

Abstract
RNA editing within the pore loop controls the pharmacology and permeation properties of ion channels formed by neuronal AMPA and kainate receptor subunits. Genomic sequences for the glutamate receptor 2 (GluR2) subunit of AMPA receptors and the GluR5 and GluR6 subunits of kainate receptors all encode a neutral glutamine (Q) residue within the channel pore that can be converted by RNA editing to a positively charged arginine (R). Receptors comprised of unedited subunits are permeable to calcium and display inwardly rectifying current-voltage relationships, because of blocking of outward current by intracellular polyamines. In contrast, receptors that include edited subunits conduct less calcium, resist polyamine block, and have relatively linear current-voltage relationships. We showed previously that cis-unsaturated fatty acids, including arachidonic acid and docosahexanoic acid, exert a potent block of native kainate receptors as well as homomeric recombinant receptors formed by transfection of heterologous cells with cDNA for the GluR6(R) subunit. Here, we show that fatty acid blockade of recombinant homomeric and heteromeric kainate receptors is strongly dependent on editing at the Q/R site. Recombinant channels that include unedited subunits exhibit significantly weaker block than channels made up of fully edited subunits. Inhibition of fully edited channels is equivalent at voltages from -70 to +40 mV and is noncompetitive, consistent with allosteric regulation of channel function.
Keywords: arachidonic acid, docosahexanoic acid, polyamines, rectification, receptor, hippocampus
Introduction
The activity of voltage- and ligand-gated ion channels can be modulated by direct interactions with cis-unsaturated fatty acids, including arachidonic acid (AA) and docosahexanoic acid (DHA), which represent major constituents of brain membrane phospholipids (Salem et al., 2001). Normally esterified in the 2 position of phosphatidylserine (PS) and phosphatidylethanolamine (PE), AA and DHA can be released as free fatty acids through the action of phospholipase A2 (Six and Dennis, 2000; Murakami and Kudo, 2002). Channels known to be susceptible to direct modulation by free AA or DHA include voltage-gated potassium channels (Honore et al., 1994), members of the two-pore domain family of channels (Fink et al., 1996; Patel et al., 2001), GABA and glycine receptor channels (Schwartz and Yu, 1992; Hamano et al., 1996), as well as NMDA (Miller et al., 1992) and kainate (Wilding et al., 1998) receptors, which form ion channels gated by the excitatory transmitter l-glutamate. For NMDA receptors (Miller et al., 1992; Nishikawa et al., 1994; Casado and Ascher, 1998) and two-pore domain channels (Patel et al., 2001), exposure to AA or DHA potentiates channel activity. In contrast, cis-unsaturated fatty acids inhibit kainate receptors (Wilding et al., 1998) and produce a variety of effects on voltage-gated K+ channels, including inhibition (Poling et al., 1996; Villarroel and Schwarz, 1996) and kinetic modulation (Honore et al., 1994; Keros and McBain, 1997; Oliver et al., 2004). The mechanisms that underlie these varied effects on channel gating are not fully understood, but they are thought to involve either a direct interaction of fatty acids with the channel protein or a local change in the lipid environment immediately surrounding the channel.
RNA editing regulates the ionic selectivity and pharmacology of ion channels formed by non-NMDA receptor subunits (Seeburg and Hartner, 2003). mRNAs for the glutamate receptor 2 (GluR2) subunit of AMPA receptors, and the GluR5 and GluR6 subunits of kainate receptors, undergo editing at a site within the channel pore that encodes a glutamine (Q) residue in the genomic sequence, but which can be edited to encode for an arginine (R) in the mature polypeptide (Sommer et al., 1991). All of the other AMPA and kainate receptor subunits encode for a Q at this location and are not susceptible to editing (Dingledine et al., 1999). The change from Q to R at the Q/R site regulates channel properties (Seeburg and Hartner, 2003). Channels composed only of subunits that contain a Q at this site are permeable to sodium, potassium, and calcium and are subject to voltage-dependent block by internal polyamines, which produces strong inward rectification of the current-voltage relationship (Dingledine et al., 1999). In contrast, the inclusion of one or more subunits that contain an R at the Q/R site reduces the single-channel conductance (Howe, 1996; Swanson et al., 1996), eliminates block by polyamines (Bowie and Mayer, 1995; Kamboj et al., 1995; Bähring et al., 1997; Mott et al., 2003), and reduces channel permeability to calcium (Egebjerg and Heinemann, 1993; Köhler et al., 1993; Burnashev et al., 1995, 1996).
In a previous study of kainate receptor modulation by AA and DHA (Wilding et al., 1998), we demonstrated strong inhibition of native receptors expressed by cultured postnatal rat hippocampal neurons and by freshly dissociated sensory neurons from early postnatal rat dorsal root ganglia. In addition, we observed a potent block of homomeric recombinant receptors formed by transient transfection of human embryonic kidney 293 (HEK 293) cells with cDNA for the GluR6 subunit. The present study extends this analysis to additional kainate receptor subunits and demonstrates that susceptibility to modulation by fatty acids exhibits a dramatic dependence on editing at the Q/R site within the channel pore. Fully edited channels are strongly inhibited by low concentrations of fatty acid, whereas channels that include unedited subunits resist inhibition, even by the maximal tolerated dose of fatty acid (50 μm).
Materials and Methods
Cell culture and transfection. HEK 293 cells were maintained in MEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (Sigma, St. Louis, MO), as described previously (Wilding et al., 1998). Cells were cotransfected with channel subunit cDNAs and with the mouse L3T4 surface antigen for identification. Subunit cDNA (1-3 μg) and L3T4 cDNA (0.5 μg) were incubated in 80 μl of OptiMEM (Invitrogen) with Superfect reagent (15 μg; Qiagen, Santa Clarita, CA) for 30 min and then diluted onto HEK 293 cells at 50-70% confluence in 600 μl of MEM plus fetal calf serum and 5 mm kynurenic acid in one well of a 12-well plate. After 3-4 h at 37°C, an additional 400 μl of medium with serum and 5 mm kynurenic acid was added. On the next day, cells were incubated for 10 min with protease XXIII (Sigma) at 1 mg/ml, dissociated, and replated at low density on 35 mm culture dishes, which had received a thin coating of nitrocellulose to promote cell attachment. Between 24 and 48 h after replating, cells were incubated for 30 min with phycoerythrin-conjugated monoclonal anti-L3T4 (BD Biosciences, San Jose, CA). Isolated cells labeled with the fluorescent antibody were targeted for recording.
Electrophysiology. Culture dishes were perfused at 1-2 ml/min with Tyrode's solution containing the following (in mm): 150 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES, pH adjusted to 7.4 with NaOH. Boralex glass recording electrodes were pulled to an open tip resistance of 1-5 MΩ when filled with internal solution containing the following (in mm): 140 Cs glucuronate, 10 EGTA, 5 CsCl, 5 MgCl2, 5 ATP, 1 GTP, and 10 HEPES, pH adjusted to 7.4 with CsOH. Whole-cell currents were recorded with an Axopatch 200A amplifier (Molecular Devices, Union City, CA), filtered at 1 kHz (-3 dB, four-pole Bessel), and digitized at 5-10 kHz. Agonist solutions were applied by local perfusion from an eight-barreled pipette as described previously (Wilding et al., 1998). For rapid solution exchange, the drug reservoirs were maintained under static air pressure (∼10 psi), and flow was controlled by computer-gated electronic valves. Fatty acid stock solutions (50 mm) were prepared in DMSO and stored in aliquots under argon gas.
Data were fit to modified Hill equations by nonlinear regression (SigmaPlot; Systat Software, Point Richmond, CA). Concentration-inhibition relationships were fit with Equation 1 as follows:
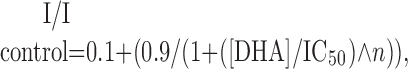 |
(1) |
where IC50 is the DHA concentration producing half-maximal inhibition, and n is the slope factor. Equation 1 provides an empirical description of antagonist potency, without implying a specific inhibition mechanism. Concentration-response relationships in the presence of DHA were fit with Equation 2 as follows:
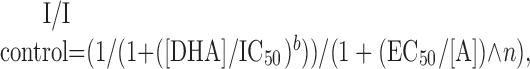 |
(2) |
where [A] and [DHA] are the agonist and DHA concentrations, respectively; EC50 and IC50 are the agonist and DHA concentrations producing half-maximal activation and inhibition, respectively; and n and b are the Hill coefficients for activation and inhibition. Measured parameters are reported as mean ± SEM, and statistical significance was assigned at p < 0.05.
Results
Differential block of GluR5 and GluR6
Our previous study of kainate receptor modulation by fatty acids (Wilding et al., 1998) demonstrated potent inhibition of recombinant homomeric GluR6(R) channels expressed in transfected HEK 293 cells. DHA concentrations as low as 0.5-1 μm produce maximal inhibition, blocking ∼80-90% of peak whole-cell current evoked by rapid applications of kainate (Fig. 1A). To facilitate analysis of fatty acid inhibition, we blocked kainate receptor desensitization by exposing transfected cells to concanavalin A (Con A) (Huettner, 1990; Wong and Mayer, 1993). As shown in Figure 1A, DHA inhibited homomeric GluR6(R) channels that had been treated with Con A, albeit with lower potency (Fig. 1C) (IC50 ∼ 2.6 μm) than that observed in our previous study without Con A treatment. To extend our analysis to additional kainate receptor subunits, we transfected HEK 293 cells with cDNA encoding the GluR5-2a(Q) subunit, which was kindly provided by Peter Seeburg (Max-Planck-Institute for Medical Research, Heidelberg, Germany) (Sommer et al., 1992). Surprisingly, DHA caused significantly less inhibition of homomeric channels formed by transfection of GluR5(Q) (Fig. 1B) than occurred in cells transfected with GluR6(R). In Con A-treated cells tested with 10 μm kainate, exposure to 15 μm DHA produced 17 ± 4% inhibition of homomeric GluR5-2a(Q) compared with 75 ± 2% block of homomeric GluR6(R).
Figure 1.
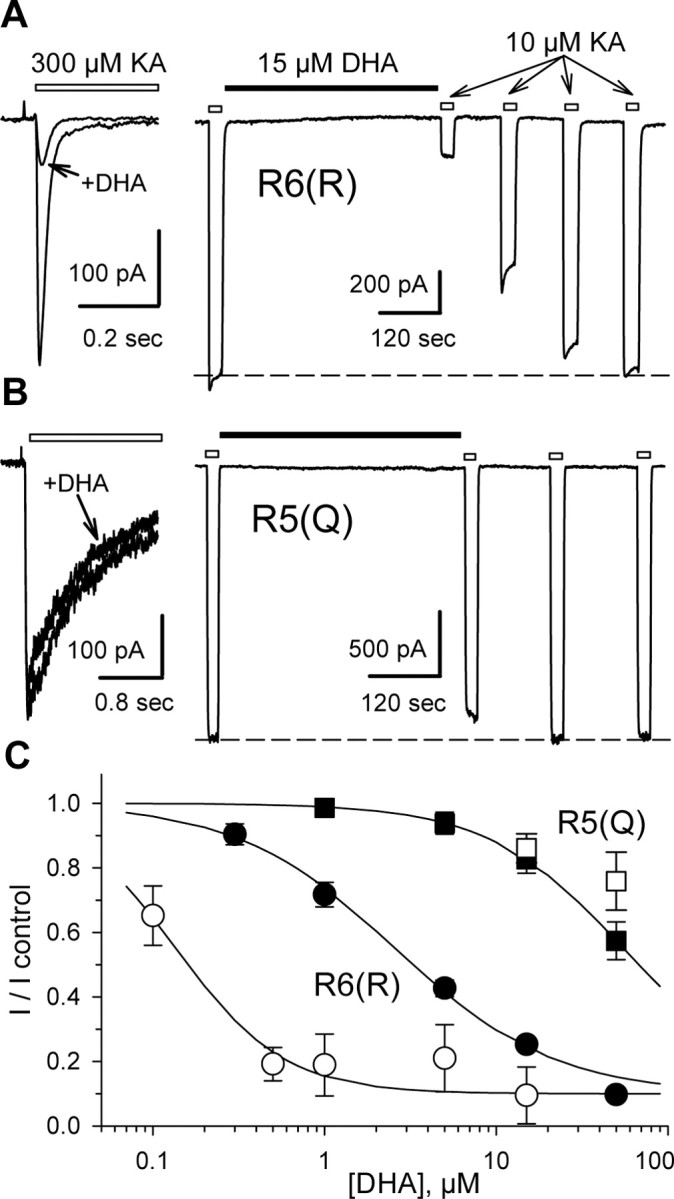
Differential block of homomeric GluR6(R) and GluR5(Q). A, B, HEK 293 cells transfected with GluR6(R) (A) or GluR5-2a(Q) (B) were tested with 300 μm kainate (KA) before (left) or 10 μm kainate after (right) treatment with Con A. Left, Superimposed whole-cell currents evoked by rapid application of kainate, indicated by the open bar, before and after exposure to 15 μm DHA. Right, Current evoked by kainate applications indicated by the open bars, before and after exposure to 15 μm DHA, as indicated by the solid bar. C, Inhibition as a function of DHA concentration. Current evoked by kainate (mean ± SEM; n = 5-15 cells per point) immediately after exposure to DHA is plotted as a fraction of the current before DHA. Smooth curves are the best fit of the following: I/I control = 0.1 + (0.9/(1 + ([DHA]/IC50)n̂)), where IC50 is the DHA concentration producing half-maximal inhibition, and n is the slope factor. R6(R) without Con A (○), IC50 = 136 nm, n = 1.4; with Con A (•), IC50 = 2.6 μm, n = 0.94. R5(Q) without Con A(□) and after Con A treatment (▪), IC50 = 55 μm; n = 1.1.
The GluR5 and GluR6 subunits display ∼75% amino acid identity, with the exact percentage depending on which alternately spliced version of GluR5 is used for comparison (Hollmann and Heinemann, 1994). This high degree of homology has facilitated efforts to identify key amino acid residues that underlie differences in physiology and/or pharmacology between these two subunits (Swanson et al., 1997). As an initial step toward defining the region, or regions, that allow for potent fatty acid inhibition of GluR6 channels, we tested four different chimeric subunits, generated by Swanson et al. (1997), that combined N- and C-terminal segments from GluR5 and GluR6. All four of these chimeric subunits code for a glutamine residue at the Q/R site, and all four of them, when expressed as homomeric receptors, displayed <50% inhibition by 15 μm DHA (Fig. 2D).
Figure 2.
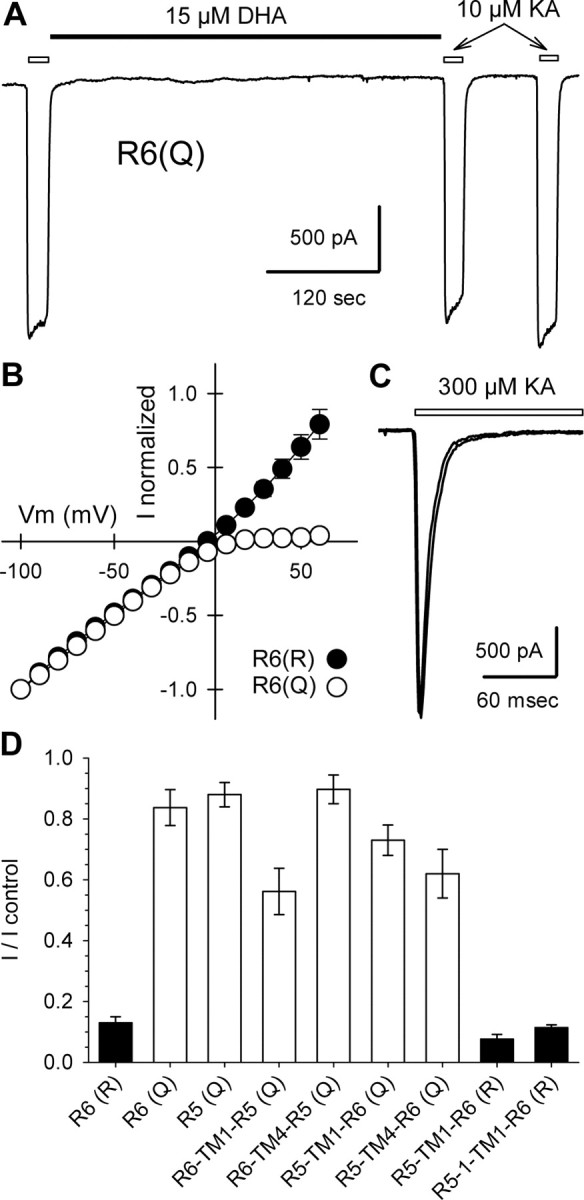
Homomeric GluR6(Q) resists inhibition. A, Current evoked by kainate applications indicated by open bars, before and after exposure to 15 μm DHA, as indicated by the solid bar. B, Normalized currents as a function of holding potential for cells transfected with R6(R) (n = 8 cells) or R6(Q) (n = 4 cells). C, Superimposed whole-cell currents evoked by rapid application of 300 μm kainate, indicated by the open bar, before and after exposure to 15 μm DHA. D, Current evoked by 1 or 10 μm kainate immediately after exposure to 15 μm DHA as a fraction of control current before DHA (3-9 cells per construct).
Homomeric GluR6(Q) resists inhibition
The failure of DHA to block the GluR5/GluR6 hybrid channel subunits suggested that susceptibility to inhibition might depend on the presence of an R at the Q/R site. Previous work has shown that GluR5(R) subunits function poorly as homomeric channels when transfected into heterologous cells (Sommer et al., 1992; Partin et al., 1993; Swanson et al., 1996), whereas homomeric GluR6 receptors produce functional channels in either the Q or R form (Egebjerg and Heinemann, 1993; Köhler et al., 1993; Partin et al., 1993). Therefore, we prepared GluR6(Q) cDNA and tested for inhibition by DHA. As shown in Figure 2, A and D, exposure to 15 μm DHA produced only minimal inhibition of homomeric GluR6(Q) receptors, compared with the strong block of GluR6(R). The effect of DHA on GluR6(Q) was weak either with (Fig. 2A) or without (Fig. 2C) previous exposure to Con A. These results demonstrate the requirement for arginine at the Q/R site of GluR6 in order for strong inhibition to occur, and they suggest that the presence of a glutamine residue at this location in GluR5-2a(Q), and in the four hybrid channel subunits, likely explains their reduced susceptibility to inhibition. To confirm this point, we generated a Q to R point mutation of the R5-TM1-R6 hybrid channel subunit. This hybrid links the N-terminal domain of GluR5-2, up to the first transmembrane domain (TM1), with the channel core and C-terminal domain of GluR6(Q) (Swanson et al., 1997). Homomeric R5-TM1-R6(R) channels were strongly blocked by DHA (Fig. 2D). We also observed strong inhibition of homomeric R5-1-TM1-R6(R) channels (Fig. 2D), which include the GluR5-1 N-terminal domain insert of 15 amino acids encoded by an alternately spliced exon (Bettler et al., 1990).
Strong block of heteromeric GluR5(R) plus GluR6(R) receptors
To test whether strong inhibition by fatty acids was restricted to homomeric GluR6(R) receptors, we transfected HEK 293 cells with cDNA for both the GluR5(R) and GluR6(R) subunits. These two subunits form heteromeric receptors (Bortolotto et al., 1999; Cui and Mayer, 1999; Paternain et al., 2000) that are sensitive to the GluR5-selective agonist (RS)-2-amino-3-(3-hydroxy-5-tertbutyl-isoxazol-4-yl)propanoic acid (ATPA). Homomeric GluR6 receptors are not affected by ATPA, and, as mentioned above, GluR5(R) subunits produce homomeric channels with only marginal functionality (Sommer et al., 1992; Partin et al., 1993; Swanson et al., 1996). Thus, activation by ATPA allows for relatively unambiguous evaluation of current through heteromeric GluR5(R) plus GluR6(R) receptors. As shown in Figure 3, application of DHA to cells cotransfected with GluR5(R) and GluR6(R) reduced the current evoked by ATPA or kainate by 70 ± 6 and 69 ± 5%, respectively. Although the stoichiometry of heteromeric GluR5(R) plus GluR6(R) receptors is not known, their strong block by DHA (Fig. 3) demonstrates that simple inclusion of GluR5 subunits in a heteromeric assembly does not preclude fatty acid inhibition.
Figure 3.
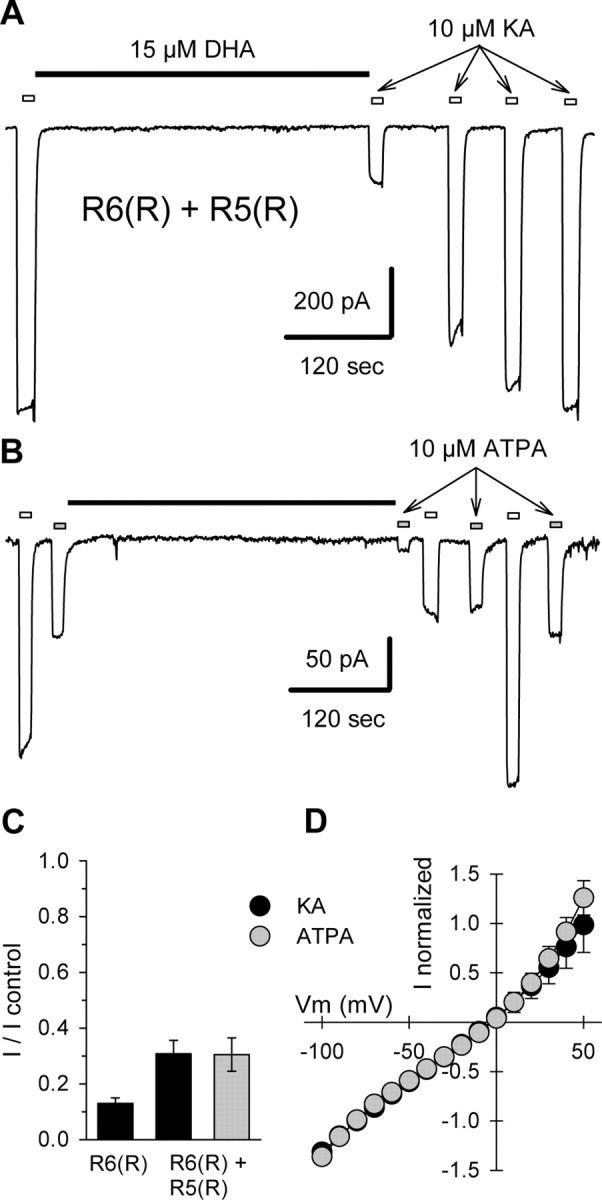
Strong block of heteromeric GluR5(R)/GluR6(R). A, Current evoked by 10 μm kainate (KA) applications indicated by open bars, before and after exposure to 15 μm DHA, as indicated by the solid bar. B, Current evoked by alternating applications of 10 μm kainate (open bars), and 10 μm ATPA (gray bars), before and after exposure to 15 μm DHA. C, Current evoked by 10 μm kainate (n = 9 cells) or 10 μm ATPA (n = 7 cells) immediately after exposure to 15 μm DHA as a fraction of control current before DHA. D, Normalized currents evoked by 10 μm kainate (n = 4 cells) or 10 μm ATPA (n = 7 cells) as a function of holding potential for cells transfected with R6(R) plus R5(R).
Weak inhibition of Q/R heteromeric receptors
Among kainate receptor subunits, only GluR5 and GluR6 can undergo editing at the Q/R site. The remaining subunits, GluR7, kainate receptor subunit 1 (KA1), and KA2, all encode for a glutamine at the homologous location with in the pore loop. To test for inhibition of receptors that included both edited and unedited subunits, we cotransfected HEK 293 cells with several different combinations of subunit cDNAs (Fig. 4). In cells that received GluR6(Q) plus GluR5(R) or GluR6(R) plus GluR5(Q), exposure to 15 μm DHA produced only 9 and 11% inhibition, respectively. In both of these cases, the currents evoked by ATPA were similarly resistant to blocking by DHA (data not shown). Moreover, currents elicited by either kainate or ATPA displayed relatively linear current-voltage relationships (Fig. 4C), suggesting that there was little contribution by homomeric unedited GluR6(Q) or GluR5(Q) channels, which exhibit inward rectification (Fig. 2 B).
Figure 4.
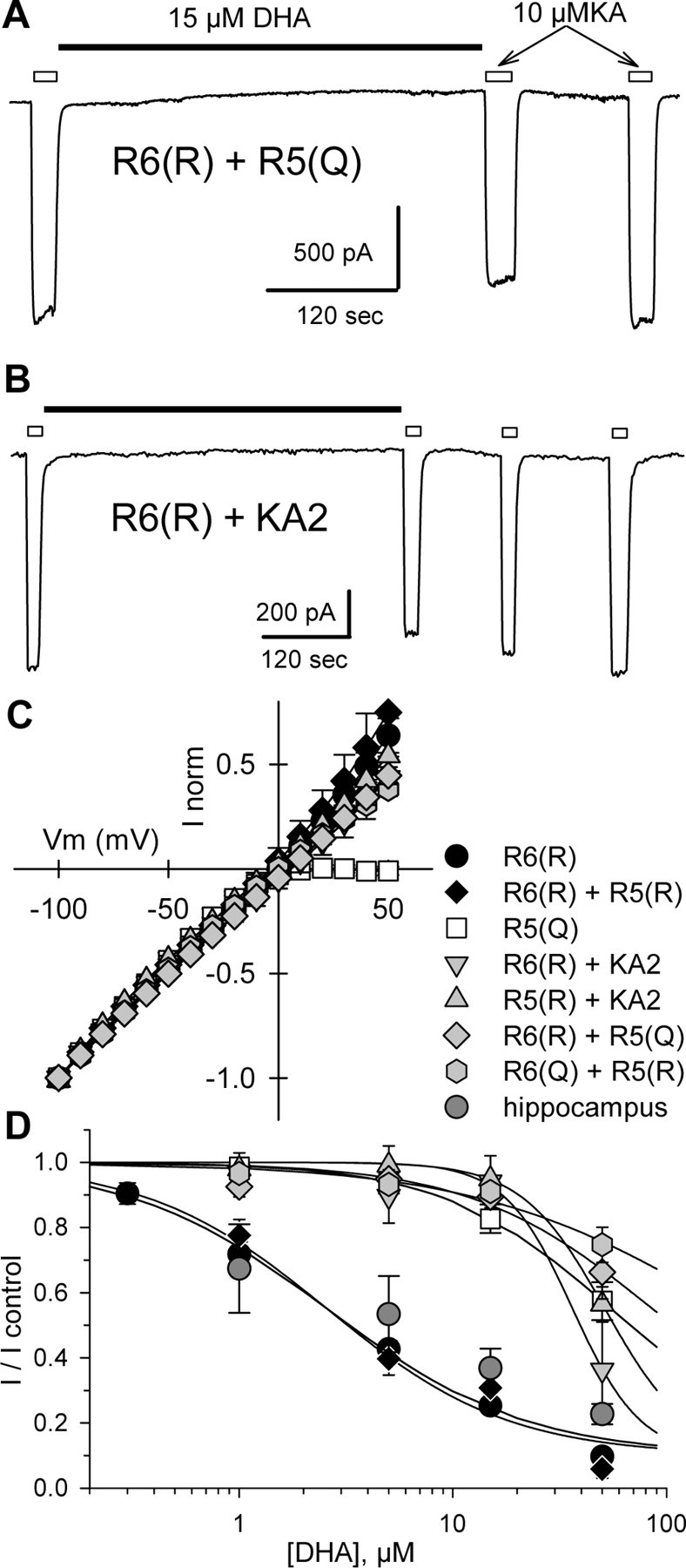
Weak inhibition of Q/R heteromeric receptors. A, B, Current evoked by 10 μm kainate applications indicated by open bars, before and after exposure to 15 μm DHA, as indicated by the solid bar. C, Normalized currents as a function of holding potential for cells transfected with R6(R) (n = 8 cells), R6(R) plus R5(R) (n = 4 cells), R5(Q) (n = 4 cells), R6(R) plus KA2 (n = 5 cells), R5(R) plus KA2 (n = 6 cells), R6(R) plus R5(Q) (n = 8 cells), or R6(Q) plus R5(R) (n = 6 cells). D, Inhibition as a function of DHA concentration. Current evoked by 10 μm kainate (n = 3-9 cells per point) immediately after exposure to DHA is plotted as a fraction of the current before DHA. Smooth curves are the best fit of the following: I/I control = 0.1 + (0.9/(1 + ([DHA]/IC50)n̂)), where IC50 is the DHA concentration producing half-maximal inhibition, and n is the slope factor. R6(R) plus R5(R), IC50 = 2.6 μm, n = 1.02; R6(R) plus KA2, IC50 = 36.4 μm, n = 2.73; R5(R) plus KA2, IC50 = 51.5 μm, n = 2.24; R6(R) plus R5(Q), IC50 = 86.1 μm, n = 1.01; R6(Q) plus R5(R), IC50 = 192.3 μm, n = 0.74. Data for R6(R) and R5(Q) are replotted from Figure 1C. Data from cultured rat hippocampal neurons (hippocampus, dark gray circles) were adapted from Wilding et al. (1998).
In addition to GluR5 plus GluR6 heteromeric channels, we also examined cells that received the KA2 subunit together with either GluR5(R) or GluR6(R). KA2 does not form functional homomeric channels but can combine with GluR5 or GluR6 into functional heteromeric receptors (Herb et al., 1992; Sakimura et al., 1992). Cells cotransfected with either GluR5(R) plus KA2 or GluR6(R) plus KA2 displayed linear current-voltage relationships and only weak inhibition by 15 μm DHA. Figure 4D shows concentration-inhibition relationships for seven different homomeric or heteromeric subunit combinations. For comparison, data points for DHA inhibition of kainate receptor-mediated current in cultured hippocampal neurons taken from our previous study (Wilding et al., 1998) have also been plotted. These graphs highlight the difference in potency observed at receptors in which all subunits are edited versus receptors that include one or more unedited subunits. Notably, our previous results in hippocampal neurons (Wilding et al., 1998) agree best with the potency of inhibition at fully edited receptors.
We also examined the effect of 15 μm DHA on cells that received equal cDNA mixtures of GluR6(Q) plus GluR6(R) and GluR5(Q) plus GluR5(R) (Fig. 5). In this case, similar to the GluR5/GluR6 heteromeric receptors just described, exposure to DHA produced only weak inhibition of whole-cell currents. It is more difficult to rule out the formation of subpopulations of channels that included only Q or only R subunits; however, we note that the linear whole-cell current-voltage relationships for both the GluR6(Q) plus GluR6(R) and GluR5(Q) plus GluR5(R) transfected cells argue against a significant contribution by fully unedited channels (Fig. 5C). Furthermore, the weakness of block in cells that received the GluR5(Q) plus GluR5(R) or GluR6(Q) plus GluR6(R) mixtures (Fig. 5D) suggests that few fully edited channels were produced, because these should have been strongly inhibited (Fig. 1).
Figure 5.
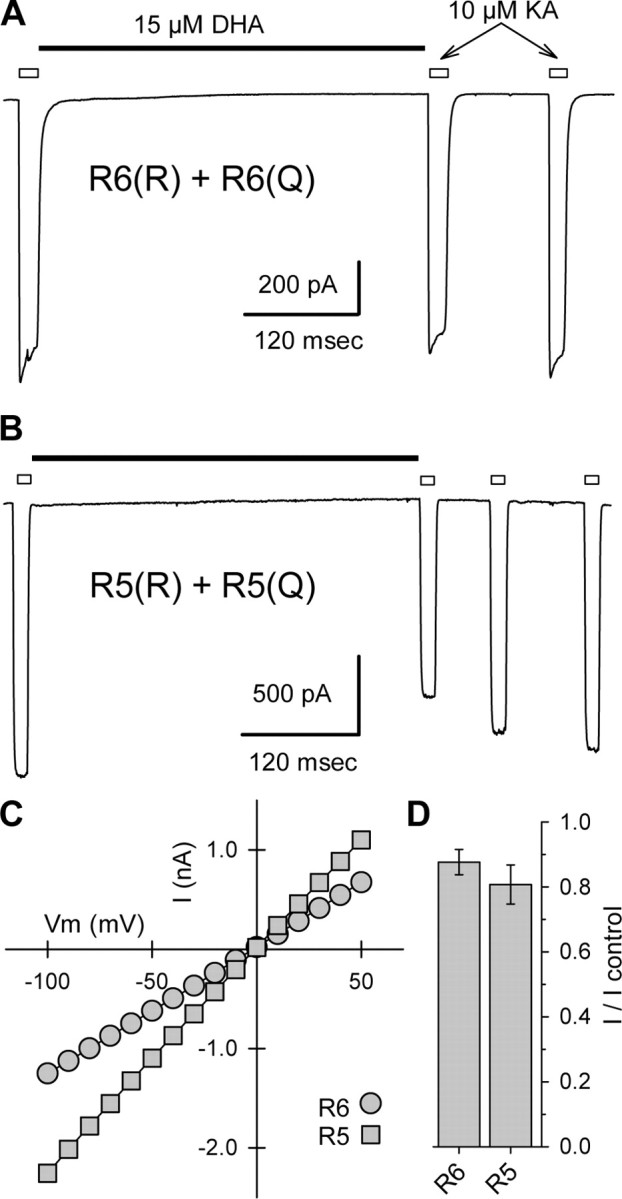
Weak inhibition of GluR6(Q/R) and GluR5(Q/R) receptors. A, B, Current evoked by 10 μm kainate applications indicated by open bars, before and after exposure to 15 μm DHA, as indicated by the solid bar. C, Currents plotted as a function of holding potential for the two cells shown in A and B. D, Current evoked by 10 μm kainate immediately after exposure to 15 μm DHA as a fraction of control current before DHA (n = 6 cells for each subunit combination).
Voltage-independent, noncompetitive inhibition
Our previous work on kainate receptor modulation by fatty acids used rapid agonist applications to test cells that had not been exposed to Con A. In those experiments, fatty acid inhibition of peak kainate-evoked current was not surmounted by increasing the agonist concentration or by depolarizing cell membrane potentials to +40 mV. Because our results in the present study implicated the Q/R site within the channel pore as a critical determinant of inhibitory potency, we examined the effect of voltage and agonist concentration more rigorously on homomeric GluR6(R) channels in cells that had been exposed to Con A. In most cells (Fig. 6A), the onset of block was well fit by the sum of two exponential decay functions, whereas a single exponential was sufficient to describe recovery from inhibition. As shown in Figure 6, there were no significant differences in the kinetics or the steady-state level of block at holding potentials of -70 and +40 mV. Thus, in contrast to block of GluR6(Q) channels by polyamines, which displays modest voltage dependence for inhibition from the extracellular solution (Bähring et al., 1997) and much stronger voltage dependence for cytoplasmic exposure (Bowie and Mayer, 1995; Kamboj et al., 1995), we observe essentially no voltage dependence to inhibition by extracellularly applied DHA. Figure 7 illustrates the dependence of inhibition on both agonist and antagonist concentration, confirming the non-competitive nature of block. Smooth curves in this figure represent the best simultaneous fit of all data points to Equation 2 (see Materials and Methods), a modified form of the Hill equation that describes fatty acid inhibition as a monotonic reduction in maximal current.
Figure 6.

Voltage-independent inhibition of R6(R). A, Currents evoked by 1 μm kainate (open bar) at -70 and +40 mV were strongly blocked by 15 μm DHA (solid bar). Superimposed lines show the best fits of the sum of two exponentials plus a constant to block onset or one exponential plus a constant to recovery from block. B, Steady-state block by 15 μm DHA at -70 mV (open bar; n = 18 cells) and +40 mV (shaded bar; n = 14 cells). C, Time constants (left) and relative amplitudes (right) for exponential fits to onset of block and recovery from block. Error bars represent SEM.
Figure 7.

Noncompetitive inhibition of R6(R). A, Currents evoked by 10 mm kainate (open bars) in control solution or by 160 nm through 160 μm kainate (shaded bars) during exposure to 15 μm DHA (solid bar). B, Current (mean ± SEM; n = 6-14 cells per point) as a faction of the control response to 10 mm kainate versus kainate concentration. R6(R) in control solution (○) or during exposure to 0.3 μm (♦), 1 μm (▪), 5 μm (▴), or 15 μm (•) DHA. Smooth curves are the best fit of all data points to Equation 2. Error bars represent SEM.
Discussion
Our results show that inhibition of kainate receptors by DHA depends on the editing status at the Q/R site within the channel pore. In contrast to the potent block of unedited channels by internal and external polyamines (Bowie and Mayer, 1995; Kamboj et al., 1995; Bähring et al., 1997; Mott et al., 2003), we find that extracellular fatty acid applications only produce strong inhibition of recombinant kainate receptor channels that are composed of fully edited subunits. Heteromeric GluR5(Q)/GluR6(R) or GluR5(R)/GluR6(Q) channels were inhibited much less than were fully edited GluR5(R)/GluR6(R) heteromeric receptors or homomeric GluR6(R) receptors. In addition, heteromeric combination of the KA2 subunit, which encodes a glutamine at the location homologous to the Q/R site, with either GluR5(R) or GluR6(R) produced channels that displayed only weak inhibition by DHA. Importantly, the potency of inhibition is nearly identical at native receptors in cultured neurons and at fully edited recombinant receptors that were exposed to Con A.
Although we only tested combinations that include three of the five known kainate receptor subunits, our results collectively provide strong evidence that inclusion of an unedited subunit dramatically weakens the ability of DHA to inhibit the channels. This conclusion is surprising for several reasons. First, our previous study documented relatively strong inhibition of whole-cell currents mediated by native kainate receptors in cultured rat hippocampal cells and in sensory neurons from dorsal root ganglia (Wilding et al., 1998). The maximum level of inhibition (∼90%) produced by 50 μm DHA, the highest concentration tolerated, was nearly identical in native receptors as in recombinant GluR6(R) receptors (Wilding et al., 1998), although the potency of block was at least 10-fold greater for GluR6(R) expressed in HEK 293 cells without Con A treatment (IC50 ∼ 136 nm) than for native receptors in neurons (IC50 ∼ 10 μm). In the present study, the concentration dependence for DHA inhibition of homomeric GluR6(R) or heteromeric GluR5(R)/GluR6(R) channels after exposure to Con A agreed well with the potency of block demonstrated previously at native receptors (Wilding et al., 1998). In contrast, blockade of recombinant channels that included a subunit encoding a glutamine at the Q/R site showed <50% inhibition by DHA (IC50 > 50 μm). Together, these results suggest that few receptors in cultured neurons include unedited subunits or that the specific subunit stoichiometry of native receptors allows for inhibition by DHA, despite the inclusion of subunits with a Q at the Q/R site. A third possibility is that interactions with auxiliary proteins, which are present in neurons but absent from HEK 293 cells, may modify the ability of DHA to regulate native kainate receptor channels, rendering native receptors with one or more subunit bearing a Q at the Q/R site more sensitive to DHA inhibition than recombinant channels in HEK 293 cells.
The strong dependence of DHA block on Q/R site editing is also surprising because the inhibition displays little or no voltage dependence (Wilding et al., 1998). Channel block by intracellular and extracellular polyamines, which preferentially affect channels lacking an R within the pore, displays significant voltage dependence (Bowie and Mayer, 1995; Kamboj et al., 1995), although dependence on voltage is weaker for inhibition by extracellular versus intracellular polyamines (Bähring et al., 1997). If penetration of the negatively charged fatty acid carboxyl group into the channel pore was required for interaction with the positively charged side chains of arginine at the Q/R site, potency of block would be expected to change as the voltage gradient along the axis of the pore varied. In fact, we see no change in the extent of block between -70 and +40 mV. These considerations suggest that, although arginine residues are required at the Q/R site in order for strong block to occur, the actual site of interaction between fatty acids and the channel may not be located within the pore. Instead, allosteric interactions at a site that does not require penetration of the charged carboxyl group of the fatty acid within the membrane field may be sufficient to render fully edited channels nonconducting but less effective at blocking conductance through channels that include one or more glutamine residues at the Q/R site. Such a mechanism predicts that, in addition to the Q/R site, there will be other structural determinants of DHA inhibition. This might explain minor differences in block among the chimeric subunits and between GluR5(R)/GluR6(R) heteromers compared with GluR6(R) alone. The relatively rapid recovery from inhibition suggests a peripheral site of action by DHA, although speedy recovery requires addition of BSA (Wilding et al., 1998) that can extract fatty acids from membranes.
Editing efficiency varies among glutamate receptor subunits. GluR2 mRNA editing is highly efficient (Seeburg and Hartner, 2003). Cells expressing high GluR2 levels exhibit AMPA receptors with low calcium permeability, whereas cells with calcium-permeable AMPA receptors produce relatively less GluR2 (Geiger et al., 1995). In contrast, GluR5 and GluR6 are edited with lower efficiency, resulting in a complex mixture of edited and unedited subunits even within individual cells (Pemberton et al., 1998). Moreover, the editing of GluR5 and GluR6 increases over the course of development (Bernard and Khrestchatisky, 1994). In rodents, unedited subunits predominate in embryonic and early postnatal cells but progressively give way to edited subunits within the first 2 weeks of life to reach the adult ratios of 50-60% GluR5(Q)/40-50% GluR5(R) and 15-30% GluR6(Q)/70-85% GluR6(R) (Sommer et al., 1991; Bernard and Khrestchatisky, 1994; Paschen et al., 1997).
In addition to controlling block by DHA and polyamines, editing at the Q/R site regulates channel conductance and ion selectivity (Seeburg and Hartner, 2003) and plays a role in subunit assembly into functional tetramers (Greger et al., 2003). Channels lacking an edited subunit exhibit large unitary conductance levels (up to 25 pS) (Swanson et al., 1996) and are permeable to monovalent cations and calcium (Burnashev et al., 1995; Swanson et al., 1996). Inclusion of edited subunits reduces conductance and calcium permeability, culminating in fully edited channels, which display the lowest estimated unitary conductance (∼200-250 nS) (Howe, 1996; Swanson et al., 1996). Ion-replacement studies indicate that channels composed only of edited (R) subunits are permeable to both cations and anions. In addition, experiments using organic cations suggest that there is little difference in the physical pore diameter of channels made from edited versus unedited subunits (Burnashev et al., 1996). Channels composed only of edited (R) subunits or only of unedited (Q) subunits both exhibit apparent pore diameters of 0.75-0.76 nm (Burnashev et al., 1996). Thus, the presence of positively charged arginine side chains appears to constitute an energetic barrier to ion transit rather than a change in physical dimensions of the narrowest region of the pore. Our analysis of the agonist and antagonist concentration dependence of inhibition is consistent with a reduction in open probability by DHA. In addition, the ability of Con A to increase steady-state open probability and decrease DHA potency suggests that DHA may reduce stability of the open state. In contrast, our results do not rule out the possibility of a change in unitary conductance. Indeed, the fact that DHA inhibition is selective for channels that already exhibit the lowest conductance suggests that an additional allosteric decrease in conductance might be a plausible mechanism of block.
DHA is a major component of brain plasma membrane phospholipids (Salem et al., 2001), representing up to 30% of PE and PS fatty acyl chains in synaptosomal (Breckenridge et al., 1972) and synaptic vesicle (Breckenridge et al., 1973) membranes. The high flexibility of unsaturated acyl chains contributes to membrane fluidity and is thought to allow for close apposition between integral membrane proteins and surrounding lipids (Eldho et al., 2003; Feller et al., 2003). Various stimuli elicit mobilization of DHA and AA as free fatty acids (Kim et al., 1999). Interestingly, analysis of synaptosomes and cultured cells suggests that astrocytes release both AA and DHA into the surrounding medium, whereas neurons preferentially release AA but retain DHA (Kim et al., 1999). The effects of cis-unsaturated fatty acids on ion channels have been proposed to involve changes in membrane curvature, comparable with mechanical deformations that affect the gating of several different ion channel types (Casado and Ascher, 1998; Patel et al., 2001). Direct contact with channels by free DHA or AA at the protein-lipid interface might also alter gating properties. Structural analysis indicates that lipid moieties cocrystallize with and are required for correct refolding and functional operation of purified bacterial KcsA channels (Valiyaveetil et al., 2002), which exhibit homology to glutamate receptor channels. Such specific contacts might be sensitive to free DHA or AA within the membrane and thereby alter channel operation.
Dietary depletion of DHA, particularly during early development, is known to change the membrane composition (Salem et al., 2001) and might consequently alter the function of kainate receptors and other sensitive channels. Free DHA and AA have direct effects on a number of different channel types. Exogenously applied AA has recently been reported to produce direct inhibition of presynaptic kainate receptors in hippocampal area CA1 (Cunha et al., 2004). In addition, exposure to exogenous DHA produces a variety of specific changes in hippocampal synaptic transmission and excitability (Itokazu et al., 2000; Young et al., 2000). Extending these studies to evaluate the role of fatty acids released from endogenous sources will require development of better pharmacological reagents to mimic or inhibit the direct actions of DHA and AA on channel function. Site-directed mutagenesis of channel subunit cDNA has yielded important insights into the structural basis for polyamine regulation of GluR6 (Panchenko et al., 2001; Mott et al., 2003). Our discovery that Q/R site editing also controls fatty acid inhibition of kainate receptors suggests that a similar approach may provide additional understanding of the mechanism of channel regulation by DHA. Such studies could provide new information about the basic operation of glutamate receptor channels (Wollmuth and Sobolevsky, 2004) and aid in efforts to identify more selective reagents to test for channel regulation by endogenous fatty acids.
Footnotes
This work was supported by National Institutes of Health Grant NS30888. We are grateful to Fernanda Laezza for critical reading of this manuscript; to Steve Heinemann, Geoffry Swanson, and Peter Seeburg for providing channel subunit cDNAs; and to David Clapham for L3T4 cDNA.
Correspondence should be addressed to James E. Huettner, Department of Cell Biology and Physiology, Washington University Medical School, 660 South Euclid Avenue, St. Louis, MO 63110. E-mail: huettner@cellbio.wustl.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259470-09$15.00/0
References
- Bähring R, Bowie D, Benveniste M, Mayer ML (1997) Permeation and block of rat GluR6 glutamate receptor channels by internal and external polyamines. J Physiol (Lond) 502: 575-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard A, Khrestchatisky M (1994) Assessing the extent of RNA editing in the TMII regions of GluR5 and GluR6 kainate receptors during rat brain development. J Neurochem 62: 2057-2060. [DOI] [PubMed] [Google Scholar]
- Bettler B, Boulter J, Hermans-Borgmeyer I, O'Shea-Greenfield A, Deneris E, Moll C, Borgmeyer U, Hollmann M, Heinemann S (1990) Cloning of a novel glutamate receptor subunit, GluR5: expression in the nervous system during development. Neuron 5: 583-595. [DOI] [PubMed] [Google Scholar]
- Bortolotto ZA, Clarke VR, Delany CM, Parry MC, Smolders I, Vignes M, Ho KH, Miu P, Brinton BT, Fantaske R, Ogden A, Gates M, Ornstein PL, Lodge D, Bleakman D, Collingridge GL (1999) Kainate receptors are involved in synaptic plasticity. Nature 402: 297-301. [DOI] [PubMed] [Google Scholar]
- Bowie D, Mayer ML (1995) Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron 15: 453-462. [DOI] [PubMed] [Google Scholar]
- Breckenridge WC, Gombos G, Morgan IG (1972) The lipid composition of adult rat brain synaptosomal plasma membranes. Biochim Biophys Acta 266: 695-707. [DOI] [PubMed] [Google Scholar]
- Breckenridge WC, Morgan IG, Zanetta JP, Vincendon G (1973) Adult rat brain synaptic vesicles. II. Lipid composition. Biochim Biophys Acta 320: 681-686. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Zhou Z, Neher E, Sakmann B (1995) Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA and kainate receptor subtypes. J Physiol (Lond) 485: 403-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnashev N, Villarroel A, Sakmann B (1996) Dimensions and ion selectivity of recombinant AMPA and kainate receptor channels and their dependence on Q/R site residues. J Physiol (Lond) 496: 165-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado M, Ascher P (1998) Opposite modulation of NMDA receptors by lysophospholipids and arachidonic acid: common features with mechanosensitivity. J Physiol (Lond) 513: 317-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C, Mayer ML (1999) Heteromeric kainate receptors formed by the coassembly of GluR5, GluR6, and GluR7. J Neurosci 19: 8281-8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA, Ribeiro JA, Malva JO (2004) Presynaptic kainate receptors modulating glutamatergic transmission in the rat hippocampus are inhibited by arachidonic acid. Neurochem Int 44: 371-379. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7-61. [PubMed] [Google Scholar]
- Egebjerg J, Heinemann SF (1993) Ca2+ permeability of unedited and edited versions of the kainate selective glutamate receptor GluR6. Proc Natl Acad Sci USA 90: 755-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldho NV, Feller SE, Tristram-Nagle S, Polozov IV, Gawrisch K (2003) Polyunsaturated docosahexaenoic vs docosapentaenoic acid-differences in lipid matrix properties from the loss of one double bond. J Am Chem Soc 125: 6409-6421. [DOI] [PubMed] [Google Scholar]
- Feller SE, Gawrisch K, Woolf TB (2003) Rhodopsin exhibits a preference for solvation by polyunsaturated docosohexaenoic acid. J Am Chem Soc 125: 4434-4435. [DOI] [PubMed] [Google Scholar]
- Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C, Lazdunski M (1996) Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J 15: 6854-6862. [PMC free article] [PubMed] [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H (1995) Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 15: 193-204. [DOI] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Kong X, Ziff EB (2003) AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40: 763-774. [DOI] [PubMed] [Google Scholar]
- Hamano H, Nabekura J, Nishikawa M, Ogawa T (1996) Docosahexanoic acid reduces GABA response in substantia nigra neuron of rat. J Neurophysiol 75: 1264-1270. [DOI] [PubMed] [Google Scholar]
- Herb A, Burnashev N, Werner P, Sakmann B, Wisden W, Seeburg PH (1992) The KA-2 subunit of excitatory amino acid receptors shows widespread expression in brain and forms ion channels with distantly related subunits. Neuron 8: 775-785. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S (1994) Cloned glutamate receptors. Annu Rev Neurosci 17: 31-108. [DOI] [PubMed] [Google Scholar]
- Honore E, Barhanin J, Attali B, Lesage F, Lazdunski M (1994) External blockade of the major cardiac delayed-rectifier K+ channel (Kv1.5) by polyunsaturated fatty acids. Proc Natl Acad Sci USA 91: 1937-1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe JR (1996) Homomeric and heteromeric ion channels formed from the kainate-type subunits GluR6 and KA2 have very small, but different, unitary conductances. J Neurophysiol 76: 510-519. [DOI] [PubMed] [Google Scholar]
- Huettner JE (1990) Glutamate receptor channels in rat DRG neurons: activation by kainate and quisqualate blockade of desensitization by Con A. Neuron 5: 255-266. [DOI] [PubMed] [Google Scholar]
- Itokazu N, Ikegaya Y, Nishikawa M, Matsuki N (2000) Bidirectional actions of docosahexaenoic acid on hippocampal neurotransmissions in vivo. Brain Res 862: 211-216. [DOI] [PubMed] [Google Scholar]
- Kamboj SK, Swanson GT, Cull-Candy SG (1995) Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. J Physiol (Lond) 486: 297-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keros S, McBain CJ (1997) Arachidonic acid inhibits transient potassium currents and broadens action potentials during electrographic seizures in hippocampal pyramidal and inhibitory interneurons. J Neurosci 17: 3476-3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler M, Burnashev N, Sakmann B, Seeburg PH (1993) Determinants of Ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: diversity by RNA editing. Neuron 10: 491-500. [DOI] [PubMed] [Google Scholar]
- Kim HY, Edsall L, Garcia M, Zhang H (1999) The release of polyunsaturated fatty acids and their lipoxygenation in the brain. Adv Exp Med Biol 447: 75-85. [DOI] [PubMed] [Google Scholar]
- Miller B, Sarantis M, Traynelis SF, Attwell D (1992) Potentiation of NMDA receptor currents by arachidonic acid. Nature 355: 722-725. [DOI] [PubMed] [Google Scholar]
- Mott DD, Washburn MS, Zhang S, Dingledine RJ (2003) Subunit-dependent modulation of kainate receptors by extracellular protons and polyamines. J Neurosci 23: 1179-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Kudo I (2002) Phospholipase A2. J Biochem (Tokyo) 131: 285-292. [DOI] [PubMed] [Google Scholar]
- Nishikawa M, Kimura S, Akaike N (1994) Facilitatory effect of docosahexaenoic acid on N-methyl-d-aspartate response in pyramidal neurones of rat cerebral cortex. J Physiol (Lond) 475: 83-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B (2004) Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science 304: 265-270. [DOI] [PubMed] [Google Scholar]
- Panchenko VA, Glasser CR, Mayer ML (2001) Structural similarities between glutamate receptor channels and K+ channels examined by scanning mutagenesis. J Gen Physiol 117: 345-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A (1993) Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin A. Neuron 11: 1069-1082. [DOI] [PubMed] [Google Scholar]
- Paschen W, Schmitt J, Gissel C, Dux E (1997) Developmental changes of RNA editing of glutamate receptor subunits GluR5 and GluR6: in vivo versus in vitro. Dev Brain Res 98: 271-280. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Lazdunski M, Honore E (2001) Lipid and mechano-gated 2P domain K(+) channels. Curr Opin Cell Biol 13: 422-428. [DOI] [PubMed] [Google Scholar]
- Paternain AV, Herrera MT, Nieto MA, Lerma J (2000) GluR5 and GluR6 kainate receptor subunits coexist in hippocampal neurons and coassemble to form functional receptors. J Neurosci 20: 196-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton KE, Belcher SM, Ripellino JA, Howe JR (1998) High-affinity kainate-type ion channels in rat cerebellar granule cells. J Physiol (Lond) 510: 401-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poling JS, Vicini S, Rogawski MA, Salem Jr N (1996) Docosahexaenoic acid block of neuronal voltage-gated K+ channels: subunit selective antagonism by zinc. Neuropharmacology 35: 969-982. [DOI] [PubMed] [Google Scholar]
- Sakimura K, Morita T, Kushiya E, Mishina M (1992) Primary structure and expression of the gamma 2 subunit of the glutamate receptor channel selective for kainate. Neuron 8: 267-274. [DOI] [PubMed] [Google Scholar]
- Salem Jr N, Litman B, Kim HY, Gawrisch K (2001) Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids 36: 945-959. [DOI] [PubMed] [Google Scholar]
- Schwartz RD, Yu X (1992) Inhibition of GABA-gated chloride channel function by arachidonic acid. Brain Res 585: 405-410. [DOI] [PubMed] [Google Scholar]
- Seeburg PH, Hartner J (2003) Regulation of ion channel/neurotransmitter receptor function by RNA editing. Curr Opin Neurobiol 13: 279-283. [DOI] [PubMed] [Google Scholar]
- Six DA, Dennis EA (2000) The expanding superfamily of phospholipase A(2) enzymes: classification and characterization. Biochim Biophys Acta 1488: 1-19. [DOI] [PubMed] [Google Scholar]
- Sommer B, Kohler M, Sprengel R, Seeburg PH (1991) RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67: 11-19. [DOI] [PubMed] [Google Scholar]
- Sommer B, Burnashev N, Verdoorn TA, Keinänen K, Sakmann B, Seeburg PH (1992) A glutamate receptor channel with high affinity for domoate and kainate. EMBO J 11: 1651-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson GT, Feldmeyer D, Kaneda M, Cull-Candy SG (1996) Effect of RNA editing and subunit co-assembly on single-channel properties of recombinant kainate receptors. J Physiol (Lond) 492: 129-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson GT, Gereau RW, Green T, Heinemann SF (1997) Identification of amino acid residues that control functional behavior in GluR5 and GluR6 kainate receptors. Neuron 19: 913-926. [DOI] [PubMed] [Google Scholar]
- Valiyaveetil FI, Zhou Y, MacKinnon R (2002) Lipids in the structure, folding, and function of the KcsA K+ channel. Biochemistry 41: 10771-10777. [DOI] [PubMed] [Google Scholar]
- Villarroel A, Schwarz TL (1996) Inhibition of the Kv4 (Shal) family of transient K+ currents by arachidonic acid. J Neurosci 16: 2522-2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding TJ, Chai YH, Huettner JE (1998) Inhibition of rat neuronal kainate receptors by cis-unsaturated fatty acids. J Physiol (Lond) 513: 331-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmuth LP, Sobolevsky AI (2004) Structure and gating of the glutamate receptor ion channel. Trends Neurosci 27: 321-328. [DOI] [PubMed] [Google Scholar]
- Wong LA, Mayer ML (1993) Differential modulation by cyclothiazide and concanavalin A of desensitization at AMPA- and kainate-preferring glutamate receptors. Mol Pharmacol 44: 504-510. [PubMed] [Google Scholar]
- Young C, Gean PW, Chiou LC, Shen YZ (2000) Docosahexaenoic acid inhibits synaptic transmission and epileptiform activity in the rat hippocampus. Synapse 37: 90-94. .0.0 [DOI] [PubMed] [Google Scholar]