

Abstract
Abnormalities in dendritic spines have long been associated with cognitive dysfunction and neurodevelopmental delay, whereas rapid changes in spine shape underlie synaptic plasticity. The key regulators of cytoskeletal reorganization in dendrites and spines are the Rho GTPases, which modify actin polymerization in response to synaptic signaling. Rho GTPase activity is modulated by multiple regulatory proteins, some of which have been found to associate with proteins localized to spines. Here, we show that the nonkinase phorbol ester receptor α1-chimerin is present in dendrites and spines, where it binds to the NMDA receptor NR2A subunit in a phorbol ester-dependent manner. α1-Chimerin contains a GTPase activating (GAP) domain, with activity toward the Rho family member Rac1. Overexpression of α1-chimerin in cultured hippocampal neurons inhibits formation of new spines and removes existing spines. This reduction in spine density is mediated by Rac1 inhibition, because it depends critically on the presence of a functional GAP domain. Conversely, depletion of α1-chimerin leads to an increase in spine density, indicating that a basal inhibition of Rac1 maintains the number of spines at a submaximal level. The ability of α1-chimerin to modulate spine number requires an interaction with the NMDA receptor, because an α1-chimerin mutant that binds weakly to NR2A fails to decrease spine density. Together, these results suggest that α1-chimerin is able to modulate dendritic spine morphology by binding to synaptic NMDA receptors and locally inactivating Rac1.
Keywords: NMDA receptors, α-chimerin, Rho GTPases, dendritic spines, phorbol ester receptors, GTPase activating proteins
Introduction
Dendritic spines are the small specialized protrusions that receive the majority of excitatory synapses in the brain (Nimchinsky et al., 2002). These structures are able to compartmentalize incoming signals, and they allow the neuron to integrate and respond to complex synaptic input patterns. The significance of these structures is underscored by the association of spine abnormalities with many cognitive disorders. Nonspecific mental retardation, Down's syndrome, Fragile X, phenylketonuria, and even child-hood malnutrition are all associated with abnormalities in dendritic spine morphology (Purpura, 1974; Ramakers, 2002). Whether a cause or a consequence, normal spine shape and density seem to be vital for normal cognitive development, a conclusion supported by a wealth of data reporting spine morphology changes in response to signals that produce long-term alterations in synaptic strength such as NMDA receptor-dependent long-term potentiation (LTP) (Bliss and Lomo, 1973; Fischer et al., 1998; Yuste and Bonhoeffer, 2001). This sensitivity of dendrite morphology to neuronal activity implies a delicate balance of opposing actin effectors at each synapse controlling spine shape in response to synaptic signaling (Matus, 2000; Okamoto et al., 2004; Kennedy et al., 2005). These opposing activities must be precisely localized and have to be responsive to multiple types of synaptic input. Significant progress has been made recently in identifying the signaling pathways responsible for transducing NMDA receptor activity into spine remodeling (Kennedy et al., 2005), but our understanding of this process is far from complete.
As the primary regulators of cytoskeletal dynamics, the Rho GTPases (Rac1, RhoA, and Cdc42) are the most likely link between NMDA receptor activity and dendritic morphologic changes (Luo, 2002). Rac1 and Cdc42 activation promote actin polymerization, increase dendritic arbor complexity, and stimulate spine formation, whereas RhoA activation leads to actin depolymerization and inhibition of both dendritic branch elongation and spine formation (Li et al., 2000). Rho GTPases are modulated by a number of guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs), which increase and decrease, respectively, the activity of their Rho target (Rossman et al., 2005). Multiple regulatory proteins for Rho family members have been found to influence dendritic spine shape and density (Govek et al., 2004; Kennedy et al., 2005; Tolias et al., 2005).
Here, we report that an inhibitory regulator of Rac1, the nonkinase phorbol ester receptor α-chimerin, binds to the NMDA receptor NR2A subunit. The two splice variants of α-chimerin both contain a protein kinase C (PKC)-like C1 phorbol ester binding domain and a Rac1 GAP domain (Ahmed et al., 1990, 1995; Areces et al., 1994). Treatment of cultured hippocampal neurons with phorbol esters resulted in translocation of α1-chimerin from the cytosol to the plasma membrane and also strengthened the interaction between α1-chimerin and NR2A. Manipulations of α1-chimerin expression levels dramatically altered spine density, and these effects required a functional GAP domain. A small C-terminal deletion that eliminates binding of α1-chimerin to NR2A no longer affected spine density.
Materials and Methods
Yeast two-hybrid screen and assay. The mouse NR2A (ϵ1) C terminus lacking the terminal four amino acids (aa 1017-1460) was subcloned into the pGBKT7 bait vector (Clontech, Cambridge, UK) using NcoI and BamHI and was used to screen a human brain yeast two-hybrid cDNA library in the pACT2 vector (Clontech). PCR was performed on positive colonies; the products were subcloned into the pGEM-T-Easy (Promega, Madison, WI) vector and sequenced. Multiple copies of the C-terminal 156 bp of α-chimerin were identified using the National Center for Biotechnology Information BLAST database. For the follow-up assay, the NR1 C terminus (amino acids 825-938), first (amino acids 1017-1158), second (amino acids 1159-1306), and third (amino acids 1305-1464) parts of the NR2A C terminus were all subcloned into pGBKT7 using NcoI and BamHI. Full-length α1-chimerin was a generous gift from Dr. M. G. Kazanietz (University of Pennsylvania School of Medicine, Philadelphia, PA) and subcloned into the pACT2 vector using the XhoI and EcoRI sites to screen α1-chimerin against the NMDA receptor C-terminal domains.
Primary hippocampal neuron culture and transfection. Hippocampi from embryonic day 18 (E18) rats were obtained from Brainbits (Spring-field, IL) and stored in Hibernate/B27 media (Brewer and Price, 1996). Cells were dissociated and plated at 40,000 cells/cm2 on poly-d-lysinecoated glass bottom culture dishes (MatTek, Ashland, MA). Neurons were cultured in a chemically defined medium of B27 plus Neurobasal as described previously (Brewer et al., 1993) and fed weekly by replacing half of the medium. Cultures were transfected with plasmid DNA using Lipofectamine 2000 (Invitrogen, Eugene, OR). For each transfection, 3 μl of Lipofectamine 2000 was added to 50 μl of Neurobasal. Plasmid DNA (1 μg) for each construct was added to a separate 50 μl of Neurobasal. Both mixtures were incubated at room temperature for 5 min. The mixtures were then combined and incubated at room temperature for 20 min. Culture media (∼2 ml) was removed, the DNA/Lipofectamine 2000 mixture was added to the cells, and the dishes were incubated at 35°C for 10 min. The original culture media was then returned to the dish, and the culture was returned to the incubator.
cDNA constructs. α1-Chimerin-cyan fluorescent protein (CFP), α1-chimerin Δ27-CFP, α1-chimerin Δ4-CFP, and α1-chimerin R179G GAP-CFP were made by PCRs from a plasmid encoding the entire coding sequence of α1-chimerin (a kind gift from Dr. Kazanietz, University of Pennsylvania School of Medicine). The PCR products were cloned into enhanced CFP (eCFP)-C1 (Clontech). α1-Chimerin in pACT2 was made by a PCR from the plasmid mentioned above. Wild-type Rac1 (wt), constitutively active Rac1 I115 and dominant-negative Rac1 N17 were a generous gift from Dr. Ann Marie Pendergast (Duke University). Rat NR2A in pcDNA3.1 was kindly provided by Dr. Robert Wenthold (National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, MD).
Antibodies. A GST-tagged α1-chimerin fragment (amino acids 156-335) was used to generate rabbit polyclonal antibodies (Covance, Princeton, NJ) and affinity purified using the original immunogen cross-linked to glutathione agarose column. The specificity of the purified antibody was tested by immunoblotting rat brain lysate. Commercial antibodies used included anti-NR2A (goat; Santa Cruz Biotechnology, Santa Cruz, CA), anti-green fluorescent protein (GFP) (mouse monoclonal; Roche Products, Welwyn Garden City, UK), anti-rabbit-HRP, anti-mouse-HRP, anti-goat-HRP (Santa Cruz Biotechnology), AF488 goat anti-mouse, and AF594 goat anti-Rbt (Invitrogen).
Immunofluorescence. For NR2A/α1-chimerin-CFP double labeling, neurons expressing α1-chimerin-CFP and full-length NR2A for 6 h were rinsed twice with Dulbecco's PBS and fixed with a 4% paraformaldehyde, 4% sucrose, and 1× PBS solution at 4°C for 15 min. Cells were then washed in PBS, permeabilized with a 0.1% Triton X-100 solution, rinsed twice with PBS, and blocked with 10% goat serum, 2% BSA, and 1× PBS for 1 h at room temperature. Cells were then incubated in rabbit anti-NR2A antibody (Calbiochem, La Jolla, CA) and anti-GFP monoclonal antibody mix (Roche Products), both diluted 1:1000 in block overnight at 4°C. They were then washed three times with block and incubated with AF488-conjugated goat anti-rabbit IgG (Invitrogen) and AF594-conjugated goat anti-mouse IgG diluted 1:1000 in block for 1 h at room temperature. Cells were washed three times in block, once in PBS, and imaged in PBS.
Immunoprecipitation. Coimmunoprecipitation from cultured rat hippocampal neurons was performed using methods adapted from Huang et al. (2001). E18 rat hippocampal neurons (Brainbits) were plated similarly to the hippocampal cultures described above except at a density of 100,000/cm3 on two poly-d-lysine-coated plastic culture dishes. At 14 d in vitro (DIV), one dish was treated with 300 nm phorbol 12-myristate 13-acetate (PMA) for a total of 4 d. The crude synaptosomal fraction was solubilized in 1% sodium deoxycholate, 50 mm Tris-HCl, pH 9.0, and 10 mm EDTA at 37°C for 15 min. An equal volume of radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, plus PIC; Sigma, St. Louis, MO) was added, and the sample was spun in a tabletop centrifuge for 30 min at 14,000 × g to remove insoluble material. Protein extracts were precleared with protein-G Sepharose beads for 2 h at 4°C. Five micrograms of anti-NR2A (Santa Cruz Biotechnology) antibody were added and incubated overnight at 4°C. The antibody and bound proteins were pulled down using protein-G Sepharose beads for 3 h at 4°C were washed three times in lysis buffer and once in PBS. Sample buffer was added, and the samples were separated using SDS-PAGE on 10% Tris-glycine gels. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes and immunoblotted as described below.
Coimmunoprecipitation from human embryonic kidney 293T (HEK293T) cells was performed after 2 d of transfection and 5 h of PMA (300 nm) treatment when appropriate. Cells were washed with PBS, scraped from the dish, and transferred to centrifuge tubes. Cells were collected in a tabletop centrifuge for 5 min at 800 × g and lysed in the 1% deoxycholate buffer described above. The lysate was sonicated with a probe sonicator, and an equal volume of RIPA buffer was added. Lysates were then treated as described above.
Immunoblotting. PVDF membranes were blocked for 1 h at room temperature with 5% nonfat dried milk in PBS. The blots were then incubated with a primary antibody (rabbit anti-α-chimerin, which recognizes both splice variants) at 1:100 or anti-GFP (mouse monoclonal; 1:1000; Roche Products) in the blocking solution. The blots were washed three times for 10 min each with PBS plus 0.1% Tween 20. Next, the blots were incubated with the proper 1:1000 diluted HRP-tagged secondary antibody (Santa Cruz Biotechnology) for 1 h at room temperature. The blots were then washed six times for 20 min each with PBS plus 1% Tween 20. Finally, the blots were exposed to SuperSignal West Pico Chemiluminescent HRP substrate (Pierce, Rockford, IL) according to the instructions and developed on x-ray film.
Image acquisition and analysis. Cells were imaged using a Nikon (Tokyo, Japan) Diaphot inverted fluorescence microscope with either a 40× or 100× oil immersion objective. A Hamamatsu (Shizuoka, Japan) cooled digital camera was used to record images, Openlab version 3.1.6 imaging software (Improvision, Lexington, MA) was used to acquire and analyze images, and Volocity version 1.5 software (Improvision) was used to render deconvolved images. Spine density was analyzed by measuring the length and number of protrusions > 0.5 μm in length on secondary and tertiary dendrites (two measurements of ∼100 μm per neuron). Five to eight neurons were imaged per experiment with two to three experiments from separate cultures performed per measurement. The length and number of spines were measured manually using Openlab Measurement software.
To test the efficacy of the short-interfering RNA (siRNA) constructs, fluorescence intensity of neuronal cell bodies was measured using ImagePro version 4.0 by drawing a polygonal area of interest (AOI) around each cell body and recording total fluorescence intensity within that AOI. The mean and SEM was calculated for each experimental condition, and a Student's t test (two-sample; equal variances) was used to evaluate statistical significance between groups.
Expression of short-interfering hairpin loop RNA constructs. The RNA-interference vector was constructed by amplifying the H1-RNase promoter from human genomic DNA by PCR with the following primers: HSH1_F, 5′-GAA TTC TTA TAG GGA GCT GAA GGG AAG GGG GTC AC-3′; and HSH1_R, 5′-AAG CTT CCG CCC TAT GGA GAT CTG TGG TCT CAT ACA-3′. This PCR product was then ligated into the pGEM-T Easy vector (Promega). The Neomycin resistance gene was obtained by digesting pcDNA3.1 with AseI. The largest fragment from this digest was ligated into the Mde1 site of the previously constructed pGem-T-EZ vector containing the H1 promoter.
An α-chimerin-hairpin sequence was ligated into the completed RNAi vector by annealing the following oligos and ligating into the BglII and HindIII sites of the vector: si_F, 5′-GAT CCC GTT CCT CTC ATC ACC TAC GAT TCA AGA GAT CGT AGG TGA TGA GAG GAA TTT TTT GGA AA-3′; and si_R, 5′-AGC TTT TCC AAA AAA TTC CTC TCA TCA CCT ACG ATC TCT TGA ATC GTA GGT GAT GAG AGG AAC GG-3′. The stem of the hairpin includes the following sequence from α-chimerin: 5′-TT CCT CTC ATC ACC TAC GA-3′.
Results
α1-Chimerin interacts with the NMDA receptor subunit NR2A
To find novel NMDA receptor interacting proteins that may be involved in signaling-dependent changes in cytoskeletal organization, we screened a human brain yeast two-hybrid cDNA library (Clontech) using, as a bait, the intracellular (C-terminal) domain of NR2A lacking the four terminal amino acids that mediate binding to postsynaptic density-95/Discs large/zona occludens-1 (PDZ) domain-containing proteins. This was done in an attempt to simplify the results of the screen by eliminating the known PDZ domain-containing binding partners for NR2A (Kornau et al., 1995; Niethammer et al., 1996). Multiple positive clones were found to contain the C-terminal domain of α-chimerin, a nonkinase phorbol ester receptor with a GTPase activation domain specific for the small GTPase Rac1 (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). To confirm the yeast two-hybrid result and narrow down the domain of NR2A responsible for α-chimerin binding, the NR2A C-terminal domain was divided into three segments (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), and each was screened against full-length α1-chimerin along with the intracellular domain of NR1 and the full-length NR2A intracellular domain. This yeast two-hybrid experiment confirmed the interaction between NR2A and α1-chimerin and localized the region responsible for binding to the central domain (residues 1159-1306) of the NR2A C terminus (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). α1-Chimerin did not bind to the C terminus of NR1 (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), which served as a negative control.
To investigate whether NR2A colocalized with α1-chimerin, rat hippocampal neurons (24 DIV) were transfected with α1-chimerin fused to eCFP and full-length wild-type NR2A and fixed 6 h later. This was done to visualize the two proteins before the effects of α1-chimerin overexpression on spine shape and density became manifest (see below). The neurons were then double immunostained and imaged (Fig. 1A). NR2A and α1-chimerin colocalized well in dendritic spines and also partially colocalized in the dendritic shaft (Fig. 1B). A similar experiment was then performed using hippocampal neurons cotransfected with α1-chimerin-CFP and NR2A fused to enhanced yellow fluorescent protein (eYFP). These neurons were imaged 24 h after transfection (Fig. 1C). Whereas most of the NR2A-YFP fluorescence localized to puncta corresponding to spines, α1-chimerin-CFP was more diffusely distributed throughout the dendrite. However, a line profile measuring fluorescence intensity along the dendrite revealed that α1-chimerin-CFP was concentrated in many of the NR2A-YFP puncta, indicating a partial colocalization of the two proteins.
Figure 1.
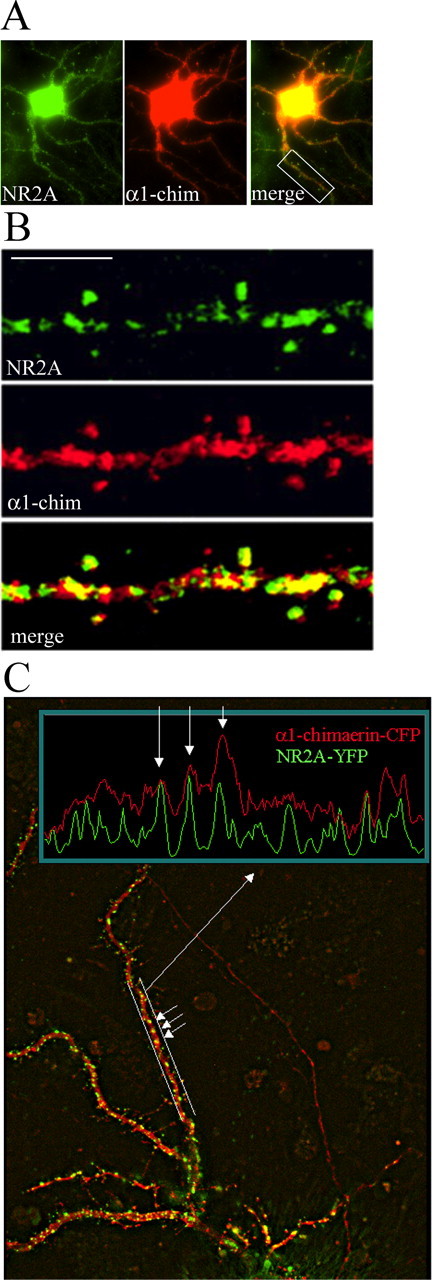
α-Chimerin and NR2A colocalize in hippocampal neurons. A, B, Cultured hippocampal neurons (24 DIV) were cotransfected with α1-chimerin (α1-chim)-CFP fusion and untagged NR2A and stained by immunofluorescence 6 h after transfection for NR2A (green) and CFP (red). The merged image shows colocalization of α1-chimerin and NR2A in dendritic spines. Scale bar, 5 μm. C, Cultured hippocampal neurons were cotransfected with α1-chimerin-CFP fusion and NR2A-YFP and imaged 24 h later. A line profile through a dendritic segment shows that increased α1-chimerin-CFP (red) fluorescence intensity correlates with increased NR2A-YFP (green) fluorescence intensity.
Phorbol ester causes translocation of α1-chimerin and enhances its interaction with NR2A
Previous work has shown that some chimerins, when overexpressed in cell lines, redistribute from the cytosol to the membrane fraction after phorbol ester treatment (Caloca et al., 2001). To determine whether phorbol ester treatment effects α1-chimerin localization, hippocampal neurons (13 DIV) were transfected with α1-chimerin-CFP and imaged the next day. Fifteen minutes after treatment with the phorbol ester PMA, the same neurons were imaged and showed a dramatic translocation of α1-chimerin-CFP from the cytosol to the plasma membrane (Fig. 2A). In a separate experiment, the time course of the PMA-mediated translocation was determined by taking images every minute, immediately after adding PMA. The results indicate that translocation of α1-chimerin to the plasma membrane is complete after 3 min of treatment with PMA (Fig. 2B).
Figure 2.
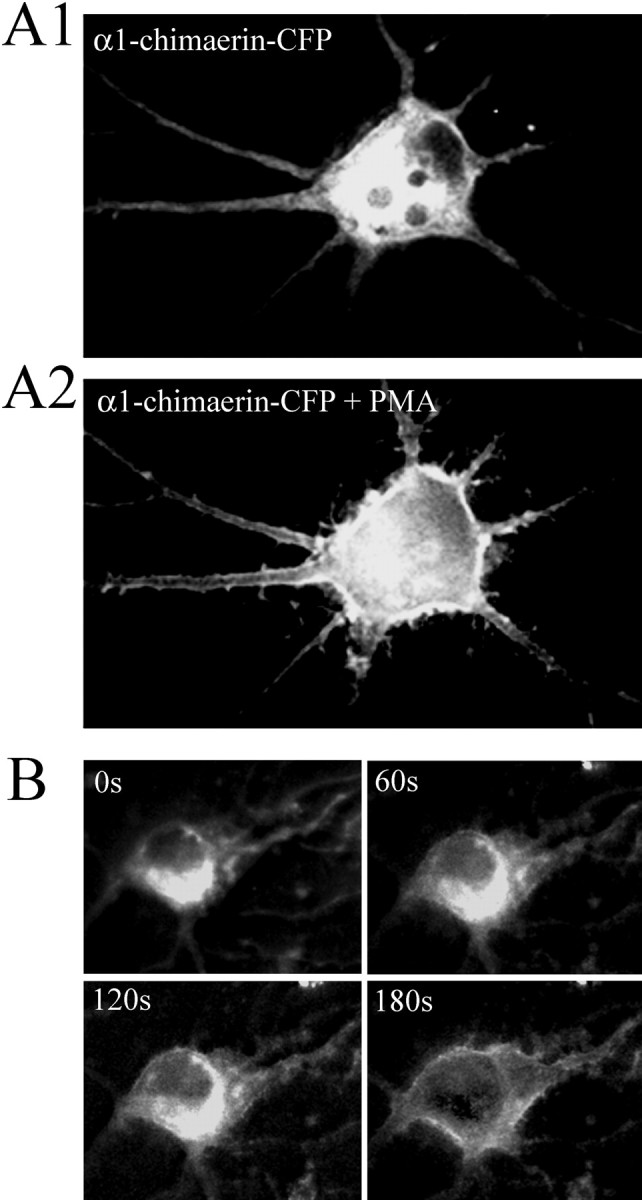
Phorbol ester translocates α1-chimerin to the plasma membrane. A, Cultured hippocampal neurons (14 DIV) transfected with α1-chimerin-CFP and imaged before (A1) and after (A2) treatment with PMA (300 nm) for 15 min. B, A time course of a 14 DIV hippocampal neuron transfected with α1-chimerin-CFP showing protein translocation after PMA (300 nm) treatment.
Because the NMDA receptor is localized to synaptic spines and PMA treatment causes α1-chimerin translocation to the dendritic plasma membrane, it seemed possible that PMA-bound α1-chimerin could have a higher affinity for NR2A. Initial coimmunoprecipitation experiments showed a strong interaction between the NR2A intracellular domain and α1-chimerin (data not shown), but when lysate from HEK293T cells cotransfected with full-length NR2A and α1-chimerin-CFP was immunoprecipitated, the interaction was very weak. However, treatment of cotransfected HEK293T cells with phorbol ester (PMA, 300 nm) greatly enhanced the stability of the NR2A/α1-chimerin complex (Fig. 3A).
Figure 3.

α-Chimerin interacts with NR2A. A, Lysates from HEK293T cells transfected with NR2A and α1-chimerin-CFP (α1-chim-CFP) were immunoprecipitated with anti-NR2A antibodies in the presence and absence of PMA (300 nm).α1-Chimerin-CFP and NR2A in the immunoprecipitates and lysates (lys) were detected by immunoblotting. PMA treatment increased the amount of α1-chimerin in the immunoprecipitate. The control lanes show the adequacy of the wash step by omitting the IP antibody. B, Association of α1-chimerin (α-Chim) and NR2A in rat hippocampal neuron synaptosomes. The solubilized crude synaptosomal fraction was immunoprecipitated with an anti-NR2A antibody in the presence and absence of PMA (300 nm). The two α-chimerin splice variants and NR2A in the immunoprecipitate and lysate were detected by immunoblotting. The binding of both α1- and α2-chimerin to NR2A was increased after PMA treatment.
The enhanced interaction between NR2A and α1-chimerin after phorbol ester treatment was also apparent in rat brain. When lysates from cultured rat hippocampal neurons were immunoprecipitated with an anti-NR2A antibody, both α1-chimerin and α2-chimerin copurified in PMA-treated neurons, whereas only a small amount of α1-chimerin pulled down in untreated neurons (Fig. 3B).
Effect of α1-chimerin mutations on NR2A binding
Because the C-terminal domain of α-chimerin was pulled out in the yeast two-hybrid screen, it seemed possible that a short C-terminal α1-chimerin deletion mutant would lose its ability to bind NR2A. To test this, two C-terminal mutants were subcloned into the eCFP-C1 vector: the first had the final four amino acids (Δ4) of the protein deleted, and the second had the terminal 27 amino acids (Δ27) removed. Also, the mutation R179G was introduced at the site previously identified to be important for the Rac1-GAP activity of chimerin (Ahmed et al., 1994; Hall et al., 2001) (Fig. 4A). These mutants were then cotransfected into HEK293T cells along with full-length NR2A. Immunoprecipitation (IP) with an anti-NR2A antibody showed that wild-type α1-chimerin, the GAP-inactive mutant (R179G), and the Δ4 mutant were all pulled down with NR2A when treated with PMA. However, binding of the Δ27 mutant was much weaker than wild-type, implicating the final 27 amino acids of α1-chimerin in its ability to bind NR2A in a phorbol ester-dependent manner (Fig. 4B). A control immunoprecipitation was performed on HEK293T cell lysate cotransfected with eCFP and NR2A showing no CFP present in the immunoprecipitate (data not shown).
Figure 4.
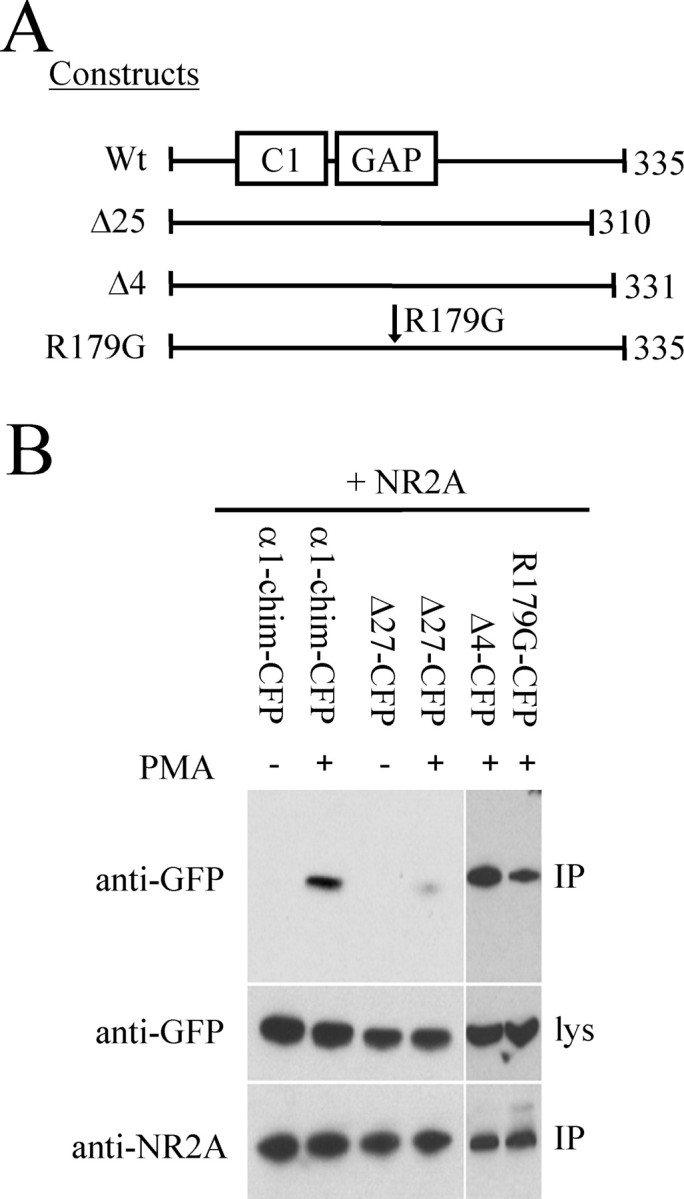
Interaction of α1-chimerin mutants with NR2A. A, Structure of α1-chimerin mutants in eCFP-C1 vector. Δ27 and Δ4 mutants are C-terminal truncations of α1-chimerin. R179G renders the Rac1 GAP domain of α1-chimerin GTPase inactive. B, Lysates from HEK293T cells transfected with NR2A, and one of the α1-chimerin mutants (α1-chim-CFP) or eCFP alone was immunoprecipitated using an anti-NR2A antibody in the presence and absence of PMA (300 nm). NR2A and the α1-chimerin mutants were detected in the immunoprecipitate and lysate (lys) by immunoblotting. The α1-chimerin Δ27 C-terminal truncation mutant loses its binding affinity for NR2A. Wt, Wild type.
α1-Chimerin overexpression decreases spine density in hippocampal neurons
Normal dendritic spine development and morphology have been shown to be dependent on the small GTPases, with Rac1 activity promoting spine formation. The α-chimerins are known to have opposing effects on Rac-dependent neurite outgrowth in neuroblastoma and PC12 cells (Hall et al., 2001), but the effect of these Rac-GAPs on the morphology of hippocampal neurons has not been studied. To determine whether α1-chimerin modifies spine development, α1-chimerin-CFP was overexpressed along with eYFP in 21 DIV hippocampal neurons and imaged 3 d later. Linear spine density was determined and compared with neurons transfected with eYFP alone. Overexpression of α1-chimerin-CFP significantly reduced spine density compared with neurons expressing YFP alone (Fig. 5A). These results suggest that α1-chimerin overexpression inhibits spine development. The effect of overexpression on already established spines was uncertain, and thus neurons were transfected with eYFP at 22 DIV and imaged to quantitate spine density. The x-y coordinates of each imaged neuron were saved, and the neurons were retransfected with α1-chimerin-CFP. Neurons that expressed both eYFP and α1-chimerin-CFP were reimaged 2 d later. These experiments indicated that α1-chimerin overexpression is able to remove existing spines (Fig. 5B).
Figure 5.
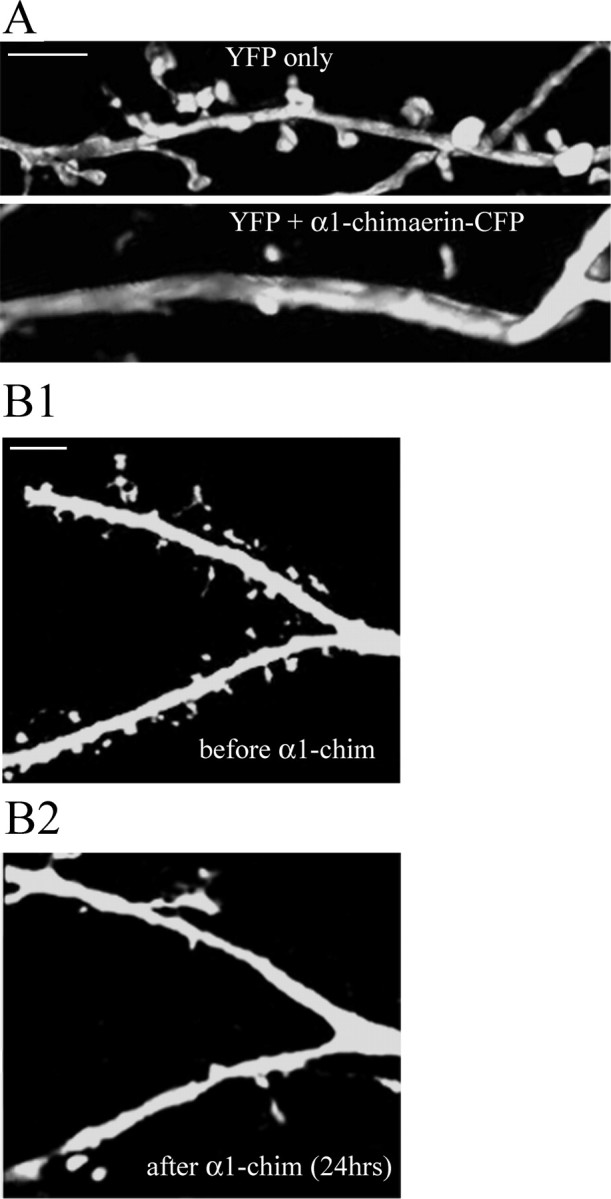
Overexpression of α1-chimerin decreases dendritic spine density. A, Cultured hippocampal neurons (21 DIV) were transfected with α1-chimerin and eYFP-N1 or eYFP-N1 alone and imaged for YFP 3 d later. Neurons overexpressing α1-chimerin had fewer spines (0.85 ± 0.0892 spines per 10 μm) than those expressing YFP alone (4.66 ± 0.28 spines per 10 μm). B, α1-Chimerin (α1-chim) overexpression reduces spine density in previously imaged neurons. Cultured hippocampal neurons were transfected with eYFP-N1 at 21 DIV and imaged, and the x, y coordinates of each neuron were recorded. At 24 DIV, the same dish of neurons was transfected with α1-chimerin-CFP, and previously characterized neurons were inspected for α1-chimerin expression. B1, Dendrite of a neuron that was transfected with eYFP-N1 at 21 DIV before overexpression of α1-chimerin. B2, The same dendrite of the neuron shown in B1, now expressing both YFP and α1-chimerin-CFP. Overexpression of α1-chimerin eliminates preexisting spines. Scale bars, 5 μm.
α1-Chimerin overexpression not only decreased spine density in normal hippocampal neurons but was also able to completely antagonize the increased spine density seen with wild-type Rac1 overexpression. Neurons (21 DIV) cotransfected with eYFP, wtRac1, and α1-chimerin had significantly lower linear spine density than those transfected with eYFP and wtRac1 alone (Fig. 6A,B). In fact, there was no significant difference in spine density between neurons expressing α1-chimerin alone (Fig. 6B) and those expressing both α1-chimerin and wtRac1. The α1-chimerin overexpression-mediated decrease in spine density was similar to the decrease in spine density observed in neurons expressing a dominant-negative form of Rac1 (Rac1 N17). Neurons overexpressing a constitutively active form of Rac1 (Rac1 I115) developed large lamellipodial protrusion along their dendrites, an activity that remained unchanged in the presence of overexpressed α1-chimerin (Fig. 6C).
Figure 6.

α1-Chimerin antagonizes the effects of Rac1 on spine density. A, Cultured hippocampal neurons (21 DIV) were transfected with eYFP, eYFP plus Rac1, eYFP plus Rac1 plus α1-chimerin, or eYFP plus dominant-negative (DN) Rac1 (N17). Neurons were imaged for YFP 3 d later. Spine density is significantly reduced in both Rac1 plus α1-chimerin and dominant-negative Rac1 (0.79 ± 0.112 spines per 10 μm). Scale bar, 5 μm. B, Quantification of spine density in neurons transfected with the constructs pictured in A. C, Hippocampal neurons (21 DIV) cotransfected with eYFP and constitutively active Rac1 (I115) developed large dendritic lamellipodia. Spines can no longer be reliably identified in these neurons. Similar lamellipodia were present when eYFP, active Rac1 (I115), and α1-chimerin were cotransfected into 21 DIV hippocampal neurons.
As reported above, the Δ27 α1-chimerin mutant had a reduced ability to bind NR2A after phorbol ester treatment. Therefore, to investigate whether the decreased spine density seen with overexpression of α1-chimerin depends on its ability to bind NR2A, the linear spine density of neurons expressing this mutant was recorded. In addition, to determine whether it is the Rac1-GAP activity of α1-chimerin that was causing a reduction of spine density, the R179G mutant was overexpressed in neurons and linear spine density recorded. The Δ4 α1-chimerin mutant, which retained the ability to form a complex with NR2A, reduced spine density to a level similar to that of wild-type α1-chimerin (Fig. 7A,B). The Δ27 mutant lacking the ability to interact with NR2A was unable to reduce spine density to the levels seen with wild-type α1-chimerin overexpression. Overexpression of the R179G GAP mutant allowed spine density to remain at control levels. These two results indicate that both the GAP activity of α1-chimerin and its ability to bind NR2A contribute to its ability to reduce spine density (Fig. 7A,B).
Figure 7.

The effects of α1-chimerin on spines depend on its Rac1 GAP activity and its ability to bind NR2A. A, Neurons were transfected with eYFP alone or with both eYFP and wild-type α1-chimerin (WT), α1-chimerin R179G GTPase activation mutant (R179G), α1-chimerin Δ4 mutant, or α1-chimerin Δ27 mutant. YFP images of representative dendrites are shown for each condition. Overexpression of the GTPase activation mutant no longer affects spine density. The Δ27 mutant, which has much lower affinity for NR2A than wild-type α1-chimerin (Fig. 4 B) also has lost its ability to reduce spine density. Scale bar, 5 μm. B, Quantification of spine density in neurons transfected with the constructs pictured to the left. Spine density is significantly reduced only for wild-type α1-chimerin and its Δ4 mutant, whereas the GAP-inactive mutation and the Δ27 deletion mutant have no effect on spine density. α1-chim-CFP, α1-Chimerin-CFP.
α1-Chimerin depletion increases spine density
Because overexpression of α1-chimerin reduced spine density, the effect of α1-chimerin depletion in hippocampal neurons was investigated. Hippocampal neurons at 10 DIV were transfected with eCFP and an siRNA-targeting α-chimerin. Neurons expressing an siRNA to α-chimerin showed a 76 ± 5% reduction in α-chimerin immunofluorescence (Fig. 8A). Neurons transfected with the siRNA-targeting α-chimerin showed an increase in spine density compared with neurons expressing a random siRNA (Fig. 8B).
Figure 8.

Downregulation of α1-chimerin increases spine density. A, Cotransfection of hippocampal neurons (10 DIV) with eCFP and α1-chimerin siRNA expression plasmid. Cells were immunostained at 14 DIV against α-chimerin. Note the decreased α-chimerin signal in the transfected neuron (arrow). B, Hippocampal neurons cotransfected with eYFP and random siRNA (left) or eYFP and α1-chimerin siRNA (right). Neurons expressing the α-chimerin-specific siRNA show increased spine density (8.31 ± 1.23 spines per 10 μm; p < 0.01) when compared with those expressing a random siRNA construct (5.56 ± 0.54 spines per 10 μm). Scale bar, 5 μm.
Discussion
The debate over whether long-term changes in synaptic strength cause, or are at least associated with, spine morphology changes appears to be almost resolved (Engert and Bonhoeffer, 1999; Fischer et al., 2000; Yuste and Bonhoeffer, 2001; Carlisle and Kennedy, 2005), but the mechanisms by which synaptic signaling is transduced into spine cytoskeletal reorganization are only now being teased apart. The Rho-GTPases are known to influence dendrite and dendritic spine morphology, and a number of Rho-GTPase regulators have been shown to affect spine density and shape (Govek et al., 2004; Carlisle and Kennedy, 2005). Recently, the Rac1-GEF, Tiam1, was shown to bind the NMDA receptor NR2B subunit and increase spine density in response to NMDA receptor activity (Tolias et al., 2005).
Here, we report that the nonkinase phorbol ester receptor and Rac1-GAP α1-chimerin is a negative regulator of spine density, an activity that depends critically on its ability to bind to the NMDA receptor subunit NR2A. α1-Chimerin inhibits the formation of new spines and can facilitate the disappearance of spines that have already formed by inactivating Rac1, a protein known to promote dendritic arbor development and dendritic spine formation.
Both α-chimerin splice variants are neuron-specific Rho-GTPase regulators with the highest concentrations of α-chimerin mRNA present in the cortex and hippocampus, suggesting they may have a role in cytoskeletal reorganization during experience-dependent synaptic plasticity. The two splice variants have been shown previously to have opposing effects on neurite outgrowth in neuroblastoma and PC12 cells. α2-Chimerin, when overexpressed in these cell lines, mimics Rac1/Cdc42 activation by promoting neurite outgrowth (Hall et al., 2001). α1-Chimerin, however, inhibits neurite outgrowth. This difference in activity depends on a functional Src homology 2 (SH2) domain in α2-chimerin (Hall et al., 2001). The α1-chimerin inhibition of neurite outgrowth in cultured cells correlates well with its effects on hippocampal neuron morphology as presented here.
Unlike the Rac1-GEF Tiam1, α1-chimerin activation may not be determined directly by NMDA receptor activity. It has no calcium responsive domain, and it is unknown whether its phosphorylation status changes during calcium entry into the spine. Because α1-chimerin inactivates Rac1 and promotes the disappearance of spines, it is possible that it plays a role in spine morphology changes associated with long-term depression, a state that has been reported to be induced in hippocampal neurons by metabotropic glutamate receptor activity through activation of mGluR1 (Oliet et al., 1997; Malenka and Bear, 2004). When mGluR1 binds glutamate, its associated heterotrimeric G-protein, Gαq, binds GTP and, through phospholipase C activation, causes phosphatidylinositol bis phosphate 2 to split into DAG and IP3. The newly formed DAG can then recruit α1-chimerin to the dendritic membrane where it binds NR2A and deactivates Rac1 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In this case, instead of α1-chimerin relying on NMDA receptor signaling to regulate its activity, NR2A acts as a molecular scaffold for α1-chimerin, providing a way to localize the GAP to the exact spine at which it needs to perform its function. Previous findings that regulation of most GAPs seems to depend more on correct localization than on covalent modification are consistent with our conclusion that α1-chimerin is regulated by translocation and binding to NR2A rather than through NMDA receptor activity-dependent phosphorylation (Moon and Zheng, 2002; Chardin, 2003).
As synapses develop, the type of NMDA receptor that predominantly populates the synapse changes (Rumbaugh and Vicini, 1999; Tovar and Westbrook, 1999; Li et al., 2002; Prybylowski and Wenthold, 2004). Before synapse formation, heteromeric NR1/NR2B-containing NMDA receptors predominate. Soon after synaptic contacts are formed, however, the number of NR2A-containing receptors at the synapse increases. It is therefore reasonable that a protein such as Tiam1, which activates Rac1 and promotes spine development, would bind to NR2B during synaptic development (Tolias et al., 2005). Likewise, it is consistent that a protein like α1-chimerin, which inactivates Rac1 leading to loss of spines, would bind NR2A and ensure that α1-chimerin is localized at excitatory synapses where, after a signaling event such as mGluR1 activation, it would cause actin depolymerization.
Phorbol ester binding to the C1-domain of α1-chimerin increases the potency of the Rac1-GAP activity of the protein (Kozma et al., 1996) and, as we have shown here, translocates the protein to the plasma membrane where it binds to NR2A. Therefore, the production of diacylglycerol in response to glutamate release may not only cause α1-chimerin to localize to NMDA receptors present in dendritic spines, but the Rac1-GAP activity of the protein would be potentiated and primed to downregulate Rac1 in the spines where it is localized. The translocation and activation of α1-chimerin by DAG therefore constitutes a mechanism by which synaptic activity causing long-term depression ultimately results in reduction in spine size, or even spine elimination.
One of the intriguing aspects of α-chimerin is the presence of a PKC-like C1 phorbol ester binding domain. Phorbol esters have traditionally been used to study PKC function with the assumption that phorbol esters were specific activators of PKC. This paradigm has slowly been changing as a number of non-PKC phorbol ester binding proteins have been discovered (Brose and Rosenmund, 2002; Kazanietz, 2002; Yang and Kazanietz, 2003). Although the appearance of these more recent phorbol ester receptors may complicate some of the data collected on PKC activity in the past, it may eventually simplify the many functions attributed to PKC, including LTP induction and NMDA receptor potentiation (Lu et al., 1999; Lan et al., 2001; Grosshans et al., 2002; Malenka and Bear, 2004).
Our results suggest that the ratio of GDP- or GTP-bound Rac1 is the major determinant of neuronal spine density. The importance of Rac1 activity as a regulator of spine density is highlighted by spine density changes observed while overexpressing wild-type Rac1 and dominant-negative Rac1. The presence of a large amount of wild-type Rac1 overwhelms the inhibitory machinery of the cell and maximizes spine density whereas the presence of dominant-negative Rac1 shifts the balance in the opposite direction and minimizes spine density. Likewise, when the GDP/GTP ratio of Rac1 is shifted by either overexpressing a Rac1 GAP like α1-chimerin or depleting constitutive α1-chimerin, spine density decreases and increases, respectively, indicating that spine number in neurons is balanced at a submaximal level by tightly controlling the amount of active Rac1.
The contribution of the α2-chimerin isoform to the observed effects on dendritic spine density has not been investigated fully. In developing rat hippocampal and cortical neurons, α2-chimerin expression slowly subsides, whereas α1-chimerin levels gradually increase. Both isoforms are present at substantial levels in the neuronal population used in these experiments, and they may therefore be acting together to inhibit dendritic spine formation. Previous studies in neuroblastoma and PC12 cells, however, showed that α2-chimerin acts as a Rac1 activator, although it contains a functional Rac1-GAP domain (Hall et al., 2001). This surprising result may have been caused by protein interactions mediated by the SH2 domain of α2-chimerin. Additional investigation of this isoform is necessary to fully elucidate the role of α-chimerin in hippocampal neurons.
Any protein that regulates cytoskeletal function in neurons may potentially play a role in human cognitive and neurodevelopmental disease by affecting spine shape and altering neuronal reaction to incoming signals. Although chimerin has not yet been connected to human disease, it is interesting that the familial autism susceptibility region of the genome with the greatest MLS score is also the region that contains the α-chimerin gene (International Molecular Genetic Study of Autism Consortium, 2001).
The intimate connection between synaptic plasticity and morphological changes in spines suggests that precise control of the shape, size, formation, and elimination of dendritic spines is vital to the formation of memory. A protein that regulates the shape and existence of spines must have a mechanism for responding to synaptic activity, a way to influence the actin cytoskeleton, and a means to find its way to the spine where it is needed. The Rac1 GAP domain of α-chimerin, its ability to bind the NMDA receptor in a phorbol ester-dependent manner, and its dependence on NMDA receptor binding to mediate changes in spine density satisfies these requirements and may help clarify the mechanism by which dendritic spines respond to synaptic activity.
Footnotes
We thank Drs. Louis Lim and Christine Hall (Institute of Neurology, London, UK) for providing α2-chimerin in pXJ40-HA and pXJ40-GFP and anti-α2-chimerin antibody. We also thank Dr. Marcelo Kazanietz (The Center for Experimental Therapeutics and Department of Pharmacology, University of Pennsylvania School of Medicine, Philadelphia, PA) for providing α1-chimerin in pLexA vector. Plasmids expressing wild-type and mutant Rac1 isoforms were a generous gift from Dr. Ann Marie Pendergast (Duke University), and a plasmid expressing NR2A was a generous gift from Dr. Robert Wenthold (Laboratory of Neurochemistry, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, MD). We thank Dr. Fernando Ribeiro-Neto (University of Pittsburgh, Pittsburgh, PA) for critical discussions.
Correspondence should be addressed to Antonius VanDongen, Department of Pharmacology and Cancer Biology, Duke University Medical Center, P.O. Box 3813, Durham, NC 27710. E-mail: vando005@mc.duke.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259488-09$15.00/0
References
- Ahmed S, Kozma R, Monfries C, Hall C, Lim HH, Smith P, Lim L (1990) Human brain n-chimaerin cDNA encodes a novel phorbol ester receptor. Biochem J 272: 767-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S, Lee J, Wen LP, Zhao Z, Ho J, Best A, Kozma R, Lim L (1994) Breakpoint cluster region gene product-related domain of n-chimaerin. Discrimination between Rac-binding and GTPase-activating residues by mutational analysis. J Biol Chem 269: 17642-17648. [PubMed] [Google Scholar]
- Ahmed S, Kozma R, Hall C, Lim L (1995) GTPase-activating protein activity of n(alpha 1)-Chimaerin and effect of lipids. Methods Enzymol 256: 114-125. [DOI] [PubMed] [Google Scholar]
- Areces LB, Kazanietz MG, Blumberg PM (1994) Close similarity of baculovirus-expressed n-chimaerin and protein kinase C alpha as phorbol ester receptors. J Biol Chem 269: 19553-19558. [PubMed] [Google Scholar]
- Bliss TVP, Lomo T (1973) Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 232: 331-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Price PJ (1996) Viable cultured neurons in ambient carbon dioxide and hibernation storage for a month. NeuroReport 7: 1509-1512. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ (1993) Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res 35: 567-576. [DOI] [PubMed] [Google Scholar]
- Brose N, Rosenmund C (2002) Move over protein kinase C, you've got company: alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci 115: 4399-4411. [DOI] [PubMed] [Google Scholar]
- Caloca MJ, Wang H, Delemos A, Wang S, Kazanietz MG (2001) Phorbol esters and related analogs regulate the subcellular localization of α2-chimaerin, a non-protein kinase C phorbol ester receptor. J Biol Chem 276: 18303-18312. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Kennedy MB (2005) Spine architecture and synaptic plasticity. Trends Neurosci 28: 182-187. [DOI] [PubMed] [Google Scholar]
- Chardin P (2003) GTPase regulation getting aRnd rock and Rho inhibition. Curr Biol 13: R702-R704. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T (1999) Dendritic spine changes associated with hippocampal long term synaptic plasticity. Nature 399: 66-70. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Knutti D, Matus A (1998) Rapid actin-based plasticity in dendritic spines. Neuron 20: 847-854. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Wagner U, Brinkhaus H, Matus A (2000) Glutamate receptors regulate actin-based plasticity in dendritic spines. Nat Neurosci 3: 887-894. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Akerman CJ, Cross JR, Van der Veken L, Van Aelst L (2004) The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat Neurosci 7: 364-372. [DOI] [PubMed] [Google Scholar]
- Grosshans DR, Clayton DA, Coultrap SJ, Browning MD (2002) LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat Neurosci 5: 27-33. [DOI] [PubMed] [Google Scholar]
- Hall C, Michael GJ, Cann N, Ferrari G, Teo M, Jacobs T, Monfries C, Lim L (2001) α2-Chimaerin, a Cdc42/Rac1 regulator, is selectively expressed in the rat embryonic nervous system and is involved in neuritogenesis in N1E-115 neuroblastoma cells. J Neurosci 21: 5191-5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Lu W, Ali DW, Pelkey KA, Pitcher GM, Lu YM, Aoto H, Roder JC, Sasaki T, Salter MW, MacDonald JF (2001) CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron 29: 485-496. [DOI] [PubMed] [Google Scholar]
- International Molecular Genetic Study of Autism Consortium (2001) A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am J Hum Genet 69: 570-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanietz MG (2002) Novel “nonkinase” phorbol ester receptors: the C1 domain connection. Mol Pharmacol 61: 759-767. [DOI] [PubMed] [Google Scholar]
- Kennedy MB, Beale HC, Carlisle HJ, Washburn LR (2005) Integration of biochemical signaling in spines. Nat Rev Neurosci 6: 423-434. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH (1995) Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269: 1737-1740. [DOI] [PubMed] [Google Scholar]
- Kozma R, Ahmed S, Best A, Lim L (1996) The GTPase-activating protein N-chimaerin cooperates with Rac1 and Cdc42Hs to induce the formation of lamellipodia and filopodia. Mol Cell Biol 16: 5069-5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS (2001) Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci 4: 382-390. [DOI] [PubMed] [Google Scholar]
- Li B, Chen N, Luo T, Otsu Y, Murphy TH, Raymond LA (2002) Differential regulation of synaptic and extra-synaptic NMDA receptors. Nat Neurosci 5: 833-834. [DOI] [PubMed] [Google Scholar]
- Li Z, Van Aelst L, Cline HT (2000) Rho GTPases regulate distinct aspects of dendritic arbor growth in Xenopus central neurons in vivo. Nat Neurosci 3: 217-225. [DOI] [PubMed] [Google Scholar]
- Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF (1999) G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci 2: 331-338. [DOI] [PubMed] [Google Scholar]
- Luo L (2002) Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol 18: 601-635. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44: 5-21. [DOI] [PubMed] [Google Scholar]
- Matus A (2000) Actin-based plasticity in dendritic spines. Science 290: 27. [DOI] [PubMed] [Google Scholar]
- Moon SY, Zheng Y (2002) Rho GTPase-activating proteins in cell regulation. Trends Cell Biol 13: 13-22. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M (1996) Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci 16: 2157-2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K (2002) Structure and function of dendritic spines. Annu Rev Physiol 64: 313-353. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y (2004) Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bi-directional plasticity. Nat Neurosci 7: 1104-1112. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA (1997) Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron 18: 969-982. [DOI] [PubMed] [Google Scholar]
- Prybylowski K, Wenthold RJ (2004) N-methyl-d-aspartate receptors: subunit assembly and trafficking to the synapse. J Biol Chem 279: 9673-9676. [DOI] [PubMed] [Google Scholar]
- Purpura DP (1974) Dendritic spine “dysgenesis” and mental retardation. Science 186: 1126-1128. [DOI] [PubMed] [Google Scholar]
- Ramakers GJ (2002) Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci 25: 191-199. [DOI] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J (2005) GEF means go: turning on Rho GT-Pases with guanine nucleotide exchange factors. Nat Rev Mol Cell Biol 6: 167-180. [DOI] [PubMed] [Google Scholar]
- Rumbaugh G, Vicini S (1999) Distinct synaptic and extrasynaptic NMDA receptors in developing cerebellar granule neurons. J Neurosci 19: 10603-10610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME (2005) The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron 45: 525-538. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL (1999) The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro J Neurosci 19: 4180-4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Kazanietz MG (2003) Divergence and complexities in DAG signaling: looking beyond PKC. Trends Pharmacol Sci 24: 602-608. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T (2001) Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci 24: 1071-1089. [DOI] [PubMed] [Google Scholar]