blot assays indicate that GR-mediated NMDAR and PKCγ expression after chronic morphine is mediated through transcriptional and translational regulation. The actions of GR through a genomic mechanism begin with activation of cytosolic GR (Neeck et al., 2002), which can be activated by circulatory corticosteroids and/or locally produced neurosteroids (Compagnone and Mellon, 2000; Plassart-Schiess and Baulieu, 2001; Vallee et al., 2001). Activation of GR requires the formation of a GR-homodimer after its dissociation from the cytosolic complex consisting of heat shock proteins (Drouin et al., 1992). A GR-homodimer serves as a transcription factor and binds to nuclear-specific DNA responsive elements, regulating gene transcription and translation for a number of cellular elements (Drouin et al., 1992). For instance, GR activation has been shown to induce the expression of a Ca2+ channel subunit in neurons of the basolateral amygdala (Karst et al., 2002) and of spinal NMDAR after peripheral nerve injury (Wang et al., 2005). The expression of spinal NMDAR and PKCγ was attenuated by a GR antisense OND in this study, lending support to a genomic mechanism of GR action. Of note is that spinal GR is mostly associated with neuronal cells because there is an abundant colocalization between spinal GR and neuronal-specific nuclear protein (a neuronal marker), as shown in a previous study (Wang et al., 2004), and among GR, NMDAR, and PKCγ, as shown in the present study. Our data also indicate that inhibition of NMDAR by MK-801 significantly reduced GR upregulation after chronic morphine, which is consistent with a reciprocal role between NMDAR and GR in the regulation of neuronal degeneration (Abraham et al., 2000; Lu et al., 2003), suggesting that the downstream response to GR may have a feedback effect on the cellular process leading to morphine-induced GR upregulation.
The regulatory role of GR in the expression of NMDAR and PKCγ may be critical to the cellular mechanisms of morphine tolerance, because either disruption of GR upregulation with a GR antisense OND or inhibition of GR activation with RU38486 blocked the development of morphine tolerance (Lim et al., 2005b,c). Moreover, our previous studies have shown that the GR-mediated effect on morphine tolerance was abolished in adrenalectomized rats, indicating that endogenous corticosteroids played a significant role in GR function after chronic morphine exposure (Lim et al., 2005b). Together with previous data indicating (1) attenuation of the development of morphine tolerance in rats after intrathecal coadministration of ddA or H89 with morphine (Lim et al., 2005a), (2) an upstream regulatory role of cAMP and cAMP-dependent PKA in morphine-induced GR upregulation (Lim et al., 2005b), and (3) interactions between cAMP and glucocorticoid effects (Kuwahara et al., 2003), the present data suggest that a genomic interaction among spinal GR, NMDAR, and PKCγ mediated by the cAMP-PKA-CREB pathway plays a critical role in the cellular mechanism of morphine tolerance. The results also suggest that a GR inhibitor such as RU38486 or agents blocking the NMDAR and PKCγ upregulation may be useful, alone or in combination, in preventing the development of opioid tolerance, an issue of significant clinical relevance (Mao, 2002; Ballantyne and Mao, 2003).
Footnotes
This work was supported by United States Public Health Service RO1 Grants DA08835, NS42661, and NS45681.
Correspondence should be addressed to Dr. Jianren Mao, Pain Research Group, Division of Pain Medicine, Wang Ambulatory Care Center 324, Massachusetts General Hospital, Harvard Medical School, 15 Parkman Street, Boston, MA 02114. E-mail: jmao@partners.org.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511145-10$15.00/0
References
- Abraham I, Harkany T, Horvath K, Veenema AH, Penke B, Nyakas C, Luiten PG (2000) Chronic corticosterone administration dose-dependently modulates Abeta(1-42)- and NMDA-induced neurodegeneration in rat magnocellular nucleus basalis. J Neuroendocrinol 12: 486-494. [DOI] [PubMed] [Google Scholar]
- Aley KO, Levine JD (1997) Different mechanisms mediate development and expression of tolerance and dependence for peripheral μ-opioid antinociception in rat. J Neurosci 17: 8018-8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann CG, Bilang-Bleuel A, De Calis S, Linthorst AC, Reul JM (2005) The selective glucocorticord receptor antagonist ORG34116 decreases immobility time in the forced swim test and affects cAMP-responsive element-binding protein phosphorylation in rat brain. Neuroendocrinology 81: 129-136. [DOI] [PubMed] [Google Scholar]
- Bailey CP, Couch D, Johnson E, Griffiths K, Kelly E, Henderson G (2003) μ-Opioid receptor desensitization in mature rat neurons: lack of interaction between DAMGO and morphine. J Neurosci 10515-10520. [DOI] [PMC free article] [PubMed]
- Ballantyne J, Mao J (2003) Opioid therapy for chronic pain. N Engl J Med 349: 1943-1953. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainestdinov RR, Lin FT, Lefkowitz RJ, Caron MG (2000) Muopioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 408: 720-723. [DOI] [PubMed] [Google Scholar]
- Cameron SA, Dutia MB (1999) Lesion-induced plasticity in rat vestibular nucleus neurons is dependent on glucocorticoid receptor activation. J Physiol (Lond) 518: 151-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso A, Di Giannuario A, Loizzo A, Pieretti S, Sorrentino L (1992) Central interaction of dexamethasone and RU-38486 on morphine antinociception in mice. Life Sci 51: PL139-PL143. [DOI] [PubMed] [Google Scholar]
- Cho K, Little HJ (1999) Effects of corticosterone on excitatory amino acid responses in dopamine-sensitive neurons in the ventral tegmental area. Neuroscience 88: 837-845. [DOI] [PubMed] [Google Scholar]
- Cintra A, Molander C, Fuxe K (1993) Colocalization of Fos- and glucocorticoid receptor-immunoreactivities is present only in a very restricted population of dorsal horn neurons of the rat spinal cord after nociceptive stimulation. Brain Res 632: 334-338. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Singer W (1990) Excitatory amino acid receptors and synaptic plasticity. Trends Pharmacol Sci 11: 290-296. [DOI] [PubMed] [Google Scholar]
- Compagnone NA, Mellon SH (2000) Neurosteroids: biosynthesis and functions of these novel neuromodulators. Front Neuroendocrinol 21: 1-56. [DOI] [PubMed] [Google Scholar]
- Coussens CM, Kerr DS, Abraham WC (1997) Glucocorticoid receptor activation lowers the threshold for NMDA-receptor-dependent homosynaptic long-term depression in the hippocampus through activation of voltage-dependent calcium channels. J Neurophysiol 78: 1-9. [DOI] [PubMed] [Google Scholar]
- Crown ED, Ye Z, Johnson KM, Xu GY, McAdoo DJ, Westlund KN, Huse-bosch CE (2005) Upregulation of the phosphorylated form of CREB in spinothalamic tract cells following spinal cord injury: relation to central neuropathic pain. Neurosci Lett 384: 139-144. [DOI] [PubMed] [Google Scholar]
- DeNicola AF, Moses DF, Gonzalez S, Orti E (1989) Adrenocorticoid action in the spinal cord: some unique molecular properties of glucocorticoid receptors. Cell Mol Neurobiol 9: 179-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diem R, Hobom M, Maier K, Weissert R, Storch MK, Meyer R, Bahr M (2003) Methylprednisolone increases neuronal apoptosis during auto-immune CNS inflammation by inhibition of an endogenous neuroprotective pathway. J Neurosci 23: 6993-7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin J, Sun YL, Tremblay S, Lavender P, Schmidt TJ, De Lean A, Nemer M (1992) Homodimer formation is rate-limiting for high affinity DNA binding by glucocorticoid receptor. Mol Endocrinol 6: 1299-1309. [DOI] [PubMed] [Google Scholar]
- Engelmann M, Landgraf R, Lorscher P, Conzelmann C, Probst JC, Holsboer F, Reul JM (1998) Downregulation of brain mineralocorticoid and glucocorticoid receptor by antisense oligodeoxynucleotide treatment fails to alter spatial navigation in rats. Eur J Pharmacol 361: 17-26. [DOI] [PubMed] [Google Scholar]
- Focking M, Holker I, Trapp T (2003) Chronic glucocorticoid receptor activation impairs CREB transcriptional activity in clonal neurons. Biochem Biophys Res Commun 304: 720-723. [DOI] [PubMed] [Google Scholar]
- Gartner H, Graul MC, Oesterreicher TJ, Finegold MJ, Henning SJ (2002) Development of the fetal intestine in mice lacking the glucocorticoid receptor (GR). J Cell Physiol 194: 80-87. [DOI] [PubMed] [Google Scholar]
- Gintzler AR, Chakrabarti S (2001) Opioid tolerance and the emergence of new opioid receptor-coupled signaling. Mol Neurobiol 21: 21-33. [DOI] [PubMed] [Google Scholar]
- Guitart X, Thompson MA, Mirante CK, Greenberg ME, Nestler EJ (1992) Regulation of cyclic AMP response element-binding protein (CREB) phosphorylation by acute and chronic morphine in the rat locus coeruleus. J Neurochem 58: 1168-1171. [DOI] [PubMed] [Google Scholar]
- He L, Fong J, von Zastrow M, Whistler JL (2002) Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell 108: 271-282. [DOI] [PubMed] [Google Scholar]
- Imai E, Miner JN, Michell JA, Yamamoto KR, Granner DK (1993) Glucocorticoid receptor-cAMP response element-binding protein interaction and the response of the phosphoenopyruvate carboxykinase gene to glucocorticoids. J Biol Chem 268: 5353-5356. [PubMed] [Google Scholar]
- Jolas T, Nestler EJ, Aghajanian GK (2000) Chronic morphine increases GABA tone on serotonergic neurons of the dorsal raphe nucleus: association with an up-regulation of the cyclic AMP pathway. Neuroscience 95: 433-443. [DOI] [PubMed] [Google Scholar]
- Karst H, Nair S, Velzing E, Rumpff-van Essen L, Slagter E, Shinnick-Gallagher P, Joels M (2002) Glucocorticoids alter calcium conductances and calcium channel subunit expression in basolateral amygdala neurons. Eur J Neurosci 16: 1083-1089. [DOI] [PubMed] [Google Scholar]
- Kuwahara S, Arima H, Banno R, Sato I, Kondo N, Oiso Y (2003) Regulation of vasopressin gene expression by cAMP and glucocorticoids in parvocellular neurons of the paraventricular nucleus in rat hypothalamic organotypic cultures. J Neurosci 23: 10231-10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Clark JD (1999) Morphine tolerance and transcription factor expression in mouse spinal cord tissue. Neurosci Lett 272: 79-82. [DOI] [PubMed] [Google Scholar]
- Lim G, Wang S, Mao J (2005a) cAMP and protein kinase A contribute to the downregulation of spinal glutamate transporters after chronic morphine. Neurosci Lett 376: 9-13. [DOI] [PubMed] [Google Scholar]
- Lim G, Wang S, Zeng Q, Sung B, Mao J (2005b) Central glucocorticoid receptors contribute to the mechanisms of morphine tolerance. Anesthesiology 102: 832-837. [DOI] [PubMed] [Google Scholar]
- Lim G, Wang S, Zeng Q, Sung B, Mao J (2005c) Evidence for a long-term influence of previous morphine exposure on morphine tolerance: a role of central glucocorticoid receptors. Pain 114: 81-92. [DOI] [PubMed] [Google Scholar]
- Lu J, Goula D, Sousa N, Almeida OF (2003) Ionotropic and metabotropic glutamate receptor mediation of glucocorticoid-induced apoptosis in hippocampal cells and the neuroprotective role of synaptic N-methyl-d-aspartate receptors. Neuroscience 121: 123-131. [DOI] [PubMed] [Google Scholar]
- Ma W, Quirion R (2001) Increased phosphorylation of cyclic AMP response element-binding protein (CREB) in the superficial dorsal horn neurons following partial sciatic nerve ligation. Pain 93: 295-301. [DOI] [PubMed] [Google Scholar]
- Ma W, Zheng WH, Powell K, Jhamandas K, Quirion R (2001) Chronic morphine exposure increases the phosphorylation of MAP kinases and the transcription factor CREB in dorsal root ganglion neurons: an in vitro and in vivo study. Eur J Neurosci 14: 1091-1104. [DOI] [PubMed] [Google Scholar]
- Ma W, Hatzis C, Eisenach JC (2003) Intrathecal injection of cAMP response element binding protein (CREB) antisense oligonucleotide attenuates tactile allodynia caused by partial sciatic nerve ligation. Brain Res 988: 97-104. [DOI] [PubMed] [Google Scholar]
- Madison DV, Malenka RC, Nicoll RA (1991) Mechanisms underlying longterm potentiation of synaptic transmission. Annu Rev Neurosci 14: 379-397. [DOI] [PubMed] [Google Scholar]
- Mao J (2002) Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain 100: 213-217. [DOI] [PubMed] [Google Scholar]
- Mao J, Regelson W, Kalimi M (1992) Molecular mechanisms of RU 486 action: a review. Mol Cell Biol 109: 1-8. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Phillips LL, Lu J, Mayer DJ (1995) Increases in protein kinase C gamma immunoreactivity in the spinal cord of rats associated with tolerance to the analgesic effects of morphine. Brain Res 677: 257-267. [DOI] [PubMed] [Google Scholar]
- Mao J, Sung B, Ji RR, Lim G (2002) Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 22: 8312-8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli M, Aouizerate B, Barrot M, Le Moal M, Piazza PV (1998) Dopamine-dependent responses to morphine depend on glucocorticoid receptors. Proc Natl Acad Sci USA 95: 7742-7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miletic G, Hanson EN, Savagian CA, Miletic V (2004) Protein kinase A contributes to sciatic ligation-associated early activation of cyclic AMP response element binding protein in the rat spinal dorsal horn. Neurosci Lett 360: 149-152. [DOI] [PubMed] [Google Scholar]
- Miyabe T, Miletic V (2005) Multiple kinase pathways mediate the early sciatic ligation-associated activation of CREB in the rat spinal dorsal horn. Neurosci Lett 381: 80-85. [DOI] [PubMed] [Google Scholar]
- Nair SM, Werkman TR, Craig J, Finnell R, Joels M, Eberwine JH (1998) Corticosterone regulation of ion channel conductances and mRNA levels in individual hippocampal CA1 neurons. J Neurosci 18: 2685-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeck G, Renkawitz R, Eggert M (2002) Molecular aspects of glucocorticoid hormone action in rheumatoid arthritis. Cytokines Cell Mol Ther 7: 61-69. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Aghajanian GK (1997) Molecular and cellular basis of addiction. Science 278: 58-63. [DOI] [PubMed] [Google Scholar]
- Oitzl M, Fluttert M, Sutanto W, de Kloet WR (1998) Continuous blockade of brain glucocorticoid receptors facilitate spatial learning and memory in rats. Eur J Neurosci 10: 3759-3766. [DOI] [PubMed] [Google Scholar]
- Olds JL, Anderson ML, McPhie DL, Ataten LD, Alkon DL (1989) Imaging of memory-specific changes in the distribution of protein kinase C in the hippocampus. Science 245: 866-869. [DOI] [PubMed] [Google Scholar]
- Pieretti S, Capasso A, Di Giannuario A, Loizzo A, Sorrentino L (1991) The interaction of peripherally and centrally administered dexamethasone and RU 38486 on morphine analgesia in mice. Gen Pharmacol 22: 929-933. [DOI] [PubMed] [Google Scholar]
- Plassart-Schiess E, Baulieu E (2001) Neurosteroids: recent findings. Brain Res Brain Res Rev 37: 133-140. [DOI] [PubMed] [Google Scholar]
- Qiu J, Wang CG, Haung XY, Chen YZ (2003) Nongenomic mechanism of glucocorticoid inhibition of bradykinin-induced calcium influx in PC12 cells: possible involvement of protein kinase C. Life Sci 72: 2533-2542. [DOI] [PubMed] [Google Scholar]
- Quirarte GL, Roozendaal B, McGaugh JL (1997) Glucocorticoid enhancement of memory storage involves noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci USA 94: 14048-14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosendaal B, Williams GL, McGaugh JL (1999) Glucocorticoid receptor activation in the rat nucleus of the solitary tract facilitates memory consolidation: involvement of the basolateral amygdala. Eur J Neurosci 11: 1317-1323. [DOI] [PubMed] [Google Scholar]
- Roosendaal B, McReynolds JJ, McGaugh JL (2003) The basolateral amygdala interacts with the medial prefrontal cortex in regulating glucocorticoid effects on working memory impairment. J Neurosci 24: 1385-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoffelmeer AN, Voorn P, Jonker AJ, Wardeh G, Nestby P, Vanderschuren LJ, De Vries TJ, Mulder AH, Tjon GH (1996) Morphine-induced increase in D-1 receptor regulated signal transduction in rat striatal neurons and its facilitation by glucocorticoid receptor activation: possible role in behavioral sensitization. Neurochem Res 21: 1417-1423. [DOI] [PubMed] [Google Scholar]
- Sheppard KA, Phelps KM, Williams AJ, Thanos D, Glass CK, Rosenfeld MG, Gerritsen ME, Collins T (1998) Nuclear integration of glucocorticoid receptor and nuclear factor-kappaB signaling by CREB-binding protein and steroid receptor co-activator-1. J Biol Chem 273: 29291-29294. [DOI] [PubMed] [Google Scholar]
- St-Hilaire M, Tremblay PO, Levesque D, Barden N, Rouillard C (2003) Effects of cocaine on c-Fos and NGFI-B mRNA expression in transgenic mice underexpressing glucocorticoid receptors. Neuropsychopharmacology 28: 478-489. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kimoto T, Tanabe N, Hattori TA, Yasumatsu N, Kawata T (2002) Corticosterone acutely prolonged N-methyl-d-aspartate receptor mediated Ca2+ elevation in cultured rat hippocampal neurons. J Neurochem 83: 1441-1451. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Shutz G (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23: 99-103. [DOI] [PubMed] [Google Scholar]
- Trujillo KA, Akil H (1991) Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science 251: 85-87. [DOI] [PubMed] [Google Scholar]
- Vallee M, Mayo W, Koob GF, Le Moal M (2001) Neurosteroids in learning and memory. Int Rev Neurobiol 46: 273-320. [DOI] [PubMed] [Google Scholar]
- Wang S, Lim G, Zeng Q, Sung B, Ai Y, Guo G, Yang L, Mao J (2004) Expression of central glucocorticoid receptors after peripheral nerve injury contributes to neuropathic pain behaviors in rats. J Neurosci 24: 8595-8605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Lim G, Zeng Q, Yang L, Sung B, Mao J (2005) Neuronal glucocorticoid receptors modulate the expression and function of spinal NMDA receptors after peripheral nerve injury. J Neurosci 25: 488-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu NJ, Bao L, Fan HP, Bao GB, Pu L, Lu YJ, Wu CF, Zhang X, Pei G (2003) Morphine withdrawal increases glutamate uptake and surface expression of glutamate transporter GLT-1 at hippocampal synapses. J Neurosci 23: 4775-4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA (1976) Chronic catheterization of the spinal subarachnoid space. Physiol Behav 17: 1031-1036. [DOI] [PubMed] [Google Scholar]
- Zeitz KP, Malmberg AB, Gilbert H, Basbaum AI (2002) Reduced development of tolerance to the analgesic effects of morphine and clonidine in PKCgamma mutant mice. Pain 94: 245-253. [DOI] [PubMed] [Google Scholar]