

Abstract
Dopamine (DA) has been postulated to play a role in the loss of dopaminergic substantia nigra (SN) neurons in Parkinson's disease because of its propensity to oxidize and form quinones and other reactive oxygen species that can alter cellular function. Moreover, DA depletion can attenuate dopaminergic cell loss in vitro. To test the contribution of DA to SN impairment in vivo, we used DA-deficient mice, which lack the enzyme tyrosine hydroxylase in dopaminergic cells, and mice pharmacologically depleted of DA by α-methyl-p-tyrosine pretreatment. Mice were treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a toxin that produces parkinsonian pathology in humans, nonhuman primates, and rodents. In contrast to in vitro results, genetic or pharmacologic DA depletion did not attenuate loss of dopaminergic neurons in the SN or dopaminergic neuron terminals in the striatum. These results suggest that DA does not contribute to acute MPTP toxicity in vivo.
Keywords: Parkinson's disease, MPTP, dopamine, neurodegeneration, substantia nigra, animal models
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disorder in the United States. The predominant motor symptoms of PD are caused by the loss of dopaminergic neurons in the substantia nigra (SN). Although the etiology of PD remains unknown, both genetic and environmental factors appear to play a role. To date, six genes associated with familial PD have been identified (α-synuclein, parkin, DJ-1, PINK1, LRRK2, and UCHL1), suggesting that molecular mechanisms underlying PD may be heterogeneous (Paisan-Ruiz et al., 2004; Vila and Przedborski, 2004; Zimprich et al., 2004). Given their widespread expression patterns, however, none of these genes can account for the selective vulnerability of dopaminergic neurons. Moreover, blockade of mitochondrial respiration via systemic rotenone delivery appears to cause specific toxicity to dopaminergic neurons despite the universal effects of rotenone (Betarbet et al., 2000; Alam and Schmidt, 2002; Sherer et al., 2003). These findings suggest that some factor(s) intrinsic to dopamine (DA) neurons contributes to the disease process.
DA can be converted rapidly to a variety of reactive oxygen species and derived quinones that can be toxic to cells (Stokes et al., 1999; Blum et al., 2001; Dryhurst, 2001). These products can damage and modify macromolecules and impair energy production (Stokes et al., 1999). Furthermore, direct intrastriatal injection of DA generates cysteine adducts (Hastings et al., 1996) that inhibit mitochondrial respiration, cause mitochondrial swelling (Berman and Hastings, 1999), and ultimately damage dopaminergic terminals (Rabinovic et al., 2000).
Given that the neurons targeted for destruction in PD are those that produce DA together with abundant evidence of oxidative stress, the selective vulnerability of DA neurons may be caused by DA itself. Additional support comes from data showing that 1-methyl-4-phenylpyridinium (MPP+), the active metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), not only affects energy metabolism but also induces DA displacement from vesicles leading to free radical production and cell death in vitro (Lotharius and O'Malley, 2000). Because MPTP toxicity is one of the best available animal models of PD, these data suggest that energy depletion as well as DA oxidation contribute to dopaminergic cell loss in PD.
To test the hypothesis that DA plays a role in MPTP toxicity in vivo, we used both genetic and pharmacologic approaches to modulate DA levels in mice. DA-deficient mice (DD mice), a genetic model of DA depletion, were generated by targeted inactivation of the tyrosine hydroxylase (Th) gene, which encodes the rate-limiting enzyme required for DA biosynthesis, specifically in dopaminergic neurons (Zhou and Palmiter, 1995). DD mice exhibit DA levels <1% of controls (CTs), whereas levels of other catecholamines are normal. Daily injection of l-3,4-dihydroxyphenylalanine (l-DOPA), the precursor of DA, circumvents the absence of TH and restores striatal DA to ∼9% of control levels. Histologically, brains of DD mice appear grossly normal and have an intact nigrostriatal pathway as indicated by
l-aromatic amino acid decarboxylase (l-AADC)-positive neurons in the SN with projections to the striatum. We also used α-methyl-d,l-p-tyrosine methyl ester (AMPT), a TH inhibitor, to pharmacologically deplete DA in normal mice before MPTP treatment. This acute DA depletion model circumvents potential developmental or compensatory changes associated with chronic DA depletion. We hypothesized that DA depletion would protect the nigrostriatal pathway from acute MPTP toxicity.
Materials and Methods
Animals and drug treatments. All experiments were conducted in accordance with guidelines established by the National Academy of Sciences overseen by each institution's animal studies committee. For the genetic experiments, DD [Th-/-, dopamine β-hydroxylase (Dbh)Th/+] mice were generated by knocking out the Th gene and knocking it back into the Dbh locus to restore norepinephrine production in noradrenergic cells while eliminating DA production in dopaminergic neurons (Zhou and Palmiter, 1995). These mice have normal levels of norepinephrine in the brain, heart, and adrenal glands. DD and CT littermates with at least one intact Th gene and Dbh gene, which is sufficient to synthesize normal levels of all catecholamines, were maintained on a mixed 129/Sv × C57BL/6 genetic background. Mice maintained at the University of Washington had ad libitum access to breeder chow (5LJ5; PMI Nutritional, Brentwood, MO) and water. Starting at ∼15 d of age, DD mice received daily intraperitoneal injections of l-DOPA (50 mg/kg; Sigma, St. Louis, MO) dissolved in 2.5 mg/ml solution of ascorbic acid in PBS (Zhou and Palmiter, 1995). Male, 8- to 12-week-old DD and CT mice were shipped to Washington University for experiments. On the day of MPTP injections, one group of DD mice received the same daily l-DOPA injection 3 h before MPTP injections (DD plus l-DOPA), whereas another group did not (DD minus l-DOPA). DA levels in the DD plus l-DOPA group were 9% of normal, and levels in the DD minus l-DOPA group were <1% of normal at the time of the first MPTP injection (Zhou and Palmiter, 1995). CT and DD mice received four intraperitoneal injections of PBS or MPTP hydrochloride (Sigma) dissolved in PBS spaced 1 h apart. The following day, all DD mice resumed daily l-DOPA treatments. Mice were killed 7 d after treatment by decapitation, and brains were removed.
For the pharmacologic DA depletion experiments, 8- to 10-week-old, male C57BL/6 mice (Charles River, Wilmington, MA) received food and water ad libitum. Mice received a single injection of AMPT (250 mg/kg, i.p.; Sigma) dissolved in PBS to deplete DA, a single injection of l-DOPA (50 mg/kg) to increase DA, or PBS alone. Mice were killed 3 or 6 h later, and striata were removed for analysis of DA levels via HPLC. Another group was injected with AMPT (250 mg/kg), l-DOPA (50 mg/kg), or saline intraperitoneally, and 3 h later, mice were injected with MPTP or PBS using the paradigm as above. Mice were killed 7 d later, and brains were collected for analysis.
Western analysis. Right striata were removed and frozen on dry ice. Samples were homogenized in lysis buffer (containing the following: 10 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 0.1% SDS, 1% Triton X-100, and 1% sodium deoxycholate) containing a complete protease inhibitor mixture (Sigma). Samples were centrifuged, and equal amounts of protein from each sample were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA). Membranes were blocked in 5% powdered milk in Tris-buffered saline containing 0.05% Tween 20 and then incubated with antibodies against l-AADC (1:300) (Zhong et al., 1995), vesicular monoamine transporter 2 (VMAT2; 1:300; Chemicon, Temecula, CA), dopamine transporter (DAT; 1:300; Chemicon), TH (1:10,000; Chemicon), synaptophysin (1:10,000; Sigma), α-synuclein (1:500; PharMingen, San Diego, CA), or actin (1:10; Developmental Studies Hybridoma Bank, Iowa City, IA) followed by incubation with horseradish peroxidase (HRP)-linked secondary antibodies (HRP anti-rabbit, 1:300, Cell Signaling Technology, Beverly, MA; HRP anti-mouse and HRP anti-rat, 1:5000, Sigma). Membranes were developed with enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ) and imaged with a Storm PhosphorImager (Molecular Dynamics, Piscataway, NJ), and band intensities were determined using Imagequant software (Amersham Biosciences).
Catecholamine measurements. For MPTP experiments, left striata were immediately homogenized in lysis buffer (0.1N perchloric acid, 0.4 mm Na2S2O5) for HPLC analysis of DA levels; DA levels were determined 3 and 6 h after AMPT or saline treatment by pooling left and right striata. Samples were diluted 1:20 into HPLC buffer (0.1 m NaH2PO4, 0.25 g/L heptanesulfonic acid, 0.08 g/L EDTA, 6% methanol, pH 2.5), separated on a catecholamine ESA (Chelmsford, MA) HR-80 column using the same HPLC buffer for the mobile phase, and analyzed by electrochemical detection (ESA). Samples were analyzed in triplicate.
Immunohistochemistry. After removal of the striatum, the remaining brain was fixed in 4% paraformaldehyde for 3 d and then cryoprotected in 20% sucrose overnight. Coronal sections (50 μm) were cut on a microtome and costained for TH (1:2000) and VMAT2 (1:200) followed by fluorescent-conjugated secondary antibodies (1:500, goat anti-mouse Alexa 488, Molecular Probes, Eugene, OR; goat anti-rabbit cyanine 3, Jackson ImmunoResearch, West Grove, PA). For cell counts, every fourth section through the SN was stained for VMAT2 or TH with 3,3′-diaminobenzidine (DAB) detection (Vector Laboratories, Burlingame, CA). Adjacent sections were stained with Nissl stain. DAB+ cells in the SN (not including the retrorubral and ventral tegmental areas) and Nissl+ cells were counted using unbiased stereological methods (Stereo Investigator; MicroBrightField, Williston, VT) (West et al., 1996). Analysis was conducted blind to genotype and treatment.
To estimate the size of neuronal soma, images were taken of VMAT2-stained sections from saline-treated CT and DD mice. Approximately 100 VMAT2+ cells from each brain were analyzed by outlining the soma using MetaMorph software (Molecular Devices, Sunnyvale, CA). Mean cell cross-sectional areas for each genotype were compared.
To estimate the size of the striatum, 100 μm coronal sections from three CT and three DD mice were stained with VMAT2. Images were captured, and the striatum was outlined using MetaMorph software. Volumes were estimated by multiplying the area by the section thickness and interval.
Synaptosomal assays. Synaptosomes were prepared from striata pooled from three mice. DD mice were not treated with l-DOPA on the day of synaptosome preparation. For uptake assays performed using synaptosomes from AMPT-pretreated mice, mice were killed 3 h after AMPT or saline injection. Briefly, striata were removed, homogenized in 0.32 m sucrose buffer using a glass-Teflon homogenizer, and centrifuged (1400 × g, 10 min, 4°C). The supernatant was saved, and the pellet was homogenized again in sucrose buffer and centrifuged (710 × g, 10 min, 4°C) to remove cellular debris and nuclei. The supernatant from the first two spins was pooled and centrifuged at high speed (13800 × g, 10 min, 4°C). The resultant pellet containing synaptosomes was resuspended in Krebs'-Ringer's solution (KRS) [containing the following (in mm): 2.5 KCl, 119 NaCl, 1.3 MgSO4, 2.5 CaCl2, 1 NaH2PO4, 26.2 NaHCO3, and 10 glucose], and the protein concentration was determined using the Bradford assay. Synaptosomes (80 μg) were loaded with 30 μm [3H]-MPP+ (specific activity, 85 Ci/mmol; American Radiolabeled Chemicals, St. Louis, MO) or [3H]-DA (specific activity, 47 Ci/mmol; Amersham Biosciences) at 37°C for 20 min. Loaded synaptosomes were washed three times by pipetting in KRS followed by centrifugation. The pellet was resuspended in KRS containing 60 mm KCl (61 mm NaCl) to measure K+-induced release of [3H]-MPP+ or [3H]-DA. Synaptosomes were washed an additional time. All steps subsequent to loading were done in 10 min at 37°C followed by centrifugation, and the radioactive activity of the supernatant from each step and the final synaptosomal pellet were determined using a scintillation counter. The amount of uptake was determined by adding the counts from the KCl, final wash, and final pellet steps. The amount of release was determined by adding the counts from the K+ and final wash steps. The assay was repeated three times using different synaptosome preparations, and results were averaged to determine a mean and variance.
Statistical analysis. The Kaplan-Meier estimator of the survival function and the nonparametric Cox proportional hazards model of the Cox regression model (PROC PHREG in SAS; University of Arizona, Tucson, AZ) (Kalbfleisch and Prentice, 1980) were used for survival analyses. Factorial ANOVA and unpaired two-tailed t test were used when appropriate (SAS; GraphPad Software, San Diego, CA).
Results
Treatment of DD mice with MPTP
DD mice (without l-DOPA) and CT mice were injected four times (1 h apart) with various doses of MPTP. All of the DD mice died within 2 d of treatment when injected with 20 or 25 mg/kg of MPTP (Fig. 1A), whereas > 60% of the CT mice survived at these doses. The Cox proportional hazards model was used to test for the effect of genotype and MPTP dose on the time to death. DD mice have a hazard ratio of 10.85 at any MPTP dose versus CT mice (p < 0.0001). A dose of 15 mg/kg MPTP was used in subsequent experiments.
Figure 1.

DD mice display altered uptake of DAT substrates. A, DD mice have decreased survival compared with CT mice in the first 2 d after MPTP treatment (sample size between 5 and 27). B, Striatal synaptosomes from DD mice take up more [3H]-MPP+ and [3H]-DA than synaptosomes from CT mice. C, DD synaptosomes released 4% more [3H]-MPP+ and 5% less [3H]-DA than CT synaptosomes after stimulation with 60 mm K+. D, Western blotting of synaptosomes reveals increased levels of DAT but not other proteins in DD striatum. synap, Synaptophysin; α-syn, α-synuclein. Three independent experiments were conducted in triplicate. Error bars represent SEM. Two-way ANOVA demonstrated significant effects as indicated: *p < 0.05; **p < 0.01; ***p < 0.001.
MPP+ and DA uptake into synaptosomes from CT and DD mice
MPP+ serves as a substrate for both the DAT and VMAT2. Acutely prepared striatal synaptosomes were used to test whether MPP+ uptake was normal in DD mice. Synaptosomes from DD mice took up three times more [3H]-MPP+ and six times more [3H]-DA than CT synaptosomes (Fig. 1B). Approximately the same proportion of transported substrate was sequestered into vesicles in DD and CT synaptosomes as measured by K+-stimulated release, although the small differences noted were significant (Fig. 1C). By Western analysis, DD synaptosomes appeared to express more DAT than CT, whereas the levels of VMAT2, synaptophysin, α-synuclein, l-AADC, and actin were the same (Fig. 1D). These results indicate that more MPP+ was present in the cytoplasm of dopaminergic terminals in DD mice compared with CT mice.
Nigrostriatal injury
To determine whether the absence of DA protects against MPTP toxicity, DD mice with and without l-DOPA treatment were tested along with littermate CT. Preliminary studies revealed that VMAT2 staining reliably labeled all TH+ neurons (Fig. 2A). Therefore, this marker was used to identify dopaminergic neurons in midbrain sections from DD and CT mice. Although DD mice had fewer VMAT2+ and Nissl+ neurons in the SN than CT mice (Fig. 2C,D), they had a larger striatal volume (28.8 ± 0.4 vs 24.4 ± 0.9 mm3; p = 0.01) but smaller brains (0.429 ± 0.003 vs 0.469 ± 0.003 g; p = 0.001). In addition, the mean cross-sectional area of VMAT2+ neurons in DD mice was significantly greater than in CT mice (591.0 ± 18.8 vs 507.4 ± 14.1 μm2; p = 0.002). Genotype had no effect on cell loss assessed with VMAT2 or Nissl staining 7 d after treatment with MPTP (15 mg/kg), and restoring dopamine in DD mice using l-DOPA also had no effect on cell loss after MPTP treatment (Fig. 2C,D).
Figure 2.
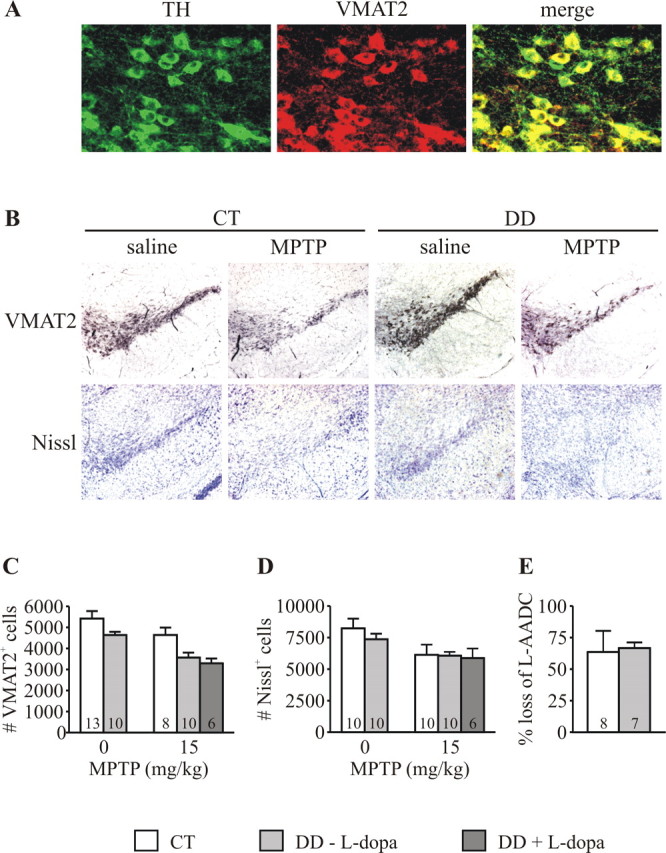
MPTP toxicity leads to similar loss of DA neuron markers in DD and CT mice. A, VMAT2 colabels all TH+ neurons in the SN. B, Loss of VMAT2+ and Nissl+ neurons as seen in the SN of CT and DD mice 7 d after MPTP treatment. C, VMAT2+ cells in the SN were counted stereologically. Two-way ANOVA demonstrated a significant effect of genotype (p = 0.004) and MPTP treatment (p = 0.004) but no evidence of an interaction, indicating that, although DD mice have fewer SN neurons, MPTP had the same effect in CT and DD mice. l-DOPA pretreatment had no effect on cell loss. D, Adjacent sections stained with Nissl were also counted stereologically. Although there was an effect of MPTP treatment on both CT and DD mice (p = 0.009), there was no evidence of a genotype effect or interaction, supporting the conclusion that MPTP had the same effect on cell loss in both CT and DD mice. l-DOPA had no effect on cell loss. E, Striatal lysates from treated CT and DD mice were analyzed by Western blotting. The loss of l-AADC was unaffected by genotype. Error bars represent SEM. The numbers within bars represent n of the experimental group.
Western analysis of striatal lysates was used to evaluate damage to dopaminergic neuron terminals. Anti-l-AADC revealed significant loss of this terminal protein, yet there was no difference between DD and CT mice (Fig. 2E). Similarly, DAT levels were decreased after treatment, but genotype had no effect on DAT loss after MPTP treatment (data not shown).
Depletion of DA with AMPT
A single AMPT injection (250 mg/kg) reduced striatal DA and metabolite levels by ∼72% at 3 and 6 h after injection into wild-type mice (Fig. 3A,B). These time points mark the first and last MPTP injection; thus, DA depletion could be maintained for the duration of MPTP injection. Higher doses of AMPT did not further deplete DA (data not shown). l-DOPA treatment (50 mg/kg) did not increase striatal DA content but did increase metabolite levels (Fig. 3A,B). Higher doses of l-DOPA (up to 120 mg/kg) had no additional effect on striatal DA or metabolite levels (data not shown).
Figure 3.

AMPT depletes striatal DA. A, DA levels were not increased by l-DOPA pretreatment but were decreased by AMPT pretreatment. B, Levels of DA metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) were increased by l-DOPA treatment and decreased by AMPT treatment. C, Radiolabeled substrate uptake in striatal synaptosomes from mice pretreated with AMPT (250 mg/kg) for 3 h versus saline-treated controls. There was a small difference in uptake of [3H]-DA but no difference in uptake of [3H]-MPP+. D, High K+-stimulated release of either [3H]-DA or [3H]-MPP+ is unaffected by pretreatment with AMPT. Error bars represent SEM. n = 3 for each treatment group in C and D. *p < 0.05; **p < 0.01; ***p < 0.001.
MPP+ and DA uptake into synaptosomes after AMPT pretreatment
Pretreatment with AMPT did not affect synaptosomal uptake of [3H]-MPP+ but did increase uptake of [3H]-DA slightly (Fig. 3C). Pretreatment did not affect 60 mm K+-stimulated release of [3H]-MPP+ or [3H]-DA (Fig. 3D), indicating that the same proportion of substrate taken up by the synaptosomes was sequestered in vesicles and K+-mediated release mechanisms were intact.
Effects of MPTP after DA depletion with AMPT
Neither AMPT nor l-DOPA treatment had any effect on survival of mice after MPTP treatment (data not shown). Likewise, these treatments had no effect on the loss of TH+ neurons in the SN (Fig. 4A) or the reduction of striatal TH levels as assessed by Western blot (Fig. 4B). There was a significant interaction between AMPT pretreatment and MPTP treatment on striatal DA levels (Fig. 4C) but not on metabolite levels (Fig. 4D). l-DOPA pretreatment did not affect the loss of DA (Fig. 4C) or metabolite levels (Fig. 4D) after MPTP treatment.
Figure 4.

DA does not contribute to acute MPTP toxicity in saline- or AMPT-treated CT mice. A, Treatment with AMPT or l-DOPA did not affect loss of TH+ neurons in the SN as determined stereologically 7 d after MPTP treatment. Factorial ANOVA comparing saline- and AMPT-pretreated groups demonstrated a significant effect of MPTP treatment (p < 0.0001) but no evidence of a pretreatment effect or interaction. B, Striatal levels of TH were equally reduced in AMPT-, l-DOPA-, and saline-pretreated mice. There was an interaction between AMPT pretreatment and MPTP treatment on DA levels (p = 0.01) (C) but not metabolite levels (D) 7 d after MPTP treatment. Treatment with l-DOPA did not affect reduction of DA (C) or metabolites (D). Values are normalized to saline-treated CT. Error bars represent SEM. The numbers within bars represent n of the experimental group. **p < 0.01.
Discussion
This study demonstrates that neither genetic nor pharmacologic DA depletion protects the nigrostriatal pathway from acute MPTP toxicity in mice. Equivalent dopaminergic neuron loss and reductions in markers for dopaminergic neuron terminal integrity were observed between DD (with DA levels <1% of CT levels) and CT mice 7 d after MPTP treatment. Moreover, increasing DA levels ∼10-fold in DD mice with l-DOPA pretreatment did not affect MPTP toxicity. Similarly, depleting striatal DA 72% with AMPT in wild-type mice did not protect dopaminergic neurons from MPTP toxicity.
DD mice provide a unique opportunity to evaluate the role of DA in a variety of behaviors and pathologies. These mice represent a conditional DA depletion because they have adequate DA for survival ∼8 h/d but are deficient for ∼16 h/d. Importantly, dopaminergic neurons are intact and appear to function normally in the absence of DA (Robinson et al., 2004). The larger size of dopaminergic neurons and striatal volume in DD mice could result from chronic l-DOPA exposure, which has been shown to affect the expression of a variety of genes, including those involved in growth, energy metabolism, and intracellular signaling (Ferrario et al., 2004). Although uptake assays in acutely prepared striatal synaptosomes do not address issues of MPTP conversion to MPP+ or clearance of the toxin, they do suggest that dopaminergic neurons in DD mice take up more toxin than neurons in CT mice. The increased uptake of both [3H]-MPP+ and [3H]-DA in DD synaptosomes probably reflects increased transport by DAT. DAT levels appear to be increased in the striatum of DD mice by Western blotting, and increased DAT expression has been associated with increased susceptibility to MPTP toxicity (Donovan et al., 1999). Striatal levels of this transporter appear normal in DD mice when assessed by ligand binding (Kim et al., 2000). However, the absence of DA could increase the substrate gradient or could affect DAT activity by covalent modification (Mortensen and Amara, 2003).
Regardless of the mechanism, acutely isolated synaptosomes from DD mice take up more MPP+ into dopaminergic terminals. Increased intracellular MPP+ would result in greater mitochondrial toxicity and, hence, might have negated any protective effects conferred on the nigrostriatal pathway by DA depletion. To address this possibility, we partially restored DA in DD mice by treating them with l-DOPA before MPTP treatment. Increasing DA levels to 9% of CT did not exacerbate MPTP toxicity in DD mice. We attempted to increase striatal DA levels to 30% of CT by using l-DOPA in combination with carbidopa (Chartoff et al., 2001). However, these doses led to stereotypy and significantly increased mortality after MPTP treatment (data not shown). Even without increased l-DOPA pretreatment, the mortality associated with toxin treatment limited our ability to evaluate the role of DA using higher doses of MPTP. This effect could be explained by greater uptake of MPP+ by sympathetic terminals in the periphery, leading to greater peripheral catecholamine release and increased cardiovascular toxicity (Giovanni et al., 1994). Additionally, increased mortality may be caused by an increase in sensitization to catecholaminergic stimulation. This is supported by previous reports demonstrating that DD mice are more sensitive to DA receptor agonists despite normal levels of DA receptors (Kim et al., 2000; Robinson et al., 2004).
To circumvent the limitations associated with MPTP treatment of DD mice, we pursued a complementary line of investigation using AMPT to acutely deplete striatal DA. In this model, no alterations in the uptake or release of MPP+ by striatal synaptosomes were observed. However, striatal DA levels could only be reduced to 28% of normal before MPTP treatment. The remaining DA could still be detrimental to dopaminergic neurons. Nevertheless, AMPT pretreatment did not ameliorate MPTP toxicity as assessed by cell counts or markers of dopaminergic terminals. We did find a significant effect of AMPT pretreatment on striatal DA levels evaluated 1 week after MPTP treatment. Because TH catalytic function is regulated by end-product inhibition (Kumer and Vrana, 1996), decreasing DA levels with AMPT pretreatment would relieve this inhibition, leading to a compensatory increase in the neurotransmitter (Fig. 4C). This increase is not observed after MPTP exposure, because MPTP inhibits TH activity by nitration (Ara et al., 1998). Therefore, the compensatory increase in TH activity observed after AMPT pretreatment would be counteracted by inhibition with MPTP. Additionally, modification of TH by catechol derivatives not only inactivates the enzyme but also converts TH to a “redox-cycling quinoprotein,” further aggravating dopaminergic damage (Kuhn et al., 1999). In contrast, our results using DD mice conclusively demonstrate that modification of TH does not significantly contribute to MPTP toxicity.
Two previous studies using chronic MPTP dosing regimens have reported no effect of AMPT pretreatment on striatal DA levels (Fuller and Hemrick-Luecke, 1985; Schmidt et al., 1985). Although these studies only depleted DA by 40% or less and did not assess other markers of nigrostriatal injury, including cell counts in the SN, they are consistent with the present findings using transgenic and pharmacologic models. Dopaminergic neurons die through different mechanisms depending on the MPTP dosing paradigm (Jackson-Lewis et al., 1995; Tatton and Kish, 1997). The chronic toxin model used in the previous studies, together with the current data using the acute MPTP paradigm, is consistent with the idea that DA oxidation does not contribute to either mechanism.
The current results challenge in vitro studies in which a similar level of DA depletion protected primary dopaminergic neurons from MPP+ (Lotharius and O'Malley, 2000). This may be because neuronal circuits are not disrupted in vivo, and synaptic activity and resulting signaling pathways remain intact. Indeed, excitotoxicity involving glutamatergic inputs has long been postulated to play a role in damage of SN neurons in PD (Greenamyre, 2001). In addition, an intact inflammatory response significantly contributes to MPTP-induced SN injury. MPTP induces a robust and chronic gliosis and microglial content correlates with neuronal death (Vila et al., 2001; Wu et al., 2002). These factors may contribute more than DA to SN injury after MPTP exposure.
We conclude that neither genetic nor pharmacologic DA depletion protects the nigrostriatal pathway from acute MPTP-induced toxicity. Although cytoplasmic DA can exacerbate MPP+-induced toxicity in vitro (Lotharius and O'Malley, 2000), the situation in vivo is more complex, and other deleterious events may predominate. The current study also points out that MPTP may not be an inclusive model of PD. In contrast with the present results, AMPT-mediated DA depletion mostly protected dopaminergic neurons from in vivo proteasomal inhibition with lactacystin (Fornai et al., 2003). Furthermore, the DA-toxicity hypothesis does not account for the preferential demise of SN dopaminergic neurons compared with other dopaminergic neurons, nor does it account for the loss of nondopaminergic cells in PD.
Footnotes
This work was supported by National Institutes of Health Grants NS39084 and MH45330 and Medical Scientist Training Program Grant T32 GM07200. We thank Bethany Sotak for maintaining the DD colony, Steven K. Harmon for technical support, and Drs. Rosalind Neuman and Hongyan Huang for statistical assistance. We also thank Drs. Eugene Johnson and David Wozniak for helpful discussions.
Correspondence should be addressed to Dr. Karen L. O'Malley, Department of Anatomy and Neurobiology, Washington University School of Medicine, Campus Box 8108, 660 South Euclid Avenue, St. Louis, MO 63110. E-mail: omalleyk@pcg.wustl.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259428-06$15.00/0
References
- Alam M, Schmidt WJ (2002) Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav Brain Res 136: 317-324. [DOI] [PubMed] [Google Scholar]
- Ara J, Przedborski S, Naini AB, Jackson-Lewis V, Trifiletti RR, Horwitz J, Ischiropoulos H (1998) Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Proc Natl Acad Sci USA 95: 7659-7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman SB, Hastings TG (1999) Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson's disease. J Neurochem 73: 1127-1137. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 3: 1301-1306. [DOI] [PubMed] [Google Scholar]
- Blum D, Torch S, Lambeng N, Nissou M, Benabid AL, Sadoul R, Verna JM (2001) Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson's disease. Prog Neurobiol 65: 135-172. [DOI] [PubMed] [Google Scholar]
- Chartoff EH, Marck BT, Matsumoto AM, Dorsa DM, Palmiter RD (2001) Induction of stereotypy in dopamine-deficient mice requires striatal D1 receptor activation. Proc Natl Acad Sci USA 98: 10451-10456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan DM, Miner LL, Perry MP, Revay RS, Sharpe LG, Przedborski S, Kostic V, Philpot RM, Kirstein CL, Rothman RB, Schindler CW, Uhl GR (1999) Cocaine reward and MPTP toxicity: alteration by regional variant dopamine transporter overexpression. Brain Res Mol Brain Res 73: 37-49. [DOI] [PubMed] [Google Scholar]
- Dryhurst G (2001) Are dopamine, norepinephrine, and serotonin precursors of biologically reactive intermediates involved in the pathogenesis of neurodegenerative brain disorders? Adv Exp Med Biol 500: 373-396. [DOI] [PubMed] [Google Scholar]
- Ferrario JE, Taravini IR, Mourlevat S, Stefano A, Delfino MA, Raisman-Vozari R, Murer MG, Ruberg M, Gershanik O (2004) Differential gene expression induced by chronic levodopa treatment in the striatum of rats with lesions of the nigrostriatal system. J Neurochem 90: 1348-1358. [DOI] [PubMed] [Google Scholar]
- Fornai F, Lenzi P, Gesi M, Ferrucci M, Lazzeri G, Busceti CL, Ruffoli R, Soldani P, Ruggieri S, Alessandri MG, Paparelli A (2003) Fine structure and biochemical mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. J Neurosci 23: 8955-8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller RW, Hemrick-Luecke SK (1985) Effects of amfonelic acid, alpha-methyltyrosine, Ro 4-1284 and haloperidol pretreatment on the depletion of striatal dopamine by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. Res Commun Chem Pathol Pharmacol 48: 17-25. [PubMed] [Google Scholar]
- Giovanni A, Sieber BA, Heikkila RE, Sonsalla PK (1994) Studies on species sensitivity to the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Part 1: Systemic administration. J Pharmacol Exp Ther 270: 1000-1007. [PubMed] [Google Scholar]
- Greenamyre JT (2001) Glutamatergic influences on the basal ganglia. Clin Neuropharmacol 24: 65-70. [DOI] [PubMed] [Google Scholar]
- Hastings TG, Lewis DA, Zigmond MJ (1996) Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci USA 93: 1956-1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Lewis V, Jakowec M, Burke RE, Przedborski S (1995) Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration 4: 257-269. [DOI] [PubMed] [Google Scholar]
- Kalbfleisch JD, Prentice RL (1980) The statistical analysis of failure time data. New York: Wiley.
- Kim DS, Szczypka MS, Palmiter RD (2000) Dopamine-deficient mice are hypersensitive to dopamine receptor agonists. J Neurosci 20: 4405-4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn DM, Arthur Jr RE, Thomas DM, Elferink LA (1999) Tyrosine hydroxylase is inactivated by catechol-quinones and converted to a redox-cycling quinoprotein: possible relevance to Parkinson's disease. J Neurochem 73: 1309-1317. [DOI] [PubMed] [Google Scholar]
- Kumer SC, Vrana KE (1996) Intricate regulation of tyrosine hydroxylase activity and gene expression. J Neurochem 67: 443-462. [DOI] [PubMed] [Google Scholar]
- Lotharius J, O'Malley KL (2000) The parkinsonism-inducing drug 1-methyl-4-phenylpyridinium triggers intracellular dopamine oxidation. A novel mechanism of toxicity. J Biol Chem 275: 38581-38588. [DOI] [PubMed] [Google Scholar]
- Mortensen OV, Amara SG (2003) Dynamic regulation of the dopamine transporter. Eur J Pharmacol 479: 159-170. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, de Munain AL, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, et al. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44: 595-600. [DOI] [PubMed] [Google Scholar]
- Rabinovic AD, Lewis DA, Hastings TG (2000) Role of oxidative changes in the degeneration of dopamine terminals after injection of neurotoxic levels of dopamine. Neuroscience 101: 67-76. [DOI] [PubMed] [Google Scholar]
- Robinson S, Smith DM, Mizumori SJ, Palmiter RD (2004) Firing properties of dopamine neurons in freely moving dopamine-deficient mice: effects of dopamine receptor activation and anesthesia. Proc Natl Acad Sci USA 101: 13329-13334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CJ, Bruckwick E, Lovenberg W (1985) Lack of evidence supporting a role for dopamine in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Eur J Pharmacol 113: 149-150. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Kim JH, Betarbet R, Greenamyre JT (2003) Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol 179: 9-16. [DOI] [PubMed] [Google Scholar]
- Stokes AH, Hastings TG, Vrana KE (1999) Cytotoxic and genotoxic potential of dopamine. J Neurosci Res 55: 659-665. [DOI] [PubMed] [Google Scholar]
- Tatton NA, Kish SJ (1997) In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labelling and acridine orange staining. Neuroscience 77: 1037-1048. [DOI] [PubMed] [Google Scholar]
- Vila M, Przedborski S (2004) Genetic clues to the pathogenesis of Parkinson's disease. Nat Med [Suppl] 10: S58-S62. [DOI] [PubMed] [Google Scholar]
- Vila M, Jackson-Lewis V, Guāegan C, Wu DC, Teismann P, Choi DK, Tieu K, Przedborski S (2001) The role of glial cells in Parkinson's disease. Curr Opin Neurol 14: 483-489. [DOI] [PubMed] [Google Scholar]
- West MJ, Ostergaard K, Andreassen OA, Finsen B (1996) Estimation of the number of somatostatin neurons in the striatum: an in situ hybridization study using the optical fractionator method. J Comp Neurol 370: 11-22. [DOI] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S (2002) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22: 1763-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong XH, Haycock JW, Shannak K, Robitaille Y, Fratkin J, Koeppen AH, Hornykiewicz O, Kish SJ (1995) Striatal dihydroxyphenylalanine decarboxylase and tyrosine hydroxylase protein in idiopathic Parkinson's disease and dominantly inherited olivopontocerebellar atrophy. Mov Disord 10: 10-17. [DOI] [PubMed] [Google Scholar]
- Zhou QY, Palmiter RD (1995) Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell 83: 1197-1209. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Muller-Myhsok B, Farrer M, Leitner P, Sharma M, Hulihan M, Lockhart P, Strongosky A, Kachergus J, Calne DB, Stoessl J, Uitti RJ, Pfeiffer RF, Trenkwalder C, Homann N, Ott E, Wenzel K, Asmus F, Hardy J, Wszolek Z, et al. (2004) The PARK8 locus in autosomal dominant parkinsonism: confirmation of linkage and further delineation of the disease-containing interval. Am J Hum Genet 74: 11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]