

Abstract
Prions represent a unique class of infectious agents in which the normal cellular prion protein (PrPC) is converted to an abnormal isoform (PrPSc), which accumulates in the brain and constitutes the major, if not the only, component of the infectious particle. Factors that still remain to be identified may facilitate the conversion of PrPC to PrPSc. In the present study, we first demonstrated that a growth factor of the neurotrophin family, brain-derived neurotrophic factor (BDNF), stimulates the formation of PrPSc in a gonadotropin-releasing hormone-secreting neuronal cell line (GT1-1 cells) infected with the Rocky Mountain Laboratory (RML) strain of scrapie as determined by Western blot analysis. We then observed that the prion-infected cells can be cleared from PrPSc by treatment with three inhibitors of mitogen-activated protein kinase kinase 1/2 (MEK1/2) [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene and 2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one, as well as α-[amino[(4-aminophenyl)thio]methylene]-2-(trifluoromethyl) benzeneacetonitrile, which passes the blood–brain barrier], a component of one of the intracellular signaling pathways activated by BDNF. The MEK1/2 inhibitors were also efficient in clearing PrPSc from prion-infected GT1-1 cells stimulated to accumulate high levels of PrPSc by enhanced serum concentrations in the medium or by the use of a serum-free neuron-specific neurobasal medium. PrPSc did not reappear in the cultures within 5 weeks after completion of treatment. We conclude that inhibitors of the MEK1/2 pathway can efficiently and probably irreversibly clear PrPSc from prion-infected cells. The MEK pathway may therefore be a suitable target for therapeutic intervention in prion diseases.
Keywords: prions, MEK inhibitors, growth factors, BDNF, neurons, MAP kinases
Introduction
Prions cause fatal neurodegenerative diseases characterized by spongiform vacuolation, synaptic alterations, nerve cell loss, and astrogliosis in the brain. These changes are associated with the deposition in the brain of an abnormal isoform of the prion protein (PrPSc), which is derived from a normal cellular prion protein (PrPC). The conversion of PrPC to PrPSc is a posttranslational process that occurs at the plasma membrane in association with caveolae-like domains or rafts or in the early endosome compartment of the cell (for review, see Prusiner, 2004). The conversion process may be facilitated by cellular factors, such as the laminin receptor (Leucht et al., 2003) and heparan sulfate (Ben-Zaken et al., 2003), as well as by yet unidentified other factors in the cells or tissues (Telling et al., 1994).
Although they are partially resistant to proteinase K (PK), prions may be degraded by cellular proteases. Thus, in a prioninfected neuroblastoma cell line, N2a, prion infectivity is cleared after treatment with an anti-PrPC antibody, which blocks formation of new PrPSc (Enari et al., 2001; Peretz et al., 2001). The lysosomal proteases cathepsin B and L are involved in clearance of PrPSc from a neuronal cell line, GT1-1 (Luhr et al., 2004b). For PrPSc to accumulate over time, its rate of formation (Borchelt et al., 1990) has to be faster than its degradation, a condition met in several neuronal cell lines. Prions accumulate in particular in neurons; however, whether special properties of neurons can affect the balance between formation and degradation of PrPSc to favor prion deposits is not known. Nerve growth factor (NGF) can induce increased expression of transcripts encoding PrPC in NGF-responsive cholinergic neurons in neonatal hamster brains (Mobley et al., 1988), and NGF is requested for PC12 cells to support scrapie replication (Rubenstein et al., 1990). Growth and differentiation of most neurons in the brain depend, however, on other growth-regulating molecules than NGF and whether such factors can influence the formation of prions has not been studied.
In the present study, we first examined whether a growth factor intrinsic to the brain can influence the amount of prion protein in prion-infected GT1-1 (ScGT1-1) cells. We choose brain-derived neurotrophic factor (BDNF), because in adult mammals the expression of this growth factor occurs primarily in the CNS (Murer et al., 2001), which is the most prominent target for prion infections. We here show that BDNF can enhance the formation of PrPSc in GT1-1 cells infected with the Rocky Mountain Laboratory (RML) strain of scrapie. Because one of the main intracellular signaling pathways stimulated by BDNF is the MEK pathway (for review, see Bibel and Barde, 2000), we then examined the effects of inhibitors of this pathway and report the unexpected and striking observation that the mitogen-activated protein kinase kinase 1/2 (MEK1/2) inhibitors 1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene (U0126) and 2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one (PD098059) efficiently cleared PrPSc from cells grown in different culture media. The MEK1/2 inhibitor α-[amino[(4-aminophenyl)thio] methylene]-2-(trifluoromethyl)benzeneacetonitrile (SL327), which can pass the blood–brain barrier (BBB), also reduced the content of PrPSc from the infected cells, whereas an inhibitor of the PI3 pathway, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY94002), had no effect.
Materials and Methods
GT1-1 cells and scrapie infection. The GT1-1 cell line is derived from a subtype of mouse immortalized hypothalamic gonadotropin-releasing hormone neurons and was a generous gift from Pamela Mellon (University of California, San Diego, CA). The medium used for cultivating the cells was DMEM 4.5 g/l glucose with GlutaMAX I (DMEM) supplemented with 5% heat-inactivated fetal bovine serum (FBS), 5% heat-inactivated horse serum (HS) and 50 U/ml penicillin–streptomycin (PEST; all obtained from Invitrogen, Paisley, UK). The cells were split at a ratio of 1:5 once a week using 1× trypsin-EDTA (Invitrogen).
Infection of GT1-1 cells with scrapie was performed in 24-well culture clusters (Corning, Corning, NY). The cells were grown in supplemented DMEM to ∼75% confluence and then incubated at 32°C with a 0.1% homogenate of mouse brains infected with the RML strain of scrapie (a gift from Stanley B. Prusiner, University of California, San Francisco, CA). After 4 d of exposure, the medium was changed, and the temperature was raised to 37°C. The presence of PK (Roche Diagnostics, Mannheim, Germany) resistant PrPSc was confirmed by Western blotting (see below) after six passages. These infected cells are referred to as ScGT1-1 cells as follows.
Treatment with BDNF and different cell culture media. GT1-1 and ScGT1-1 cells were seeded on 35 mm cell-culture dishes (Corning) coated with poly-l-lysine (Sigma, St. Louis, MO) and grown in DMEM containing 10% serum (HS and FBS; ratio, 1:1; used in all experiments). The day after seeding, the cultures were rinsed in PBS. For treatment with BDNF, cultures were incubated overnight in DMEM containing only 1% serum. BDNF (Preprotech, Rocky Hill, NY) was then added to the cell cultures at concentrations of 50–200 ng/ml in DMEM containing 1% serum. Other cultures were grown in DMEM containing either 1 or 10% serum or in Neurobasal medium supplemented with B27 and 2 mm l-glutamine (NB; all obtained from Invitrogen). All media contained 50 U/ml PEST. Cells were harvested for Western blot analysis after treatment for 4 d.
Treatment with MEK and PI3 kinase inhibitors. The following inhibitors of MEK1/2 were used: UO126 (Promega, Madison, WI), PD098059 (Sigma-Aldrich Chemie, Steinhem, Germany), and SL327 (Calbiochem, Darmstadt, Germany). LY294002 (Promega) was used as an inhibitor of PI3 kinase. All inhibitors were dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich Chemie) and stored at –20°C before use. The dose–response to UO126 was tested at concentrations of 2–8 μm in DMEM containing 1% serum (2 d of treatment). Treatments with U0126 in DMEM containing 1 or 10% serum or in NB were maintained from 2 d to 4 weeks. Control cultures were treated with DMSO. Cells treated with U0126 for longer periods of time were split at a ratio of 1:4. Treatment of cells with PD098059 (5–10 μm, 2 weeks), SL327 (4–6 μm, 2 weeks), and LY294002 (2 μm, 3–4 d) was performed in DMEM with 1% serum.
Treatment with leupeptin and pentosan polysulfate. Leupeptin hydrochloride (leupeptin; Sigma) and pentosan polysulfate (PPS; Sigma) were dissolved in PBS and added to the cell cultures at a concentration of 15 μm and 5 μg/ml, respectively, in 2 ml of DMEM containing 1% serum.
Cell counts and cell death markers. To evaluate effects of the treatments on cell survival, the number of cells both floating in the media (which was not replaced during the 4 d incubation periods) and attached to the culture dishes (after their mechanical detachment) was counted in a Bürker chamber. In addition, cultures were incubated with Hoechst 33342 stain (5 μg/ml) and propidium iodide (0.5 μg/ml) for 10 min and thereafter fixed in 10% formalin (Merck, Darmstadt, Germany) to determine the number of apoptotic and necrotic cells, respectively, in cultures exposed to the various treatments.
Western immunoblot. The cells were lysed on ice in lysis buffer (10 mm Tris-HCl, pH 8, 150 mm NaCl, 0.5% sodium deoxycholate, and 0.5% Triton X-100) without previous rinsing to include loosely attached cells. Nuclei and large debris were removed by centrifugation for 1 min at 16,000 × g. The protein concentration was determined using the Bio-Rad (Hercules, CA) Bradford protein assay and spectrophotometry (Ultrospec plus; Pharmacia LKB, Cambridge, UK) at 595 nm according to the instructions of the manufacturer, and all samples were standardized to contain the same amount of protein. The lysates were then split into two parts. One part was treated with PK (20 μg/ml) at 37°C for 30 min, and the reaction was stopped by incubation with 3 mm phenylmethylsulfonyl fluoride (Sigma) at room temperature for 10 min. The remaining part of the lysate was not PK treated. All samples were boiled for 10 min in SDS sample buffer. Electrophoresis was performed using NuPAGE 12% Bis-Tris gels with morpholinepropanesulfonic acid (Sigma) SDS running buffer. Proteins were transferred to Immobilon-PSQ Transfer membranes (Millipore, Bedford, MA) at 35 V for 2 h and blocked in 5% bovine serum albumin (BSA; Sigma) for 30 min. PrP was detected with recombinant Fab HuM-D13 (1 μg/ml; InPro Biotechnology, San Francisco, CA), followed by a secondary goat anti-human Fab2–peroxidase-conjugated antibody (0.16 μg/ml; Pierce, Rockford, IL). Labeling was detected with ECL plus (Amersham Biosciences, Little Chalfont, UK). In experiments using leupeptin, this protease inhibitor was also added to the Western blot lysis buffer of all samples (15 μm) to correct for any possible inhibition of PK by residual leupeptin in the cells.
Optical densities of the bands were determined on a GelDoc 2000 using the software Quantity One, version 4.2.2 (Bio-Rad). Statistics used were Student's t test in GraphPad Prism (GraphPad Software, San Diego, CA). All samples were normalized to the mean of the control samples for each experiment.
Immunofluorescence. Cells grown on 35 mm cell-culture dishes (Corning) were fixed in 10% formalin (Merck) for 30 min, permeabilized with 0.1% Triton X-100 (Sigma) in PBS for 5 min and treated with 3 m guanidinium thiocyanate (GdnSCN; Merck-Schuchardt, Hohenbrunn, Germany) for 5 min, for detection of PrPSc (Taraboulos et al., 1990). After blocking with 5% BSA for 40 min, the cells were incubated overnight at 4°C with the primary antibodies diluted in PBS containing 5% BSA. The monoclonal primary antibodies used were as follows: Fab HuM-D13 (3.5 μg/ml) and anti-neuron-specific class III β-tubulin (2 μg/ml; Babco, Richmond, CA). The cells were then incubated with secondary antibodies for 40 min at room temperature. The secondary antibodies used were as follows: Cy3-conjugated donkey anti-human IgG (7.5 μg/ml; Jackson ImmunoResearch, West Grove, PA), and Cy2-conjugated donkey anti-mouse IgG (6.5 μg/ml; Jackson ImmunoResearch). The cells were rinsed in PBS with 1% NH4Cl (Sigma), mounted in glycerol with 2.5% 1,4-diazabicyclo[2.2.2]octane (Sigma), and coverslipped.
To determine the percentage of infected cells after various treatments, cells with clear punctate PrP labeling after GdnSCN treatment were counted in micrographs from three random visual fields of each culture (objective, 20×) under coded numbers.
Results
BDNF increases the formation of PrPSc in ScGT1-1 cells
The levels of PrPSc in ScGT1-1 cells grown in DMEM containing 1% serum were increased after treatment with BDNF for 3–4 d (Fig. 1A). Quantification of PK-resistant PrP showed a twofold increase in PrPSc after BDNF treatment (1.934 ± 0.2855) compared with untreated controls that were normalized to 1 ± 0.06336 (p < 0.005, Student's t test, five experiments with duplicate cultures treated with either 100 or 200 ng/ml BDNF). To determine whether this increase in PrPSc reflects an increase in its formation or a decrease in its degradation, the effects of BDNF treatment were analyzed in the presence of inhibitors of formation, PPS, and degradation, leupeptin. No effect by BDNF on PrPSc levels was seen after treatment in combination with PPS (Fig. 1B, lanes 3 and 6), whereas in the presence of leupeptin, BDNF caused an increase in PrPSc levels, compared with treatment with leupeptin alone (Fig. 1B, lanes 2 and 5). BDNF treatment had no effect on the levels of PrPC in uninfected GT1-1 cells (Fig. 1C). Thus, BDNF appears to increase the formation of PrPSc.
Figure 1.

Western blots showing levels of PrP after BDNF treatment. A, ScGT1-1 cells treated with BDNF for 4 d; the gel on the left shows the total amount of PrP, and the gel on the right shows PK-resistant PrP. B, ScGT1-1 cells treated with BDNF (200ng/ml), leupeptin, or PPS or in combinations. C, Uninfected GT1-1 cells treated with BDNF. In all experiments, the cells were grown in DMEM containing 1% serum.
Effect of different culture media on the levels of PrPSc
To determine whether factors other than BDNF can induce increased levels of PrPSc, the effect of different cell culture media on the levels of PrPC and PrPSc in GT1-1 and ScGT1-1 cells was studied. First, the cells grown in DMEM containing 1% serum were compared with cells grown in DMEM containing 10% serum (HS:FBS; ratio, 1:1, as above). The data show a concentration-dependent increase in the level of PrPSc (Fig. 2A) but not in PrPC (Fig. 2B). To simulate the microenvironment in the CNS tissues, in which no serum is present, we also tested the effect of the serum-free Neurobasal medium, supplemented with B27 (which does not contain BDNF) and l-glutamine. This medium induced high levels of PrPSc in the ScGT1-1 cells, as seen in the Western blots (Fig. 2A), whereas the levels of PrPC were unaltered (Fig. 2B). Quantification of PK-resistant PrP showed a significant decrease in PrPSc levels after cultivation in DMEM with 1% serum (0.3751 ± 0.1006) compared with controls grown in 10% serum, which were normalized to 1 ± 0.00874 (p < 0.0001, Student's t test; nine samples from seven different sets of experiments) and a twofold increase in PrPSc levels after cultivation in NB (2.009 ± 0.2160) compared with DMEM with 10% serum, which were normalized to 1 ± 0.03799 (p = 0.0001, Student's t test; nine samples from five different sets of experiments). The cells in NB medium showed a marked increase in PrPSc positive punctate as compared with cells in DMEM containing 1% serum by immunofluorescence, but the percentage of immunopositive cells, ∼80%, remained the same (data not shown). The cells could be maintained in the NB medium for 5 d; longer incubation in this medium leads to extensive cell death.
Figure 2.

Effect of different cell culture media on levels of PrP. A, B, Western blots showing levels of PrP in ScGT1-1 (A) and GT1-1 (B) cells cultured in DMEM with 1 or 10% serum or in Neurobasal medium (NB).
The number of cells attached to the dishes at the end of the 4 d incubation was similar in cultures grown in NB medium and DMEM containing 10% serum (0.8 to 1.4 × 106 and 0.7 to 1.1 × 106 cells/dish, respectively), but the number of cells floating in the media was higher in cultures maintained in the NB medium (1.3 to 1.5 × 105 and 0.2 to 0.4 × 105 cells/dishes, respectively). The number of apoptotic (pyknotic or fragmented nuclei, as seen by Hoechst staining) cells with or without secondary necrotic changes (stained red with propidium iodide) attached to the dishes was increased in NB medium compared with DMEM containing 10% serum (14 and 3%, respectively). There was a lower number of cells attached to the dishes in cultures grown in DMEM containing 1% serum compared with cultures grown in DMEM with 10% serum (0.5 to 0.9 × 106 cells/dish and 1.0 to 1.1 × 106, respectively), and the number of cells floating in the medium was higher (0.5 to 0.6 × 105 and 0.2 × 105 cells/dish, respectively). The number of apoptotic cells attached to the dishes was higher in DMEM containing 1% serum compared with DMEM containing 10% serum (13 and 3%, respectively).
Inhibition of MEK, but not PI3 kinase, clears PrPSc from cultured cells
In the subsequent set of experiments, we determined whether the MEK and the PI3 kinase pathways, which are two main signaling pathways in neurons activated by BDNF, can affect the levels of PrPSc in the cells (Bibel and Barde, 2000).
For this purpose, we first exposed ScGT1-1 cells grown in DMEM containing 1% serum to the MEK inhibitor U0126 (2 μm) for 5 d. This treatment reduced PrPSc to undetectable levels (Fig. 3A). The levels of PrPSc could also be reduced when U0126 was added to cells grown in DMEM containing 10% serum, or in NB (Fig. 3B); however, in these media, longer incubation times and/or higher concentrations of U0126 (4 μm) were needed. The number of cells attached to the dishes in untreated and U0126-treated cultures was similar (1.1 to 1.8 × 106 cells/dish and 1.1 to 1.4 × 106 cells/dish, respectively), as were the number of cells floating in the medium (0.8 × 105 cells/dish and 0.7 to 1.0 × 105 cells/dish, respectively). There was also no increase in the proportion of apoptotic cells after treatment. Thus, because the vast majority of cells were infected, and no increased loss of cells was seen after treatment, the effect of the drug on PrPSc cannot be the result of a selective killing of infected cells.
Figure 3.

Western blots showing levels of PrP after treatment with MEK1/2 inhibitor U0126. A, ScGT1-1 cells treated with the MEK inhibitor U0126 (2 μm) in DMEM with 1% serum for 5 d. B, ScGT1-1 cells treated with U0126 (4 μm) in DMEM with 10% serum or in NB for 5 d. C, ScGT1-1 cells treated with U0126 (2 μm) in combinations with leupeptin or PPS for 4 d. D, Uninfected GT1-1 cells treated with U0126 (2 μm) in DMEM with 1% serum for 5 d.
PrPSc did not reappear in the GT1-1 cells up to 5 weeks after completion of a 4 week U0126 (2 μm) treatment in DMEM containing 1% serum (data not shown). This indicates that the effect of treatment may be irreversible.
To analyze whether the clearance of PrPSc after U0126 treatment was attributable to decreased formation or increased degradation of PrPSc, we then exposed the ScGT1-1 cells to the drug in combination with PPS or leupeptin, as described above. After combined treatment with U0126 and leupeptin, the increase in the levels of PrPSc seen after treatment with leupeptin alone (Fig. 3C) did not occur (Fig. 3C). In contrast, when PPS was used in combination with U0126, the level of PrPSc was reduced (Fig. 3C), compared with treatment with PPS alone (Fig. 3C). No reduction in the levels of PrPC in GT1-1 cells could be seen after treatment with U0126 (Fig. 3D). This suggests that U0126 acts by preventing the synthesis of PrPSc.
Approximately 80% of the infected, untreated cells showed punctate immunolabeling, with the Fab HuM-D13 antibody in their cytoplasm after guanidine treatment. This punctate labeling appeared only after denaturation with guanidine and was not seen in uninfected cells. The immunopositivity, therefore, most likely represent PrPSc (Taraboulos et al., 1990). After treatment with U0126 for 3 d, a few punctate was seen in a few cells, whereas no immunolabeling remained after treatment for 5 d (Fig. 4).
Figure 4.
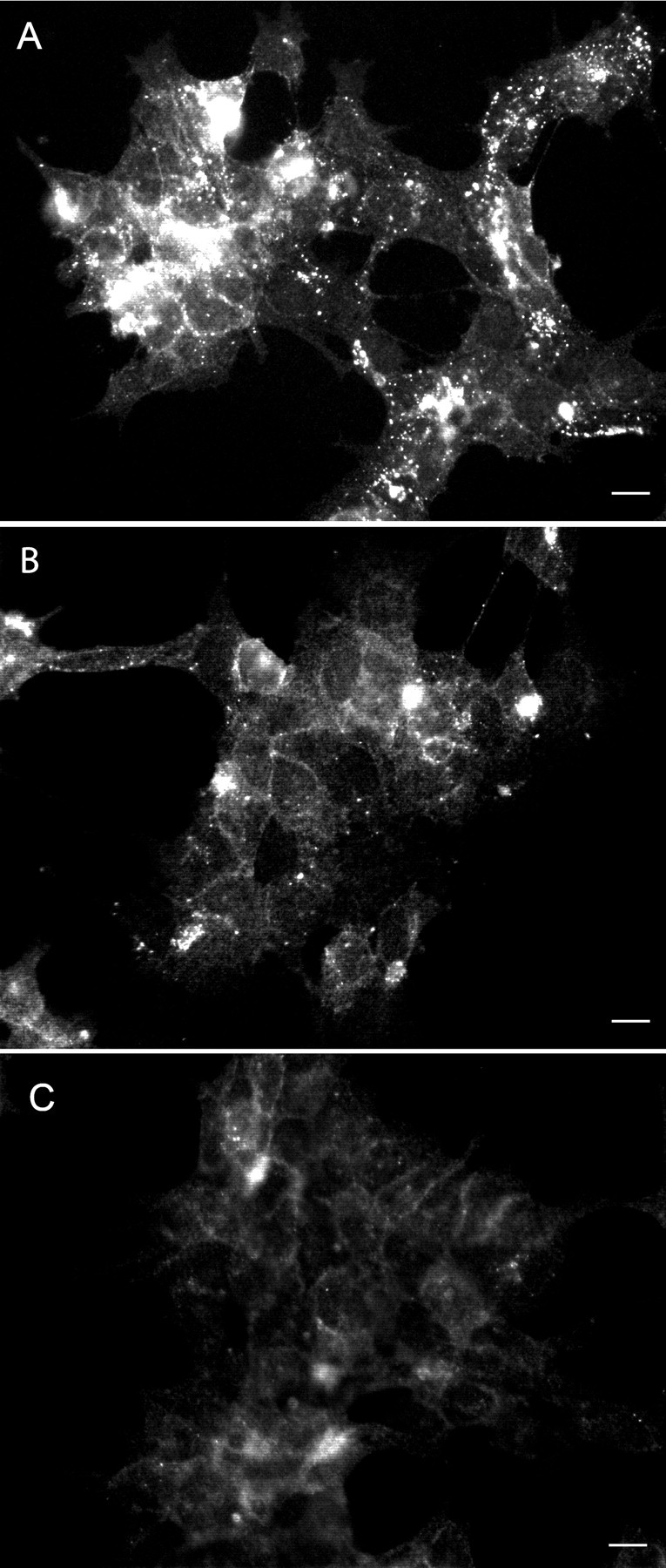
A–C, PrPSc immunofluorescence of untreated ScGT1-1 cells (A) and U0126 (2 μm)-treated ScGT1-1 cells (B, C) for 3 d (B) and 5 d (C). Cells were exposed to guanidine thiocyanate before immunolabeling with Fab Hum-D13. Note the punctate immunolabeling in the cytoplasm in the untreated cells (A), which disappears after treatment (B, C). Scale bars, 20 μm.
Treatment of the cells with PD098059 (10–20 μm) and SL327 (4–6 μm) for 14 and 17 d, respectively, also reduced the levels of PrPSc in ScGT1-1 cells maintained in DMEM containing 1% serum (Fig. 5A,B).
Figure 5.

A, B, Western blots showing levels of PrP after treatment with the MEK inhibitors PD098059 and SL327. ScGT1-1 cells cultivated in DMEM with 1% serum and treated with the MEK-inhibitor PD098059(A; 10–20 μm) for 14 d are shown; the MEK-inhibitor SL327(B; 4–6 μm) for 17 d is shown.
Treatment with the PI3 kinase inhibitor LY294002 did not affect the levels of either PrPC or PrPSc in GT1-1 and ScGT1-1 cells (data not shown).
Discussion
In the present study, we observed that BDNF caused an increase in levels of PrPSc, but not of PrPC, and that treatment with inhibitors of the intracellular MEK/extracellular signal-regulated kinase 1/2 (ERK1/2) pathway could clear PrPSc from prion-infected GT1-1 cells. MEK inhibitors were efficient in the clearance of PrPSc from cells grown in various culture media.
The experiments, using treatment with BDNF in combination with the polyanion PPS or leupeptin, indicate that the enhanced PrPSc levels reflect an increase in formation rather than a decrease in degradation of the protein. Thus, when formation of PrPSc was blocked by PPS (Caughey and Raymond, 1993) during the BDNF treatment, no effect on PrPSc levels was seen, which makes it unlikely that the BDNF effect is attributable to a decrease in PrPSc degradation. In contrast, when degradation of PrPSc was inhibited with leupeptin (Luhr et al., 2004a), BDNF caused an increase in PrPSc levels compared with treatment with leupeptin alone. This indicates that the BDNF-induced increase in PrPSc levels reflects an increase in PrPSc formation. Because BDNF treatment of the uninfected GT1-1 cells, in contrast to observations on NGF treatment of neuronal precursor cell lines (Kuwahara et al., 2000), had no effect on the levels of PrPC, it is likely that the increased formation of PrPSc reflects an enhanced rate of conversion of PrPC to PrPSc. The PK-resistant PrP does not represent a misfolded, protease resistant protein that may appear as a consequence of metabolic disturbances (Yedidia et al., 2001), because no PK-resistant PrPs were seen in uninfected GT1-1 cells after treatment.
BDNF mediates its effects via activation of three main intracellular signaling pathways, the MEK/ERK1/2, the PI3 kinase, and the phospholipase C-γ pathways (Bibel and Barde, 2000). In our experiments, inhibition of PI3 kinase had no effect on the level of PrPSc, which is in contrast to the clearance of PrPSc caused by inhibition of MEK1/2. Several intracellular signaling pathways operate via sequential activation of kinases and are collectively known as mitogen-activated protein kinase signaling cascades. The MEK pathway, which has been investigated in the present study, involves activation of MEK1/2 and subsequently of ERK1/2 via phosphorylation of these kinases (Rubinfeld and Seger, 2004). The MEK/ERK1/2 cascade plays a major role in cell proliferation and differentiation and is activated by growth factors, hormones, and neurotransmitters (Rubinfeld and Seger, 2004). Our experiments, using inhibitors of formation or degradation of PrPSc in combination with the MEK-inhibitor U0126, indicate that the effect is attributable to an inhibited formation of PrPSc rather than an enhanced degradation of the protein. Provided that U0126 does not stimulate an unknown leupeptin-independent pathway of PrPSc degradation, this suggests that U0126 acts by preventing the synthesis of PrPSc. Because treatment with U0126 had no effect on the levels of PrPC, it is likely that the inhibited formation of PrPSc is because of interference with the rate of conversion of PrPC to PrPSc in the cells. One possible mechanism for such an inhibited conversion may be that blocking the MEK pathway activated a factor that could interfere with prion formation. Alternatively, the inhibitor could have caused a decreased expression of a factor that stimulates formation of PrPSc. The existence of such a factor, protein X, involved in prion formation is suggested from experiments using mice expressing a chimeric human/mouse PrP transgene, or a human transgene, infected with a human adapted prion strain (Telling et al., 1995). Protein X has not yet been isolated, but PrPC binds via residues located at the C terminus to this protein (Kaneko et al., 1997) and to PrPSc via a central domain (Telling et al., 1994, 1995). Alternatively, the MEK inhibitors may have affected the availability of accessory molecules, such as heparan sulfate (Schonberger et al., 2003), that are involved in the conversion of PrPC into PrPSc, or the trafficking of both PrPC and PrPSc, leading to their redistribution into a compartment nonpermissive for PrPSc formation (Gilch et al., 2001), or have modified the compartments (e.g., rafts) to decrease their support for PrPSc formation (Taraboulos et al., 1995).
The MEK inhibitors cleared PrPSc from ScGT1-1 cells maintained not only in DMEM with different serum concentrations but also in cells grown in the NB medium. The ScGT1-1 cells did not survive longer in this medium than in supplemented DMEM; in contrast, they showed a greater death rate and did not survive for >5 d. The increased levels of PrPSc observed in the ScGT1-1 cells grown in NB medium therefore does not reflect accumulation in cells with enhanced life span, as suggested recently for scrapie-infected N2a cells cultivated in the same medium (Bate et al., 2004).
The finding of an effect of MEK inhibitors in ScGT1-1 cells maintained in various culture media indicates that clearance of PrPSc by the inhibitors is a generalized phenomenon. A number of antiprion substances, with different modes of action, have been described (Prusiner et al., 2004; Weissmann and Aguzzi, 2005). Some of these antiprion compounds induce cellular clearance of prions by directly targeting PrPSc (branched polyamines) (Supattapone et al., 1999; Winklhofer and Tatzelt, 2000; Supattapone et al., 2001). There are other compounds, such as Congo red and its analogs, that presumably overstabilize PrPSc and thereby block the partial denaturation that is needed for the conversion of PrPC to PrPSc (Caughey and Race, 1992; Caspi et al., 1998). Related compounds (i.e., sulfated polyanions) bind PrPSc directly and may inhibit binding of endogenous sulfated glycosaminoglycans to PrPSc, a binding that may facilitate the conversion (Ehlers and Diringer, 1984; Caughey and Raymond, 1993; Caughey et al., 1994; Brimacombe et al., 1999). In this respect, synthetic heparan mimetic biopolymers are particularly effective in inhibiting the conversion of PrPC to PrPSc (Schonberger et al., 2003). The interaction between PrPC and PrPSc can also be blocked by the following: anti-PrP antibodies (Enari et al., 2001; Peretz et al., 2001), anti-PrP aptamers (Proske et al., 2002), and β-sheet breaker peptides (Soto et al., 2000). Cp60 and its derivates may inhibit the interaction between PrPC and protein X (Perrier et al., 2000), and lovastatin and squalestatin may interfere with PrPSc formation by changing the lipid composition of the rafts (Taraboulos et al., 1995; Bate et al., 2004). A tyrosine kinase c-Abl inhibitor, 4-[(4-methyl-1-piperazinyl)methyl]-N-[4-methyl-3-[[4-(3-pyridinyl)-2-yrimidinyl]amino]-phenyl] benzamide, acts most likely by increasing lysosomal degradation of PrPSc (Ertmer et al., 2004). Finally, there are treatments that aim at removing the substrate for prion formation, PrPC, by inhibiting its synthesis (small interfering RNA) (Tilly et al., 2003; Luhr et al., 2004a) or blocking its transport to the plasma membrane (Gilch et al., 2001).
Several of these compounds, however, do not cross the BBB, and most are toxic and not suitable candidates for treatment of prion diseases. Interest has therefore been paid to the acridine derivative quinacrine, which passes the BBB and clears PrPSc as well as the infectivity from scrapie-infected N2a cells by as yet not understood mechanisms (Korth et al., 2001). This drug is the subject of clinical trials with, until now, variable results (Barret et al., 2003; Nakajima et al., 2004), and its effect in vivo in scrapie-infected mice is still under investigation (Yung et al., 2004). It is therefore interesting to note that new, relatively nontoxic MEK inhibitors have been developed that are undergoing clinical trials for certain forms of tumors. MEK inhibitors have also been designed to pass the BBB, and presently we observed that one such inhibitor, SL327, could reduce the levels of PrPSc in ScGT1-1 cells. Our findings therefore not only demonstrate that the MEK intracellular signaling pathway may be involved in regulating formation of PrPSc but also that inhibitors of this pathway could potentially be of therapeutic value in prion diseases.
In conclusion, our experiments have shown that BDNF, as well as factors in sera and serum-free media, can increase the formation of PrPSc in scrapie-infected GT1-1 cells and that inhibitors of MEK1/2 can reduce levels of PrPSc, and one of them may even possibly irreversibly clear the infection from the cells. The MEK/ERK1/2 pathway is therefore involved in PrPSc formation and may be a potential therapeutic target for antiprion drug design.
Footnotes
This work was supported by Swedish Research Council Grant 04480, United States Army, and European Commission Grant QLK2-CT-2002-81628. Elin Nordström is the recipient of a fellowship from Fondation Internationale Carrefour.
Correspondence should be addressed to Krister Kristensson, Department of Neuroscience, Retzius väg 8, Karolinska Institutet, SE-171 77 Stockholm, Sweden. E-mail: krister.kristensson@neuro.ki.se.
DOI:10.1523/JNEUROSCI.2349-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258451-06$15.00/0
References
- Barret A, Tagliavini F, Forloni G, Bate C, Salmona M, Colombo L, De Luigi A, Limido L, Suardi S, Rossi G, Auvre F, Adjou KT, Sales N, Williams A, Lasmezas C, Deslys JP (2003) Evaluation of quinacrine treatment for prion diseases. J Virol 77: 8462–8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate C, Langeveld J, Williams A (2004) Manipulation of PrPres production in scrapie-infected neuroblastoma cells. J Neurosci Methods 138: 217–223. [DOI] [PubMed] [Google Scholar]
- Ben-Zaken O, Tzaban S, Tal Y, Horonchik L, Esko JD, Vlodavsky I, Taraboulos A (2003) Cellular heparan sulfate participates in the metabolism of prions. J Biol Chem 278: 40041–40049. [DOI] [PubMed] [Google Scholar]
- Bibel M, Barde YA (2000) Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev 14: 2919–2937. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Scott M, Taraboulos A, Stahl N, Prusiner SB (1990) Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured cells. J Cell Biol 110: 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimacombe DB, Bennett AD, Wusteman FS, Gill AC, Dann JC, Bostock CJ (1999) Characterization and polyanion-binding properties of purified recombinant prion protein. Biochem J 342: 605–613. [PMC free article] [PubMed] [Google Scholar]
- Caspi S, Halimi M, Yanai A, Sasson SB, Taraboulos A, Gabizon R (1998) The anti-prion activity of Congo red. Putative mechanism. J Biol Chem 273: 3484–3489. [DOI] [PubMed] [Google Scholar]
- Caughey B, Race RE (1992) Potent inhibition of scrapie-associated PrP accumulation by congo red. J Neurochem 59: 768–771. [DOI] [PubMed] [Google Scholar]
- Caughey B, Raymond GJ (1993) Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J Virol 67: 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B, Brown K, Raymond GJ, Katzenstein GE, Thresher W (1994) Binding of the protease-sensitive form of PrP (prion protein) to sulfated glycosaminoglycan and congo red. J Virol 68: 2135–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers B, Diringer H (1984) Dextran sulphate 500 delays and prevents mouse scrapie by impairment of agent replication in spleen. J Gen Virol 65: 1325–1330. [DOI] [PubMed] [Google Scholar]
- Enari M, Flechsig E, Weissmann C (2001) Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci USA 98: 9295–9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertmer A, Gilch S, Yun SW, Flechsig E, Klebl B, Stein-Gerlach M, Klein MA, Schatzl HM (2004) The tyrosine kinase inhibitor STI571 induces cellular clearance of PrPSc in prion-infected cells. J Biol Chem 279: 41918–41927. [DOI] [PubMed] [Google Scholar]
- Gilch S, Winklhofer KF, Groschup MH, Nunziante M, Lucassen R, Spielhaupter C, Muranyi W, Riesner D, Tatzelt J, Schatzl HM (2001) Intracellular re-routing of prion protein prevents propagation of PrP(Sc) and delays onset of prion disease. EMBO J 20: 3957–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, Cohen FE, Prusiner SB (1997) Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci USA 94: 10069–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korth C, May BC, Cohen FE, Prusiner SB (2001) Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci USA 98: 9836–9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara C, Kubosaki A, Nishimura T, Nasu Y, Nakamura Y, Saeki K, Matsumoto Y, Onodera T (2000) Enhanced expression of cellular prion protein gene by insulin or nerve growth factor in immortalized mouse neuronal precursor cell lines. Biochem Biophys Res Commun 268: 763–766. [DOI] [PubMed] [Google Scholar]
- Leucht C, Simoneau S, Rey C, Vana K, Rieger R, Lasmezas CI, Weiss S (2003) The 37 kDa/67 kDa laminin receptor is required for PrP(Sc) propagation in scrapie-infected neuronal cells. EMBO Rep 4: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhr KM, Nordstrom EK, Low P, Ljunggren HG, Taraboulos A, Kristensson K (2004a) Scrapie protein degradation by cysteine proteases in CD11c+ dendritic cells and GT1-1 neuronal cells. J Virol 78: 4776–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhr KM, Nordstrom EK, Low P, Kristensson K (2004b) Cathepsin B and L are involved in degradation of prions in GT1-1 neuronal cells. NeuroReport 15: 1663–1667. [DOI] [PubMed] [Google Scholar]
- Mobley WC, Neve RL, Prusiner SB, McKinley MP (1988) Nerve growth factor increases mRNA levels for the prion protein and the beta-amyloid protein precursor in developing hamster brain. Proc Natl Acad Sci USA 85: 9811–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murer MG, Yan Q, Raisman-Vozari R (2001) Brain-derived neurotrophic factor in the control human brain, and in Alzheimer's disease and Parkinson's disease. Prog Neurobiol 63: 71–124. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Yamada T, Kusuhara T, Furukawa H, Takahashi M, Yamauchi A, Kataoka Y (2004) Results of quinacrine administration to patients with Creutzfeldt-Jakob disease. Dement Geriatr Cogn Disord 17: 158–163. [DOI] [PubMed] [Google Scholar]
- Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, Dwek RA, Burton DR, Prusiner SB (2001) Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 412: 739–743. [DOI] [PubMed] [Google Scholar]
- Perrier V, Wallace AC, Kaneko K, Safar J, Prusiner SB, Cohen FE (2000) Mimicking dominant negative inhibition of prion replication through structure-based drug design. Proc Natl Acad Sci USA 97: 6073–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proske D, Gilch S, Wopfner F, Schatzl HM, Winnacker EL, Famulok M (2002) Prion-protein-specific aptamer reduces PrPSc formation. Chembiochem 3: 717–725. [DOI] [PubMed] [Google Scholar]
- Prusiner SB (2004) An introduction to prion biology and diseases. In: Prion biology and diseases, Ed 2 (Prusiner SB, ed), pp 1–87. Cold Spring Harbor, NY: Inglis J.
- Prusiner SB, May BCH, Cohen FE (2004) Therapeutic approaches to prion diseases. In: Prion biology and diseases, Ed 2 (Prusiner SB, ed), pp 961–1014. Cold Spring Harbor, NY: Inglis J.
- Rubenstein R, Scalici CL, Papini MC, Callahan SM, Carp RI (1990) Further characterization of scrapie replication in PC12 cells. J Gen Virol 71: 825–831. [DOI] [PubMed] [Google Scholar]
- Rubinfeld H, Seger R (2004) The ERK cascade as a prototype of MAPK signaling pathways. In: MAP kinase signaling protocols (Seger R, ed), pp 1–28. Totowa, NJ: Humana. [DOI] [PubMed]
- Schonberger O, Horonchik L, Gabizon R, Papy-Garcia D, Barritault D, Taraboulos A (2003) Novel heparan mimetics potently inhibit the scrapie prion protein and its endocytosis. Biochem Biophys Res Commun 312: 473–479. [DOI] [PubMed] [Google Scholar]
- Soto C, Kascsak RJ, Saborio GP, Aucouturier P, Wisniewski T, Prelli F, Kascsak R, Mendez E, Harris DA, Ironside J, Tagliavini F, Carp RI, Frangione B (2000) Reversion of prion protein conformational changes by synthetic beta-sheet breaker peptides. Lancet 355: 192–197. [DOI] [PubMed] [Google Scholar]
- Supattapone S, Nguyen H-OB, Cohen FE, Prusiner SB, Scott MR (1999) Elimination of prions by branched polyamines and implications for therapeutics. Proc Natl Acad Sci USA 96: 14529–14534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supattapone S, Wille H, Uyechi L, Safar J, Tremblay P, Szoka FC, Cohen FE, Prusiner SB, Scott MR (2001) Branched polyamines cure prion-infected neuroblastoma cells. J Virol 75: 3453–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboulos A, Serban D, Prusiner SB (1990) Scrapie prion proteins accumulate in the cytoplasm of persistently infected cultured cells. J Cell Biol 110: 2117–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB, Avraham D (1995) Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol 129: 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling GC, Scott M, Hsiao KK, Foster D, Yang SL, Torchia M, Sidle KC, Collinge J, DeArmond SJ, Prusiner SB (1994) Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci USA 91: 9936–9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB (1995) Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83: 79–90. [DOI] [PubMed] [Google Scholar]
- Tilly G, Chapuis J, Vilette D, Laude H, Vilotte JL (2003) Efficient and specific down-regulation of prion protein expression by RNAi. Biochem Biophys Res Commun 305: 548–551. [DOI] [PubMed] [Google Scholar]
- Weissmann C, Aguzzi A (2005) Approaches to therapy of prion diseases. Annu Rev Med 56: 321–344. [DOI] [PubMed] [Google Scholar]
- Winklhofer KF, Tatzelt J (2000) Cationic lipopolyamines induce degradation of PrPSc in scrapie-infected mouse neuroblastoma cells. Biol Chem 381: 463–469. [DOI] [PubMed] [Google Scholar]
- Yedidia Y, Horonchik L, Tzaban S, Yanai A, Taraboulos A (2001) Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J 20: 5383–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung L, Huang Y, Lessard P, Legname G, Lin ET, Baldwin M, Prusiner SB, Ryou C, Guglielmo BJ (2004) Pharmacokinetics of quinacrine in the treatment of prion disease. BMC Infect Dis 4: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]