

Abstract
Amyloid-β (Aβ) has been implicated in memory loss and disruption of synaptic plasticity observed in early-stage Alzheimer's disease. Recently, it has been shown that soluble Aβ oligomers target synapses in cultured rat hippocampal neurons, suggesting a direct role of Aβ in the regulation of synaptic structure and function. Postsynaptic density-95 (PSD-95) is a postsynaptic scaffolding protein that plays a critical role in synaptic plasticity and the stabilization of AMPA (AMPARs) and NMDA (NMDARs) receptors at synapses. Here, we show that exposure of cultured cortical neurons to soluble oligomers of Aβ1-40 reduces PSD-95 protein levels in a dose- and time-dependent manner and that the Aβ11-40-dependent decrease in PSD-95 requires NMDAR activity. We also show that the decrease in PSD-95 requires cyclin-dependent kinase 5 activity and involves the proteasome pathway. Immunostaining analysis of cortical cultured neurons revealed that Aβ treatment induces concomitant decreases in PSD-95 at synapses and in the surface expression of the AMPAR glutamate receptor subunit 2. Together, these data suggest a novel pathway by which Aβ triggers synaptic dysfunction, namely, by altering the molecular composition of glutamatergic synapses.
Keywords: phosphorylation, glutamate receptor, metabotropic glutamate receptor, AMPA receptor, synaptic plasticity, amyloid β, Aβ peptide, NMDA receptor, PSD-95, proteasome, cdk5
Introduction
Insoluble amyloid-β (Aβ1-40 and Aβ1-42) fibrils, generated from the cleavage of amyloid precursor protein, constitute the extracellular senile plaques that typify the brains of patients with Alzheimer's disease (AD); plaque formation is a protracted process. However, recent studies have shown that soluble, low-molecular-weight (8-24 kDa) Aβ oligomers, referred to as amyloid-derived diffusible ligands, may be responsible for initiating neuronal dysfunction (Lambert et al., 1998; Walsh et al., 2002; Gong et al., 2003) (for review, see Mattson, 2004). Soluble Aβ peptides can be detected, albeit in low amounts, in normal brain (Lue et al., 1999) in which they seem to play a physiological role (Wilquet and De Strooper, 2004). Notwithstanding that neuronal loss is seen in later stages of AD, an emerging view is that synaptic failure is a key pathogenic factor in the disease (Selkoe, 2002). Strong evidence links reductions in synaptic density with severity of dementia (DeKosky and Scheff, 1990; Coleman and Yao, 2003), and, interestingly, changes in synaptic density have been recorded in terminal, distal dendrites in the frontal cortex at very early stages of disease (Coleman et al., 2004). Thus, impairment in synaptic function and plasticity might be an early event in the pathogenesis of Alzheimer's disease (Oddo et al., 2003; Walsh and Selkoe, 2004; Wang et al., 2004).
Postsynaptic density-95 (PSD-95) is an abundant postsynaptic scaffolding protein that plays an important role in synapse maturation and synaptic plasticity (El-Husseini et al., 2000). PSD-95 is implicated in the assembly of many components of the PSD, including downstream signaling molecules and cytoskeletal linker proteins (McGee and Bredt, 2003; Kim and Sheng, 2004). Palmitoylation of PSD-95 is required for the synaptic localization and clustering of PSD-95 (Christopherson et al., 2003; McGee and Bredt, 2003). Morabito et al. (2004) have shown that T19 and S25 residues in the N-terminus of PSD-95 are phosphorylated in a cyclin-dependent kinase 5 (cdk5)-dependent manner and that inhibition of cdk5 in neurons results in larger PSD-95 clusters. These observations suggest that cdk5 regulates the number/density of PSD-95 clusters at synapses (Morabito et al., 2004).
PSD-95 interacts directly with NMDA receptors (NMDARs), modulating their channel properties (Iwamoto et al., 2004; Lin et al., 2004) and posttranslational processing (Dong et al., 2004) and stabilizing them at synapses (Niethammer et al., 1996; Roche et al., 2001; Lavezzari et al., 2003; Li et al., 2003; Lin et al., 2004). Recent studies have shown that PSD-95 interacts indirectly with AMPA receptors (AMPARs) through the transmembrane protein stargazin (L. Chen et al., 2000; Schnell et al., 2002) and regulates the trafficking and localization of AMPARs at synapses (McGee and Bredt, 2003). Activity-mediated internalization of AMPARs is inhibited by PSD-95 and requires the binding of PSD-95 to stargazin (L. Chen et al., 2000; Schnell et al., 2002). Furthermore, overexpression of PSD-95 enhances AMPAR-mediated synaptic transmission and occludes long-term potentiation (LTP) (Stein et al., 2003; Ehrlich and Malinow, 2004). Colledge et al. (2003) demonstrated that, in response to NMDAR activation, PSD-95 is ubiquitinated and rapidly removed from synaptic sites by proteasome-dependent degradation. Mutations that block PSD-95 ubiquitination prevent NMDA-induced AMPAR endocytosis, suggesting a key role for PSD-95 degradation in the induction of long-term depression (LTD). The importance of PSD-95 in cognitive processes is attested by the observation that PSD-95 knock-out mice show impaired spatial learning abilities (Migaud et al., 1998) and that PSD-95 is driven in synapses during experience-induced plasticity (Ehrlich et al., 2004).
Recently, Lacor et al. (2004) demonstrated that Aβ oligomers bind to synaptic sites that are immunopositive for PSD-95. Given the importance of PSD-95 in synaptic function, we examined its possible involvement in Aβ-associated changes in glutamatergic synapses. Our results suggest that degradation of PSD-95 contributes to alterations in synaptic plasticity that are observed in the pathogenesis of Alzheimer's disease.
Materials and Methods
Drugs and peptides. Aβ1-40 was from American Peptides (Sunnyvale, CA); Aβ1-40 peptide was dissolved in DMSO at 2 mm and then diluted 1:10 in sterile PBS, vortexed for 30 min [at room temperature (RT)], and centrifuged at 15,000 × g at 4°C for 1 h; the supernatant (∼200 μm) was aliquoted (25 μl) and snap frozen at -20°C. Unless stated differently, aliquots were diluted in culture medium to a final concentration of 10 μm immediately before use. The predominant aggregates in such preparations are reported to be low N-oligomers (mainly monomeric to tetrameric) (Walsh et al., 1997; Bitan et al., 2001, 2003; Stine et al., 2003). Although the possibility that our preparation contained a minor amount of protofibrils cannot be excluded, it should be noted that fibrillogenesis requires longer incubation times and higher concentrations (>10 μm) of the peptide (O'Nuallain et al., 2004; Wogulis et al., 2005).
NMDA, (±)-verapamil, chloroquine, and KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)amino-N-(4-chlorocinnamyl)-N-methylbenzylamine) were purchased from Sigma (Deisenhofen, Germany). Bicuculline, MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate], ifenprodil, 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX), E4CPG [(RS)-α-ethyl-4-carboxyphenylglycine], and CPPG [(RS)-α-cyclopropyl-4-phosphono-phenyl-glycine] were from Tocris Cookson (Bristol, UK). 6-Aminopyridine-sulfonamide (PNU 112455A; cdk2/5 inhibitor), 2,4-dibenzyl-5-oxothiadiazolinidine-3-thione [TDZT; glycogen synthase kinase-3β (GSK-3β) inhibitor], and MG132 (z-Leu-Leu-Leu-al) (proteasome inhibitor) were purchased from Calbiochem (La Jolla, CA). U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto) butadiene] (extracellular signal-regulated kinase 1/2 inhibitor) was from Cell Signaling Technology (Beverly, MA).
Primary cell culture. Cortical primary cell cultures were prepared by dissociating frontal cortical tissue from postnatal day 4 (P4) Wistar rats (Charles River Wiga, Sulzfeld, Germany) with trypsin [0.05% in Earle's balanced salt solution (EBSS) with 0.3% bovine serum albumin (BSA); Invitrogen, Eggenstein, Germany]. Neurons were isolated from trypsinized frontal cortex tissue after centrifugation on a one-step BSA (4%) density gradient (Crochemore et al., 2005). Frontal cortex was isolated by dissection, after complete removal of meninges. Isolated brain tissue was triturated mechanically, digested with trypsin (0.05% in EBSS), triturated gently, and transferred to Neurobasal/B27 medium containing 1% fetal calf serum (FCS) and 0.2% BSA at 37°C. Triturated tissue was filtered through a sterile nylon mesh (30 mm pore size) and centrifuged at 200 × g (20°C, 5 min) before resuspension in Neurobasal/B27 medium containing 0.1% ovomucoid/0.005% DNase I (Worthington, Freehold, NJ). Aliquots of this suspension were stratified on 4% BSA containing 0.1% ovomucoid and centrifuged at 70 × g (20°C, 5 min). Cells were plated in Neurobasal/B27 medium containing basic fibroblast growth factor (10 ng/ml), glutamax I (0.5 mm), and kanamycin (100 mg/ml) (all from Invitrogen) onto gelatin/poly-d-lysine-coated glass coverslips at a density of 450,000 cells/mm2. Cultures were incubated at 37°C, under 5% CO2/95% air and 90% relative humidity. Half of the culture medium was renewed every 3 d. Experiments were started 7 d after seeding [7 d in vitro (DIV)].
SK-N-MC cell line cultures. Human neuroblastoma SK-N-MC cells were maintained at 37°C (5% CO2/95% air) in DMEM containing 10% FCS (Invitrogen) and 1% kanamycin. Cells (200 × 103) were seeded on six-well plates 1 d before use.
Transfection. SK-N-MC cells were transfected with 500 ng of DNA per well using jet-PEI (Polytransfection, Illkirch, France), as described previously (Tirard et al., 2004). After 24 h, cells were fed with fresh medium before application of drugs.
Western blot. Cells were lysed by brief sonication in 100 mm Tris-HCl, 250 mm NaCl, 1 mm EDTA, 5 mm MgCl2, 1% NP-40, a mixture of protease inhibitors (Complete Protease Inhibitor; Roche, Mannheim, Germany), and a phosphatase inhibitor mixture (Phosphatase Inhibitor Cocktails I and II; Sigma), before centrifugation. Cleared lysates were resolved by electrophoresis on 8% acrylamide gels and transferred onto nitrocellulose membranes. Membranes were blocked in PBS containing 5% nonfat milk powder and 0.2% Tween-20 and incubated with the following antibodies: anti-PSD-95 (1:6000; Upstate Biotechnology, Lake Placid, NY), anti-synapsin I (1:500; Chemicon, Temecula, CA), anti-cdk5 (1:1000; Biomol, Plymouth Meeting, PA), anti-GAP-43 (1:5000; Chemicon), and anti-actin (1:10,000; Chemicon) or anti-β-tubulin (1: 2000; Oncogene Sciences, Uniondale, NY). Antigens were revealed by enhanced chemiluminescence (Amersham Biosciences, Freiburg, Germany) after incubation with appropriate horseradish peroxidase-IgG conjugates (Amersham Biosciences); blots were scanned and quantified using TINA 3.0 bioimaging software (Raytest, Straubenhardt, Germany). Linearity was routinely checked during semiquantification of all blots. All values were normalized and expressed as percentages of control; in pharmacological experiments, percentages were calculated as Aβ treated versus Aβ untreated. Each set of numerical data shown were obtained from three to five independent sets of experiments, with three replicates in each run.
DNA constructs. Wild-type PSD-95 and triple alanine (T19A, S25A, S35A) mutant PSD-95 were in pcDNA3 expression vectors (Morabito et al., 2004). Wild-type PSD-95 and PSD-95ΔPEST [PSD-95 mutant in which the PEST motif (amino acids 13-23) was deleted] in pGW1-cytomegalovirus expression vectors were kindly provided by Dr. Marcie Colledge (Vollum Institute, Portland, OR).
Immunofluorescence. Cells were fixed in ice-cold 4% paraformaldehyde (5 min), rinsed in PBS, and permeabilized in PBS containing 0.1% Triton X-100 and 5% horse serum (at RT). All reagents and incubations were in PBS containing 0.1% Triton X-100 and 3% BSA. Coverslips were blocked (10% horse serum in PBS) and incubated overnight (4°C) with anti-PSD-95 (1:500). After thorough washing, coverslips were incubated with biotinylated goat anti-mouse IgG (1:500; Molecular Probes, Eugene, OR) for 1 h (at RT) and rinsed in PBS before incubation with FITC-conjugated streptavidin (diluted 1:500; Molecular Probes). To double stain for synapsin I, cells were incubated (overnight, 4°C) with rabbit anti-synapsin I (1:500; Chemicon), rinsed in PBS, and incubated at RT (1 h) in biotinylated goat anti-rabbit IgG (1:500). Coverslips were stained with Texas Red-conjugated streptavidin (1:500; Molecular Probes). For glutamate receptor subtype 2 (GluR2) surface immunostaining, cells were washed with PBS (at RT) and incubated (40 min, on ice) with anti-GluR2 antibody (1:150; Chemicon) diluted in PBS. After washing, cells were fixed in ice-cold 4% paraformaldehyde, pH 7.4, blocked with PBS containing 10% horse serum, incubated (overnight, 4°C) with biotinylated goat anti-mouse IgG (1:500; Molecular Probes) diluted in PBS containing 10% horse serum, rinsed, and stained with FITC-conjugated streptavidin (1:500; Molecular Probes). Coverslips were mounted with Vectashield (Vector Laboratories, Burlingame, CA). Optical section images and stacks of images from fluorescence-labeled cells were obtained with a confocal laser scanning microscope (LSM 510; Zeiss, Jena, Germany) using a plan apochromat 63×/1.4 numerical aperture oil lens. PSD-95 immunostaining was monitored in 200 puncta within 12 randomly chosen dendrites from four neurons (triplicate specimens). To evaluate puncta density, images were thresholded at the arbitrary value of 50, and puncta density was expressed as puncta number per 100 μm. Immunofluorescence intensity of manually selected PSD-95 synaptic puncta was evaluated on synapsin I-labeled samples using Scan-Image software (Scion, Frederick, MD) after subtracting background intensity value in each region of interest. For evaluating surface-bound AMPAR immunostaining, 225 positive puncta were measured within 10 randomly chosen dendrites from five neurons (triplicate determinations), with fluorescence intensity and puncta density being quantified as described above.
Statistical analysis. All data are depicted as mean ± SD from three to five independent experiments. Data were analyzed for statistical significance using ANOVA and appropriate post hoc tests (Student-Keuls or Kruskal-Wallis multiple comparison procedures, as appropriate) in which p < 0.05 was set as the minimum level of significance.
Results
Aβ1-40 decreases levels of PSD-95 but not of other synaptic proteins
Synthetic oligomerized Aβ1-42 and Aβ1-40, as well as natural Aβ oligomers, were recently shown to disrupt LTP induction (Q. S. Chen et al., 2000; Walsh et al., 2002; Wang et al., 2002; Kamenetz et al., 2003; Wang et al., 2004; Klyubin et al., 2004). To investigate the pathway(s) through which Aβ might impair synaptic plasticity, we monitored the expression of several synaptic proteins in rat primary cortical neuronal cultures. Neurons were exposed to 10 μm freshly-dissolved Aβ1-40 for 15, 60, or 120 min. (Fig. 1A), and proteins were detected by Western blot analysis of cell lysates. Levels of synapsin I, AMPAR subunit GluR2, GAP-43, cdk-5, and calcium/calmodulin-dependent kinase II (CaMKII) protein expression were unaltered by Aβ treatment at all time points studied. Neither actin nor tubulin expression were affected by these treatments. In contrast, application of Aβ resulted in a 53.3 ± 6.5% decrease in PSD-95 levels at 60 min (p < 0.05), and PSD-95 levels remained significantly lower (76 ± 10.9%; p < 0.05) after 120 min when compared with untreated cultures (100%). We next examined whether the Aβ effects on PSD-95 expression occurred in a dose-dependent manner. Treatment of cortical neurons with 100 nm, 1 μm, or 10 μm Aβ for 60 min resulted in PSD-95 levels that were, respectively, 81.9 ± 3.6, 73.2 ± 3.4, and 51 ± 5.5% of those found in controls (Fig. 1B). Pairwise comparisons between all groups revealed significant differences (p < 0.05), and linear regression analysis of the dose-response curve yielded an r of 0.879 (p = 0.007).
Figure 1.
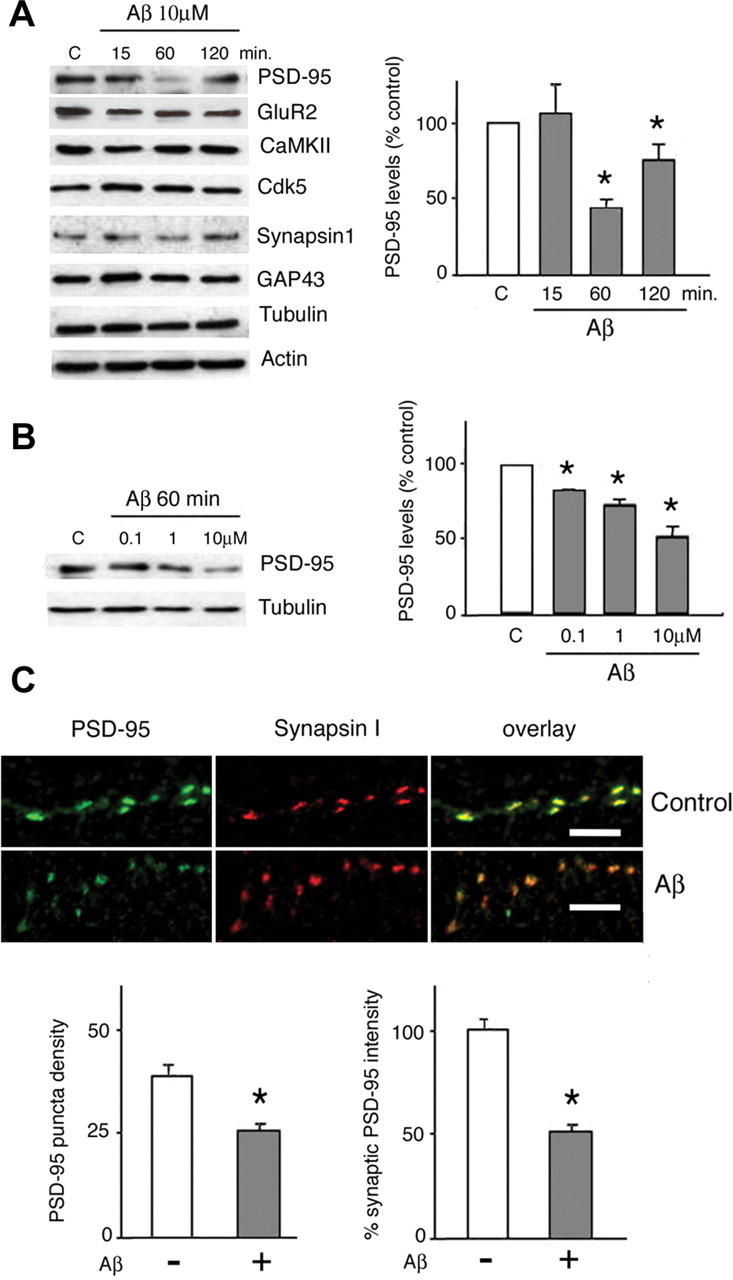
PSD-95 levels are reduced after Aβ treatment in rat primary cortical neurons. A, Aβ selectively downregulates PSD-95 levels in a time-dependent manner, without altering the expression of other synaptic proteins. Cells were exposed to 10 μm soluble Aβ1-40 (see Materials and Methods) for between 15 and 120 min before analysis by Western blot. As shown in the histogram, semiquantitative evaluation of PSD-95 levels, normalized against tubulin, shows that Aβ treatment led to a significant reduction of PSD-95 expression (53.3 ± 6.5% of control levels; p < 0.05) within 60 min; despite a trend to recover by 120 min after treatment, PSD-95 expression remained significantly lower than in controls (76 ± 10.9%; p < 0.05). Analysis of levels of the synaptic proteins GluR2, GAP-43, synapsin I, cdk5, and CaMKII revealed no significant effect of Aβ over the treatment duration. Actin and tubulin levels were not influenced by the experimental manipulations. B, Dose-dependent effects of Aβ on PSD-95 levels. Neurons were exposed to Aβ (0.1-10 μm) for 1 h before they were analyzed for levels of PSD-95 expression in Western blot assays. One representative Western blot is shown, and the semiquantitative data from three independent experiments are shown in the histogram. Levels of PSD-95, normalized with respect to tubulin levels, are shown as means ± SD. Asterisks indicate significant changes from untreated control cells (p < 0.05). The dose-response curve had r = 0.879 (p = 0.007). C, Aβ effect on synaptic PSD-95. Primary rat cortical neurons were treated with 10 μm Aβ for 1 h, fixed, and immunostained for synapsin I and PSD-95. Synaptic sites were identified as synapsin I-positive puncta. Puncta density in vehicle-treated control cultures was 36 ± 2.4 puncta/100 μm; after Aβ treatment, the density of PSD-95 immunoreactive puncta was 24.6 ± 2.1/100 μm (p < 0.001). Numerous synaptic sites (identified by synapsin staining) showed prominent decreases in PSD-95 fluorescence after Aβ treatment; fluorescence intensity of synaptic PSD-95 was reduced after Aβ treatment (49.4 ± 2.4% compared with 100 ± 6.7% in controls; p < 0.01, p < 0.05; n = 200). Scale bars, 5 μm. All numerical data represent mean ± SD.
PSD-95 is predominantly localized at synapses in which it plays an important role in activity-dependent remodeling of neuronal connections (Okabe et al., 1999; Ehlers, 2003). To assess whether synaptic PSD-95 is affected by Aβ treatment, cortical neurons (7 DIV) were treated with soluble Aβ (10 μm, 60 min) and immunostained for synapsin I (a marker of presynaptic terminals) and PSD-95. Quantitative immunofluorescence analysis of confocal images was conducted blind, as described by Colledge et al. (2003). Treatment with Aβ did not affect fluorescence intensity of synapsin I-immunoreactive puncta (89.9 ± 25.4% of control). However, as Figure 1C shows, Aβ treatment decreased both the density of synaptic PSD-95 puncta (36 ± 2.4 vs 24.6 ± 2.1 puncta/100 μm in vehicle-treated and Aβ-treated cultures, respectively; p < 0.05) and the intensity of synaptic PSD-95-labeled puncta (49.4 ± 2.4 compared with 100 ± 6.7% in controls; p < 0.01). These observations indicate that Aβ targets PSD-95 in a sizeable subset of synapses.
Aβ-induced downregulation of PSD-95 depends on NMDAR activity and is Ca2+ dependent
Aβ peptides modulate glutamate receptor activity (Ye et al., 2004), and NMDAR activity leads to decreased PSD-95 protein levels (Colledge et al., 2003). To test the role of NMDARs in the effects of Aβ on PSD-95 levels, P4 cortical neurons were pretreated (1 h) with either the NMDAR antagonist MK-801 (10 μm) or the NR2B-specific antagonist ifenprodil (10 μm) before being exposed to Aβ. Subsequent Western blot analysis of cell lysates revealed that both MK-801 and ifenprodil inhibited Aβ-induced downregulation of PSD-95 levels (106.1 ± 3.1 and 115.4 ± 13.4%, respectively; PSD-95 levels in untreated controls, 100%) (Fig. 2A), indicating that NMDAR activation is required for manifestation of the Aβ effects. Consistent with this observation, treatment with 10 μm NMDA (1 h) and Aβ did not significantly alter PSD-95 levels compared with Aβ alone. Next, because NMDAR activation results in Ca2+ influx, we tested the importance of extracellular Ca2+ for Aβ-induced downregulation of PSD-95. No significant changes in PSD-95 were detected when neurons were treated with Aβ under Ca2+-free conditions (106.5 ± 19.1 vs 50 ± 10.5% reduction of PSD-95 levels when Aβ was added in standard medium) (Fig. 2B). Verapamil (100 μm) did not block the effects of Aβ, indicating that Ca2+ influx through L-type voltage-dependent calcium channels is not involved (data not shown).
Figure 2.
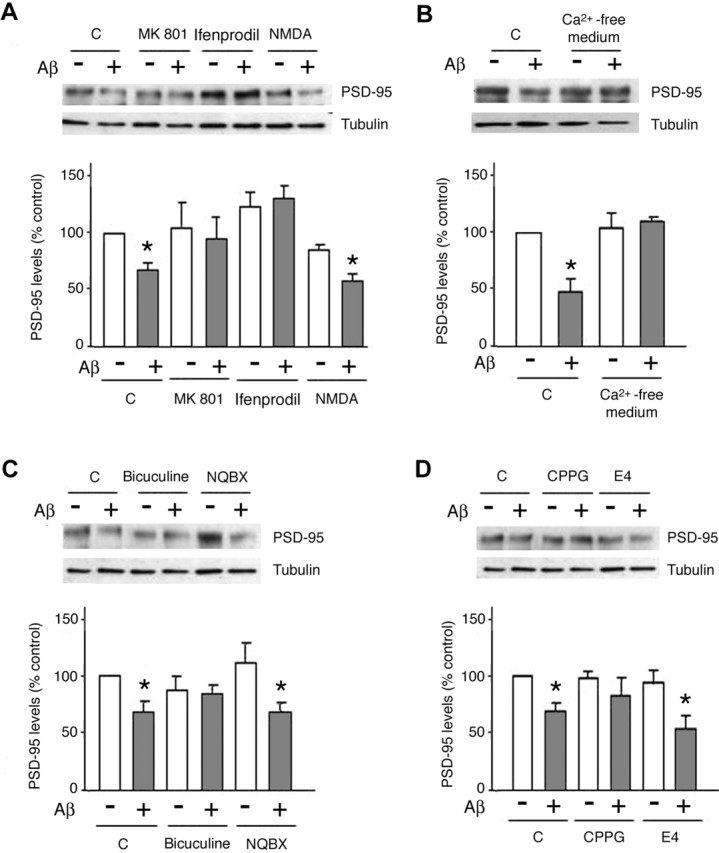
Aβ-induced PSD-95 downregulation requires NMDAR activity and calcium influx. A, Aβ-induced decrease in PSD-95 levels requires NMDAR activity. Cells treated with Aβ showed a significant decrease in PSD-95 levels (67.5 ± 6.7%; p < 0.05 vs untreated cells). Treatment (1 h) of cells with MK-801 (10 μm) or ifenprodil (10 μm) prevented the Aβ-induced decrease in PSD-95 levels, whereas NMDA (10 μm) did not influence the effects of Aβ (59 ± 5%; p < 0.05 compared with nontreated cells). C, Control. B, PSD-95 downregulation by Aβ is a calcium-dependent process. Neurons exposed to Aβ (10 μm; 1 h) under calcium-free conditions did not show a reduction in PSD-95 levels compared with untreated control cells. C, The PSD-95 down-regulating actions of Aβ are not dependent on AMPAR and are attenuated by bicuculline. Neurons were pretreated (1 h) with NBQX (20 μm), bicuculline (40 μm), or vehicle before exposure to Aβ (10 μm; 1 h). NBQX proved ineffective in counteracting Aβ action (61.9 ± 9.1%), whereas bicuculline attenuated the effects of Aβ on PSD-95 levels (81.4 ± 15%; p < 0.05 vs Aβ alone). D, The Aβ-induced decrease in PSD-95 levels is blocked in the presence of a metabotropic II/III receptor antagonist. Neurons were pretreated (1 h) with the mGluRI/II antagonist E4CPG (E4) (10 μm), the mGluRII/III antagonist CPPG (10 μm), or vehicle before treatment with Aβ (10 μm) for an additional 1 h. E4CPG did not alter the actions of Aβ on PSD-95, whereas CPPG significantly antagonized the Aβ effect (81.4 ± 15%; p > 0.05 vs response to Aβ only). Asterisks indicate significant changes from untreated control cells (p < 0.05). All data are given as mean ± SD.
Glutamate receptors other than NMDARs have also been shown to regulate PSD-95 levels; thus, whereas AMPAR activation results in reduced PSD-95 levels (Bingol and Schuman, 2004), activation of the metabotropic glutamate receptor mGluR1 increases PSD-95 levels (Todd et al., 2003). Pretreatment (1 h) of neurons with the AMPAR antagonist NBQX (20 μm) did not interfere with the ability of Aβ to reduce PSD-95 levels (Aβ, 56.8 ± 7.6% vs Aβ plus NBQX, 61.9 ± 9.1%) (Fig. 2C); likewise, blockade of group I mGluRs with E4CPG (10 μm) did not alter the ability of Aβ to reduce PSD-95 levels (49.8 ± 15%) (Fig. 2D). Interestingly, treatment of neurons with CPPG (10 μm), an antagonist of metabotropic glutamate receptors mGluRII/III, significantly attenuated the effects of Aβ on PSD-95 levels (81.4 ± 15%) (Fig. 2D). Because mGluRII/III negatively regulate glutamate release, we next examined the effects of Aβ under conditions of increased glutamatergic drive by applying the GABAA receptor antagonist bicuculline (40 μm). Bicuculline treatment significantly attenuated the Aβ-induced decrease in PSD-95 levels (97.7 ± 9.6%) (Fig. 2C), suggesting that increased excitatory activity can override Aβ-dependent regulation of PSD-95 levels activity; it also suggests that mGluRII/III can exert a modulatory effect in this process.
cdk5 activity is required for Aβ-induced decreases in PSD-95
PSD-95 is a target of mitogen-activated protein kinase (MAPK) (Sabio et al., 2004) and cdk5 (Morabito et al., 2004). cdk5 and its activators p35 and p39 are localized postsynaptically (Humbert et al., 2000; Niethammer et al., 2000; Morabito et al., 2004) and can regulate GSK3β (Morfini et al., 2004). Furthermore, NMDAR activation can lead to activation of cdk5 (Kerokoski et al., 2004), and cdk5 can also be activated by Aβ1-40 peptide (Cruz et al., 2004). To evaluate the role of various kinases in the Aβ-induced reduction in PSD-95 levels, we pharmacologically inhibited the activity of kinases putatively involved in the regulation of PSD-95 using human neuroblastoma cell line (SK-N-MC) for screening; this cell line expresses MAPK, cdk5, CaMKII, and GSK3β (data not shown) and functional NMDA receptors (Pizzi et al., 2002; Deiva et al., 2004). Because SK-N-MC cells display low-to-undetectable PSD-95 levels, we transiently transfected these cells with wild-type PSD-95 plasmid. Neither transfections of PSD-95 nor pharmacological treatments affected endogenous levels of the kinases studied (data not shown). Exposure of cells expressing heterologous PSD-95 to Aβ (10 μm, 1 h) resulted in a significant reduction in PSD-95 levels (58.31 ± 8.2% of controls; p < 0.05) (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material); the magnitude of changes was comparable with those observed in Aβ-treated primary neurons (compare Figs. 1A, 2A). Treatment with inhibitors of CaMKII (KN-93), GSK-3β (TDZT), and MAP kinase kinase 1/2 (U0126) did not alter the effect of Aβ on PSD-95 (54.2 ± 7, 57.4 ± 29, and 65.2 ± 6.2%, respectively). In contrast, pharmacological inhibition of cdk5 with 10 μm PNU 112455A prevented Aβ-induced downregulation of PSD-95 (105.4 ± 22%) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
To further investigate the role of the cdk5-dependent phosphorylation of PSD-95 in mediating the effect of Aβ, we transiently expressed the triple alanine mutant of full-length PSD-95 (T19A, S25A, S35A) (Morabito et al., 2004) in the human neuroblastoma cell line (SK-N-MC). Transient transfection of these cells with wild-type PSD-95 plasmid and exposure to Aβ (10 μm, 1 h) resulted in a significant reduction in PSD-95 levels (58.31 ± 8.2% of controls; p < 0.05) (Fig. 3B); the magnitude of changes was comparable with those observed in Aβ-treated primary neurons (Fig. 3A). Whereas Aβ led to a significant decrease in the expression levels of wild-type PSD-95 (57.9 ± 5.6%), this treatment did not influence mutant PSD-95 levels (107.7 ± 11.2%) (Fig. 3B), suggesting that the integrity of the N-terminal domain of PSD-95 is required for PSD-95 downregulation by Aβ.
Figure 3.
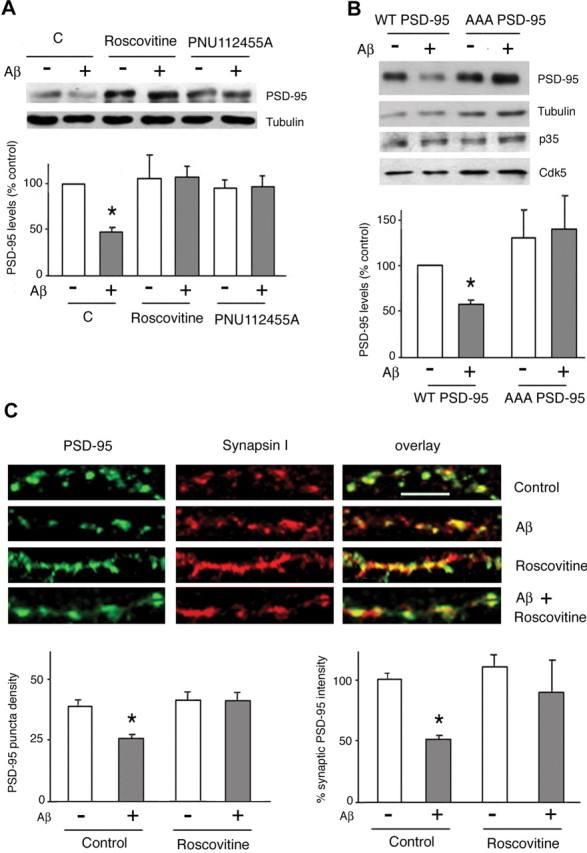
Aβ-induced downregulation of synaptic PSD-95 requires cdk5 activity. A, cdk5 activity is necessary for Aβ effects in cortical neurons. Primary rat cortical neurons were pretreated (1 h) with roscovitine (15 μm) or PNU 112455A (10 μm), or vehicle (DMSO) before exposure to Aβ (10 μm; 1 h). As shown by Western blotting, both inhibitors abrogated the ability of Aβ to reduce PSD-95 levels. C, Control. B, Aβ cannot downregulate PSD-95 levels when SK-N-MC cells are transiently transfected with the T19A, S25A, S35A phosphorylation mutant of PSD-95. Cells transfected with wild-type PSD-95 and expressing p35/cdk5 responded to Aβ with the expected decrease in PSD-95 levels (57.9 ± 5.6% of controls; p < 0.05). C, The Aβ effects on synaptic PSD-95 are blocked by roscovitine, an inhibitor of cdk5 activity. Primary rat cortical neurons were pretreated (1 h) with roscovitine (15 μm) or vehicle (DMSO) before exposure to Aβ (10 μm; 1 h). After fixation, neurons were immunostained for synapsin I and PSD-95. Roscovitine alone did not show a significant effect on the density of PSD-95 puncta or their fluorescence intensity but inhibited the effects of Aβ on these parameters. Scale bar, 5 μm. Asterisks indicate significant changes from untreated control cells (p < 0.05).
To further substantiate cdk-5 involvement in Aβ-induced PSD-95 downregulation, rat primary cortical neurons were treated with two structurally unrelated cdk5 inhibitors, roscovitine (15 μm) and PNU 112455A (10 μm), or vehicle (DMSO), for 1 h before the addition of Aβ peptide (10 μm) for 1 h. Both inhibitors prevented Aβ-induced downregulation of PSD-95 levels (104.1 ± 16.8 and 99.9 ± 8.2%, respectively) (Fig. 3A), indicating that cdk5 activity is critical in the regulation of PSD-95 by Aβ in primary neurons. To confirm a role for cdk5 in the downregulation of the synaptic pool of PSD-95, cortical neurons were pretreated with roscovitine (15 μm) or vehicle before exposure to Aβ peptide (10 μm, 1 h) and subsequent immunostaining for PSD-95 and synapsin I. Image analysis revealed that roscovitine markedly attenuated the effect of Aβ on synaptic PSD-95 puncta density (35.3 ± 4.7 puncta/100 μm after roscovitine plus Aβ vs 24.6 ± 2.1 puncta/100 μm after Aβ treatment alone; p < 0.05) and intensity (90.1 ± 12.4% after roscovitine plus Aβ; 49.4 ± 2.4% after Aβ alone; and 100 ± 6.7% in treatment-free conditions; p < 0.05), whereas neither DMSO nor roscovitine alone significantly altered synaptic PSD-95 puncta density and intensity (Fig. 3C); however, cdk5 inhibition increased the size of PSD-95 puncta (cf. Morabito et al., 2004). Together, these experiments strongly implicate cdk5 in mediating the effect of Aβ on the regulation of synaptic PSD-95 levels.
Involvement of proteasome pathway in Aβ-induced reduction of PSD-95 levels
The proteasome has recently emerged as a key regulator of PSD protein composition and turnover (Ehlers, 2003). Activation of NMDARs and AMPARs is followed by PSD-95 degradation by the proteasome (Colledge et al., 2003; Bingol and Schuman, 2004). Having shown that Aβ-induced PSD-95 downregulation depends on previous NMDAR activation, we were prompted to analyze whether the pathway downstream of Aβ involves the proteasome-dependent degradation. To this end, SK-N-MC cells expressing heterologous wild-type PSD-95 were incubated with MG132 (0.1 μm), an inhibitor of the proteasome, chloroquine (100 μm), a lysosome inhibitor, or vehicle (DMSO), before treatment with Aβ (10 μm, 1 h). Analysis of cell lysates by Western blot revealed that MG132 treatment strongly attenuated the effect of Aβ on PSD-95 levels (control, 100%; Aβ, 65.2 ± 9.8%; MG132, 104.6 ± 13.7%), whereas PSD-95 levels after chloroquine treatment did not differ significantly (71.7 ± 6%) from those found in cells treated with Aβ alone (Fig. 4 A). Consistent with these observations, pretreatment (1 h) of cortical neurons with MG132 before the addition of Aβ (10 μm) abolished Aβ-induced reductions in PSD-95 expression (data not shown), providing additional evidence that the proteasome pathway is involved in the regulation of PSD-95 by Aβ.
Figure 4.

The proteasome pathway is implicated in the regulation of PSD-95 by Aβ. A, PSD-95-transfected cultures were pretreated (1 h) with a proteasome inhibitor (MG132; 0.1 μm), a lysosome inhibitor (chloroquine; 100 μm), or vehicle (DMSO) before treatment with Aβ (10 μm; 1 h). MG132 treatment prevented Aβ-induced reductions in PSD-95, whereas chloroquine did not exert a significant influence on the actions of Aβ. C, Control. B, The PEST consensus sequence in PSD-95 is necessary for the Aβ-induced effects. SK-N-MC cultures that had been transfected with either wild-type PSD-95 or the ΔPEST deletion mutant of PSD-95 were exposed to Aβ (10 μm; 1 h). Whereas Aβ led to a reduction in PSD-95 levels in wild-type transfected cells (53.6 ± 19.8%; p < 0.05 vs non-Aβ-treated controls), PSD-95 levels were unchanged in cells expressing the ΔPEST mutant. Asterisks indicate significant changes from untreated control cells (p < 0.05).
Ubiquitinylation is a posttranslational modification that targets proteins to the proteasome (DiAntonio and Hicke, 2004). The N-terminal domain of PSD-95 contains a PEST motif that is essential for its ubiquitination (Colledge et al., 2003). To determine whether the PEST motif is important in the regulation of PSD-95 by Aβ, SK-N-MC cells were transfected with a mutant of PSD-95 (PSD-95ΔPEST) that lacks the PEST sequence. Control cells that were transfected with wild-type PSD-95 responded to Aβ with a significant reduction in PSD-95 protein levels (53.6 ± 19.8%), whereas cells expressing PSD-95ΔPEST were not affected by Aβ treatment (102.6 ± 5.8%) (Fig. 4B). Thus, the integrity of the N-terminal domain of PSD-95 is essential for the effect of Aβ on PSD-95.
Aβ decreases the surface expression of AMPARs
Trafficking and synaptic targeting of AMPARs are important determinants of synaptic strength (LTP or LTD) (Bredt and Nicoll, 2003). Previous studies have shown that PSD-95 regulates the dynamics of AMPARs through interactions with stargazin (Schnell et al., 2002); whereas PSD-95 overexpression drives AMPARs into synapses (Ehrlich et al., 2004), PSD-95 degradation is followed by endocytosis of AMPARs (Colledge et al., 2003). To assess whether the downregulation of PSD-95 induced by Aβ also affects the synaptic localization of AMPARs, we analyzed the surface expression of GluR2, an AMPAR subunit, in cultured cortical neurons (7 DIV) that had been exposed to Aβ for 1 h. In agreement with the results of Western blot analysis, we observed that Aβ treatment did not significantly change the intensity of total GluR2 immunostaining in permeabilized neurons (103.2 ± 3.6%; data not shown). In contrast, Aβ treatment induced a marked reduction in the density of surface GluR2-positive puncta (21.2 ± 6.54 vs 55.5 ± 7.2 puncta/100 μm in controls; p < 0.001) (Fig. 5A,B) and intensity of puncta immunostained for surface GluR2 (38.6 ± 11.6 vs 100 ± 13.5% in controls; p < 0.001) (Fig. 5A,C). Furthermore, we found that pretreatment with the cdk5 inhibitor roscovitine (15 μm) abolished the effect of Aβ on the density (53.5 ± 8.2 vs 54 ± 12.8 puncta/100 μm in roscovitine only-treated cells) and intensity of surface GluR2 (99.5 ± 13.1% versus roscovitine alone) (Fig. 5A-C). Together, these results show that Aβ treatment of neurons results in a decrease in the surface expression of GluR2 in a cdk5-dependent manner.
Figure 5.

Aβ reduces the expression of surface AMPARs. Primary rat cortical neurons were immunostained for the AMPAR subunit of the GluR2; permeabilization steps were excluded to ensure labeling of surface receptors. A, B, The density of GluR2-positive puncta (expressed as the number of puncta per 100 μm) was reduced after Aβ treatment (21.2 ± 6.5 vs 55.5 ± 7.2; p < 0.001). A, C, Aβ treatment led to a significant reduction in the intensity of immunofluorescence of GluR2 puncta (38.6 ± 11.6%; p < 0.001; 225 puncta analyzed). Pretreatment with roscovitine (15 μm) abrogated the effects of Aβ on these parameters (p < 0.05) but had no effect of its own. Scale bar, 5 μm. Asterisks indicate significant changes from untreated control cells (p < 0.05).
Discussion
Aβ peptides play an unequivocal role in AD, being prominent components of senile plaques. Although Aβ1-42 is required for plaque formation (McGowan et al., 2005) and can induce synaptic dysfunction (Wang et al., 2004), we focused our investigations on Aβ1-40, the most abundant Aβ peptide in the healthy and AD-afflicted brain. Although the physiological functions of Aβ1-40 are a matter of some conjecture (Kamenetz et al., 2003), this peptide has been strongly implicated in synaptic loss in AD patients (cf. Lue et al., 1999; McLean et al., 1999) and impaired LTP in murine models (Klyubin et al., 2004). Moreover, the correlation between plaque burden and cognitive impairment (Guillozet et al., 2003) and between Aβ1-42 and synaptic loss in humans are rather weak (Lue et al., 1999). Recent studies in animals have established links between natural, as well as synthetic, soluble Aβ oligomers and cognitive impairment (Richardson et al., 2003; Cleary et al., 2005), and Aβ oligomers have been shown to induce disruption of LTP (Q. S. Chen et al., 2000; Walsh et al., 2002; Kamenetz et al., 2003; Klyubin et al., 2004; Wang et al., 2004) but not LTD (Wang et al., 2002) induction. These observations suggest that excitatory synapses might be the early targets of soluble Aβ, a view supported by evidence that oligomerized Aβ can bind to synaptic sites, namely, PSD-95-containing postsynaptic sites (Lacor et al., 2004). In the present study, cortical neurons were treated with soluble Aβ1-40 so as to obtain a preparation containing low-molecular-weight oligomers (Walsh et al., 1997; Bitan et al., 2001, 2003; Stine et al., 2003); it is unlikely that our preparation contained a significant amount of Aβ1-40 fibrils (O'Nuallain et al., 2004; Wogulis et al., 2005).
Based on the aforementioned studies, we here addressed the possibility that soluble Aβ1-40 exerts its effects on synaptic plasticity by regulating the molecular composition and stability of excitatory synapses. We focused our studies on PSD-95, a key player in the organization, function, and plasticity of excitatory synapses (Ehrlich and Malinow, 2004; Kim and Sheng, 2004). The importance of PSD-95 in cognitive processes is attested by the observation that PSD-95 knock-out mice show impaired learning abilities (Migaud et al., 1998). The dynamic manner in which PSD-95 levels are regulated contributes to the key role of the protein in synaptic plasticity: brief NMDAR activation induces rapid PSD-95 proteasomal degradation, an event accompanied by AMPAR internalization (Colledge et al., 2003).
Experiments in this study indicate that soluble Aβ induces a decrease in PSD-95 levels in a time- and dose-dependent manner, without altering the expression of the presynaptic protein synapsin I, the AMPAR subunit GluR2, and the kinases CaMKII and cdk5. These findings indicate that PSD-95 is a specific target of Aβ. Interestingly, the dose of Aβ that affects PSD-95 is within the range that blocks LTP (Kamenetz et al., 2003; Wang et al., 2004), suggesting a correlation between the reduction of PSD-95 levels and the inhibition of LTP. The Aβ effects occurred within 1 h, i.e., within a period shown previously to be required for the manifestation of NMDAR- and AMPAR-dependent alterations in PSD-95 turnover (Colledge et al., 2003; Bingol and Schuman, 2004). We observed that, after initial downregulation, PSD-95 levels returned to control levels within 2 h of application of Aβ; although proteolysis of Aβ cannot be ruled out, it is plausible that adaptive mechanisms (cf. Turrigiano, 1999; Todd et al., 2003) are recruited over time.
The ionotropic glutamate receptors AMPARs and NMDARs have been implicated in the regulation of PSD-95. Although AMPARs contribute to the regulation of PSD-95 degradation (Bingol and Schuman, 2004), the AMPAR antagonist NBQX failed to modulate the effect of Aβ on PSD-95 in the present study. Activation of NMDARs was reported previously to result in reduced PSD-95 levels (Colledge et al., 2003). In investigations of the relationship between NMDAR activation and Aβ-induced downregulation of PSD-95, we found that the specific NMDAR inhibitor MK-801 abolishes the effect of Aβ, indicating that NMDAR activity is prerequisite for the Aβ effects to occur. Similar observations were made with ifenprodil, an NR2B subunit-specific antagonist; the latter results most likely reflect the relative abundance of NR2B subunit in the early postnatal brain (Liu et al., 2004). Furthermore, we demonstrated the Ca2+ dependency of the regulation of PSD-95 protein levels by Aβ, consistent with a role of NMDAR activity in mediating the Aβ effects (Fig. 2A,B).
Our study included an analysis of the role of other modulators of glutamatergic activity in the regulation of PSD-95 levels. Interestingly, class I mGluR activation leads to increased PSD-95 levels (Todd et al., 2003), and the mGluR5 (a class I mGluR) has been implicated in Aβ-induced disruption of LTP (Wang et al., 2004). Activation of the predominantly postsynaptic class I mGluRs has been linked to long-term synaptic plasticity, including LTP induction. However, because E4CPG failed to modify the effects of Aβ, mGluRI does not appear to be required for Aβ-induced PSD-95 degradation in our experimental setting. In contrast, treatment of neurons with CPPG, an antagonist of mGluRII/III, significantly inhibited the effects of Aβ on PSD-95 levels, suggesting that class I and class II/III mGluRs play different roles in the response to Aβ. mGluRII/III are mainly presynaptic and negatively regulate glutamate release (Grassi et al., 2002). Increased synaptic activity and glutamate receptor activation (after blockade of mGluRII/III or of GABAA receptors) efficiently blocked the effects of Aβ on PSD-95 levels. These results suggest that increased synaptic activity can override or prevent Aβ-induced PSD-95 degradation. Our findings that increased excitatory synaptic activity can attenuate the actions of Aβ appear to be at odds with the fact that NMDAR activation failed to block Aβ-induced PSD-95 degradation. It should be noted, however, that an ensemble of glutamate receptors and downstream signal transduction pathways is activated when glutamatergic synaptic activity is increased (e.g., after treatment with mGluRII/III antagonists or bicuculline) and that these may influence PSD-95 levels (cf. Colledge et al., 2003). Also, it should be recalled that, depending on the intensity and timing of synaptic activity, NMDAR can result in the expression of either LTP or LTD (Malenka and Bear, 2004); thus, it is conceivable that strong excitatory drive blocks the downregulation of PSD-95 levels by Aβ, whereas weak stimulation of NMDARs is inadequate in this respect. Our results are consistent with the findings that prolonged increases in excitatory synaptic activity upregulate PSD-95 levels (Ehlers, 2003), whereas transient NMDAR activation can induce PSD-95 degradation (Colledge et al., 2003); we suggest that maximal reductions in PSD-95 levels occur when Aβ is applied at 10 μm, resulting in an occlusion of additional NMDA effects.
The serine-threonine kinase cdk5 plays a prominent role in the development of the nervous system (Dhavan and Tsai, 2001) and has been implicated in the pathogenesis of AD (Cruz and Tsai, 2004). Recent studies have shown that PSD-95 is phosphorylated in a cdk5-dependent manner (Morabito et al., 2004). In the present study, we identify a novel role for cdk5 in the regulation of Aβ-induced effects on PSD-95 levels. We show that inhibition of cdk5 by roscovitine inhibits the effect of Aβ on PSD-95 protein levels and that levels of PSD-95 do not decline after Aβ treatment of cultured cells expressing the triple alanine mutant form of PSD-95 (T19A, S25A, S35A), which lacks phosphorylation sites. Together, these experiments clearly implicate cdk5 in the regulation of synaptic PSD-95 protein levels and establish an important novel connection between cdk5 activity and the effects of Aβ on the molecular composition of glutamatergic synapses.
Proteins are degraded in the cell through activation of either the proteasome or lysosome. The posttranslational tagging of proteins with ubiquitin represents a major mechanism through which proteins are targeted for proteasomal degradation (DiAntonio and Hicke, 2004). The proteasome pathway plays a major role in synaptic protein turnover (Ehlers, 2003), and PSD-95 is ubiquitinated at synapses after brief activation of NMDARs (Colledge et al., 2003). In our study, the proteasome inhibitor MG132 markedly attenuated the ability of Aβ to downregulate PSD-95, indicating a role for the proteasome pathway in regulating the effect of Aβ. Moreover, Aβ failed to downregulate levels of the PEST deletion mutant of PSD-95 in transiently transfected cells. Because this mutant lacks both the ubiquitination motif and one of the cdk5 phosphorylation sites (T19), failure to induce the downregulation of the PEST PSD-95 mutant and the nonphosphorylatable triple alanine mutant form of PSD-95 indicates that the integrity of the N-terminal domain of PSD-95 is essential for the Aβ-dependent regulation of PSD-95. Because chloroquine failed to prevent Aβ-induced downregulation of PSD-95, lysosome-mediated degradation is clearly not involved in the regulation of PSD-95 levels by Aβ.
PSD-95, which is predominantly localized at synapses, regulates the expression of NMDAR and AMPAR expression at postsynaptic membranes. Our study indicates that Aβ treatment of cortical cultured neurons leads to a significant decrease in the density and intensity of synaptic puncta positive for PSD-95; this finding is consistent with previous observations showing that PSD-95-positive synaptic puncta are targeted by Aβ peptides (Lacor et al., 2004). PSD-95 is coupled to AMPARs through the stargazin family of proteins (Schnell et al., 2002). Decreased PSD-95 levels result in decreased AMPAR localization at synapses (Colledge et al., 2003), whereas PSD-95 overexpression drives AMPAR incorporation into the postsynaptic membrane (Ehrlich et al., 2004). Our immunocytochemical studies of cortical cultured neurons indicate that Aβ treatment results in downregulation of the expression of surface GluR2 (an AMPAR subunit), without changing the levels of total GluR2, as measured by Western blotting and immunostaining. Given the importance of PSD-95 in the synaptic stabilization of AMPARs, it is likely that the decrease in surface expression of GluR2 is attributable to an increase in GluR2 endocytosis, following the downregulation of synaptic PSD-95. Interestingly, cotreatment with roscovitine prevents the downregulation of GluR2 surface expression by Aβ, implicating cdk5 in the regulation of the molecular composition and function of synapses in AD.
Both synthetic Aβ1-42 and naturally secreted Aβ oligomers were recently shown to rapidly (within 1 h) induce endocytosis of NMDARs in rat cortical neurons (Snyder et al., 2005). Because PSD-95 binding is known to prevent NMDAR endocytosis (Roche et al., 2001), we tentatively suggest that Aβ-induced PSD-95 degradation might contribute to NMDAR endocytosis and that PSD-95 degradation itself may result from Aβ-induced dissociation of PSD-95 from NMDARs.
The results presented here are consistent with the view that exposure of neurons to Aβ peptides alters the availability of PSD-95 and, most likely, dendritic spine morphology. The regulation of these proteins may constitute a physiological role for Aβ peptides, and genetic or other predisposing factors that result in increased production of soluble Aβ are likely to trigger pathogenic mechanisms leading to AD. The Aβ-dependent reduction in synaptic PSD-95, which requires NMDAR activity, Ca2+ influx, cdk5 activity, and the proteasome pathway, eventually leads to internalization of AMPARs. Because AMPARs are key players in synaptic function, their surface downregulation could constitute a crucial mechanism for the synaptic derangement induced by Aβ. The results of the present study, demonstrating that soluble Aβ can interfere with postsynaptic function, provide substantial support to the view that soluble Aβ may be responsible for the cognitive impairments observed in early-phase AD patients (Walsh and Selkoe, 2004).
Footnotes
F.R. was partly supported by the Max Planck Institute of Psychiatry (Munich, Germany). M.T. and J.L. were Max Planck Society stipend holders. M.M. is supported by National Institutes of Health/National Institute on Drug Abuse Grant 1R01DA019451. Dieter Fischer and Jutta Waldherr provided dedicated technical assistance, and Carola Hetzel provided administrative help. We thank Dr. M. Colledge for kindly providing plasmids, and Drs. Spiros Efthimiopoulos and Emilio Jirillo for helpful comments.
M. Tirad's present address: Max Planck Institute of Experimental Medicine, D-37075 Göttingen, Germany.
J. Lu's present address: Department of Neurology, Harvard Medical School, Boston, MA 02115.
Correspondence should be addressed to O. F. X. Almeida, Max Planck Institute of Psychiatry, Kraepelinstrasse 2, 80804 Munich, Germany. E-mail: osa@mpipsykl.mpg.de.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511061-10$15.00/0
References
- Bingol B, Schuman E (2004) A proteasome-sensitive connection between PSD-95 and GluR1 endocytosis. Neuropharmacology 47: 755-763. [DOI] [PubMed] [Google Scholar]
- Bitan G, Lomakin A, Teplow DB (2001) Amyloid β protein oligomerization prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J Biol Chem 276: 35176-35184. [DOI] [PubMed] [Google Scholar]
- Bitan G, Vollers SS, Teplow DB (2003) Elucidation of primary structure elements controlling early amyloid beta-protein oligomerization. J Biol Chem 278: 34882-34889. [DOI] [PubMed] [Google Scholar]
- Bredt D, Nicoll R (2003) AMPA receptor trafficking at excitatory synapses. Neuron 40: 361-379. [DOI] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA (2000) Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 408: 936-943. [DOI] [PubMed] [Google Scholar]
- Chen QS, Kagan BL, Hirakura Y, Xie CW (2000) Impairment of hippocampal long-term potentiation by Alzheimer beta peptides. J Neurosci Res 60: 65-72. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Sweeney NT, Craven SE, Kang R, El-Husseini AE, Bredt D (2003) Lipid- and protein-mediated multimerization of PSD-95: implication for receptor clustering and assembly of synaptic protein networks. J Cell Sci 16: 3213-3219. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH (2005) Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 8: 79-84. [DOI] [PubMed] [Google Scholar]
- Coleman PD, Yao PJ (2003) Synaptic slaughter in Alzheimer's disease. Neurobiol Aging 24: 1023-1027. [DOI] [PubMed] [Google Scholar]
- Coleman PD, Federoff H, Kurlan R (2004) A focus on synapse for neuroprotection in Alzheimer disease and other dementias. Neurology 63: 1155-1162. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg KL, Lu H, Bear MF, Scott JD (2003) Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40: 595-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crochemore C, Lu J, Wu Y, Liposits Z, Sousa N, Holsboer F, Almeida OFX (2005) Direct targeting of hippocampal neurons for apoptosis by glucocorticoids is reversible by mineralocorticoid receptor activation. Mol Psychiatry 10: 790-798. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tsai LH (2004) Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends Mol Med 10: 452-458. [DOI] [PubMed] [Google Scholar]
- Deiva K, Geeraerts T, Salim H, Leclerc P, Hery C, Hugel B, Freyssinet JM, Tardieu M (2004) Fractalkine reduces N-methyl-d-aspartate-induced calcium flux and apoptosis in human neurons through extracellular signal-regulated kinase activation. Eur J Neurosci 20: 3222-3232. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severitiy. Ann Neurol 27: 457-464. [DOI] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH (2001) A decade of cdk-5. Nat Rev Neurosci 2: 749-759. [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Hicke L (2004) Ubiquitin-dependent regulation of the synapse. Annu Rev Neurosci 27: 223-246. [DOI] [PubMed] [Google Scholar]
- Dong YN, Waxman EA, Lynch DR (2004) Interactions of postsynaptic density-95 and the NMDA receptor 2 subunit controls calpain-mediated cleavage of the NMDA receptor. J Neurosci 24: 1035-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD (2003) Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6: 231-242. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R (2004) Postsynaptic density 95 controls AMPA receptor incorporation during long term potentiation and experience driven synaptic plasticity. J Neurosci 24: 916-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS (2000) PSD-95 involvement in maturation of excitatory synapses. Science 290: 1364-1368. [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL (2003) Alzheimer's disease-affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA 100: 10417-10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi S, Frondaroli A, Pettorossi VE (2002) Different metabotropic glutamate receptors play opposite roles in synaptic plasticity of the rat medial vestibular nuclei. J Physiol (Lond) 543: 795-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillozet AL, Weintraub S, Mash DC, Mesulam MM (2003) Neurofibrillary tangles, amyloid and memory in aging and mild cognitive impairment. Arch Neurol 60: 729-736. [DOI] [PubMed] [Google Scholar]
- Humbert S, Lanier LM, Tsai LH (2000) Synaptic localization of p39, a neuronal activator of cdk5. NeuroReport 11: 2213-2216. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Yamada Y, Hori K, Watanabe Y, Sobue K, Inui M (2004) Differential modulation of NR1-NR2A and NR1-NR2B subtypes of NMDA receptor by PDZ domain-containing proteins. J Neurochem 89: 100-108. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R (2003) APP processing and synaptic function. Neuron 37: 925-937. [DOI] [PubMed] [Google Scholar]
- Kerokoski P, Suuronen T, Salminen A, Soininen H, Pirttila T (2004) Both N-methyl-d-aspartate (NMDA) and non-NMDA receptors mediate glutamate-induced cleavage of the cyclin-dependent kinase 5 (cdk5) activator p35 in cultured rat hippocampal neurons. Neurosci Lett 368: 181-185. [DOI] [PubMed] [Google Scholar]
- Kim E, Sheng M (2004) PDZ domain proteins of synapse. Nat Neurosci Rev 5: 771-781. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Cullen WK, Fadeeva JV, Anwyl R, Selkoe DJ, Rowan MJ (2004) Soluble arctic amyloid beta protein inhibits hippocampal long-term potentiation in vivo. Eur J Neurosci 19: 2839-2846. [DOI] [PubMed] [Google Scholar]
- Lacor P, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Kraft GA, Klein WL (2004) Synaptic targeting by Alzheimer's related amyloid oligomers. J Neurosci 24: 10191-10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft CA, Klein WL (1998) Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 95: 6448-6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Lee R, Roche KW (2003) Differential binding of the AP-2 adaptor complex and PSD-95 to the C-terminus of the NMDA receptor subunit NR2B regulates surface expression. Neuropharmacology 45: 729-737. [DOI] [PubMed] [Google Scholar]
- Li B, Otsu Y, Murphy TH, Raymond LA (2003) Developmental decrease in NMDA receptor desensitization associated with shift to synapse and interaction with postsynaptic density-95. J Neurosci 23: 11244-11254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Skeberdis VA, Francesconi A, Bennett MV, Zukin RS (2004) Postsynaptic density protein-95 regulates NMDA channel gating and surface expression. J Neurosci 24: 10138-10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XB, Murray KD, Jones EG (2004) Switching of NMDA receptor 2A and 2B subunits at thalamic and corical synapses during early postnatal development. J Neurosci 24: 8885-8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J (1999) Soluble amyloid-β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 155: 853-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44: 5-21. [DOI] [PubMed] [Google Scholar]
- Mattson M (2004) Pathways toward and away from Alzheimer's disease. Nature 430: 631-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee AW, Bredt DS (2003) Assembly and plasticity of the glutamatergic postsynaptic specialization. Curr Opin Neurobiol 13: 111-118. [DOI] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T (2005) Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 47: 191-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL (1999) Soluble pool of Abeta amyloid as a determinant of severiy of neurodegeneration in Alzheimer's disease. Ann Neurol 46: 860-866. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charleswoth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RGM, Morrison JH, O'Dell TJ, Grant SGN (1998) Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396: 433-439. [DOI] [PubMed] [Google Scholar]
- Morabito M, Sheng M, Tsai LH (2004) Cyclin dependent kinase 5 phosphorylates the N-terminal domain of postsynaptic density protein PSD-95 in neurons. J Neurosci 24: 865-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST (2004) A novel cdk-5 dependent pathway for regulating gsk3 activity and kinesin-driven motility in neurons. EMBO J 23: 2235-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M (1996) Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci 16: 2157-2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee MS, Morabito M, Tsai LH (2000) NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 28: 697-711. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple transgenic model of Alzheimer disease with plaques and tangles: intracellular A-beta and synaptic dysfunction. Neuron 39: 409-421. [DOI] [PubMed] [Google Scholar]
- Okabe S, Kim HD, Miwa A, Kuriu T, Okado H (1999) Continual remodelling of postsynaptic density and its regulation by synaptic activity. Nat Neurosci 2: 804-811. [DOI] [PubMed] [Google Scholar]
- O'Nuallain B, Williams AD, Westermark P, Wetzel R (2004) Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem 279: 17490-17499. [DOI] [PubMed] [Google Scholar]
- Pizzi M, Boroni F, Bianchetti A, Moraitis C, Sarnico I, Benarese M, Goffi F, Valerio A, Spano P (2002) Expression of functional NR1/NR2B-type NMDA receptors in neuronally differentiated SK-N-SH human cell line. Eur J Neurosci 16: 2342-2350. [DOI] [PubMed] [Google Scholar]
- Richardson JC, Kendal CE, Anderson R, Priest F, Gower E, Soden P, Gray R, Topps S, Howlett DR, Lavender D, Clarke NJ, Barnes JC, Haworth R, Stewart MG, Rupniak HT (2003) Ultrastructural and behavioural changes precede amyloid deposition in a transgenic model of Alzheimer's disease. Neuroscience 122: 213-228. [DOI] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ (2001) Molecular determinants of NMDA receptor internalization. Nat Neurosci 4: 794-802. [DOI] [PubMed] [Google Scholar]
- Sabio G, Reuver S, Feijoo C, Hasegawa M, Thomas GM, Centeno F, Kuhlendahl S, Leal-Ortiz S, Goedert M, Garner C, Cuenda A (2004) Stress- and mitogen-induced phosphorylation of the synapse associated protein SAP90/PSD-95 by activation of SAPK3/p38gamma and ERK1/ERK2. Biochem J 380: 19-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA (2002) Direct interaction between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA 99: 13902-13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D (2002) Alzheimer's disease is a synaptic failure. Science 298: 789-791. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida GC, Paul S, Moran T, Choi EY, Nairns AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P (2005) Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci 8: 1051-1058. [DOI] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, Nicoll RA (2003) Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci 23: 5503-5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine BW, Dahlgren KN, Krafft GA, LaDu MJ (2003) In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J Biol Chem 278: 11612-11622. [DOI] [PubMed] [Google Scholar]
- Tirard M, Jasbinsek J, Almeida OFX, Michaelidis TM (2004) The manifold actions of the protein inhibitor of activated STAT proteins on the transcriptional activity of mineralocorticoid and glucocorticoid receptors in neural cells. J Mol Endocrinol 32: 825-841. [DOI] [PubMed] [Google Scholar]
- Todd PK, Mack KJ, Malter JS (2003) The fragile X mental retardation protein is required for type I metabotropic glutamate receptor dependent translation of PSD-95. Proc Natl Acad Sci USA 24: 14374-14378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GC (1999) Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci 22: 221-227. [DOI] [PubMed] [Google Scholar]
- Walsh D, Selkoe DS (2004) Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 44: 181-193. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB (1997) Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem 272: 22364-22372. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe (2002) Naturally secreted oligomers of amyloid protein potently inhibit hippocampal long-term potentiation. Nature 426: 535-539. [DOI] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL (2002) Soluble oligomers of beta-amyloid (1-42) inhibit long-term potentiation but not long term depression in rat dentate gyrus. Brain Res 924: 133-140. [DOI] [PubMed] [Google Scholar]
- Wang QW, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R (2004) Block of long-term potentiation by naturally occurring secreted and synthetic amyloid-peptide in hippocampal slices is mediated by activation of the kinases c-jun terminal kinases, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci 24: 3370-3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilquet V, De Strooper B (2004) Amyloid-beta precursor protein processing in neurodegeneration. Curr Opin Neurobiol 14: 582-588. [DOI] [PubMed] [Google Scholar]
- Wogulis M, Wright S, Cunningham D, Chilcote T, Powell K, Rydel RE (2005) Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. J Neurosci 25: 1071-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C, Walsh D, Selkoe D, Hartley DM (2004) Amyloid beta protein induced electrophysiological changes are dependent on aggregation state: N-methyl-d-aspartate (NMDA) versus non-NMDA receptor/channel activation. Neurosci Lett 366: 320-325. [DOI] [PubMed] [Google Scholar]