

Abstract
Based on the Torpedo acetylcholine receptor structure, Unwin and colleagues (Miyazawa et al., 2003; Unwin, 2005) hypothesized that the transduction of agonist binding to channel gate opening involves a “pin-into-socket” interaction between αV46 at the tip of the extracellular β1-β2 loop and the transmembrane M2 segment and M2-M3 loop. We mutated to cysteine the aligned positions in the 5-HT3A and 5-HT3B subunit β1-β2 loops K81 and Q70, respectively. The maximal 5-HT-activated currents in receptors containing 5-HT3A/K81C or 5-HT3B/Q70C were markedly reduced compared with wild type. Desensitization of wild-type currents involved fast and slow components. Mutant currents desensitized with only the fast time constant. Reaction with several methanethiosulfonate reagents potentiated currents to wild-type levels, but reaction with other more rigid thiol-reactive reagents caused inhibition. Single-channel conductances of wild type, K81C, and K81C after modification were similar. We tested the proximity of K81C to the M2-M3 loop by mutating M2-M3 loop residues to cysteine in the K81C background. Disulfide bonds formed in 5-HT3A/K81C/A304C and 5-HT3A/K81C/I305C when coexpressed with 5-HT3B. We conclude that in the resting state, K81 is not in a hydrophobic pocket as suggested by the pin-into-socket hypothesis. K81 interacts with the extracellular end of M2 and plays a critical role in channel opening and in the return from fast desensitization. We suggest that during channel activation, β1-β2 loop movement moves M2 and the M2-M3 loop so that the M2 segments rotate/translate away from the channel axis, thereby opening the lumen. Recovery from fast desensitization requires the interaction between K81 and the extracellular end of M2.
Keywords: ion channel, SCAM, acetylcholine, GABA, serotonin, cross-linking
Introduction
Serotonin 5-HT3 receptors (5-HT3R) are members of the Cys loop receptor superfamily of ligand-gated ion channels (Reeves and Lummis, 2002) that includes the GABAA, glycine, and nicotinic acetylcholine receptors (AChR). 5-HT3R antagonists are used clinically for the prevention and amelioration of nausea and emesis (Costall and Naylor, 2004). The receptors are allosteric, pentameric proteins that couple agonist binding to transmembrane channel gating. The ability to create functional chimeras between AChR and 5-HT3R implies that ligand binding induces similar conformational changes in all superfamily members (Eiselé et al., 1993). Agonists such as serotonin bind in the extracellular domain at the interface between two subunits, causing the binding site to contract (Karlin, 2002). The extracellular domain structure can be inferred from the atomic resolution structures of the homologous acetylcholine binding protein (AChBP) (Brejc et al., 2001; Celie et al., 2004). The ion channel is lined by the M2 transmembrane segments (Reeves et al., 2001; Panicker et al., 2002) that in the 4 Å resolution AChR structure form an inner ring of helices separated from the lipid bilayer by an outer ring of helices formed by the M1, M3, and M4 segments (Miyazawa et al., 2003; Unwin, 2005). The interface between the extracellular and transmembrane domains (TMDs) involves two major interactions: (1) two extracellular domain loops: the β1-β2 loop (loop 2) and the Cys loop (loop 7) interact with the extracellular end of M2 and the M2-M3 loop; and (2) the pre-M1 segment that extends into the M1 transmembrane segment (see Fig. 1a) (Brejc et al., 2001; Miyazawa et al., 2003). Although interactions between these loops have been linked to channel gating (Kash et al., 2003, 2004a; Bouzat et al., 2004; Schofield et al., 2004), it is not clear precisely how these interactions influence gating.
Figure 1.

a, Homology model of a 5-HT3A homopentameric receptor, viewed from the plane of the membrane. One subunit is highlighted in red for clarity. Green and blue regions show the β1-β2loop and M2-M3 loop, respectively. The dashed lines indicate the approximate limits of the lipid bilayer. b, Portion of a ClustalW multiple sequence alignment of the putative β1-β2 loop in several neurotransmitter-gated ion channels. The vertical arrow marks residues that align with AChR αV46, the residue proposed to be critical for the pin-into-socket mechanism for gating. The asterisks indicate absolutely conserved residues, and the dots indicate partially conserved positions. nACh, Nicotinic ACh.
Based on these structures, hypotheses for the molecular mechanisms that couple ligand binding to channel opening in all members of the superfamily have been generated (Absalom et al., 2004; Lester et al., 2004; Unwin, 2005). Unwin and colleagues (Miyazawa et al., 2003) proposed the “pin-into-socket” hypothesis that states that in the resting state, AChR αV46, at the tip of the extracellular domain β1-β2 loop, is buried in a hydrophobic pocket formed at the top of M2. They proposed that agonist binding-induced displacement of the binding site loops B and C is transmitted via the inner β-sheet to pull the β1-β2 loop away from the membrane. This may either release the M2 segments allowing channel opening or, alternatively, form a pivot by which M2 is moved via force applied to it by the Cys loop. This hypothesis is consistent with the “conformational wave” observed on ligand binding (Grosman et al., 2000; Chakrapani et al., 2004).
In this study, we tested the generality of the pin-into-socket mechanism by mutating the 5-HT3R β1-β2 loop residue K81, aligned with AChR αV46, to cysteine. We monitored the functional effect of cysteine substitution on channel gating and conductance, the effects of covalent modification with thiol-reactive reagents, and disulfide cross-linking to engineered cysteines in the M2-M3 loop.
Materials and Methods
Reagents. (2-(Trimethylammonium)ethyl) methanethiosulfonate bromide (MTSET+), (2-aminoethyl) methanethiosulfonate hydrobromide (MTSEA+), sodium (2-sulfonatoethyl) methanethiosulfonate (MTSES-), and MTS 5(6)-carboxytetramethylrhodamine (MTS-TAMRA) were purchased from Biotium (Hayward, CA). Benzophenone-4-carboxamidocysteine methanethiosulfonate (BPMTS) and p-chloromercuribenzene sulfonate (pCMBS-) were purchased from Toronto Research Chemicals (North York, Ontario, Canada). Methyl methanethiosulfonate (MMTS) was purchased from Sigma (St. Louis, MO). All other reagents, unless specified otherwise, were purchased from Sigma or Fisher Scientific (Hampton, NH).
Site-directed mutagenesis of ligand-gated ion channel subunits. Mutagenesis of full-length cDNAs encoding the mouse 5-HT3A(s) and 5-HT3B subunits, the rat GABAA α1 and β1 subunits in the pGEMHE vector, and the mouse AChR α1 subunit in the pSP64T vector was performed using the Quikchange mutagenesis kit (Stratagene, La Jolla, CA) following the manufacturer's instructions. Mutagenic primers were obtained from Sigma, and sequences are available on request. All plasmid constructs were sequenced to confirm the presence of the desired mutations and to rule out a secondary mutation. Wild-type (WT) and mutant 5-HT3R subunit cDNAs were subcloned into pcDNA3.1 (Invitrogen, Carlsbad, CA) for expression in mammalian cells. An additional round of mutagenesis was performed on the 5-HT3A construct to introduce the set of mutations R437Q/R441D/R445A (Kelley et al., 2003), and the constructs were resequenced as above.
Expression and electrophysiological recording in Xenopus oocytes. In vitro RNA transcription, preparation, and injection of Xenopus oocytes were performed as described previously (Horenstein and Akabas, 1998). Currents were recorded under two-electrode voltage clamp from oocytes perfused continuously at 5 ml min-1 with Ca2+-free Frog Ringer buffer (in mm: 115 NaCl, 2.5 KCl, 1.8 MgCl2, and 10 HEPES, pH 7.5 with NaOH) using equipment and procedures described previously (Horenstein and Akabas, 1998). The perfusion chamber volume was 200 μl. Agarose cushion electrodes were filled with 3 m KCl and had a resistance of <2 MΩ. The ground electrode was connected to the bath by a 3 m KCl/agar bridge. The holding potential was maintained at -80 mV, unless specified otherwise. EC50 values were determined by nonlinear least-squares fits to the Hill equation as described previously (Reeves et al., 2001). The irreversible effects of reaction with thiol-reactive reagents were measured as described previously (Reeves et al., 2001). Briefly, pairs of test pulses at a saturating 5-HT concentration (50 μm) were applied, followed by washout for 3 min and reaction for 1 min with a saturating concentration of thiol-reactive reagent, as indicated. After a 3 min washout, an additional pair of test pulses of 50 μm 5-HT were applied, and the effect of reaction was quantitated as follows:
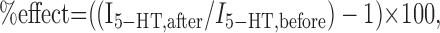 |
where I5-HT, after is the mean peak current of the 5-HT test pulses after thiol-reactive reagent application and I5-HT, before is the mean peak current of the initial 5-HT applications. The data for the effect of each reagent on mutant receptors were compared with that obtained with WT receptors using one-way ANOVA (Prism 3.0; GraphPad Software, San Diego, CA). The effects of reaction of the thiol-reactive reagents with GABAA and AChR were determined using a similar protocol. We also used this procedure to characterize the effect of oxidation by copper phenanthroline (Cu:phen; 100:200 μm) and reduction with dithiothreitol (DTT; 10 mm) on the receptors.
The rates of reaction of thiol-reactive reagents with substituted-cysteine mutant receptors were determined as described previously (Reeves et al., 2001). Briefly, a test pulse of 5-HT (50 μm) was applied, followed by a 3 min washout period and five to eight short (10-20 s) pulses of methanethiosulfonate (MTS) reagent (0.1-20 μm) in the absence or presence of 5-HT, alternating with test pulses of 5-HT. The peak currents induced by the 5-HT test pulses were normalized to the first test pulse, plotted as a function of the cumulative duration of MTS reagent application, and fitted with a single exponential function. The second-order rate constant was calculated by dividing the pseudo-first-order rate constant obtained from the exponential fit by the MTS reagent concentration. For a given mutant, second-order rate constants were independent of the MTS reagent concentration used.
Expression in human embryonic kidney 293 cells. Human embryonic kidney cells (HEK293T; American Tissue Culture Collection, Manassas, VA) were grown at 37°C in DMEM supplemented with 10% fetal calf serum, 2.5 mm l-glutamine, 100 IU of penicillin, and 170 μm streptomycin in an atmosphere of 5% CO2/95% air. Cells were seeded in 100 mm plates at a density of 1.2-1.5 × 106 cells and transfected 24 h later for 12 h using the calcium phosphate precipitation technique (Chen and Okayama, 1988) with 5-7 μg of plasmid DNA. Constructs containing the mutant receptor subunits were cotransfected with empty pXOON plasmid (Jespersen et al., 2002), which encodes a neomycin-enhanced green fluorescent protein (GFP) fusion protein for the visual identification of expression in transfected cells. Cells were washed with PBS and detached with trypsin before reseeding at low density in 35 mm polylysine-treated dishes that were mounted directly on the stage of an inverted microscope (Zeiss IM; Zeiss, Thornwood, NY) for patch-clamp experiments 24-48 h later.
Single-channel and whole-cell electrophysiology. Pipettes were pulled from thick-walled borosilicate glass, coated with Sylgard, and fire-polished to a resistance of 10-14 MΩ (single-channel recording) or 2-4 MΩ (whole-cell recording) when filled with the internal solution. For outside-out configuration and whole-cell recording, the pipette contained the following (in mm): 130 CsCl, 10 EGTA, 10 glucose, 2 MgCl2, 2 CaCl2, 2 K-ATP, 0.2 Tris-GTP, and 10 HEPES, pH 7.3 adjusted with CsOH. The bath solution for outside-out configuration and whole-cell recording was as follows (in mm): 140 NaCl, 2.8 KCl, and 10 HEPES, pH 7.3 adjusted with NaOH. The pipette solution for inside-out configuration contained the following (in mm): 140 NaCl, 2.8 KCl, 2 MgCl2, 0.1 CaCl2, 2 EGTA, 10 glucose, and 10 HEPES, pH 7.3 adjusted with NaOH. The bath solution for inside-out configuration was as follows (in mm): 140 CsCl, 2 MgCl2, 0.1 CaCl2, 1.1 EGTA, and 10 HEPES, pH 7.3 adjusted with CsOH. The transfected cells chosen for the experiments had similar GFP fluorescence intensity. The 5-HT currents were recorded using the outside-out and the inside-out configuration of the patch-clamp technique. In all configurations, a holding potential of -60 mV was used. Cells were perfused continuously with external solution at a rate of 2 ml min-1 via a gravity-driven, dual-barrel perfusion system. For the outside-out and whole-cell configurations, 50 μm 5-HT dissolved in the bath solution was applied to the patch using the second barrel. In the inside-out configuration, the same concentration of 5-HT was diluted in the pipette solution. Currents were low-pass filtered at 8 kHz (eight-pole Bessel filter) and acquired at 20 kHz using Pulse software interfaced with an EPC-9 amplifier (HEKA, Darmstadt, Germany). When applicable, cells were treated before patching with 20 μm MTSEA+ or MTSET+ dissolved in the bath solution (of the outside-out configuration) for 3 min. Plates were then washed thoroughly with the bath solution and used within 30 min of the treatment.
Homology modeling of the 5-HT3R subunit structure. We generated a homology model of the 5-HT3A subunit transmembrane segments based on the cryo-electron microscopic structure of the Torpedo AChR TMD [Protein Data Bank (PDB) code 1OED] (Miyazawa et al., 2003) using a web-based structure prediction service (SWISS-MODEL) (Schwede et al., 2003) (see Fig. 1). The resulting model was combined with a 5-HT3A subunit extracellular domain (ECD) model (Reeves et al., 2003) in Deep-view 3.7b (Swiss Institute of Bioinformatics). The domains were aligned electronically by minimizing the distance between α-carbons for the single residue common to both domains (P211 from the ECD structure). The relative orientation of the five subunits in each domain was preserved. The figures were rendered in POV-Ray version 3.6 (Persistence of Vision Pty. Ltd., Williamtown, Victoria, Australia).
Computer modeling of MTS-modified cysteine flexibility. We generated a potential energy function for rotation around the S-S bond created by the covalent reaction of each MTS reagent with cysteine using quantum chemical calculations (Dewar et al., 1985; Stewart, 1990). MTS-modified cysteines were energy minimized using semi-empirical PM3-Hamiltonian calculations (Spartan '04; Wavefunction, Irvine, CA). After rotation of a starting conformation through 180° in 10 steps, the free energy difference between the calculated conformational isomer with the lowest and highest energy was as follows: MTSES-, 25 kJ mol-1; MTSET+, 36 kJ mol-1; MTSEA+, 24 kJ mol-1; MMTS, 25 kJ mol-1; MTS-TAMRA, 39 kJ mol-1; BPMTS, 26 kJ mol-1. Without steric hindrance from neighboring parts of the receptor protein, all rotations should be possible at room temperature (<84 kJ mol-1).
Results
Mutation of the 5-HT3R β1-β2 loop
Alignment of the 5-HT3R sequences with those of AChBP, AChR, and other members of the superfamily identified K81 (5-HT3A subunit) and Q70 (5-HT3B subunit) as the residues aligned with the β1-β2 loop residues V44 (AChBP) and αV46 (AChR), both identified previously as the residue critical for the pin-into-socket mechanism (Fig. 1b) (Miyazawa et al., 2003). On expression of homopentameric 5-HT3A/K81C receptors in Xenopus oocytes, we observed a 25-fold decrease in the peak current response to a saturating concentration of 5-HT compared with WT homopentamers (Fig. 2a). A similar decrease was noted for heteropentameric receptors containing the 5-HT3B/Q70C mutant and WT 5-HT3A. However, heteropentameric receptors containing the 5-HT3A/K81C subunit with WT 5-HT3B subunits only displayed a 4.7-fold decrease compared with WT heteropentamers. The basis for the difference in the effect of the mutations on the heteropentameric receptors is uncertain, especially because the subunit stoichiometry is unknown. Coexpression of the 5-HT3A/K81C and 5-HT3B/Q70C subunits produced a 63-fold decrease in peak current compared with WT heteropentamers, suggesting a synergistic effect of the mutations when coexpressed, compared with the homopentameric mutant, which also contains five cysteines at this position. Small (threefold to fivefold) rightward shifts in the dose-response relationship were also observed in receptors containing either or both of the mutant subunits (Fig. 2b).
Figure 2.

a, Mean peak currents recorded at a holding potential of -80 mV from 5-HT3R containing the K81C and/or Q70C mutations, expressed in Xenopus oocytes. b, EC50 values for 5-HT for WT and mutant 5-HT3R, as in a. c, Time constants of desensitization (τ) in the presence of 5-HT (50 μm) for WT and mutant 5-HT3R, as in a. Error bars indicate SEM; n = 6. *p < 0.05, significantly different from WT (ANOVA). Note that the y-axis has a log10 scale in a and c.
We also noted that the apparent rate of desensitization of the mutant receptors was increased (Figs. 2c, 3a). This increase was ∼19-fold for mutant 5-HT3R, whether homopentameric or heteropentameric.
Figure 3.

a, Representative traces showing the effect of reaction with MTSEA+ on currents recorded at a holding potential of -80 mV from 5-HT3A/K81C homopentamers expressed in Xenopus oocytes. b, Sample traces showing an experiment to determine the rate of reaction of MTSEA+ with 5-HT3A/K81C homopentamers expressed in Xenopus oocytes and a plot of the peak currents from this experiment. The second-order rate of reaction is calculated from the nonlinear least squares fit shown on the graph. c, Representative traces showing currents obtained on application of 5-HT to HEK293T cells expressing either WT 5-HT3A or 5-HT3A/K81C subunits, each along with WT 5-HT3B subunits, and the effect of reaction with MTSET+. Note that in contrast to data obtained in Xenopus oocytes, desensitization could be fit by a double exponential, giving rate constants for both fast and slow desensitization.
To test the generality of the effects that we observed on other Cys-loop receptors, we also created mutants in GABAA and AChR subunits at the AChBP V44-aligned positions. After expression in Xenopus oocytes, mutant α1β1 GABAA receptors, containing either or both of the α1H82C or β1V53C mutations, activated normally in response to saturating GABA concentrations. The mean peak current and GABA EC50 value were not significantly different from WT receptors (data not shown), in agreement with published results (Kash et al., 2004b). However, we did note that the apparent rate of desensitization of GABAA receptors containing mutant subunits was increased; the time constant of desensitization was 35.9 ± 3.4 and 47.9 ± 6.9 s for α1H82Cβ1 and α1β1V53C, respectively, compared with 74.5 ± 4.5 s for WT α1β1 receptors (Student's t test; p < 0.05). In contrast, ACh receptors formed by expression of αV46Cβγδ did not produce measurable current in response to ACh concentrations up to 1 mm. Although the overall mechanism of transduction between different Cys-loop receptor families may be similar, the details of the specific residues involved and the effects of mutating them appear to be different. Whether this is attributable to issues of different subunit stoichiometry or problems in sequence alignment are uncertain.
Modification of K81C and Q70C mutants by the thiol-reactive reagent MTSEA +
The pin-into-socket hypothesis states that the α1V46 residue is in a hydrophobic pocket formed at the extracellular end of the M2 segment and the M2-M3 loop (Miyazawa et al., 2003). Having established that receptors containing the 5-HT3A K81C mutation were functional, we tested the water-surface accessibility of the cysteine by its ability to react with the MTS reagent MTSEA+. This reagent was selected for the initial test because a cysteine residue that has been modified by MTSEA+ resembles the structure and charge of the original lysine residue in the WT receptor. MTS reagents react 5 × 109 times faster with a deprotonated thiolate (S-) than with the protonated thiol (SH), and only cysteines that are, at least transiently, on the water-accessible protein surface will ionize (Karlin and Akabas, 1998). Figure 3a shows a sample trace from one of these experiments. Test pulses of 5-HT were applied to oocytes expressing unmodified 5-HT3A/K81C homopentameric receptors to establish an initial peak current. This was followed by a 1 min application of 1 mm MTSEA+. After washout of the reagent, additional test pulses revealed an irreversible increase in peak current. The magnitude of this increase was 269 ± 29% (n = 3) over unmodified homopentameric receptors, yielding a peak current level about half that of WT homomeric receptors. After modification, the apparent rate of desensitization was decreased by 367 ± 18% (n = 3).
We measured the rate of reaction of MTSEA+ with K81C mutants by repeated (10-20 s) applications of MTSEA+ (0.1-20 μm) in the absence of 5-HT and fitted the normalized peak current to a single exponential (Fig. 3b). This yielded a second-order rate constant of 2.27 ± 0.26 × 105 m-1s-1 (n = 5). When this experiment was repeated with MTSEA+ applications in the presence of 5-HT, the rate of reaction increased to 4.02 ± 0.64 × 105 m-1s-1 (n = 4; p < 0.05; Student's t test). There was no significant difference in the magnitude of potentiation induced by MTSEA+ in the presence or absence of 5-HT (data not shown). The reactivity of MTSEA+ with K81C suggests that in 5-HT3A homopentameric receptors, at least some of the K81C residues (and therefore, we infer, K81 in the WT receptor) are not buried in hydrophobic pockets but rather are water accessible and may project to one side of the M2-M3 loop. In addition, the rate of reaction with K81C is several orders of magnitude higher than MTS reagent reaction rates measured with most channel-lining engineered cysteines to date (Reeves et al., 2001; Bera et al., 2002).
The environment of K81C probed by multiple MTS reagents
To characterize the electrostatic environment of K81C, we exposed the mutant receptors to either a neutral MTS reagent, MMTS, or a negatively charged reagent, MTSES-. Both reagents irreversibly modified K81C-containing receptors and potentiated the currents by a similar magnitude to that found for MTSEA+ modification. The rate of reaction of MMTS with 5-HT3A/K81C homopentamers was 2400 ± 230 m-1s-1 and with MTSES- was 74 ± 17 m-1s-1. This drastic decrease in the rate of reaction of K81C with neutral and negatively charged reagents suggests that there is a negative electrostatic potential in the region surrounding K81C in the resting state of the receptor. We speculate that this is because of the proximity of E80, which, by analogy with the AChR model (Unwin, 2005), should, along with K81C, straddle the M2-M3 loop (Lester et al., 2004). The ability of MTS reagents with positive, negative, or no charge to restore the current magnitude and receptor kinetics of K81C-5-HT3A receptors suggests that charge at this position is not an important factor for signal transduction but rather the size of the residue at position 81 is important. Cysteine is too small to function effectively, but the addition of even an S-CH3 by MMTS is sufficient to restore essentially normal interactions and thus signal transduction between the extracellular β1-β2 loop and the membrane-spanning domain.
We tested the ability of several additional sulfhydryl-reactive reagents with differing sizes and conformational flexibility to potentiate the currents of K81C-containing receptors (Fig. 4). All reagents tested irreversibly modified 5-HT3A/K81C homopentameric receptors, as shown in Figure 4a. Five of seven reagents tested (MTSEA+, MTSET+, MMTS, MTSES-, MTS-TAMRA) caused potentiation of the peak current response to 5-HT recorded from 5-HT3A/K81C homopentameric receptors (Fig. 4a). There was no correlation between these reagents in terms of size or charge, as may be seen from Figure 4b, which depicts a representation of cysteine residues after modification by each MTS reagent.
Figure 4.
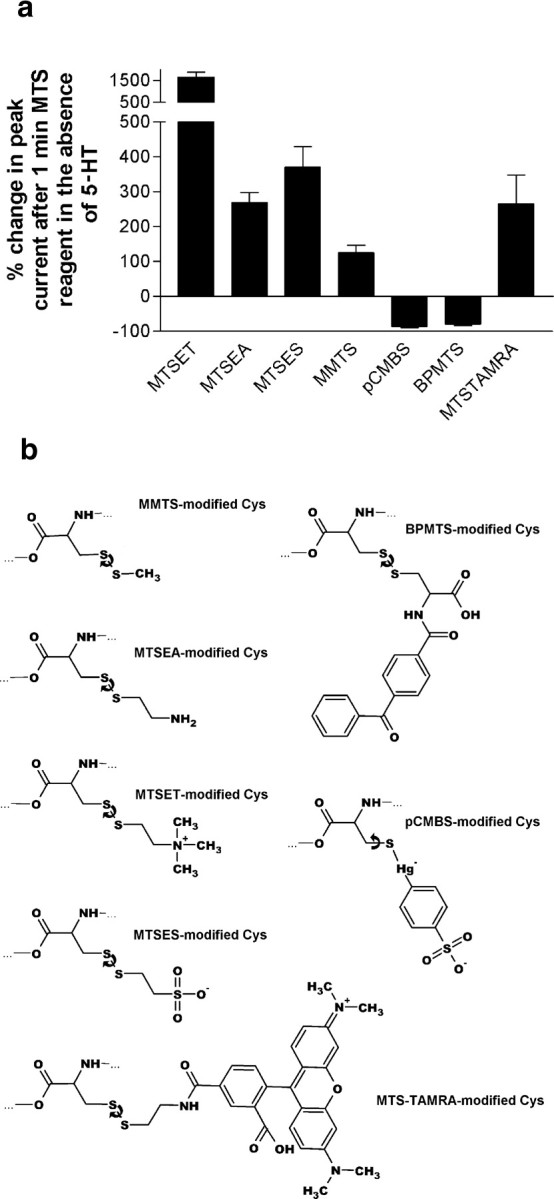
a, Modification of 5-HT3A/K81C homopentamers by various thiol-reactive reagents irreversibly effects peak current. Currents were recorded from injected Xenopus oocytes at a holding potential of -80 mV. The responses to pairs of saturating test pulses of 5-HT (50 μm) were averaged before and after reaction with each reagent. Error bars indicate SEM; n = 3. b, Chemical structures of the products formed by reacting cysteine with each thiol-reactive reagent used in this study. The cysteinyl group is oriented similarly to the left in each case. The curved arrows denote in each case the bond with which free energy calculations were performed.
Of note, MTSET+ modification restored 5-HT3/K81C receptor peak current to levels similar to WT receptors. This suggests that the decrease in peak current seen for K81C-containing receptors was not attributable to a decrease in expression level of the mutant but rather to changes in open probability. Consistent with this, as shown below, the single-channel conductance was not altered by mutation of K81 to cysteine or modification of the cysteine by MTS reagents.
Two reagents, pCMBS- and BPMTS, caused almost complete inhibition of 5-HT-induced current (Fig. 4a). There are several differences between the groups added to cysteine by pCMBS- and BPMTS compared with those added by the five reagents that potentiated currents. First, for pCMBS-, the S-Hg-C angle in pCMBS--modified cysteine is almost 180° (164, 174, and 177° in PDB entries 1HJ1, 1HDK, and 1BH9, respectively). This near-linear geometry significantly reduces the conformational flexibility of pCMBS--modified cysteine. Second, energetic calculations showed that despite the differences in the moieties added by the MTS reagents, for rotation around the S-S bond, there is not a significant difference between the MTS reagents used. There is, however, a potentially significant difference between BPMTS-modified cysteine and cysteines modified by the other MTS reagents. After reaction with MTSES-, MTSET+, MTSEA+, or MTS-TAMRA, the link between the disulfide-bonded sulfur (Sδ) and the prosthetic group is a 1,2-disubstituted ethylene group (Fig. 4b). The tetrahedral structure of the two sp3-hybridized methylene groups gives this linker considerable conformational flexibility. In contrast, for BPMTS-modified cysteine, the second carbon bears two substituents, namely the carboxamidobenzophenone and the carboxy group. Steric clash attributable to the extra substituent on this carbon may limit the conformational flexibility of the Cys-BP side chain within the protein and thus interfere with the interactions necessary for signal transduction to the M2-M3 loop.
We examined whether application of sulfhydryl reagents to the GABAA or ACh receptor cysteine mutants would have significant functional effects on gating. For the GABAA mutants α1H82Cβ1 and α1β1V53C, there were no significant differences in receptor function after a 1 min application of 2.5 mm MTSEA+ or 1 mm MTSET+ (data not shown). Similarly, after a 1 min application of 0.5 mm pCMBS-, 2.5 mm MTSEA+, or 10 mm MMTS, no currents were detected from oocytes expressing AChR αV46Cβγδ receptors in response to ACh (20-500 μm) (data not shown).
5-HT3R desensitization kinetics
The experiments described above (Fig. 2) showed that when expressed in Xenopus oocytes, the K81C mutation altered the apparent rate of desensitization compared with WT 5-HT3A receptors. Fast time-scale changes in receptor kinetics cannot be resolved using whole-cell recordings from Xenopus oocytes, because their large size places a practical limit of a few seconds on the speed of solution exchange around the cell. Therefore, we performed similar experiments using whole-cell patch-clamp recording from HEK293 cells transiently transfected with the WT 5-HT3B subunit and either the WT 5-HT3A or the K81C mutant subunits (Fig. 3c). Mean peak currents were as follows: for WT 5-HT3A/5-HT3B receptors, 3520 ± 560 pA (n = 4); for 5-HT3A/K81C/5-HT3B receptors, 310 ± 63 pA (n = 4); for 5-HT3A/K81C/5-HT3B receptors after treatment with MTSET+, 3830 ± 460 pA (n = 4). Thus, as in oocytes, the K81C mutation significantly reduced the 5-HT-induced current, and modification of the cysteine by MTSET+ restored the currents to WT levels.
Interestingly, although agonist-induced desensitization of whole-cell currents recorded from Xenopus oocytes could be fit by a single exponential (Figs. 2c,3a), the best fit for desensitization of WT 5-HT3A/5-HT3B receptor currents recorded from HEK293 cells was a double exponential (Fig. 3c). This is consistent with previously reported desensitization kinetics for WT 5-HT3A receptors (Mott et al., 2001). The double-exponential fit of the WT 5-HT3A/5-HT3B receptor desensitization currents yielded “fast” and “slow” desensitization time constants of 167 ± 35 and 4841 ± 432 ms (n = 7) with relative amplitudes of 65 ± 2 and 35 ± 2%, respectively. These are similar to the time constants reported for homomeric WT 5-HT3A receptors (Mott et al., 2001). In contrast, desensitizing currents recorded from 5-HT3A/K81C/5-HT3B receptors were best fit by a single exponential with a time constant of 194 ± 17 ms (n = 6), similar to the fast time constant in WT heteromeric receptors. After treatment with MTSET+, desensitization of 5-HT3A/K81C/5-HT3B receptor currents was again best fit by a double exponential with time constants of 244 ± 16 and 2515 ± 407 ms (n = 6) and relative amplitudes of 73 ± 3 and 27 ± 3%, respectively. Thus, it appears that the K81C mutation has two effects on desensitization kinetics. Fast desensitization predominates, and slow desensitization is no longer observed. This could occur if the rate of return from the fast desensitized state is significantly reduced, so that on the time scale of these experiments, this state becomes a trap for activated channels in much the same way as the slow desensitized state is a trap for WT receptors.
Using simple kinetic schemes, the 10-fold decrease in current observed for the K81C mutant receptors cannot be accounted for by the observed differences in the rates of desensitization. This implies that the K81C mutation is having a second effect on channel kinetics. A significant reduction in the channel opening rate for the K81C mutant would be consistent with the observed decrease in peak current. Given the limitation of the solution exchange time in our whole-cell patch-clamp perfusion system, we could not resolve any differences in activation kinetics of the mutant and WT receptors. The mean time-to-peak current was as follows: for WT 5-HT3A/5-HT3B receptors, 108 ± 9.9 ms; for 5-HT3A/K81C/5-HT3B receptors, 122 ± 12 ms; for 5-HT3A/K81C/5-HT3B receptors after treatment with MTSET+, 131 ± 23 ms. There was no significant difference between these times for activation and the time for solution exchange for our apparatus (122 ± 17 ms; n = 2). For the K81C mutant, the rate of fast desensitization cannot completely account for the observed decrease in the peak current magnitude. Given the time-to-peak current of 122 ms in our apparatus, with a time constant for fast desensitization of 167 ms, only 43% of receptors could open and desensitize during the rise time, yet the peak current of the K81C receptors was reduced by an order of magnitude. The remainder of the decrease in peak current may be accounted for by a reduced rate of entry (β) into the open state in K81C receptors compared with WT.
Single channel analysis of K81C-containing receptors
We attempted to characterize the effect of mutation and modification of K81C at the single-channel level. Single-channel currents from homopentameric 5-HT3A are too small to be recorded directly. Therefore, we used constructs containing the R437Q/R441D/R445A(QDA) mutations in the cytoplasmic loop that were previously shown to enable single-channel recording from 5-HT3A homopentamers (Kelley et al., 2003). We initially attempted to record single-channel events in the cell-attached mode, to measure the open probability of 5-HT3A channels under high (1 mm) agonist concentrations. However, we could not reliably detect channels from cells transfected with either 5-HT3A WT(QDA) or K81C(QDA)-containing receptors. The application of 5-HT to outside-out patches, however, allowed us to detect single channels from HEK293 cells transfected with these constructs (Fig. 5). Immediately after 5-HT application, multiple channels opened but the currents rapidly decayed, so that individual channel openings could be distinguished. As expected from the whole-cell results, the initial peak current after 5-HT application was much larger for WT(QDA) and K81C(QDA) treated with MTSET+ than for K81C(QDA). We obtained enough events to measure a predominant single-channel conductance of 38 ± 7.7 pS for WT(QDA) receptors (Fig. 5a) and 40 ± 7.7 pS for K81C(QDA) receptors (Fig. 5b). Therefore, the reduction in the peak current observed with the K81C-mutant whole-cell currents was not caused by a decrease in single-channel conductance. Pretreatment of cells expressing 5-HT3/K81C(QDA) for 3 min with 20 μm MTSET+ or MTSEA+ allowed recording from modified 5-HT3/K81C receptors (Fig. 5c). Modification by either MTSET+ or MTSEA+ did not significantly alter single-channel conductance (41 ± 7.5 and 38 ± 7.3 pS, respectively) compared with WT(QDA) or untreated K81C(QDA) receptors. Of note, bursts containing multiple opening and closing events were observed in the records for WT(QDA) and MTS-modified K81C(QDA) (Fig. 5a,c, insets) In contrast, openings of K81C(QDA) channels had one or, at most, a few opening and closing events per burst (Fig. 5b, inset). This is consistent with a reduced opening rate for the K81C channels, such that the opening rate was comparable to or slower than the fast desensitization rate.
Figure 5.

Currents recorded from HEK293 cells transiently transfected with constructs coding for 5-HT3A mutant receptors. Each trace shows the response of an outside-out patch to 50 μm 5-HT. The calibration bars refer to the full traces. The selected sections of each trace, indicated by the gray lines, are shown below at an expanded time scale. Displayed to the right of each trace is an all-points histogram of the expanded portion of the recording. QDA, The presence in all transfected subunits of the R437Q/R441D/R445A set of mutations in the intracellular M3-M4 loop, known to elevate the single-channel conductance of 5-HT3A receptors to directly measurable levels. 5-HT3A/WT/QDA (a), 5-HT3A/K81C/QDA (b), and 5-HT3A/K81C/QDA (c), after treatment of the cells for 3 min with 20 μm MTSET+, are shown.
Disulfide cross-linking scan of the extracellular end of M2 and the M2-M3 loop
K81C, at the tip of the β1-β2 loop, should be in close proximity to the extracellular end of M2 and the M2-M3 loop, based on a 5-HT3A subunit homology model (Fig. 6a) constructed using the atomic coordinates from AChBP (PDB code I19B) and the Torpedo AChR TMD (PDB code 1OED). We tested this hypothesis using disulfide cross-linking experiments (Horenstein et al., 2005). We created eight double-cysteine mutants by mutating the residues T303 (25′) to I310 (32′), one at a time to cysteine in the presence of the K81C mutation (Fig. 6c). The residues T303 (25′) to G306 (28′) are part of the extracellular end of M2 that extends above the membrane surface (Bera et al., 2002) and T307 to I310 form the proximal portion of the M2-M3 loop (Miyazawa et al., 2003).
Figure 6.

a, Section of a homology model of the 5-HT3A receptor subunit, focused on the junction between the extracellular domain (top) and membrane domain (bottom). Note the predicted proximity of K81 to the extracellular portion of the M2 segment. b, Sample traces from an experiment demonstrating inhibition of the double mutant 5-HT3A/K81C/A304C by oxidizing conditions (Cu:phen, 2 min) and relief of inhibition by reducing conditions (DTT, 3 min), indicating the reversible formation of a disulfide bond. c, Selected members of the Cys loop superfamily of ion channels aligned in the region between transmembrane segments M2 and M3. Blue letters indicate residues mutated to cysteine in this study. Underlined residues were found to cross-link with the β1-β2 loop in this study. Red letters indicate positions implicated previously in coupling binding to channel gating in other studies (Lynch et al., 1997; Kash et al., 2003; Sala et al., 2004). Residue numbers refer to positions from the beginning of the predicted protein precursor.
No 5-HT-induced currents were observed from oocytes injected with mRNA for any of the double mutant 5-HT3A subunits alone (data not shown). However, when coexpressed with the WT 5-HT3B subunit, small 5-HT-induced currents, ranging from 15 to 145 nA, were recorded from oocytes expressing either 5-HT3A/K81C/A304C or 5-HT3A/K81C/G306C. We tested the effects of oxidation and reduction on oocytes expressing these double mutant constructs. Oxidation by 100:200 μm Cu:phen in the absence of 5-HT, inhibited the current from 5-HT3A/K81C/A304C/5-HT3B-expressing oocytes. The 5-HT currents were restored after subsequent reduction with 10 mm DTT (Fig. 6b) (n = 3). This suggested that we could form and break a disulfide bond at this position. DTT reversal of Cu:phen-induced inhibition was not observed when the same protocol was applied to receptors containing either the K81C or A304C mutations alone (data not shown). Oxidation and reduction had no significant effects on currents from oocytes expressing 5-HT3A/K81C/G306C/5-HT3B.
For the other six double-cysteine mutants that did not give currents, we assayed for the presence of spontaneously formed disulfide bonds by treating the oocytes with 10 mm DTT for 3 min. For one of these mutants, 5-HT3A/K81C/I305C/5-HT3B, after DTT treatment, 5-HT-activated currents were observed. Application of 100:200 μm Cu:phen eliminated these 5-HT currents, and they were restored after application of 10 mm DTT. DTT can reduce disulfide bonds and can also chelate heavy metals. To ensure that the effect of DTT was attributable to reduction and not to chelation of contaminating heavy metals from the buffer, we tested whether the metal chelator EGTA could also potentiate currents. EGTA cannot reduce disulfide bonds. In contrast to DTT, a 3 min application of 1 mm EGTA did not cause the appearance of currents in this mutant. These results are consistent with the presence of a spontaneously formed disulfide bond between K81C and I305C. It should be noted that the spontaneous formation of disulfide bonds indicates that mildly oxidizing conditions as a result of ambient oxygen in the buffer were sufficient to promote disulfide bond formation. It is not fundamentally different from disulfide bond formation in the more oxidizing environment created by Cu:phen. Spontaneous disulfide bond formation is simply a measure of the relative propensity to form disulfide bonds and suggests that K81C collides more frequently with I305C than with A304C or in a more favorable orientation.
Discussion
Role of K81 in 5-HT3R channel gating
The coupling mechanism that transduces agonist binding into channel gating in nicotinic ACh and 5-HT3R should be similar, because chimeras between the extracellular and transmembrane domains of these receptors form functional channels (Eiselé et al., 1993; Bouzat et al., 2004). To test the generality of the pin-into-socket mechanism for the transduction of agonist binding to channel gating proposed by Unwin and colleagues (Miyazawa et al., 2003), we mutated to cysteine the 5-HT3A and 5-HT3B β1-β2 loop residues K81 and Q70 aligned with the AChR “pin” αV46 (Fig. 1b). 5-HT activated K81C-containing channels, but the macroscopic currents were significantly reduced compared with WT (Figs. 2, 3). The reduction in current in K81C-expressing cells was not attributable to a change in single-channel conductance (Fig. 5) or to a decrease in expression level, because after modification by MTS reagents, currents were restored to WT levels (Fig. 3). Desensitization was also significantly different between WT and mutant currents. In HEK cells, WT currents desensitized with two time components, one fast (∼170 ms) and the one slow (∼4800 ms). This implies the existence of at least two desensitized states. Currents from K81C-expressing cells desensitized with only the fast time constant. To explain the effects of the K81C mutation on peak currents and desensitization, we infer that the mutation altered two aspects of channel kinetics. The change from two components of desensitization in WT to one in the mutant occurred either because mutant channels did not return from the fast desensitized state or because entry into the slow desensitized state, from which the rate of return is very low, was accelerated to a rate comparable with entry into the fast desensitized state. Although we favor the former explanation, we cannot distinguish these possibilities experimentally. However, neither of these changes can completely explain the magnitude of the decrease in peak current amplitudes. Thus, we infer that an additional aspect of channel gating was altered. The most parsimonious explanation would be that the entry rate into the open state,β, is reduced in K81C channels. Because of limitations of solution exchange times with our gravity-fed perfusion system, we could not measure a difference in activation rates of WT and K81C whole-cell currents. However, examination of single-channel bursts was consistent with a reduced opening rate in K81C channels given the similar rates of fast desensitization that we measured. The number of reopenings per burst was smaller in K81C channels compared with WT and MTSET+-modified K81C channels (Fig. 5, insets); however, because of the extent of desensitization, we were unable to obtain sufficient numbers of bursts to quantitatively analyze this. Thus, we infer that in 5-HT3R, K81 plays an important role in the channel-opening process and in the return from the fast desensitized state.
Does K81 function as a pin-into-socket? The structural environment of K81C
K81 does not seem to be in a hydrophobic pocket as stated in the pin-into-socket hypothesis. Cysteine substituted for K81 was readily modified by MTS reagents at rates consistent with it being significantly water accessible. MTSEA+ reaction rates with K81C, both in the absence and presence of 5-HT, were considerably faster than rates measured with nearby M2-M3 loop residues (Bera et al., 2002) or with residues in the extracellular half of M2 (Reeves et al., 2001; Goren et al., 2004). The high rates were attributable in part to local electrostatic effects but indicated that K81C was water accessible, because the MTS reagents react 5 × 109 times faster with an ionized thiolate than with a thiol, and only water-accessible thiols will ionize to any significant extent (Roberts et al., 1986). Thus, we infer that K81C, and therefore K81 in the WT 5-HT3R, is not buried in a hydrophobic pocket as stated in the pin-into-socket hypothesis.
An interaction between the β1-β2 loop and the extracellular end of M2 in 5-HT3R
Although it is not buried in a hydrophobic pocket, K81 is sufficiently close to the extracellular end of M2 to allow disulfide bond formation between K81C and both A304C (26′) and I305C (27′). 5-HT3A/K81C/I305C/5-HT3B mutant receptors formed spontaneous disulfide bonds that completely inhibited currents, but this inhibition was relieved after reduction. An inhibitory but DTT-reversible disulfide bond also formed in the 5-HT3A/K81C/A304C double mutant. The spontaneous formation of the K81C-I305C disulfide bond suggests that the collision frequency is higher between these residues than between K81C and A304C. In both cases, disulfide bond formation implies that the α-carbons of K81C and either A304C or I305C can approach to within 5.5 Å in the resting state (Horenstein et al., 2005). Similar interactions between charged residues in the M2-M3 loop and in loop 2 of GABAA (Kash et al., 2003), glycine (Absalom et al., 2003), and ACh (Sala et al., 2004) receptors have been described previously (Fig. 6c). These interactions were detected by disulfide cross-linking and/or charge pair reversal mutations and were inhibitory to channel function when formed. These results, in conjunction with our finding that cross-linking of the 5-HT3A β1-β2 loop and the M2 segment is inhibitory, suggest that simple linkage of these two regions is not sufficient for channel gating and that normal channel gating may require movement of these loops relative to each other.
The interaction with the extracellular end of M2 appears to be steric because uncharged and negatively charged MMTS and MTSES- were able to restore to K81C wild-type-like currents and gating behavior (Figs. 3, 4a). It appears that the cysteine side chain in K81C is two or three atoms too short to mediate an effective interaction with the extracellular end of M2, because even MMTS, which adds an S-CH3 onto cysteine, was sufficient to restore function. Furthermore, there appears to be significant space in the vicinity, probably in the channel lumen, to allow the tetramethylrhodamine of MTS-TAMRA to move into a position where it does not significantly affect channel gating, because MTS-TAMRA also restored WT function. The interaction between K81 and the M2 segment is not completely permissive, because two reagents, pCMBS- and BPMTS, caused almost complete inhibition of channel function.
We demonstrated previously that the extracellular end of the GABAA βM2 segment (the homolog of ACh α-like subunits) is more mobile than the αM2 segment (Horenstein et al., 2005) and that the accessibility of the M2-M3 loop increases after activation of the receptors (Lynch et al., 2001; Bera et al., 2002). This could be accounted for by the unmasking of the M2-M3 loop by β1-β2 loop movement during gating (Unwin, 2005).
A possible role for the β1-β2 loop in 5-HT3R gating
At least three extracellular domain loops interact with the membrane-spanning domain and may be involved in normal receptor transduction (Bouzat et al., 2004). In the AChR resting state structure, the β1-β2 loop and Cys loop straddle the extracellular end of M2 and the M2-M3 loop (Unwin, 2005). During the conformational wave initiated by agonist binding, the β1-β2 loop and Cys loop (Φ∼ 0.8) move before the M2-M3 loop (Φ∼ 0.6) (Grosman et al., 2000; Chakrapani et al., 2004) and not simultaneously with it as would be expected for a pin-into-socket coupling mechanism (Unwin, 2005). Our data support a model in which the β1-β2 loop and possibly the Cys loop move after ligand binding. This moves the N-terminal portion of the M2-M3 loop or the extracellular end of M2, inducing the rotation/translation of the M2 segments away from the channel axis. This breaks the weak bonds of the hydrophobic ring that are proposed to form the gate region, resulting in channel opening (Unwin, 2005). A subsequent conformational change in the membrane-spanning domain results in fast desensitization of the channel. Recovery from the desensitized state may require the β1-β2 loop to reform its interaction with the M2-M3 loop and return the M2-M3 loop to its resting position. Consistent with this model, the M2 19′ residue near the extracellular membrane surface appears to return to its resting state position before recovery from desensitization is complete (Dahan et al., 2004). In our mechanism, a certain amount of molecular bulk at K81 and other αV46-aligned residues could maintain the interaction between the β1-β2 loop and the M2-M3 loop, allowing rapid recovery from the fast desensitized state. However, rigid groups attached to K81C, such as pCMBS-, would be more likely to inhibit channel opening, because they may hinder the movement of the β1-β2 loop relative to the M2-M3 loop required for channel opening.
Desensitization of 5-HT3R has been shown previously to depend on the subunit composition, the binding site occupancy, and the primary structure of the M2 transmembrane segment (Yakel, 1996; Mott et al., 2001; Hapfelmeier et al., 2003). Because Cys-loop receptor desensitization can be important in vivo for the modulation of synaptic responses (Quick and Lester, 2002), the lack of conservation at αV46-aligning residues may indicate that this position is a candidate for evolutionary tuning of the divergent functional responses of different Cys loop receptors.
Footnotes
This work was supported in part by National Institutes of Health Grants NS30808 and GM61925 (M.H.A.) and in part by a fellowship from the Deutsche Forschungsgemeinschaft (M.J.). We thank Dr. David Sharp for cell culture facilities. We thank Maya Chatav and Rachel Berkowitz for expert technical assistance. We thank Jeff Horenstein, Amal Bera, Paul Riegelhaupt, Nicole McKinnon, Eric Goren, and David Liebelt for helpful discussions and comments on this manuscript.
Correspondence should be addressed to Dr. Myles Akabas, Department of Physiology and Biophysics, Albert Einstein College of Medicine of Yeshiva University, Bronx, NY 10461. E-mail: makabas@aecom.yu.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259358-09$15.00/0
References
- Absalom NL, Lewis TM, Kaplan W, Pierce KD, Schofield PR (2003) Role of charged residues in coupling ligand binding and channel activation in the extracellular domain of the glycine receptor. J Biol Chem 278: 50151-50157. [DOI] [PubMed] [Google Scholar]
- Absalom NL, Lewis TM, Schofield PR (2004) Mechanisms of channel gating of the ligand-gated ion channel superfamily inferred from protein structure. Exp Physiol 89: 145-153. [DOI] [PubMed] [Google Scholar]
- Bera AK, Chatav M, Akabas MH (2002) GABAA receptor M2-M3 loop secondary structure and changes in accessibility during channel gating. J Biol Chem 277: 43002-43010. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Gumilar F, Spitzmaul G, Wang HL, Rayes D, Hansen SB, Taylor P, Sine SM (2004) Coupling of agonist binding to channel gating in an ACh-binding protein linked to an ion channel. Nature 430: 896-900. [DOI] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK (2001) Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411: 269-276. [DOI] [PubMed] [Google Scholar]
- Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK (2004) Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41: 907-914. [DOI] [PubMed] [Google Scholar]
- Chakrapani S, Bailey TD, Auerbach A (2004) Gating dynamics of the acetylcholine receptor extracellular domain. J Gen Physiol 123: 341-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CA, Okayama H (1988) Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques 6: 632-638. [PubMed] [Google Scholar]
- Costall B, Naylor RJ (2004) 5-HT3 receptors. Curr Drug Targets CNS Neurol Disord 3: 27-37. [DOI] [PubMed] [Google Scholar]
- Dahan DS, Dibas MI, Petersson EJ, Auyeung VC, Chanda B, Bezanilla F, Dougherty DA, Lester HA (2004) A fluorophore attached to nicotinic acetylcholine receptor beta M2 detects productive binding of agonist to the alpha delta site. Proc Natl Acad Sci USA 101: 10195-10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar MS, Zoebisch EG, Healy EF, Stewart JJP (1985) The development and use of quantum mechanical molecular models. J Am Chem Soc 107: 3902-3909. [Google Scholar]
- Eiselé J-L, Bertrand S, Galzi J-L, Devillers-Thiéry A, Changeux J-P, Bertrand D (1993) Chimaeric nicotinic-serotonergic receptor combines distinct ligand binding and channel specificities. Nature 366: 479-483. [DOI] [PubMed] [Google Scholar]
- Goren EN, Reeves DC, Akabas MH (2004) Loose protein packing around the extracellular half of the GABAA receptor beta1 subunit M2 channel-lining segment. J Biol Chem 279: 11198-11205. [DOI] [PubMed] [Google Scholar]
- Grosman C, Zhou M, Auerbach A (2000) Mapping the conformational wave of acetylcholine receptor channel gating. Nature 403: 773-776. [DOI] [PubMed] [Google Scholar]
- Hapfelmeier G, Tredt C, Haseneder R, Zieglgansberger W, Eisensamer B, Rupprecht R, Rammes G (2003) Co-expression of the 5-HT3B serotonin receptor subunit alters the biophysics of the 5-HT3 receptor. Biophys J 84: 1720-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horenstein J, Akabas MH (1998) Location of a high affinity Zn2+ binding site in the channel of alpha1beta1 gamma-aminobutyric acid A receptors. Mol Pharmacol 53: 870-877. [PubMed] [Google Scholar]
- Horenstein J, Riegelhaupt P, Akabas MH (2005) Differential protein mobility of the gamma-aminobutyric acid, type A, receptor alpha and beta subunit channel-lining segments. J Biol Chem 280: 1573-1581. [DOI] [PubMed] [Google Scholar]
- Jespersen T, Grunnet M, Angelo K, Klaerke DA, Olesen SP (2002) Dual-function vector for protein expression in both mammalian cells and Xenopus laevis oocytes. Biotechniques 32: 536-538. [DOI] [PubMed] [Google Scholar]
- Karlin A (2002) Emerging structure of the nicotinic acetylcholine receptors. Nat Rev Neurosci 3: 102-114. [DOI] [PubMed] [Google Scholar]
- Karlin A, Akabas MH (1998) Substituted-cysteine accessibility method. Methods Enzymol 293: 123-145. [DOI] [PubMed] [Google Scholar]
- Kash TL, Jenkins A, Kelley JC, Trudell JR, Harrison NL (2003) Coupling of agonist binding to channel gating in the GABAA receptor. Nature 421: 272-275. [DOI] [PubMed] [Google Scholar]
- Kash TL, Dizon MJ, Trudell JR, Harrison NL (2004a) Charged residues in the beta2 subunit involved in GABAA receptor activation. J Biol Chem 279: 4887-4893. [DOI] [PubMed] [Google Scholar]
- Kash TL, Kim T, Trudell JR, Harrison NL (2004b) Evaluation of a proposed mechanism of ligand-gated ion channel activation in the GABAA and glycine receptors. Neurosci Lett 371: 230-234. [DOI] [PubMed] [Google Scholar]
- Kelley SP, Dunlop JI, Kirkness EF, Lambert JJ, Peters JA (2003) A cytoplasmic region determines single-channel conductance in 5-HT3 receptors. Nature 424: 321-324. [DOI] [PubMed] [Google Scholar]
- Lester HA, Dibas MI, Dahan DS, Leite JF, Dougherty DA (2004) Cys-loop receptors: new twists and turns. Trends Neurosci 27: 329-336. [DOI] [PubMed] [Google Scholar]
- Lynch JW, Rajendra S, Pierce KD, Handford CA, Barry PH, Schofield PR (1997) Identification of intracellular and extracellular domains mediating signal transduction in the inhibitory glycine receptor chloride channel. EMBO J 16: 110-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JW, Han NL, Haddrill J, Pierce KD, Schofield PR (2001) The surface accessibility of the glycine receptor M2-M3 loop is increased in the channel open state. J Neurosci 21: 2589-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa A, Fujiyoshi Y, Unwin N (2003) Structure and gating mechanism of the acetylcholine receptor pore. Nature 423: 949-955. [DOI] [PubMed] [Google Scholar]
- Mott D, Erreger K, Banke T, Traynelis S (2001) Open probability of homomeric murine 5-HT3A serotonin receptors depends on subunit occupancy. J Physiol (Lond) 535 2: 427-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicker S, Cruz H, Arrabit C, Slesinger PA (2002) Evidence for a centrally located gate in the pore of a serotonin-gated ion channel. J Neurosci 22: 1629-1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Lester RA (2002) Desensitization of neuronal nicotinic receptors. J Neurobiol 53: 457-478. [DOI] [PubMed] [Google Scholar]
- Reeves DC, Lummis SC (2002) The molecular basis of the structure and function of the 5-HT3 receptor: a model ligand-gated ion channel [review]. Mol Membr Biol 19: 11-26. [DOI] [PubMed] [Google Scholar]
- Reeves DC, Goren EN, Akabas MH, Lummis SCR (2001) Structural and electrostatic properties of the 5-HT3 receptor pore revealed by substituted cysteine accessibility mutagenesis. J Biol Chem 276: 42035-42042. [DOI] [PubMed] [Google Scholar]
- Reeves DC, Sayed MF, Chau PL, Price KL, Lummis SC (2003) Prediction of 5-HT3 receptor agonist-binding residues using homology modeling. Biophys J 84: 2338-2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DD, Lewis SD, Ballou DP, Olson ST, Shafer JA (1986) Reactivity of small thiolate anions and cysteine-25 in papain toward methyl methanethiosulfonate. Biochemistry 25: 5595-5601. [DOI] [PubMed] [Google Scholar]
- Sala F, Mulet J, Sala S, Gerber S, Criado M (2004) Charged amino acids of the N-terminal domain are involved in coupling binding and gating in alpha 7 nicotinic receptors. J Biol Chem 280: 6642-6647. [DOI] [PubMed] [Google Scholar]
- Schofield CM, Trudell JR, Harrison NL (2004) Alanine-scanning mutagenesis in the signature disulfide loop of the glycine receptor alpha 1 subunit: critical residues for activation and modulation. Biochemistry 43: 10058-10063. [DOI] [PubMed] [Google Scholar]
- Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31: 3381-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JJP (1990) MOPAC—a semiempirical molecular orbital program. J Comput Aided Mol Des 4: 1-45. [DOI] [PubMed] [Google Scholar]
- Unwin N (2005) Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J Mol Biol 346: 967-989. [DOI] [PubMed] [Google Scholar]
- Yakel JL (1996) Desensitization of 5-HT3 receptors expressed in Xenopus oocytes: dependence on voltage and primary structure. Behav Brain Res 73: 269-272. [DOI] [PubMed] [Google Scholar]