

Abstract
Intercellular signaling dynamics critically influence the functional roles that the signals can play. Small lipids are synthesized and released from neurons, acting as intercellular signals in regulating neurotransmitter release, modulating ion channels on target cells, and modifying synaptic plasticity. The repertoire of biological effects of lipids such as endocannabinoids (eCBs) is rapidly expanding, yet lipid signaling dynamics have not been studied. The eCB system constitutes a powerful tool for bioassaying the dynamics of lipid signaling. The eCBs are synthesized in, and released from, postsynaptic somatodendritic domains that are readily accessible to whole-cell patch electrodes. The dramatic effects of these lipid signals are detected electrophysiologically as CB1-dependent alterations in conventional synaptic transmission, which therefore serve as a sensitive reporter of eCB actions. We used electrophysiological recording, photolytic release of caged glutamate and a newly developed caged AEA (anandamide), together with rapid [Ca2+]i measurements, to investigate the dynamics of retrograde eCB signaling between CA1 pyramidal cells and GABAergic synapses in rat hippocampus in vitro. We show that, at 22°C, eCB synthesis and release must occur within 75-190 ms after the initiating stimulus, almost an order of magnitude faster than previously thought. At 37°C, the time could be <50 ms. Activation of CB1 and downstream processes constitute a significant fraction of the total delay and are identified as major rate-limiting steps in retrograde signaling. Our findings imply that lipid messenger dynamics are comparable with those of metabotropic neurotransmitters and can modulate neuronal interactions on a similarly fast time scale.
Keywords: anandamide, metabotropic glutamate, calcium, photolysis, GABA, endocannabinoid
Introduction
The dynamic properties of intercellular signaling systems greatly influence the ways in which cells affect each other. Moreover, signaling dynamics impose critical constraints on models of the underlying signaling mechanisms. Small lipid molecules, including platelet-activating factor, arachidonic acid, prostaglandins (for review, see Bazan, 2003), and endocannabinoids (eCBs) (for review, see Alger, 2002; Freund et al., 2003), constitute a major class of intercellular messengers. However, there is little information on the dynamics of lipid signal mobilization. This reflects partly the difficulty in capturing and measuring tiny quantities of lipid messengers, and partly the absence of a well established model system that offers sufficient experimental control over the lipid release process. Lipid signals are not stored in vesicles but are generated from membrane-bound precursors by enzymatic activity initiated in response to cellular stimuli, such as a rise in [Ca2+]i, or the activation of biochemical cascades. Lipids are released across the plasma membrane. Rapid kinetic measurements have not been the focus of previous investigations, and the maximum speed with which lipid messengers can be produced is essentially unknown.
Recently, eCBs have emerged as putative retrograde intercellular messengers in the mammalian CNS (Kreitzer and Regehr, 2001a,b; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). The two major eCBs, arachidonoylethanolamide [anandamide (AEA)] and 2-arachidonoyl glycerol (2-AG), both derivatives of arachidonic acid (Iversen, 2003) (for review, see Freund et al., 2003) are agonists at the G-protein-coupled receptor (GPCR), CB1. Both AEA and 2-AG can be released from postsynaptic principal cell somata and dendrites, and their release is stimulated by a marked rise in [Ca2+]i. Release of eCB can also be caused by activation of GPCRs including dopamine (Giuffrida et al., 1999), metabotropic glutamate (Maejima et al., 2001; Varma et al., 2001), or muscarinic acetylcholine receptors (Kim et al., 2002; Ohno-Shosaku et al., 2003) and can persist for many minutes. The metabotropic glutamate receptor (mGluR)- and muscarinic acetylcholine receptor-dependent pathways are not associated with a marked rise in [Ca2+]i (Maejima et al., 2001; Kim et al., 2002), and can be triggered even in the presence of high intracellular concentrations of Ca2+ chelators, although they may nevertheless be sensitive to ambient [Ca2+]i (Hashimotodani et al., 2005). Whether these separate pathways operate on the same time scales is not known. The kinetic properties of the eCB-mediated responses will critically constrain the candidate biochemical pathways.
The eCB system constitutes a powerful tool for bioassaying the dynamics of lipid signaling. The eCBs are synthesized in, and released from, postsynaptic somatodendritic domains that are readily accessible to whole-cell patch electrodes. The dramatic effects of these lipid signals are detected electrophysiologically as CB1-dependent alterations in conventional synaptic transmission, which therefore provide a sensitive means of bioassaying eCB actions. Indeed, the first estimates of the speed of intercellular lipid signaling were provided by the study of depolarization-induced suppression of inhibition (DSI) (Pitler and Alger, 1994; Wilson et al., 2001). In the present study, by combining whole-cell voltage-clamp, [Ca2+]i measurements, and photorelease of caged glutamate and a novel, caged AEA, we investigated the dynamics of eCB signaling in hippocampal slices.
Materials and Methods
Preparation of acute hippocampal slices
Hippocampal slices were obtained mainly from 4- to 6-week-old male Sprague Dawley rats. In some experiments, as noted, mice (25-40 g) in which the CB1 gene had been inactivated (Ledent et al., 1999) were used. All experiments were performed in accordance with the guidelines set forth by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine. After the animals were deeply anesthetized with halothane and decapitated, the hippocampi were removed and sectioned into 400-μm-thick slices in ice-cold saline using a vibratome (Technical Products International, St. Louis, MO). The slices were maintained at room temperature in an interface holding chamber in a humidified atmosphere saturated with 95% O2/5% CO2 for at least an hour before being transferred to the submersion-type chamber (Nicoll and Alger, 1981) where the experiments were performed at 30 ± 1°C.
Preparation of organotypic slice cultures
Organotypic hippocampal slice cultures were prepared either as roller-tube cultures (Gähwiler et al., 1998) or as interface cultures on semiporous membranes (Stoppini et al., 1991). Hippocampi were dissected from 5- or 6-d-old CO2-anesthetized rat or CB1-/- mouse pups, and cut into 375-μm-thick transverse slices using a McIlwain tissue chopper (Brinkmann Instruments, Westbury, NY). Slices were attached to polylysine-coated glass coverslips in 20 μl of chicken plasma coagulated with thrombin. Coverslips were placed into culture tubes with 750 μl of serum-containing medium and incubated in a roller-drum at 36°C. Alternatively, slices were placed on porous, transparent membranes attached to the bottom of a round polystyrene chamber and kept in six-well tissue culture plates. Organotypic slices prepared with the roller-tube method were x-irradiated at the time of explantation and treated overnight with antimitotics to reduce the proliferation of glial cells. Slice cultures were held in a 36°C incubator with 5% CO2 and maintained in vitro for > 14 d before performing experiments to allow for synaptic maturation.
Cultures were perfused at ∼1 ml/min with control saline containing the following (in mm): 137 NaCl, 2.8 KCl, 2.5 CaCl2, 2.5 MgCl2, 23.2 NaHCO3, 0.4 NaH2PO4, pH to 7.2 with HEPES, and 5.6 glucose at room temperature (20-22°C) in a closed recirculating system. Caged glutamate (Ncm-Glu) (1 mm) or caged AEA (200 μm) was included in the extracellular saline as indicated in the relevant text or figure.
Electrophysiology
Current-clamp recordings from CA1 pyramidal cells in acute slices were obtained with sharp, glass microelectrodes (50-150 MΩ) filled with 3 m KCl. Reliable spontaneous IPSP activity (seen in ∼80% of all recorded cells) was observed after a 5-20 min bath application of carbachol (CCh), 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (NBQX), and dl-2-amino-5-phosphonopentanoic acid (AP5). We refer to this IPSC activity as “spontaneous” as is conventional, because it is persistent and does not require additional stimulation to evoke it. “Theta burst” trains of action potentials, each comprising five action potentials elicited by positive, constant-current pulses (each 5 or 10 ms in duration) were delivered via the microelectrode at 50 Hz. Repetitive bursts were delivered at 200 ms intervals (i.e., at 5 Hz) to elicit DSI. Signals were digitized at 5 kHz with an analog-to-digital interface (Digidata 1200A; Molecular Devices, Palo Alto, CA) and analyzed with pClamp 9.0 software (Molecular Devices).
The intracellular whole-cell patch pipette solution used in slice-cultured cells contained the following (in mm): 90 CsCH3SO3, 50 CsCl, 1 MgCl2, 2 Mg-ATP, 0.3 Tris-GTP, 0.2 Cs4BAPTA, 10 HEPES, and 5 QX-314 (lidocaine N-ethyl bromide), pH 7.20 with CsOH, and 295 mOsm. In some experiments, 90 mm CsCH3SO3 was replaced by 20 mm Cs4BAPTA (Invitrogen, Eugene, OR) to block rises in [Ca2+]i. Seal resistance was routinely > 1 GΩ. Recordings were made with an Axoclamp 2B amplifier and collected through a Digidata-1200A Interface (Molecular Devices). Analog signals were recorded, digitized at 5 kHz, low-pass filtered at 2 kHz, and subsequently analyzed using Clampfit (Clampfit 9; Molecular Devices) and Mini Analysis Program (version 6.0.1; Synaptosoft, Decatur, GA).
Monosynaptic evoked IPSCs (eIPSCs) were elicited by 100-μs-long extracellular stimulus pulses (150-250 μA) delivered with concentric bipolar stimulating electrodes (David Kopf Instruments, Tujunga, CA), or with a low resistance (1-2 MΩ) patch pipette filled with extracellular saline, placed in the stratum oriens between CA3 and CA1, 0.5-1 mm apart from the recording site. To isolate monosynaptic eIPSCs, ionotropic glutamate receptor blockers NBQX (10 μm) and AP5 (50 μm) were present in the bath solution throughout all experiments. In experiments that focused on spontaneous IPSPs (sIPSPs) or spontaneous IPSCs (sIPSCs), CCh (500 nm or 5 μm, as noted) was added to the bathing solution. The lower concentration of CCh was used in cultured slices because they are prone to generating paroxysmal sIPSC activity in 5 μm CCh, which was used for acute slices. Water-based stock solutions of CCh, 2S-2-amino-2-(1S,2S-2-carboxycycloprop-1-yl)-3-(xanth-9-yl)propanoic acid (LY341495) (Tocris, Ballwin, MO), or DMSO-based stock solutions of N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251) (Tocris), and R-(+)-(2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl)-(1-naphthalenyl)methanone monomethanesulfonate (WIN55212-2) (Tocris) were added to the bath solution and perfused into the recording chamber when needed. The final concentration of DMSO was 0.01% (v/v). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Ca2+ imaging
Ca2+ -dependent fluorescence of fluo-3 (50 μm in the whole-cell pipette) was collected by a photomultiplier tube (PMT) (H5783; Hamamatsu, Hamamatsu City, Japan) and recorded concurrently with IPSCs. A filter set suitable for fluo-3 (Chroma 41025; exciter HQ470/40×; emitter HQ515/30m; dichroic Q495LP) was used in conjunction with a Nikon (Melville, NY) Eclipse 600 upright microscope (40× 0.8 numerical aperture objective). The output of the PMT was low-pass filtered at 2 kHz (Frequency Devices, Haverhill, MA), digitized at 5 kHz (Digidata 1322; Molecular Devices), and displayed by pClamp9 (Molecular Devices) using Clampex. Trials were analyzed off-line using Origin software (OriginPro 7; OriginLab, Northampton, MA). Data from three to four trials were averaged. To correct fluorescence baseline drift caused by gradual photobleaching, two 3 s segments (immediately before photolytic or electrical stimulation and at the end of each fluorescence record) were fit by a single exponential, which was used as the baseline. Details of the optics used for focal photolysis are given below.
Chemical synthesis and characterization of caged AEA General.
Unless otherwise specified, reagents and solvents of American Chemical Society grade or better were purchased from Sigma-Aldrich, Advanced ChemTech (Louisville, KY), Fisher Scientific (Pittsburgh, PA), or VWR International (West Chester, PA), and were used without additional purification. UV-visible absorption spectra were recorded on a Cary 300 Bio spectrophotometer (Varian Instruments, Walnut Creek, CA). Proton nuclear magnetic resonance (NMR) spectra were recorded on either a 300 MHz (QE-300; General Electric, Fairfield, CT) or a 500 MHz spectrometer (INOVA 500; Varian Instruments) and were referenced to tetramethylsilane (δ = 0). For reporting NMR spectra, the following descriptors are used: singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br); underlining within a molecular fragment indicates the group of protons to which a spectral resonance is assigned. HPLC was performed on a system fitted with a photodiode array detector (model 600; Waters Associates, Milford, MA). Fast-atom bombardment (FAB) high-resolution mass spectrometry (HRMS) was performed at the analytical facility in the Department of Biochemistry at Michigan State University (East Lansing, MI).
Ethyl 2-(3-formyl-4-nitrophenoxy)acetate (1). Under dry N2, anhydrous K2CO3 (2.90 g; 21 mmol) was added to a solution of 5-hydroxy-2-nitrobenzaldehyde (5.01 g; 30 mmol) and ethyl bromoacetate (6.01 g; 36 mmol) in dry acetonitrile (50 ml). The reaction mixture was stirred at room temperature for 24 h. After filtration to remove solids, the filtrate was concentrated and purified by column chromatography on silica gel (hexane/ethyl acetate, 95:5 → 80:20) to yield compound 1 (all compounds are numbered as in Fig. 1 A) as yellow needles [6.84 g (90%); melting point (m.p.), 58-59°C]. 1H NMR (300 MHz) δ was as follows: 1.32 (t, 3H, CH3), 4.30 (q, 2H, CH2CH3), 4.78 (s, 2H, OCH2CO), 7.18 (d, 1H, J = 9.0 Hz, aromatic, H-6), 7.31 (s, 1H, aromatic, H-2), 8.16 (d, 1H, J = 8.8 Hz, aromatic, H-5), and 10.5 (s, 1H, CHO).
Figure 1.

Synthesis and photochemical release mechanism of caged AEA. A, Synthesis and structures of the caged AEA molecules, N-CmNb-arachidonoylethanolamide (4b) and N-(mPEG-CmNb)-arachidonoylethanolamide (6). B, Photorelease of AEA from caged AEA. Light absorption by caged AEA generates the short-lived aci-nitro intermediate, which decomposes into the spent cage and free AEA.
(2-tert-Butoxyethyl)-(5-ethoxycarbonylmethoxy-2-nitrobenzyl)amine (2a). Starting material 1 (0.506 g; 2.0 mmol), (2-t-butoxyethyl)amine (0.281 g; 2.4 mmol; CSP Pharmaceuticals, San Diego, CA), and sodium acetate (0.164 g; 2.0 mmol) were added to 1,2-dichloroethane (10 ml). The mixture was stirred for 1 h under dry N2. NaBH(CH3CO2)3 (1.06 g; 5 mmol) was then added in one portion and stirring was continued at room temperature overnight (∼18 h). The mixture was diluted with CH2Cl2 (20 ml), and the reaction was quenched with 10 ml of saturated NaHCO3 solution. The organic phase was separated, washed with saturated NaHCO3 (10 ml; two times) and brine (15 ml), and dried over anhydrous MgSO4. Flash chromatography on silica gel (hexane/ethyl acetate, 90:10 → 65:35:1% Et3N) yielded compound 2 as a pale yellow oil [0.465 g (65.6%)]. 1H NMR (300 MHz) δ was as follows: 1.2 (s, 9H, C(CH3)3), 1.31 (t, 3H, CH3), 2.04 (br s, 1H, NH), 2.76 (t, 2H, CH2OBu-t), 3.49 (t, 2H, NCH2CH2O), 4.14 (s, 2H, CH2Ar), 4.30 (q, 2H, OCH2CH3), 4.72 (s, 2H, OCH2CO), 6.85 (d, 1H, J = 8.8 Hz, aromatic), 7.27 (s, 1H, aromatic), and 8.10 (d, 1H, J = 9.0 Hz, H ortho to NO2).
N-(2-tert-Butoxyethyl)-N-(5-ethoxycarbonylmethoxy-2-nitrobenzyl) arachidonoylamide (3a). To prepare arachidonoyl chloride, oxalyl chloride (0.32 g; 0.22 ml; 2.5 mmol) was added to a solution of arachidonic acid (0.5 g; 1.64 mmol) in anhydrous CH2Cl2 (15 ml); a drop of dry dimethylformamide (DMF) was added to initiate the reaction. The mixture was stirred at room temperature for 1 h, and then evaporated under reduced pressure to yield an oily residue. To ensure complete removal of trapped HCl and residual oxalyl chloride, the residue was twice redissolved in dry CH2Cl2 (10 ml) and evaporated to dryness under reduced pressure. The final product was dissolved in dry CH2Cl2 to make 5.0 ml of solution in a volumetric flask.
Under dry N2, arachidonoyl chloride solution in CH2Cl2 (4 ml; 1.3 mmol) was added dropwise by syringe to a solution of amine 2a (0.425 g; 1.2 mmol) and Et3N (0.243 g; 0.334 ml; 2.4 mmol) in dry CH2Cl2 (10 ml). The resulting mixture was stirred at room temperature for 2 h, during which the amine was completely converted to the amide, as verified by TLC on silica gel plates (hexane/EtOAc, 7:3). The reaction mixture was diluted with CH2Cl2 (20 ml), and saturated NaHCO3 solution (10 ml) was added. The organic phase was separated, washed with saturated NaHCO3 (10 ml; two times) and brine (15 ml), and dried over anhydrous MgSO4. Purification by flash chromatography on silica gel (hexane/EtOAc, 95:5 → 85:15) yielded a viscous yellow oil [0.665 g (86.5%)]. 1H NMR (300 MHz) δ was as follows: 0.88 (t, 3H, CH3 of aliphatic chain), 1.15 (s, 9H, C(CH2)3), 1.30 (m, 9H, 3 × CH2 + OCH2CH3), 1.72 (m, 2H, CH2), 2.05 (m, 3H, CH2 + 0.5 × CH2CO), 2.18 (m, 2H, CH2), 2.57 (t, 1H, 0.5 × CH2CO), 2.80 (m, 6H, 3 × CH2), 3.42-3.65 (m, 4H, CH2CH2OBu-t), 4.28 (q, 2H, OCH2Me), 4.65 (s, 2H, OCH2CO), 5.02, 5.13 (s, 2H, CH2Ar), 5.37 (m, 8H, 4 × CH = CH), 6.75-6.95 (m, 2H, aromatic), and 8.22 (d, 1H, H ortho to NO2).
N-(2-tert-butoxyethyl)-N-(5-carboxymethoxy-2-nitrobenzyl)arachidonoyl amide (4a). Under N2, compound 3a (1.0 mmol) was dissolved in MeOH (3 ml) and cooled in an ice-water bath for 5 min. A solution of 1N NaOH (2.0 ml; 2 mmol) was added dropwise to the stirred methanolic solution over 3 min. Stirring was continued in a room temperature water bath for 1 h, whereupon the solution became clear and the hydrolysis was complete (as verified by TLC on silica gel plates). The reaction mixture was chilled in an ice-water bath, neutralized with 1N HCl (2.1 ml; 2.1 mmol), and extracted with EtOAc (10 ml; two times) and ether (10 ml; two times). The organic extracts were pooled, washed with brine (20 ml; two times), and dried over anhydrous MgSO4. Removal of solvent under vacuum yielded a pale yellow oil [0.520 g (84.9%)]. 1H NMR (300 MHz) δ was as follows: 0.88 (t, 3H, aliphatic CH3), 1.15 (s, 9H, C(CH2)3), 1.29 (m, 6H, 3 × CH2), 1.72 (m, 2H, CH2), 2.00-2.30 (m, 5H, 2 × CH2 + 0.5 × CH2CO), 2.63 (t, 1H, 0.5 × CH2CO), 2.80 (m, 6H, 3 × CH2), 3.40-3.65 (m, 4H, CH2CH2OBu-t), 4.67 (s, 2H, OCH2CO), 5.04, 5.18 (s, 2H, CH2Ar), 5.40 (m, 8H, 4 × CH = CH), 6.75-6.95 (m, 2H, aromatic), 7.80 (br s, 1H, COOH), and 8.24 (d, 1H, ortho to NO2). HRMS (FAB) was as follows: m/z calculated for C35H52N2O7, 613.3853 (M + H+), found 613.3854.
N-(2-tert-butoxyethyl)-N-[5-(mPEG-aminocarbonylmethoxy)-2-nitrobenzyl]-arachidonoylamide (5). A solution of mPEG-NH2 [molecular weight (MW), 5000 Da; Nektar Therapeutics, San Carlos, CA] (2.5 g; 5 mmol) in dry CH2Cl2 (10 ml) was dried with anhydrous MgSO4 for 24 h. After removal of MgSO4 by filtration, the solution was concentrated to one-half volume and diluted with dry CH2Cl2 to 10.0 ml in a volumetric flask. A stirred mixture of acid 4 (0.307 g; 0.5 mmol) and HOBt (1-hydroxybenzotriazole) (66 mg; 0.49 mmol) in dry CH2Cl2 (10 ml) was chilled in an ice-water bath. Then, DCC (1,3-dicyclohexylcarbodiimide) (107 mg; 0.52 mmol) was added in one portion. After 30 min, a 5 ml aliquot of the solution of mPEG-NH2 (1.25 g; 0.25 mmol) in dry CH2Cl2 (5 ml) was added by syringe. Stirring was maintained at room temperature for 24 h. To remove precipitates, the reaction mixture was filtered through a 2 cm bed of Celite, which was then washed with 5 ml of CH2Cl2. The combined filtrate was diluted with anhydrous ethyl ether to yield a pale yellow powder (1.14 g). The powder was redissolved in dry CH2Cl2 (3 ml), and the solution was added to anhydrous ethyl ether (120 ml) to precipitate the polymer conjugate a second time. The precipitate was recrystallized from CH2Cl2-diethyl ether to yield the polymer conjugate 5 (0.53 g; 42.5%).
N-[5-(mPEG-aminocarbonylmethoxy)-2-nitrobenzyl]arachidonoylamide, caged AEA (6). A solution of compound 5 (0.47 g) in dry CH2Cl2 (5 ml) was chilled in an ice-water bath; trifluoroacetic acid (5 ml) was then added in one portion. The mixture was stirred at room temperature for 24 h. The residue remaining after solvent removal under reduced pressure was dissolved in CH2Cl2 (2 ml), and the polymer conjugate was precipitated by adding anhydrous diethyl ether (100 ml). Recrystallization of the precipitate from CH2Cl2-diethyl ether yielded the caged AEA 6 as a yellow powder [0.37 g (79.4%)].
(5-Ethoxycarbonylmethoxy-2-nitrobenzyl)-(2-hydroxyethyl)amine (2b). Compound 1 (0.506 g; 2.0 mmol), 2-aminoethanol (0.147 g; 2.4 mmol), and sodium acetate (0.164 g; 2.0 mmol) were added to 1,2-dichloroethane (10 ml); the mixture was magnetically stirred for 1 h under dry N2. Then NaBH(CH3CO2)3 (1.06 g; 5 mmol) was added in one portion, and stirring was continued at room temperature overnight (∼18 h). The mixture was diluted with CH2Cl2 (20 ml), and the reaction was quenched with 10 ml of saturated NaHCO3 solution. The organic phase was separated, washed with saturated NaHCO3 (10 ml; two times) and brine (15 ml), and dried over anhydrous MgSO4. After purification by flash chromatography on silica gel (hexane/ethyl acetate, 80:20 → 1:1:1% Et3N), amine 2b was obtained as a pale yellow solid [0.22 g (36.8%); m.p. 68-69°C]. 1H NMR (500 MHz; CDCl3) δ was as follows: 1.31 (t, 3H, CH3), 2.08-2.24 (br s, 2H, NH + OH), 2.82 (t, 2H, CH2OH), 3.69 (t, 2H, NCH2CH2O), 4.11 (s, 2H, CH2Ar), 4.28 (q, 2H, OCH2CH3), 4.72 (s, 2H, OCH2CO), 6.85 (d, 1H, J = 8.8 Hz, aromatic H-4), 7.17 (s, 1H, aromatic H-6), and 8.08 (d, 1H, J = 8.9 Hz, H ortho to NO2).
N-(2-hydroxyethyl)-N-(5-ethoxycarbonylmethoxy-2-nitrobenzyl)-arachidonoylamide (3b). To a solution of arachidonic acid (0.23 g; 0.74 mmol) and oxalyl chloride (0.14 g; 0.10 ml; 1.1 mmol) in anhydrous CH2Cl2 (5 ml), a drop of dry DMF was added. The reaction mixture was treated precisely as described in the procedure for compound 3a. The product arachidonoyl chloride was dissolved in dry CH2Cl2 to make 5.0 ml of solution in a volumetric flask.
To a solution of amine 2b (0.22 g; 0.74 mmol) and Et3N (0.22 g; 0.31 ml; 2.2 mmol) in dry CH2Cl2 (5 ml) under dry N2, 5 ml (0.74 mmol) of the solution of arachidonoyl chloride in CH2Cl2 was added dropwise by syringe. The reaction mixture was stirred at room temperature for 2 h, during which time amine 2b was completely converted to amide, as verified by TLC on silica gel plates (hexane/ethyl acetate, 7:3). The reaction mixture was diluted with CH2Cl2 (20 ml) and treated with saturated NaHCO3 solution (5 ml). The organic phase was separated, washed with saturated NaHCO3 (5 ml) and brine (10 ml), and dried over anhydrous MgSO4. After purification by flash chromatography on silica gel (hexane/ethyl acetate, 95:5 → 80:20), amide 3b was obtained as a yellow viscous oil [0.12 g (27.7%)]. 1H NMR (300 MHz) δ was as follows: 0.88 (t, 3H, aliphatic CH3), 1.22-1.42 (m, 9H, 3 × CH2 + COOCH2CH3), 1.72 (m, 2H, CH2), 2.05 (m, 3H, CH2 + 0.5 × CH2CO), 2.20 (m, 2H, CH2), 2.56 (t, 1H, 0.5 × CH2CO), 2.65-2.90 (m, 6H, 3 × CH2), 3.08 (br s, 1H, OH), 3.50-3.86 (m, 4H, NCH2CH2), 4.27 (q, 2H, OCH2Me), 4.72 (s, 2H, OCH2CO), 5.00, 5.04 (s, 2H, CH2Ar), 5.37 (m, 8H, 4 × CH = CH), 6.80-6.98 (m, 2H, aromatic), and 8.26 (d, 1H, ortho to NO2). HRMS (FAB) was as follows: m/z calculated for C33H48N2O7, 585.3540 (M + H+), found 585.3554 (M + H+).
N-(2-hydroxyethyl)-N-(5-carboxymethoxy-2-nitrobenzyl)-arachidonoyl amide (4b). Under N2, compound 3b (18 mg; 0.03 mmol) was dissolved in 50 μl of MeOH, and the solution was chilled in an ice-water bath. NaOH solution (1N; 45 μl; 0.045 mmol) was added, and the reaction mixture was maintained with periodic agitation at room temperature for 1 h. The mixture became homogeneous and TLC on silica gel plates (hexane/ethyl acetate, 7:3) confirmed completion of hydrolysis. The solution was chilled in an ice-water bath, neutralized with 1N HCl (60 μl; 0.06 mmol), and extracted with CH2Cl2 (5 ml; two times). The combined organic extract was washed with brine (5 ml; two times), dried over anhydrous MgSO4, and evaporated under reduced pressure to yield free acid 4b as a yellow oil [14.1 mg (84%)].
Quantum yield of photolysis. A pulsed Nd:YAG laser (QuantaRay GCR-18S; Spectra Physics, Mountain View, CA), at 1 Hz repetition rate, was used as the photolysis light source. A pair of harmonic separators (BSR-355-1025; CVI Laser, Albuquerque, NM) was used to isolate the third harmonic emission (355 nm) from the fundamental (1064 nm) and second harmonic (532 nm) emissions. A standard chemical actinometer (potassium ferrioxalate) (Hatchard and Parker, 1956) was used to confirm the stability of the laser output (1.4% drift over 2.5 h) and to verify that energy output was linearly related to the number of laser pulses (> 99.8% linearity from 1 to 60 pulses). A solution of compound 3b in acetonitrile was prepared to have an absorbance > 2.5 at 355 nm. In all experiments, we ensured that <20% of the caged compound was consumed by photolysis. This was confirmed by HPLC analysis using 4-nitrophenol as an internal standard: equal amounts of 4-nitrophenol were added to the photolyzed and unphotolyzed samples of compound 3b, and the extent of consumption of compound 3b was determined by numerical integration of corresponding peaks in the HPLC (de Mayo and Shizuka, 1976). Photolysis experiments were performed in triplicate; HPLC analysis for each replicate experiment was repeated three times.
Kinetics of flash photolysis. The kinetics of flash photolysis was studied on a kinetic spectrophotometer (LP920; Edinburgh Instruments, Livingston, UK). A solution of compound 4b was prepared in a 1:1 mixture of 0.1 m sodium phosphate buffer, pH 7.41, and methanol to have an absorbance > 2.0 (in a 1 cm cuvette) at 355 nm, the wavelength of the photolytic light. The air-equilibrated solution was stirred magnetically at room temperature. Photolysis of the sample was effected by a 8.6 ns [full width at half-maximum (FWHM)], 100 mJ pulse of 355 nm third harmonic light from a Nd-YAG laser (Quanta-Ray GCR-18S). A pair of harmonic separators (BSR-355-1025; CVI Laser) was used to remove residual fundamental (1064 nm) and second harmonic (532 nm) laser emissions from the photolysis beam. A probe beam from a stabilized xenon arc lamp was focused so that the focal point coincided with the region of maximal photolysis in the sample. After passing through the sample, the probe beam was dispersed through a monochromator. Modulation of the probe beam intensity by the transient photochemical intermediate species was monitored at 430 nm by a fast photomultiplier tube. The intensity-versus-time data were converted to an absorbance-versus-time trace. Nonlinear least-squares analysis allowed exponential decay times to be extracted from the data. Data in the initial 45 ns window contain artifactual contributions from scattering of the laser pulse and instabilities in the high-sensitivity detection circuitry, and are not included in data analysis.
Photolysis of caged glutamate and caged AEA
Photolysis of caged glutamate (Ncm-Glu) and caged AEA was accomplished using previously published methods (Wei et al., 2001; Cai et al., 2004). The multiline UV emission of an argon ion laser (wavelength, 333-364 nm; BeamLok 2065-7S; Spectra Physics) was launched into a 100 μm diameter fused silica optical fiber (OZ Optics, Westbrook, Ontario, Canada). The exit end of the fiber was projected onto a conjugate of the field diaphragm plane along the epifluorescence path of the microscope. After establishing whole-cell recording, the laser spot (∼15-20 μm diameter) was centered on the soma of the patched cell. The laser output was gated by a shutter (NM Laser Products, Sunnyvale, CA) to generate photolytic flashes (100 ms duration). In our experimental setup, there is an instrumentation delay of 1.4 ms from triggering the UV laser flash to a laser signal measured by the PMT.
Data analysis
All data are presented as mean ± SEM. Student's paired and unpaired t test or repeated-measures ANOVAs were used as needed. To estimate the time constant of the exponential change in sIPSP or sIPSC frequency, we averaged the response frequency between 2 and 3 s after stimulus onset and defined this average to be the minimal frequency value. All frequency values were then normalized between the minimum frequency and the control IPSP/C frequency, which correspond to 0 and 100%, respectively, on the graphs. Nonlinear least-squares curve fitting of flash photolysis kinetic data were performed through Origin software (OriginLab).
Results
Synthesis and characterization of caged AEA
Caged AEA was synthesized as outlined in Figure 1A. Details of the synthetic procedures are described in Materials and Methods. We prepared two versions of caged AEA: one in which a PEG [poly(ethyleneglycol)] moiety is covalently attached to the cage to confer high aqueous solubility on the caged AEA (Fig. 1A, compound 6), and for comparison, one in which the cage bears a single carboxyl group (Fig. 1A, compound 4b). In the caged molecules, the cage is covalently attached to the amide nitrogen atom in AEA, because structure-activity studies have shown that covalent modification of the amide nitrogen abolishes both cannabinoid activity and susceptibility to lipase cleavage (Sheskin et al., 1997; Lang et al., 1999).
The photochemical process through which AEA is generated from caged AEA is shown in Figure 1B. The UV-visible absorption spectrum of compound 6, presented as extinction coefficient (ϵ) versus wavelength, is shown in Figure 2A (solid curve). As expected for a caged molecule, the spectrum changed markedly when the sample was photolyzed by UV light (Fig. 2A, dashed curve). The spectroscopic properties of compound 4b were similar to those of compound 6 except that peak absorption occurred at a slightly longer wavelength (314 vs 303 nm for compound 6); the prephotolysis and postphotolysis spectra of compound 4b are supplied as supplemental Figure 1 (available at www.jneurosci.org as supplemental material). Photolysis of caged compound released bona fide AEA, as verified by HPLC analysis of the photolysate, with authentic, never-caged AEA acting as reference standard (data not shown). The quantum efficiency for photorelease of AEA (Q) on photolysis was determined to be 0.062 ± 0.003 (n = 3).
Figure 2.

Photolysis of caged AEA monitored by UV-visible absorption spectroscopy. A, The UV-visible absorption spectra, shown as solid and dashed lines, respectively, were acquired before and after photolysis of caged AEA 6 for 60 s with 75 mW of the UV emission from an argon ion laser. Solvent was 0.1 m sodium phosphate buffer, pH 7.4. Extinction coefficient (ϵ) values are reported to facilitate read-out of ϵ at any wavelength. B, Photolysis kinetics measured by transient absorbance spectral change after laser flash photolysis of caged AEA 6. The absorbance at 470 nm of a solution of caged AEA in 0.1 m sodium phosphate, pH 7.4, was monitored. At time 0, a pulse of 355 nm light (8.6 ns FWHM; ∼30 mJ) was delivered to the sample. In the top panel, gray points are experimental data (average of 16 replicate measurements), and the solid black curve is the nonlinear least-squares single-exponential fit to the data. The exponential time constant of the fitted decay curve is τ = 27.2 ± 0.7 μs. The residuals of the fit are shown in the bottom panel.
For examining the kinetics of photolysis, we used transient absorption spectroscopy to monitor the short-lived, photolytically generated aci-nitro intermediate (Fig. 1B, species in brackets) (Yip et al., 1985; Schupp et al., 1987; Zhu et al., 1987; Yip et al., 1991), which has a decay that is commonly taken to be concomitant with cleavage of the caging group and release of product (Walker et al., 1988; McCray and Trentham, 1989). The kinetic spectroscopy trace for laser flash photolysis of compound 6 in 0.1 m sodium phosphate buffer, pH 7.4, is shown in Figure 2B. Photolysis by a single 8.6 ns pulse of 355 nm light gave rise to a transient absorption that decayed with single-exponential time course, showing exponential time constant τ = 27.2 ± 0.7 μs (equivalent to a half-life of t1/2 = 18.9 ± 0.5 μs). Thus, photorelease of AEA from compound 6 is expected to be complete in <100 μs after flash photolysis. In comparison, photolysis of compound 4b, which does not bear the 5000 MW poly(ethyleneglycol) moiety, was even more rapid, proceeding with exponential time constant τ = 0.106 ± 0.001 μs. The kinetic spectroscopy trace for compound 4b is supplied as supplemental Figure 2 (available at www.jneurosci.org as supplemental material).
Dynamics of DSI in adult acute slices
We began by analyzing the DSI dynamics in the CA1 region of acute adult hippocampal slices using from one to five theta burst trains of action potentials recorded under current-clamp conditions to initiate DSI. Each theta burst consisted of 5 positive DC pulses delivered at 50 Hz (i.e., five action potentials occurred within 100 ms). When more than one theta burst was used, they were triggered at 5 Hz. We induced sIPSPs or sIPSCs by bath-applying 5 μm carbachol (Pitler and Alger, 1992; Martin and Alger, 1999), because these events occur continuously at a higher frequency, and therefore provide better temporal resolution, than evoked synaptic responses.
Figure 3, A and B, shows the effect of one (top panels) or three (bottom panels) theta bursts on sIPSP frequency. The transient suppression of sIPSPs is DSI. To examine the onset of DSI in more detail, we reduced the sIPSP bin sizes from 1 s to 200 ms and reanalyzed the data from 0 to 3 s after the action potential train. Figure 3C shows that, after a single theta burst train, there was no effect on sIPSPs for the first bin after the train (i.e., ∼300 ms after the start of the train), but thereafter the frequency decreased nearly to zero over the next 1-2 s with a time course that was well fit by a single exponential having a time constant of τ = 416 ms. Extrapolating the exponential to the control level (100% baseline IPSP frequency) gave a value of 343 ms, which we took as the minimal onset time for DSI (tDSI) after a single train. When data obtained after one to five theta burst trains were analyzed in the same way as in Figure 3C, and the data were grouped across cells (n = 10), a very similar exponential decay function fit the combined data (τ = 345 ms) (Fig. 3D) as for one train (τ = 416 ms). Likewise, tDSI determined from multiple- or single-train experiments was the same (351 vs 343 ms, respectively). We conclude that a single process is set into motion by one train, and its dynamic properties are not altered by additional stimulation. In addition, there is a minimal latency between the start of action potential stimulation and the onset of eCB effects; thereafter, the effects grow larger with an exponential time constant of ∼400 ms.
Figure 3.

Dynamics of DSI in adult acute slices. A, DSI of sIPSPs induced by theta burst patterns of action potentials. Raw traces from a CA1 pyramidal cell recorded with a sharp microelectrode filled with 3 m KCl (see Materials and Methods); sIPSPs appear as upward deflections of 2-10 mV. In the top trace, a single theta burst train (5 action potentials, each induced by 5-ms-long DC pulse injected through the recording electrode) occurred at the point of the large upward deflection; in the bottom trace, three theta burst trains (200 ms interburst intervals) were used. The temporary reduction in sIPSPs occurring after each theta train is DSI. B, Analysis of several trials of sIPSPs as in corresponding rows of A (top panel, 1 theta burst; bottom panel, 3 theta bursts). Shown are total sIPSPs over all trials binned in 1 s intervals as described in Materials and Methods. The onset of theta burst trains occurred at time 0. C, Onset of sIPSP suppression. Shown are total sIPSPs occurring immediately after a single theta train which occurred at time 0, rebinned in 200 ms bins (same experiment shown in top part of B). In the first bin (ending at 300 ms), there was no suppression of sIPSPs, and thereafter, the development of DSI is well fit by a single exponential with a time constant of 416 ms. Note in this and other similar figures that the y-axis represents the percentage of the control sIPSP/C frequency (see Materials and Methods); hence negative values do not imply negative frequencies. D, Combined data from experiments (n = 10 cells) in which from one to five theta trains were given (each symbol represents data obtained after a different number of trains). The best-fitting exponential through the combined data are similar to the fit in C. E-G, Recovery from DSI has a similar time course after either one (E), three (F), or five (G) theta trains.
Brown et al. (2003) proposed that the enhancement in DSI produced by an increasing amount of synaptic stimulation reflects augmentation in the amount of eCB released. Alternatively, the rate of eCB termination could have been altered. However, we found that the recovery time constant, ∼5 s, did not increase with increasing stimulation (one, three, or five trains) (Fig. 3E-G), supporting the conclusion that increased eCB release accounts for the prolongation of DSI in our experiments.
Components of the Ca2+-induced eCB signal process
The analysis in Figure 3 distinguishes two kinetic parameters of DSI: time-to-onset (tDSI) and the exponential time constant of DSI development. The quantity tDSI must include the time from the start of the postsynaptic pyramidal cell depolarization and Ca2+ influx through to activation of the presynaptic CB1 on the interneurons. The pyramidal cell steps include time for [Ca2+]i to rise sufficiently to initiate eCB synthesis (“tCa”), and the time for eCB synthesis, release, and transit to CB1 (teCB). The presynaptic interneuron steps comprise time for activation of CB1 and downstream steps (tCB1). These parameters can be related as follows:
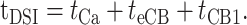 |
(1) |
We want to determine teCB, which cannot be measured directly. We can, however, estimate teCB from Equation 1 if the other values are known. To measure tCa and tCB1, we applied optical methods to organotypic slices (Gähwiler et al., 1998), which are thinner and thus more suitable for optical approaches than are acute slices. DSI occurs in organotypic slices (Martin and Alger, 1999); however, the cultured slice chamber is not heated, and these experiments were done at room temperature, ∼22°C, rather than 30°C. Because eCB signaling is temperature dependent (Kreitzer et al., 2002), we first repeated the experiments shown in Figure 3, in acute slices at 22°C (n = 5 slices) (data not shown). As expected, at the lower temperature, DSI was slowed: tDSI at 22°C was 409 ms (vs 351 ms at 30°C), and a single exponential with a time constant of 714 ms fit the group data for the rise-to-peak DSI (vs 345 ms at 30°C). Using these data in combination with the van't Hoff equation, ln(τT1/τT2) = (-Ea/R)[T2-1 - T1-1], where R = 1.987 cal · mol-1 · K-1 is the gas constant and τT1 and τT2 are time constants determined at temperatures T1 and T2 (in kelvins), we can estimate an activation energy, Ea = 16.2 kcal · mol-1, or, equivalently, a Q10 (= Rate37°C/Rate27°C) of 2.4, for the growth-to-peak DSI. All subsequent experiments were done in organotypic cultured slices at room temperature.
Estimating tCa
To estimate tCa, we detected changes in [Ca2+]i by including the fluorescent Ca2+ indicator, fluo-3, in the recording pipette and using a PMT to capture the fluorescence signal. The PMT signal reflected the volume-averaged [Ca2+]i rise within the recorded cell. We measured Ca2+ signals and DSI concomitantly to determine the minimal duration of the Ca2+ signal associated with detectable DSI (Fig. 4). A 50 ms step depolarization produced DSI in ∼33% of the cells (n = 6) and an associated peak [Ca2+]i increase (ΔF/F0) of 28 ± 5.5%. We took this as the threshold for DSI induction. In the same cells, 10 or 20 ms steps produced essentially no DSI, although a 20 ms step resulted in peak ΔF/F0 of 26 ± 3.2%. For voltage step durations > 100 ms, the Ca2+ response peaked at the same time as, or shortly before, the end of the pulse. For voltage step durations ≤100 ms, the peak of the Ca2+ response occurred slightly after the end of the steps, probably reflecting Ca2+ release from intracellular stores (Nakamura et al., 2002) (M. Isokawa and B. E. Alger, unpublished observations), although imperfect voltage control or slow closure of the Ca2+ channels might also contribute. Mean times-to-peak of the Ca2+ signals were 35.5 ± 6.4 and 60.0 ± 4.0 ms for the 20 and 50 ms voltage steps, respectively. Because peak ΔF/F0 in these cases did not differ significantly (p > 0.05), it appears that detectable eCB production requires > 35 ms, with 60 ms being sufficient in approximately one-third of the cells. We suggest that ∼60 ms is a reasonable estimate of tCa.
Figure 4.

Calcium dependence of DSI. A, A 50 ms voltage step resulted in minimal DSI in a pyramidal cell. In this neuron, the sIPSC frequency before the voltage step was 6.65 Hz (averaged over 32 s before the step) and 5.55 Hz after the step (averaged over 20 s after the step). This represents a 16.6% reduction in sIPSC frequency. The accompanying Ca2+ transient (fluo-3 signal) was measured with a PMT and is shown immediately below and, on a faster time scale, in C. The peak of the transient occurred after the end of the voltage step. B, A 1 s voltage step evoked pronounced DSI. The frequency before the step was 6.43 Hz and was reduced to 1.75 Hz after the step (averaged over 20 s after the step) (i.e., a 72.8% reduction in sIPSC frequency). The Ca2+ signal for the 1 s step was substantially larger than in A and peaked at the end of the voltage step (D). Ca2+ transients in C and D were normalized to facilitate visual comparison. The dotted lines in C mark the terminations of the 50 ms voltage step and peak of the Ca2+ transient (determined from the trace after a smoothing procedure). In D, the dotted line shows that the peak of the Ca2+ transient occurred at the end of the 1 s voltage step.
We also observed that, with 1 s voltage steps, the rise in the PMT signal was linear for the first 171.4 ± 18.3 ms (n = 6) (data not shown) after the onset of a voltage step, in accord with predictions based on the [Ca2+]i measurements made by Brenowitz and Regehr (2003). This permits an independent estimate of tCa to be made (see Discussion).
Measuring tCB1 with “caged” AEA
To assess the contribution of CB1 activation and downstream steps to tDSI, we developed a novel caged form of AEA (Fig. 1) that is biologically inert and highly hydrophilic and thus can be bath-applied without affecting the hippocampal slices, but that releases bona fide AEA in situ when illuminated by a focally delivered flash of UV light. Caged AEA by itself has no effect on cell membrane properties (data not shown) or synaptic responses (see below).
The UV flash alone had no effect on CCh-induced sIPSCs (Fig. 5C). The sIPSC frequencies before and after the flash were 2.29 ± 0.47 and 2.32 ± 0.38 Hz, respectively (n = 8; p > 0.8). Caged AEA by itself did not affect sIPSCs. The sIPSC frequencies before and after wash-in of caged AEA were 2.29 ± 0.47 and 2.21 ± 0.36 Hz, respectively (n = 8; p > 0.6). However, in the presence of caged AEA (200 μm), the UV flash transiently reduced sIPSC frequency (n = 8) (Fig. 5D). We then tested the effect of the CB1 antagonist, AM251 (3 μm) (Fig. 5, compare E, F). Although AM251 slightly reduced the sIPSC frequency (1.27 ± 0.06 Hz; n = 9; vs 1.67 ± 0.03 Hz in control), it fully blocked the ability of photoreleased AEA to suppress sIPSC frequency. These results show that the effect of photoreleased AEA is mediated by CB1 activation.
Figure 5.

Dynamics of sIPSC suppression by photorelease of AEA in cultured hippocampal slices. A, Schematic structure of functionally caged AEA. B, Photorelease of caged AEA, present at 200 μm, by a 100 ms UV laser flash (given at jagged arrow) transiently suppresses sIPSCs (downward deflections) recorded under whole-cell voltage clamp from a pyramidal cell. Ionotropic glutamate receptors were blocked by AP-5 and NBQX. sIPSC binning was done as in Figure 1. C, A UV laser flash alone in the absence of caged AEA did not affect sIPSCs. D, After wash-in of caged AEA (same cells as in C), a laser flash was followed by transient sIPSC suppression. E, F, The CB1 antagonist AM251 (3 μm) prevents photoreleased AEA from affecting sIPSCs. The same cells in E and F are shown. All data in this and subsequent figures were obtained at room temperature (∼22°C).
AEA photorelease did not instantaneously suppress sIPSCs. The combined data from 17 cells were reasonably well fit by an exponential that decayed with a time constant of 551 ms (Fig. 6). By extrapolating the exponential back to 100% (the control frequency), we infer that detectable activation of CB1 responses requires 162 ms.
Figure 6.

Time course of suppression of inhibition induced by AEA photorelease. A, Grouped data from all cells tested with caged AEA (n = 17) showing suppression of sIPSCs. Each time point is the average from 17 cells; error bars were omitted for clarity (the SEs for the individual time points varied from 11.2 to 19.8% of the means). B, Dynamics of AEA-induced suppression of sIPSCs during the initial 3 s after the UV laser flash (data from A, rebinned in 200 ms intervals). No change in sIPSC occurrence is evident at the first time bin after the flash (ending at 200 ms), but thereafter the data are fit by an exponential function that has a time constant of 551 ms. Error bars indicate SE.
The data in Figures 3, 4, 5, 6, in combination with Equation 1 with tDSI taken as 409 ms, allow an estimate of 187 ms for teCB, the time required for eCB synthesis, release, and access to the presynaptic cannabinoid receptors at 22°C. The data in Figure 6 also suggest that the time-to-peak DSI is governed mainly by the dynamics of the G-protein-coupled CB1 receptor, because the time constants of the single exponentials that characterize the increase in DSI are all essentially identical with the time constant of IPSC suppression caused by photolytically released AEA.
Dynamics of mGluR-induced eCB release in cultured hippocampal slices
Activation of mGluRs suppresses IPSCs through a mechanism that resembles DSI (Morishita et al., 1998; Morishita and Alger, 1999). It appears that group I mGluR activation stimulates release of eCBs from hippocampal pyramidal cells (Varma et al., 2001) and cerebellar Purkinje cells (Maejima et al., 2001), because this effect is blocked by CB1 antagonists and is absent in slices from CB1-/- mice (Varma et al., 2001). The mGluR effect differs from DSI in that it cannot be prevented by very high intracellular concentrations of the Ca2+ chelator, BAPTA (Maejima et al., 2001; Kim et al., 2002). These and other differences between DSI and GPCR-induced eCB release (Kim et al., 2002) suggest that the dynamics of GPCR-dependent and Ca2+-dependent eCB release might not be the same. We explored this issue using caged glutamate.
The caged glutamate, Ncm-Glu (0.5 or 1 mm), was bath-applied and allowed to equilibrate in a slice. Photolytic uncaging of Ncm-Glu to release glutamate is intrinsically fast and occurs on the microsecond time scale (t1/2 < 4.5 μs) (S. Muralidharan and J. P. Y. Kao, unpublished observations). In slice preparations, glutamate photorelease occurs more slowly than in a cuvette, because UV flashes of much lower power must be used. In slices, photoreleased glutamate evokes AMPA receptor-mediated responses in pyramidal cells (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) with a time-to-onset of ≤4 ms (n = 13), which encompasses the time required for glutamate photorelease, binding, and activation of AMPA receptors. Therefore, times-to-onset significantly longer than 4 ms must reflect the kinetics of downstream signaling steps. In subsequent experiments, glutamate photorelease was done in the presence of ionotropic glutamate receptor blockers.
In control experiments, we found that the magnitude of DSI of evoked IPSCs and the suppression of IPSCs by photolysis of caged glutamate were not significantly different (Fig. 7A,B). Simultaneous fluorescence measurements show that, whereas DSI is associated with a sharp rise in [Ca2+]i (Fig. 7C), the photorelease of glutamate is not (Fig. 7D). Group data on IPSC suppression and Ca2+ transients are summarized in Figure 7, E and F, respectively (n = 6). The mGluR antagonist, LY341495, which blocks all mGluRs at a concentration of 100 μm (Fitzjohn et al., 1998), prevented the suppression of eIPSCs by photoreleased glutamate without significantly affecting DSI (Fig. 7G,H) or the Ca2+ transient associated with DSI (Fig. 7I,J). Group data on the effect of LY341495 on IPSC suppression and Ca2+ transients are summarized in Figure 7, K and L, respectively (n = 4). These results confirm that the suppression of IPSCs after photolysis of caged glutamate was attributable to the activation of metabotropic glutamate receptors. In CB1-/- mice, DSI is absent, and mGluR agonists do not suppress eIPSCs (Varma et al., 2001). As expected, photolytic activation of mGluRs did not suppress eIPSCs in CB1-/- mice (n = 3 slices from three mice) (data not shown).
Figure 7.

Photoreleased glutamate activates mGluR and induces eCB-dependent responses in cultured hippocampal cells. Each downward deflection represents an evoked GABAA synaptic response. Caged glutamate, Ncm-Glu (500 μm), was in the bath. A, Transient suppression of eIPSCs evoked by 1 s voltage steps to 0 mV on two DSI trials. B, Two trials from the same cell in which 100 ms UV laser flashes were given. C, D, Simultaneous Ca2+ indicator fluorescence measurements for single DSI or glutamate photorelease trials. E, F, Grouped data showing IPSC suppression and Ca2+ transient magnitude from cells treated as in A and B. All data in A-F were obtained from the same six cells. G, Both DSI and glutamate photorelease reduce evoked IPSCs. H, Addition of the mGluR antagonist LY341495 (100 μm) to the bath prevented the photolytic glutamate responses without significantly affecting DSI. I, J, Corresponding Ca2+ signals produced by voltage steps and photoreleased glutamate before (I) and after (J) perfusion of LY341495. K, L, Grouped data (n = 4) for control (open bars) and LY341495 (gray bars) conditions; different cells than A-F. Error bars indicate SE. Jagged arrows represent laser flashes.
Dynamics of mGluR-dependent eCB suppression of sIPSCs in cultured hippocampal slices
We then determined the dynamic components of mGluR-induced eCB response on sIPSC frequency in pyramidal cells (Fig. 8A). The mean onset latency, duration, and magnitude of the IPSC suppression caused by uncaged glutamate (Fig. 8B) were similar to that caused by uncaged AEA (Fig. 6A). The grouped data for suppression of IPSCs by photolytically activated mGluR was fit by an exponential that decayed with a time constant of 486 ms (Fig. 8C). There was no reduction in sIPSCs for 236 ms (determined by extrapolation of the exponential fit to the control sIPSC level). If the time-to-onset of IPSC suppression caused by the mGluR-induced eCB process (tmGluRSI) is described by the following:
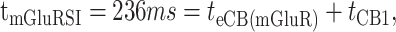 |
(2) |
Figure 8.

Dynamics of mGluR-dependent eCB suppression of sIPSCs in cultured hippocampal slices. A, Recording from a pyramidal cell illustrates the reduction of sIPSC frequency after flash photorelease of caged glutamate. B, Time course of sIPSC suppression by photoreleased glutamate; sIPSC frequency binned and analyzed as in Figures 3 and 6 (n = 12). Each time point is the average from 12 cells; error bars were omitted for clarity (the SEs for the individual time points varied from 7.9 to 15.8% of the means). C, sIPSCs rebinned during the initial 3 s after the UV laser flash in the same set of cells as in B. No change in sIPSC occurrence is evident at the first time bin after the flash (ending at 200 ms), but thereafter the data are fit by an exponential function that has a time constant of 486 ms. Error bars indicate SE. Jagged arrows represent laser flashes.
where teCB(mGluR) is the time for activation of the mGluR-dependent eCB synthesis and release, and tCB1 is 162 ms (Fig. 6), then teCB(mGluR) would be 74 ms.
Discussion
This is the first detailed attempt to determine the minimal time required for activation of an intercellular neuronal lipid messenger system. We conclude that eCBs, and by extension similar lipid messengers, can be mobilized and evoke responses as quickly as conventional metabotropic, GPCR-coupled neurotransmitters can. Hence, lipids are not relegated to homeostatic processes, or slowly activating forms of regulation, but rather can affect neuronal excitability in moment-to-moment information processing. The data are also important for a complete understanding of the biochemical mechanisms of lipid signal mobilization, in particular endocannabinoid synthesis and action, because of the temporal constraints that they place on these mechanisms.
A great deal of evidence shows that an eCB, probably 2-AG or AEA, is released as a retrograde signal and mediates the phenomenon called DSI (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001) and other effects (for review, see Alger, 2002). DSI represents a powerful bioassay for the rapid action of eCBs. Previous studies of DSI (Pitler and Alger, 1994; Wilson et al., 2001; Hampson et al., 2003; Fortin et al., 2004) suggested that the time-to-onset of eCB responses ranged between 1 and 6 s. Our experiments suggest that the delay (tDSI)is shorter, ∼400 ms, and presumably experimental factors account for the differences. [Ca2+]i measurements and photorelease of caged AEA permitted us to distinguish the components of the total signal delay that are attributable to eCB synthesis and release from those that involve cannabinoid receptor activation and downstream steps.
Our major parameter was the time from the start of the stimulus used to initiate eCB signaling (either the voltage step used to open voltage-gated calcium channels or the UV flash) to the onset of the eCB-dependent sIPSC suppression. Synthesis and release of eCB must occur at least as quickly as this, and may be faster if additional intermediate steps are significantly time-consuming. We reasoned that one such step might be the rise in [Ca2+]i required to initiate eCB synthesis and release. The suggestion that a minimal duration of stimulation is required for DSI induction has previous empirical support. For example, a 50-ms-long voltage step from -70-0 mV produces just-detectable DSI in cells in acute slices recorded with whole-cell pipettes containing 0.1 mm BAPTA (Lenz and Alger, 1999). We found that to induce just-detectable DSI, Ca2+ signals must be elevated for 35-60 ms, which we designated tCa. We infer that tCa represents the time necessary to initiate the eCB synthesis and release process. Detection of cellular [Ca2+]i changes with a PMT necessarily involves “volume averaging,” and it is possible that the magnitude or kinetics of local [Ca2+]i changes near the membrane where the eCBs are presumably produced, will differ from this global value.
Brenowitz and Regehr (2003) found that [Ca2+]i increases linearly with voltage steps up to 1.5 s in duration, reaching a maximal concentration of 60 μm [Ca2+]i at that point. Our PMT signals were linear for at least 100 ms after the start of the voltage step (data not shown). A linear increase in [Ca2+]i for 100 ms would result in [Ca2+]i of ∼4 μm (60 μm/1.5 s = 4 μm/0.1 s). Wang and Zucker (2001) measured DSI as a function of [Ca2+]i and found that DSI would be > 50% maximal when [Ca2+]i is 4 μm, and that an increase in [Ca2+]i of 2 μm would produce detectable DSI. This concentration should be achieved within ∼50 ms after the beginning of our voltage steps, consistent with our present measurements. The concordance among these various results suggests that the time required for Ca2+-dependent steps in the postsynaptic cell contributes significantly (∼60 ms) to the latency-to-onset of eCB effects.
Activation of CB1s and their downstream effectors (tCB1) accounted for much of the eCB response onset latency. Rapid photorelease of caged compounds was critical to refining the estimates for eCB synthesis and release. For example, Mackie and Hille (1992), using a rapid perfusion system that delivered WIN55212-2 to cultured NG108-15 cells within 1 s, observed that the N-type Ca2+ current was suppressed with a time constant of ∼8 s. Photolytic release of AEA in cultured slices suppressed sIPSCs with a delay of 162 ms at 22°C. The high affinity of AEA for CB1 (52 nm) (Devane et al., 1992) suggests that the time for AEA binding to CB1 is likely to be a negligible fraction of tCB1. Equilibration of caged AEA within the slices before photolysis means that AEA photorelease will occur in situ in the immediate vicinity of CB1, so the time for diffusion of AEA to CB1 should be negligible. Supporting this conclusion, we found that uncaged glutamate evoked AMPA responses that began ≤4 ms after the UV flash. Hence the bulk of the time from the photolytic flash to sIPSC suppression by AEA reflects steps downstream from CB1 activation. CB1 is a heptahelical GPCR, and our measured response latency is compatible with that of other GPCR-mediated responses. For example, Mott et al. (1999) measured the kinetics of GABAB-mediated currents in cells in brain slices and found a mean onset latency of 30 ms, but their measurements were made at 32-34°C. Canepari et al. (2001) show that photolytically releasing glutamate onto Purkinje cells (at 32°C) produces a transient mGluR1-mediated inward current with an onset latency of ∼90 ms. The difference between these values and ours is no doubt temperature dependent. Delmas et al. (2004) report that GPCR-mediated suppression of M-current in sympathetic ganglion cells begins after 250 ms at 20°C.
Combining the values of tCa and tCB1 allows us to estimate that the actual time required for eCB synthesis and release at 22°C is in the 75-190 ms range, depending on whether the initiating signal is a GPCR or Ca2+. Note that our bioassay was sensitive to sIPSC suppression of ≥15% (i.e., eCB concentrations too low to produce such a decrease would have been undetectable). If eCBs are released before our detection threshold is reached, then the upper and lower limits of our estimated time range for eCB synthesis and release would decrease. However, even if 190 ms are required for eCB mobilization, this would be much less than previously assumed for a lipid signal process. Finally, eCB effects are very temperature sensitive (Kreitzer and Regehr, 2001b), and hence at physiological temperatures, eCB synthesis and release will take place faster than our estimates at 22°C. For example, if eCB mobilization should have a Q10 of 2.4, as does the growth of DSI, then, at 37°C, eCB synthesis and release would occur in the 20-50 ms range.
For simplicity, we assumed that eCBs are produced in the postsynaptic pyramidal cell and travel to the presynaptic, CB1-bearing interneurons. This model, although widely accepted, is still only indirectly supported; conceivably release of an unknown messenger from the pyramidal cell triggers the production of eCBs in the interneuron where they access CB1 directly. This possibility does not diminish the significance of our experiments, which focused on the dynamics of the eCB system. Indeed, if eCBs were generated in the interneurons, then, because of the time required for transmission of the unknown signal, eCB mobilization and action would be even more rapid than our data imply.
For technical reasons, AEA is more amenable to chemical caging than is 2-AG, and so we began with AEA. It is still not certain which eCB is actually the messenger for the eCB effects in hippocampus. Increasing evidence suggests that the GPCR actions are probably mediated by 2-AG (Stella et al., 1997; Kim and Alger, 2004; Hashimotodani et al., 2005). Nevertheless, conclusions regarding functional eCB kinetics may not depend significantly on the difference. AEA has 2-25 times higher binding affinity (Ki) for CB1 (Mechoulam et al., 1995; Sugiura et al., 1995) depending on assay conditions but is only a partial agonist at CB1 (for review, see Hillard, 2000). However, 2-AG is present in tissue at concentrations from 170-1000 times higher than AEA (Stella et al., 1997; Sugiura and Waku, 2000) and is a full agonist at CB1. If 2-AG were to bind and activate CB1 more slowly than AEA, then correcting for this difference in tCB1 would actually leave less time for 2-AG synthesis and release (see Eq. 1). Given the present uncertainties, it is unlikely that our conclusions would have been markedly altered had caged 2-AG been available.
Although the eCBs can facilitate (Carlson et al., 2002) or produce long-term changes in synaptic strength (Gerdeman et al., 2002; Robbe et al., 2002; Chevaleyre and Castillo, 2003; Ronesi et al., 2004), they also act on shorter time scales in producing DSI and depolarization-induced suppression of excitation (Alger, 2002). Indeed, eCB release, detected as DSI, is produced by short trains of action potentials (Fig. 3) (Pitler and Alger, 1992; Fortin et al., 2004) or synaptic responses (Brown et al., 2003), and this has suggested that rapid neuromodulation may be a major function of these lipids.
The present study demonstrates that the time courses of mGluR-dependent eCB responses and DSI are both fast. Evidently, despite the different biochemical cascades involved (Hashimotodani et al., 2005), both pathways can rapidly generate eCBs. Probably the molecular machineries for both processes reside near the plasma membrane where they can be readily activated.
Footnotes
This work was supported by Public Health Service Grants RO1 NS30219 and RO1 DA14625 (B.E.A.) and GM56481 (J.P.Y.K.) and a University of Maryland School of Medicine Bressler Fund Award (T.H.). C.G.R. was supported by National Institutes of Health Postdoctoral Training Grant T32 NS07375. We thank Drs. Scott Thompson and Masako Isokawa for allowing us to use their hippocampal slice cultures. We thank Catherine Ledent for giving us the CB1 knock-out mice. D.H.B., T.H., and C.G.R. conducted the physiological studies in B.E.A.'s laboratory. J.Z. and S.M. synthesized and tested the caged compounds in J.P.Y.K.'s laboratory.
Correspondence should be addressed to Dr. Bradley E. Alger, Department Physiology, Program in Neuroscience, University of Maryland School of Medicine, 655 West Baltimore Street, Baltimore, MD 21201. E-mail: balger@umaryland.edu.
T. Heinbockel's present address: Department of Anatomy, Howard University College of Medicine, Washington, DC 20059.
D. H. Brager's present address: Center for Learning and Memory, University of Texas at Austin, Austin, TX 78712-0132.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259449-11$15.00/0
T.H. and D.H.B. contributed equally to this work.
References
- Alger BE (2002) Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol 68: 247-286. [DOI] [PubMed] [Google Scholar]
- Bazan NG (2003) Synaptic lipid signaling: significance of polyunsaturated fatty acids and platelet-activating factor. J Lipid Res 44: 2221-2233. [DOI] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG (2003) Calcium dependence of retrograde inhibition by eCBs at synapses onto Purkinje cells. J Neurosci 23: 6373-6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SP, Brenowitz SD, Regehr WG (2003) Brief presynaptic bursts evoke synapse-specific retrograde inhibition mediated by endogenous cannabinoids. Nat Neurosci 6: 1048-1057. [DOI] [PubMed] [Google Scholar]
- Cai X, Liang CW, Muralidharan S, Kao JP, Tang CM, Thompson SM (2004) Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron 44: 351-364. [DOI] [PubMed] [Google Scholar]
- Canepari M, Papageorgiou G, Corrie JE, Watkins C, Ogden D (2001) The conductance underlying the parallel fibre slow EPSP in rat cerebellar Purkinje neurones studied with photolytic release of l-glutamate. J Physiol (Lond) 533: 765-772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson G, Wang Y, Alger BE (2002) Endocannabinoids facilitate that induction of LTP in the hippocampus. Nat Neurosci 5: 723-724. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo P (2003) Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of eCBs in regulating excitability. Neuron 38: 461-472. [DOI] [PubMed] [Google Scholar]
- Delmas P, Crest M, Brown DA (2003) Functional organization of PLC signaling microdomains in neurons. Trends Neurosci 27: 41-47. [DOI] [PubMed] [Google Scholar]
- De Mayo P, Shizuka H (1976) Measurements of reaction quantum yields. In: Creation and detection of excited states, Vol 4 (Ware WR, ed), pp 139-215. New York: Marcel Dekker. [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258: 1946-1949. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Bortolotto ZA, Palmer MJ, Doherty AJ, Ornstein PL, Schoepp DD, Kingston AE, Lodge D, Collingridge GL (1998) The potent mGlu receptor antagonist LY341495 identifies roles for both cloned and novel mGlu receptors in hippocampal synaptic plasticity. Neuropharmacol 37: 1445-1458. [DOI] [PubMed] [Google Scholar]
- Fortin DA, Trettel J, Levine ES (2004) Brief trains of action potentials enhance pyramidal neuron excitability via eCB-mediated suppression of inhibition. J Neurophysiol 92: 2105-2112. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D (2003) Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83: 1017-1066. [DOI] [PubMed] [Google Scholar]
- Gähwiler BH, Thompson SM, McKinney RA, Debanne D, Robertson RT (1998) Organotypic slice cultures of neural tissue. In: Culturing nerve cells, Ed 2 (Banker G, Goslin K, eds), pp 461-498. Cambridge, MA: MIT.
- Gerdeman GL, Ronesi J, Lovinger DM (2002) Postsynaptic eCB release is critical to long-term depression in the striatum. Nat Neurosci 5: 446-451. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D (1999) Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci 2: 358-363. [DOI] [PubMed] [Google Scholar]
- Hampson RE, Zhuang S, Weiner JL, Deadwyler SA (2003) Functional significance of cannabinoid-mediated, depolarization-induced suppression of inhibition (DSI) in the hippocampus. J Neurophysiol 90: 55-64. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin HS, Kano M (2005) Phospholipase Cβ serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron 45: 257-268. [DOI] [PubMed] [Google Scholar]
- Hatchard CG, Parker CA (1956) A new sensitive chemical actinometer. II. Potassium ferrioxalate as a standard chemical actinometer. Proc R Soc Lond A 235: 518-536. [Google Scholar]
- Hillard CJ (2000) Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat 61: 3-18. [DOI] [PubMed] [Google Scholar]
- Iversen L (2003) Cannabis and the brain. Brain 126: 1252-1270. [DOI] [PubMed] [Google Scholar]
- Kim J, Alger BE (2004) Inhibition of cyclooxygense-2 potentiates retrograde eCB signaling effects in hippocampus. Nat Neurosci 7: 697-698. [DOI] [PubMed] [Google Scholar]
- Kim J, Isokawa M, Ledent C, Alger BE (2002) Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci 22: 10182-10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001a) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29: 717-727. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001b) Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J Neurosci 21: RC174(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Carter AG, Regehr WG (2002) Inhibition of interneuron firing extends the spread of eCB signaling in the cerebellum. Neuron 34: 787-796. [DOI] [PubMed] [Google Scholar]
- Lang W, Qin C, Lin S, Khanolkar AD, Goutopoulos A, Fan P, Abouzid K, Meng Z, Biegel D, Makriyannis A (1999) Substrate specificity and stereoselectivity of rat brain microsomal anandamide amidohydrolase. J Med Chem 42: 896-902. [DOI] [PubMed] [Google Scholar]
- Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, Bohme GA, Imperato A, Pedrazzini T, Roques BP, Vassart G, Fratta W, Parmentier M (1999) Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science 283: 401-404. [DOI] [PubMed] [Google Scholar]
- Lenz RA, Alger BE (1999) Calcium dependence of depolarization-induced suppression of inhibition in rat hippocampal CA1 pyramidal neurons. J Physiol (Lond) 521: 147-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K, Hille B (1992) Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci USA 89: 3825-3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M (2001) Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31: 463-475. [DOI] [PubMed] [Google Scholar]
- Martin LA, Alger BE (1999) Muscarinic facilitation of the occurrence of depolarization-induced suppression of inhibition in rat hippocampus. Neuroscience 92: 61-71. [DOI] [PubMed] [Google Scholar]
- McCray JA, Trentham DR (1989) Properties and uses of photoreactive caged compounds. Annu Rev Biophys Biophys Chem 18: 239-270. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin F, Bayewitch M, Barg J, Vogel Z (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 1: 83-90. [DOI] [PubMed] [Google Scholar]
- Morishita W, Alger BE (1999) Evidence for endogenous excitatory amino acids as mediators in DSI of GABAAergic transmission in hippocampal CA1. J Neurophysiol 82: 2556-2564. [DOI] [PubMed] [Google Scholar]
- Morishita W, Kirov SA, Alger BE (1998) Evidence for metabotropic glutamate receptor activation in the induction of depolarization-induced suppression of inhibition in hippocampal CA1. J Neurosci 18: 4870-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott DD, Li Q, Okazaki MM, Turner DA, Lewis DV (1999) GABAB-receptor-mediated currents in interneurons of the dentate-hilus border. J Neurophysiol 82: 1438-1450. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Lasser-Ross N, Nakamura K, Ross WN (2002) Spatial segregation and interaction of calcium signaling mechanisms in rat hippocampal CA1 pyramidal neurons. J Physiol (Lond) 543: 465-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Alger BE (1981) A simple chamber for recording from submerged brain slices. J Neurosci Methods 4: 153-156. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M (2001) Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29: 729-738. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Matsui M, Fukudome Y, Shosaku J, Tsubokawa H, Taketo MM, Manabe T, Kano M (2003) Postsynaptic M1 and M3 receptors are responsible for the muscarinic enhancement of retrograde eCB signalling in the hippocampus. Eur J Neurosci 18: 109-116. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE (1992) Cholinergic excitation of GABAergic interneurons in the rat hippocampal slice. J Physiol (Lond) 450: 127-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitler TA, Alger BE (1994) Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G protein involvement in a presynaptic mechanism. Neuron 13: 1447-1455. [DOI] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ (2002) Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA 99: 8384-8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi J, Gerdeman GL, Lovinger DM (2004) Disruption of eCB release and striatal long-term depression by postsynaptic blockade of eCB membrane transport. J Neurosci 24: 1673-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schupp H, Wong WK, Schnabel W (1987) Mechanistic studies of the photorearrangement of o-nitrobenzyl esters. J Photochem 36: 85-97. [Google Scholar]
- Sheskin T, Hanus L, Slager J, Vogel Z, Mechoulam R (1997) Structural requirements for binding of anandamide-type compounds to the brain cannabinoid receptor. J Med Chem 40: 659-667. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D (1997) A second endogenous cannabinoid that modulates long-term potentiation. Nature 388: 773-778. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37: 173-182. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Waku K (2000) 2-Arachidonylglycerol and the cannabinoid receptors. Chem Phys Lipids 108: 89-106. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K (1995) 2-Arachidonylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun 215: 89-97. [DOI] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE (2001) Metabotropic glutamate receptors drive the eCB system in hippocampus. J Neurosci 21: RC188(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JW, Reid GP, McCray JA, Trentham DR (1988) Photolabile 1-(2-nitrophenyl)ethyl phosphate esters of adenine nucleotide analogs. Synthesis and mechanism of photolysis. J Am Chem Soc 110: 7170-7177. [Google Scholar]
- Wang J, Zucker RS (2001) Photolysis-induced suppression of inhibition in rat hippocampal CA1 pyramidal neurons. J Physiol (Lond) 533: 757-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei DS, Mei YA, Bagal A, Kao JP, Thompson SM, Tang C-M (2001) Compartmentalized and binary behavior of terminal dendrites in hippocampal pyramidal neurons. Science 293: 2272-2275. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signaling at hippocampal synapses. Nature 410: 588-592. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA (2001) Presynaptic specificity of eCB signaling in the hippocampus. Neuron 31: 453-462. [DOI] [PubMed] [Google Scholar]
- Yip RW, Sharma DK, Giasson R, Gravel D (1985) Photochemistry of the o-nitrobenzyl system in solution: evidence for singlet-state intramolecular hydrogen abstraction. J Phys Chem 89: 5328-5330. [Google Scholar]
- Yip RW, Wen YX, Gravel D, Giasson R, Sharma DK (1991) Photochemistry of the o-nitrobenzyl system in solution: identification of the biradical intermediate in the intramolecular rearrangement. J Phys Chem 95: 6078-6081. [Google Scholar]
- Zhu QQ, Schnabel W, Schupp H (1987) Formation and decay of nitronic acid in the photorearrangement of o-nitrobenzyl esters. J Photochem 39: 317-332. [Google Scholar]