

Abstract
Neuregulin-1 (NRG-1) has been identified genetically as a schizophrenia susceptibility gene, but its function in the adult brain is unknown. Here, we show that NRG-1β does not affect basal synaptic transmission but reverses long-term potentiation (LTP) at hippocampal Schaffer collateral→CA1 synapses in an activity- and time-dependent manner. Depotentiation by NRG-1β is blocked by two structurally distinct and selective ErbB receptor tyrosine kinase inhibitors. Moreover, ErbB receptor inhibition increases LTP at potentiated synapses and blocks LTP reversal by theta-pulse stimuli. NRG-1β selectively reduces AMPA, not NMDA, receptor EPSCs and has no effect on paired-pulse facilitation ratios. Live imaging of hippocampal neurons transfected with receptors fused to superecliptic green fluorescent protein, as well as quantitative analysis of native receptors, show that NRG-1β stimulates the internalization of surface glutamate receptor 1-containing AMPA receptors. This novel regulation of LTP by NRG-1 has important implications for the modulation of synaptic homeostasis and schizophrenia.
Keywords: neuregulin, depotentiation, ErbB receptor, LTP, LTD, schizophrenia, superecliptic GFP
Introduction
Long-term potentiation (LTP) and long-term depression (LTD) at glutamatergic synapses between Schaffer collaterals and CA1 pyramidal neurons (hereafter denoted CA1 synapses) are excellent models of bidirectional synaptic plasticity underlying complex processes such as learning, memory, and cognition (Bliss and Collingridge, 1993; Albright et al., 2000). Whereas LTP can be induced throughout postnatal development, LTD is difficult to induce in older animals, suggesting that additional mechanisms bidirectionally regulate synaptic plasticity in the adult. LTP at adult CA1 synapses is depotentiated in acute hippocampal slices and in freely moving rodents by brief theta-pulse stimuli (TPS) and physiological stimuli, if delivered shortly after LTP induction (Huang and Hsu, 2001). Therefore, LTP, LTD, and depotentiation can serve as opposing mechanisms that maintain a dynamic range of synaptic responses and plasticity during development and in the adult (Abraham and Bear, 1996; Wagner and Alger, 1996; Nicoll and Malenka, 1999; Malenka and Bear, 2004).
Our present knowledge of neuregulin-1 (NRG-1) as a trophic and differentiation factor in the peripheral nervous system (Fischbach and Rosen, 1997; Adlkofer and Lai, 2000) and CNS (Buonanno and Fischbach, 2001; Corfas et al., 2004) is restricted mostly to early development. Although NRG-1 and its ErbB tyrosine kinase receptors (ErbB 1-4) are expressed highly in the adult rodent and human brain (Garcia et al., 2000; Huang et al., 2000; Gerecke et al., 2001; Chaudhury et al., 2003; Law et al., 2004), little is known about their functions. NRG-1 is expressed highly in CA3 pyramidal neurons that project to CA1 (Law et al., 2004), accumulates at various central synapses including the hippocampal CA1 molecular layer (Chaudhury et al., 2003; Law et al., 2004), and is processed and released at synapses in an activity-dependent manner (Loeb et al., 2002). In adult brain, ErbB and NMDA receptors (NMDARs) colocalize at glutamatergic postsynaptic sites and interact with PDZ (postsynaptic density-95/Discs large/zona occludens-1)-domain scaffolding proteins (Garcia et al., 2000; Huang et al., 2000). Based on these findings, we proposed that NRG signaling might rapidly modulate synaptic plasticity (Garcia et al., 2000; Buonanno and Fischbach, 2001), and initial work by Huang et al. (2000) supports this notion. In this study, we demonstrate that NRG-1β depotentiates LTP by reducing surface AMPA receptor (AMPAR) expression.
Materials and Methods
Materials. Human NRG-1β1 (amino acids 176-246) and NRG-1α2 (amino acids 177-241) epidermal growth factor (EGF)-like domain peptides (R & D Systems, Minneapolis, MN) were stabilized in 0.1% BSA, and ErbB receptor inhibitors 4-[(3-bromophenyl)amino]-6-(methyl-amino)-pyrido[3,4-d]pyrimidine (PD158780) and 4-[3(bromophenyl)-amino]-6-acrylamidoquinazoline (PD168393) (Calbiochem, La Jolla, CA) were dissolved in DMSO. Antibodies were against glutamate receptor 1 (GluR1) (Calbiochem) and NMDAR subunit 1 (NR1) (Pharmigen, Carlsbad, CA), and secondary goat antisera were conjugated to cyanine 3 (Cy3) (Jackson ImmunoResearch, West Grove, PA) and Alexa Fluor 488 (Molecular Probes, Eugene, OR).
Electrophysiology. Transverse hippocampal slices (300 μm) were prepared from 4- to 5-week-old C57BL/6 male mice and transferred to a submerged recording chamber continuously perfused at 2 ml/min (30°C) with artificial CSF (ACSF) containing (in mm) 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 25 glucose with 10 μm bicuculline (Tocris Cookson, Ballwin, MO) as described previously (Hoffman et al., 2002). Field and whole-cell patch-clamp recordings were performed with glass microelectrodes (6-7 MΩ) filled with internal solution containing the following (in mm): 145 K-gluconate, 10 HEPES, 5 ATP-Mg, 0.2 GTP-Na, and 0.6 EGTA, at pH 7.2 using KOH. Schaffer collateral/commissural fibers were stimulated at 0.05 Hz (0.1 ms, 20-40 μA) using a borosilicate two-barrel stimulation electrode (silver wire) filled with oxygenated ACSF recording solution. Baseline field EPSP (fEPSP) slopes were set to 40-50% of maximum responses and recorded for at least 10 min after obtaining a stable baseline. LTP was induced according to Lin et al. (2002) using theta-burst stimuli (TBS) consisting of five trains delivered at 0.2 Hz (train, five stimuli at 0.1 ms, 100 Hz). Field EPSPs from distinct synaptic pathways were measured using two stimulation pathways (S1 and S2) tested for independence by cross-facilitation (Wagner and Alger, 1995), and depotentiation was elicited by TPS (5 Hz, 1 min) as described by Staubli and Chun (1996). Paired-pulse facilitation (PPF) was recorded from CA1 pyramidal neurons using whole-cell current-clamp mode by stimulating at interpulse intervals of 50, 100, 150, and 200 ms. The AMPAR and NMDAR components of evoked EPSCs were measured in whole-cell voltage-clamp mode (Multiclamp 700A amplifier; Molecular Devices, Union City, CA) using holding potentials of -70 and +40 mV, respectively, as described by Lei and McBain (2004). NMDAR EPSCs were measured 50 ms after stimulus, a time when AMPARs are mostly deactivated. Slices showing <130% LTP were excluded from analyses. Recorded data were filtered at 3 kHz, sampled at 10 kHz using pClamp, and analyzed with Clampfit (Molecular Devices). Results are presented as mean ± SEM and analyzed for significance using Student's t test.
Live imaging in neurons. Dissociated hippocampal neuronal cultures, essentially glial free, were prepared from embryonic day 19 rat pups and maintained in defined media as reported previously (Brewer et al., 1993; Longart et al., 2004). Chemically induced LTP (chemLTP) was induced with 200 μm glycine and control cultures maintained in APV during the entire experiment, as described previously (Liao et al., 2001; Lu et al., 2001). Neurons were cotransfected at 10-12 d in vitro (DIV) with GluR2 and superecliptic green fluorescent protein (seGFP)-tagged GluR1 using Lipofectamine 2000 (Invitrogen, San Diego, CA) and imaged 24-72 h later. Cells were perfused with glycine-containing HEPES-buffered balanced salt solution (HBBSS) [containing the following (in mm): 25 HEPES, pH 7.4, 119 NaCl, 2.5 KCl, 10 NaH2PO4, 2 CaCl2, and 30 glucose] to induce chemLTP and then continuously perfused with HBBSS containing either NRG-1β (1 nm, 10 min) or NRG-1β plus 10 μm PD158780. Coverslips were maintained at 35°C with continuous perfusion (0.4 ml/min). Surface GluR1-seGFP fluorescence was quenched with acidic MES buffer [containing the following (in mm): 25 MES, pH 5.5, 119 NaCl, 2.5 KCl, 2 CaCl2, and 30 glucose] to measure the proportion of surface receptors. Images were collected every 5 s using a Zeiss (Jena, Germany) Axiovert 200 microscope (63×; numerical aperture, 1.4) coupled to a spinning disk scanhead (Ultraview RS; PerkinElmer, Wellesley, MA) with an argon-krypton laser (15 mW, 488 excitation line) and a cooled Hamamatsu (Bridgewater, NJ) CCD camera using 400 ms exposures and two-by-two binning. Fluorescence was determined in 20 μm segments of neuronal processes taken at 10 μm distance from the soma. Measurements over time were obtained using NIH ImageJ.
Immunofluorescence analysis of native surface AMPARs and NMDARs. Cells (18-22 DIV) were treated for 20 min with 0.5 nm NRG-1β with or without a 10 min of PD158780 (10 μm) preincubation. Cells were fixed with 4% paraformaldehyde-PBS for 20 min. Surface AMPARs (GluR1) and NMDARs (NR1) were detected under nonpermeabilizing conditions with antibodies against their extracellular domains and visualized with Alexa488- and Cy3-conjugated secondary antibodies using a Zeiss LSM-500 confocal microscope. Total fluorescence in 25 μm of dendritic regions of interest (ROI) from 10 neurons per treatment (n = 5) were analyzed for each experiment using MetaMorph (Molecular Devices). Cell bodies and proximal segments of dendrites were excluded.
Results
NRG-1β reverses the expression of LTP
To determine whether and how the NRG-1/ErbB signaling pathway regulates synaptic transmission and plasticity, we recorded fEPSPs from adult mouse hippocampal slices perfused with NRG-1β peptide. This short peptide encodes the active EGF-like domain necessary and sufficient for ErbB receptor activation (Buonanno and Fischbach, 2001). We found that perfusion with 0.1 or 1 nm NRG-1β did not modify basal synaptic transmission (Fig. 1A). Next, we analyzed the effects of 0.1 nm NRG-1β on LTP expression by perfusing slices with the peptide 20 min after TBS (Fig. 1B). This treatment rapidly and quantitatively reversed LTP. Field EPSP slopes were significantly reduced, compared with control slices, by the end of the 10 min of NRG-1β application (117.1 ± 6.5 vs 138.4 ± 9.8%; p < 0.05). Maximal depotentiation was attained by 20 min after the onset of peptide perfusion (104.7 ± 7.1 vs 141.5 ± 9.7%; p < 0.001). Importantly, NRG-1β never affected basal synaptic transmission or reversed LTP below pre-TBS levels, indicating that its action is stimulus dependent.
Figure 1.

NRG-1β depotentiates LTP at CA1 synapses. The black bars indicate time of NRG-1 treatment. A, Basal synaptic transmission is not modified by 0.1 nm (•; n = 7) or 1.0 nm (○; n = 7) NRG-1β. B, Effects of 0.1 nm NRG-1β (•) or vehicle (○) on LTP analyzed 20 min after TBS (arrow). Values are normalized to mean fEPSP slopes recorded 10 min before TBS (n = 10). Inset, Representative fEPSPs (10 traces averaged) taken before the TBS (1), after stabilization of LTP expression (2), and after perfusion with 0.1 nm NRG-1β (3). Calibration: 5 ms, 1 mV. C, No effect on LTP expression observed with 1 nm NRG-1α isoform (▴; n = 5) or vehicle (▵; n = 5). D, PD158780 (10 μm; gray bar) blocks LTP depotentiation by NRG-1β and augments its expression (n = 7; *p < 0.05). Data are expressed as mean ± SEM.
NRG-1 effects are splice variant selective and blocked by ErbB receptor inhibitors
We used a different NRG-1 isoform and two structurally distinct ErbB receptor inhibitors to determine the selectivity and specificity of the NRG-1β effect. NRG-1α, a splice variant that is similar to NRG-1β but activates ErbB receptors with lower efficacy (Jones et al., 1999), failed to depotentiate LTP even at 1.0 nm (Fig. 1C). Therefore, NRG-dependent depotentiation is selective for β splice variants, which are the isoforms highly expressed in brain. Next, we tested PD158780 and PD168393, highly specific and nontoxic membrane-permeable ErbB receptor antagonists that block autophosphorylation. Incubation of slices with 10 μm PD158780 efficiently blocked NRG-1β-mediated depotentiation (Fig. 1D); the same results were obtained with PD168393 (supplemental material, available at www.jneurosci.org). Interestingly, LTP slightly increased after PD158780 addition (123.8 ± 1.7 before vs 138.8 ± 0.8% after inhibitor addition; p < 0.05) but had no effect on basal synaptic transmission, suggesting that it blocked endogenous ErbB signaling at potentiated synapses.
ErbB receptor inhibition blocks depotentiation by TPS
Although TPS depotentiate LTP in the adult, it is generally accepted that their efficacy decrease with the time elapsed after TBS (after 6 min, <50% efficacious) and that LTP is difficult to reverse 30 min after induction (Staubli and Chun, 1996; Huang and Hsu, 2001). We perfused slices with NRG-1β at 30 and 50 min after LTP induction to test whether NRG-induced depotentiation is also time dependent. We found that NRG-1β could reverse LTP when perfused up to 30 min after TBS (Fig. 2A) (**p < 0.01) but was ineffective after 50 min (Fig. 2B). The limited time window of NRG-1 effectiveness suggests the stabilization of LTP expression by mechanisms that render it unresponsive to NRG-1 action (i.e., receptor, protein translation, morphological changes).
Figure 2.

NRG-1β-mediated LTP reversal shares similarities with activity-dependent depotentiation. A, B, NRG-1β reverses LTP in a time-dependent manner. Insets as in Figure 1. Slices were perfused with NRG-1β for 30 min (A) and 50 min (B) after TBS (arrow) (n = 7 and 8, respectively). C, D, TPS-induced depotentiation is blocked by ErbB receptor inhibition. C, TPS (arrowhead) blocks LTP induced in the S1 pathway (•) and has no effect on the S2 pathway (○) (n = 5). D, PD158780 (10 μm; gray bar) was added immediately after S1 stimulation (•) (n = 6). Data are expressed as mean ± SEM.
We next tested whether the NRG/ErbB signaling pathway is important for activity-induced depotentiation by TPS. A two-pathway stimulation protocol was used to test for homosynaptic effects of TPS on LTP (see Materials and Methods). In mouse hippocampal slices, as reported in the rat (Staubli and Chun, 1996), TPS reversed LTP nonreversibly when delivered 3 min after TBS (Fig. 2C, S1). The TPS used had no effect on basal synaptic transmission (data not shown) or on LTP expression in the control pathway (Fig. 2C, S2). In contrast, TPS could not reverse LTP when slices were perfused with PD158780 immediately after TBS (Fig. 2D, S1). As expected, a transient decrease in the magnitude of normalized fEPSPs was observed shortly after TPS delivery, but by 30 min after TBS, fEPSPs were not significantly different from those induced in the control pathway (Fig. 2D, S2). Together, our results indicate that the NRG/ErbB signaling pathway modulates synaptic potentiation levels in vivo in an activity-dependent manner.
NRG-1 selectively reduces AMPAR-dependent EPSCs at potentiated synapse
To determine the locus of NRG-1 action, we analyzed the contributions of presynaptic and postsynaptic mechanisms. Whole-cell current-clamp recordings were used to measure PPF ratios (expressed as EPSP2/EPSP1) in pyramidal neurons 5 min before TBS and at 10 and 50 min after TBS. NRG-1β was perfused 20-30 min after TBS. Although EPSP amplitudes recorded in potentiated slices (10 min after TBS) were higher than those before TBS and after NRG-1 treatment (Fig. 3A), there were no significant differences in PPF ratios in potentiated slices at the four interpulse intervals measured before or after NRG-1 treatment (Fig. 3B). These results indicate that NRG-1 is unlikely to reverse LTP by a presynaptic mechanism.
Figure 3.

NRG-1β acts postsynaptically on AMPA receptors to reverse LTP. For both experiments, NRG-1β was perfused for 10 min starting 20 min after TBS. A, B, PPF ratios are not modified by NRG-1β. A, Representative traces of the first (P1) and second (P2) evoked EPSPs at 50 ms interpulse intervals recorded before, and at 10 and 50 min after, TBS. Calibration: 10 ms, 1 mV. B, PPF ratios from potentiated slices stimulated at 50-200 ms intervals before (○) and after (•) NRG-1β perfusion (n = 12). C, D, NRG-1β selectively reduces AMPAR EPSCs in potentiated slices. C, Representative EPSCs recorded at holding potentials of-70 and +40 mV measured -5, 15, 30, and 50 min relative to TBS. Calibration: 10 ms, 100 pA. NMDAR EPSCs were measured at 50 ms (dotted lines) to reduce contribution of AMPAR currents. D, Quantification of voltage-clamp results: AMPAR (□) and NMDAR (▪). Comparisons are between AM-PAR EPSCs at 15 min to EPSCs at 30 and 50 min (n = 10; *p < 0.05; **p < 0.001). Data are expressed as mean ± SEM.
Whole-cell voltage-clamp recordings were used to determine the glutamate receptor subtype affected by NRG-1. As shown in sample traces recorded from a CA1 principal neuron held at -70 mV (Fig. 3C), increases in AMPAR EPSCs 15 min after TBS were reversed after NRG-1β perfusion (20-30 min after TBS). In contrast, NMDAR EPSCs were not affected appreciably by either TBS or NRG-1 treatment. This result was confirmed by measuring NMDAR EPSCs at +40 mV in presence of 10 μm CNQX, which blocked any residual AMPAR currents (supplemental material, available at www.jneurosci.org). At 15 min after LTP induction, AMPAR and NMDAR EPSCs were 152.7 ± 9.9 and 105.2 ± 4.3% of baseline, respectively (Fig. 3D). At 30 and 50 min after TBS, NRG-1β treatment decreased AMPAR EPSCs to 126.8 ± 9.7 and 109.8 ± 4.9% of baseline. At 50 min, AMPAR EPSCs were indistinguishable from those recorded 5 min before TBS (Fig. 3C,D, -5 min). No significant changes in NMDAR EPSCs were observed at any time after NRG-1 addition (Fig. 3D; supplemental material, available at www.jneurosci.org). Together, our results suggested that NRG-1β, signaling via ErbB receptors, acts postsynaptically to downregulate AMPAR function and to depotentiate LTP.
Visualization of NRG-1-induced internalization of transfected and native GluR-1-containing AMPARs
To analyze the cellular mechanisms that underlie NRG-dependent depotentiation, we used an in vitro model of chemLTP that is NMDAR dependent and routinely results in an approximately twofold increase in surface GluR1-containing AMPARs (Liao et al., 2001; Lu et al., 2001; Pickard et al., 2001). Hippocampal neurons were cotransfected with untagged GluR2 and a GluR1 construct fused at its N-terminal extracellular domain to seGFP to monitor in real time its surface expression (Ashby et al., 2004), exploiting the pH sensitivity of seGFP that renders it ∼20-fold more fluorescent at the cell surface (pH ∼ 7.4) than in endocytic vesicles (pH ∼ 5.5). Most of the fluorescence in processes of transfected neurons after chemLTP is attributable to surface seGFP-GluR1-containing AMPARs, as judged by the dramatic reduction of fluorescence after exposure to pH 5.5 recording medium (Fig. 4A,B). Next, we analyzed the acute effects of NRG-1β on chemLTP expression. NRG-1β caused a significant reduction in fluorescence at receptor clusters and in neurites (Fig. 4A, top), starting ∼2 min after the onset of perfusion, as shown in the line graphs of the entire recording session (Fig. 4B, left). Five minutes after NRG-1β application (Fig. 4C, left), the levels of surface GluR1 receptors were reduced significantly (68.6 ± 5.3%) relative to those in chemLTP cultures (set to 100%). The NRG-1β-induced reduction of surface seGFP-GluR1 requires ErbB receptor activation, because the effect was blocked by PD158780 (Fig. 4A-C, right). Fluorescence levels after NRG plus PD158780 treatment were not significantly different from chemLTP cultures (90.9 ± 5.0 vs 100%).
Figure 4.
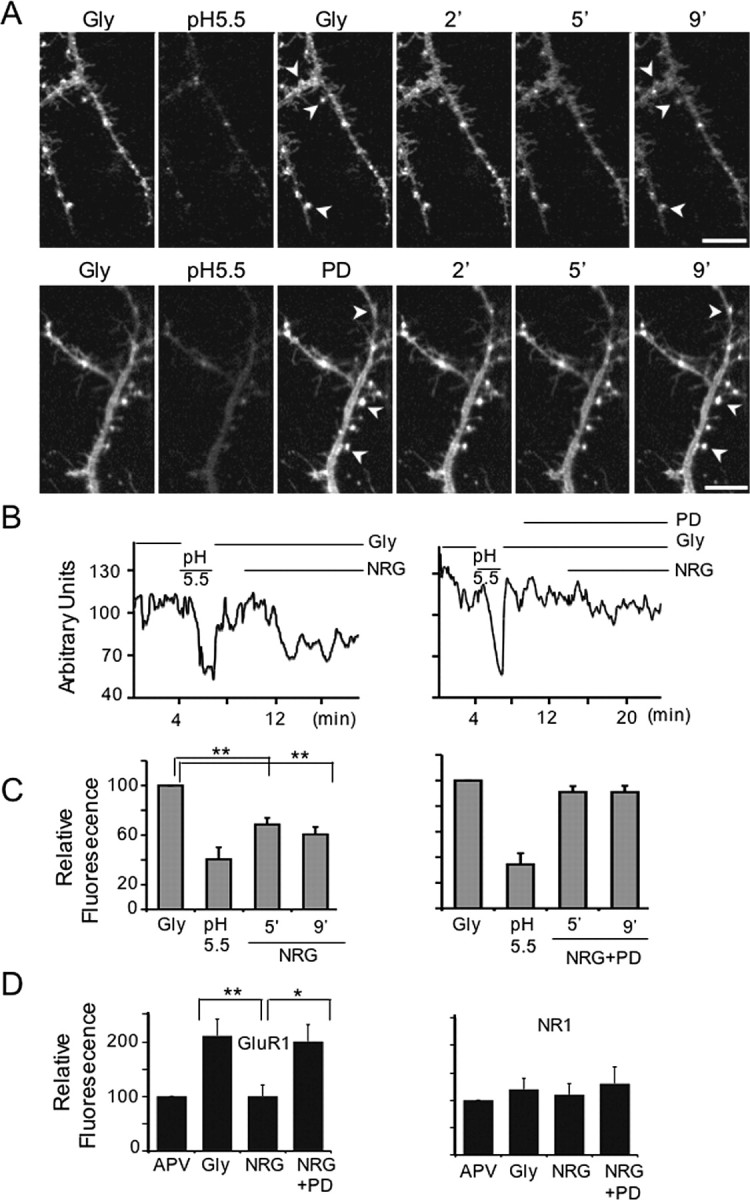
Effects of NRG-1 on transfected and native surface GluR1 AMPARs in hippocampal neurons. A, Representative micrograph series of neuronal processes imaged after chemLTP induction (Gly), pH 5.5 incubation, and 2, 5, and 9 min of treatment with NRG-1β (top) or NRG1 plus PD158780 (bottom). Arrowheads indicate the same areas before and during treatment. Scale bar, 4 μm. B, Representative line graphs of recording sessions of cultures perfused with NRG-1 (left) or NRG-1 plus PD158780 (right). The horizontal bars indicate duration of treatments. C, Quantitative analysis of seGFP-GluR1 surface expression treated with NRG-1β (left; N = 6; n = 20 ROI) or NRG-1β plus PD158780 (right; N = 5; n = 17 ROI). The intensities per 20 μm of neurite process were averaged from random ROI over 14 frames (70 s). D, Relative levels of native surface GluR1-containing AMPARs and NMDARs detected by dual immunofluorescence to GluR1 and NR1 subunits, respectively. Expression was quantified by measuring fluorescence intensities in 25 μm of dendrite (N = 5; n = 60-90 ROI per treatment). Values are normalized to APV-treated cultures (APV). Data for C and D represent the mean ± SEM (*p < 0.05; **p < 0.01; one-way ANOVA). Gly, Glycine; PD, PD158780.
We used dual-fluorescence immunocytochemistry as a complementary approach to analyze the effects of NRG-1 on surface levels of native AMPA and NMDA receptors (see Materials and Methods). As shown in Figure 4D (images are in supplemental material, available at www.jneurosci.org), chemLTP induction by glycine increased surface expression of GluR1-containing AMPARs (213 ± 29%) but not of NMDARs (120 ± 18%). Treatment of chemLTP cultures with NRG-1β reduced surface GluR1-containing AMPARs (106 ± 15%) to pre-chemLTP levels (Fig. 4D, left), and this was blocked by PD158780 (195 ± 26%). No changes in NMDAR surface levels were observed for any treatment (Fig. 4D, right), indicating that NRG-1β promotes the selective removal of surface AMPARs.
Discussion
To our knowledge, this work represents the first demonstration of how the NRG/ErbB signaling pathway regulates activity-dependent synaptic plasticity. Our results are consistent with those of Huang et al. (2000), who showed that the continuous perfusion of slices with NRG-1 before, during, and after TBS blocked LTP at CA1 synapses without affecting NMDAR currents. However, their experiments did not address whether NRG-1 blocks LTP induction or expression nor the specificity of the NRG-1 isoforms, the requirement of ErbB receptor signaling, or the cellular/molecular mechanisms underlying NRG-1 action (i.e., effects on AMPAR EPSCs were not measured). Our results indicate that NRG-1β depotentiates LTP at CA1 synapses by selectively reducing AMPAR EPSCs, which, together with our chemLTP experiments, strongly suggests that NRG-1 reverses LTP in vivo by promoting the internalization of GluR1-containing AMPARs. While this work was under review, Gu et al. (2005) reported that NRG-1 reduces whole-cell NMDAR currents in acutely dissociated and cultured prefrontal cortex pyramidal neurons by ∼15% in response to NMDA application but has no effect on AMPAR currents. The observation that NRG-1 regulates distinct components of the glutamatergic pathway in cortical (Gu et al., 2005) and hippocampal (Huang et al., 2000; this work) pyramidal neurons, suggests that NRG-1 elicits responses that vary between neuronal types or ages possibly by recruiting different downstream signaling pathways.
It is interesting to note that the effects of NRG-1/ErbB signaling on glutamatergic transmission are quite unique and differ dramatically from those of other factors signaling via tyrosine kinase receptors. Brain-derived neurotrophic factor, integrins, and ephrins facilitate or augment LTP (Kramar et al., 2004), and insulin depresses basal glutamatergic transmission at CA1 synapses independently of activity. In contrast, the NRG-1/ErbB pathway shares several properties with TPS-dependent depotentiation: its effects are activity dependent (no effect on basal transmission), it functions in the adult, and it shows a time dependence relative to the onset of LTP induction. The observation that ErbB receptor inhibitors block LTP depotentiation by NRG-1β and by TPS, and increase LTP expression at potentiated synapses, suggests that this pathway can function in the adult brain to depotentiate recently activated synapses that are not strongly reinforced. It is conceivable that the NRG/ErbB pathway preserves a dynamic range of synaptic responses and plasticity, similar to the proposed function of LTD (Abraham and Bear, 1996; Montgomery and Madison, 2004).
Many groups have replicated the original finding that NRG-1 is a candidate gene for schizophrenia (Stefansson et al., 2004). Of the 10 genetically identified putative susceptibility genes, NRG-1 is considered among the stronger candidates based on biological plausibility (Buonanno and Fischbach, 2001; Corfas et al., 2004) and genetic criteria (Harrison and Weinberger, 2005). What are the implications of our findings for a possible role of the NRG-1/ErbB signaling pathway in schizophrenia? Two studies have reported increased levels of NRG-1 transcripts in prefrontal cortex (Hashimoto et al., 2004) and peripheral leukocytes (Petryshen et al., 2005) of patients with schizophrenia. However, it is unknown whether these represent primary or compensatory changes, or whether they result in different NRG-1 protein levels. Although the long-term effects of changes in NRG-1 levels on glutamatergic function are impossible to predict a priori, it is plausible that they could modify postsynaptic responses to glutamate and general excitability. For example, changes in NRG-1 signaling could result in modified surface AMPAR expression and consequently affect directly or indirectly NMDAR function. Interestingly, pharmacological and neuropathological studies indicate that glutamatergic transmission and/or plasticity are reduced in the hippocampus and associated pathways of affected individuals (Harrison, 2004), consistent with the schizophrenia hypoglutamatergic function hypothesis (Tsai and Coyle, 2002). Thus, it is plausible that imbalances in NRG-1/ErbB signaling modify the dynamic range of glutamatergic synapses, consequently altering the complex network activity that underlies the cognitive processes affected in schizophrenia.
Footnotes
We thank Dr. Gero Miesenböck for providing superecliptic GFP. We are also grateful to Drs. Chris McBain and Phillip Nelson for critical reading of this manuscript. The invaluable assistance of Louis Dye and Vincent Schram from the National Institute of Child Health and Human Development (NICHD) Imaging Facility and Olabisi Odutayo is greatly appreciated. We thank the NICHD Intramural Program for its continued support.
Correspondence should be addressed to Dr. Andrés Buonanno, Section on Molecular Neurobiology, National Institutes of Health, Building 35, Room 2C-1000, Bethesda, MD 20892-3714. E-mail: buonanno@helix.nih.gov.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259378-06$15.00/0
O.-B.K. and M.L. contributed equally to this work.
References
- Abraham WC, Bear MF (1996) Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci 19: 126-130. [DOI] [PubMed] [Google Scholar]
- Adlkofer K, Lai C (2000) Role of neuregulins in glial cell development. Glia 29: 104-111. [DOI] [PubMed] [Google Scholar]
- Albright TD, Kandel ER, Posner MI (2000) Cognitive neuroscience. Curr Opin Neurobiol 10: 612-624. [DOI] [PubMed] [Google Scholar]
- Ashby MC, Ibaraki K, Henley JM (2004) It's green outside: tracking cell surface proteins with pH-sensitive GFP. Trends Neurosci 27: 257-261. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31-39. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ (1993) Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res 35: 567-576. [DOI] [PubMed] [Google Scholar]
- Buonanno A, Fischbach GD (2001) Neuregulin and ErbB receptor signaling pathways in the nervous system. Curr Opin Neurobiol 11: 287-296. [DOI] [PubMed] [Google Scholar]
- Chaudhury AR, Gerecke KM, Wyss JM, Morgan DG, Gordon MN, Carroll SL (2003) Neuregulin-1 and erbB4 immunoreactivity is associated with neuritic plaques in Alzheimer disease brain and in a transgenic model of Alzheimer disease. J Neuropathol Exp Neurol 62: 42-54. [DOI] [PubMed] [Google Scholar]
- Corfas G, Roy K, Buxbaum JD (2004) Neuregulin 1-erbB signaling and the molecular/cellular basis of schizophrenia. Nat Neurosci 7: 575-580. [DOI] [PubMed] [Google Scholar]
- Fischbach GD, Rosen KM (1997) ARIA: a neuromuscular junction neuregulin. Annu Rev Neurosci 20: 429-458. [DOI] [PubMed] [Google Scholar]
- Garcia RA, Vasudevan K, Buonanno A (2000) The neuregulin receptor ErbB-4 interacts with PDZ-containing proteins at neuronal synapses. Proc Natl Acad Sci USA 97: 3596-3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerecke KM, Wyss JM, Karavanova I, Buonanno A, Carroll SL (2001) ErbB transmembrane tyrosine kinase receptors are differentially expressed throughout the adult rat central nervous system. J Comp Neurol 433: 86-100. [DOI] [PubMed] [Google Scholar]
- Gu Z, Jiang Q, Fu AK, Ip NY, Yan Z (2005) Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. J Neurosci 25: 4974-4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ (2004) The hippocampus in schizophrenia: a review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology (Berl) 174: 151-162. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR (2005) Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry 10: 40-68. [DOI] [PubMed] [Google Scholar]
- Hashimoto R, Straub RE, Weickert CS, Hyde TM, Kleinman JE, Weinberger DR (2004) Expression analysis of neuregulin-1 in the dorsolateral prefrontal cortex in schizophrenia. Mol Psychiatry 9: 299-307. [DOI] [PubMed] [Google Scholar]
- Hoffman DA, Sprengel R, Sakmann B (2002) Molecular dissection of hippocampal theta-burst pairing potentiation. Proc Natl Acad Sci USA 99: 7740-7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Hsu KS (2001) Progress in understanding the factors regulating reversibility of long-term potentiation. Rev Neurosci 12: 51-68. [DOI] [PubMed] [Google Scholar]
- Huang YZ, Won S, Ali DW, Wang Q, Tanowitz M, Du QS, Pelkey KA, Yang DJ, Xiong WC, Salter MW, Mei L (2000) Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron 26: 443-455. [DOI] [PubMed] [Google Scholar]
- Jones JT, Akita RW, Sliwkowski MX (1999) Binding specificities and affinities of egf domains for ErbB receptors. FEBS Lett 447: 227-231. [DOI] [PubMed] [Google Scholar]
- Kramar EA, Lin B, Lin CY, Arai AC, Gall CM, Lynch G (2004) A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J Neurosci 24: 5151-5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law AJ, Shannon Weickert C, Hyde TM, Kleinman JE, Harrison PJ (2004) Neuregulin-1 (NRG-1) mRNA and protein in the adult human brain. Neuroscience 127: 125-136. [DOI] [PubMed] [Google Scholar]
- Lei S, McBain CJ (2004) Two loci of expression for long-term depression at hippocampal mossy fiber-interneuron synapses. J Neurosci 24: 2112-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Scannevin RH, Huganir R (2001) Activation of silent synapses by rapid activity-dependent synaptic recruitment of AMPA receptors. J Neurosci 21: 6008-6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Brucher FA, Colgin LL, Lynch G (2002) Long-term potentiation alters the modulator pharmacology of AMPA-type glutamate receptors. J Neurophysiol 87: 2790-2800. [DOI] [PubMed] [Google Scholar]
- Loeb JA, Hmadcha A, Fischbach GD, Land SJ, Zakarian VL (2002) Neuregulin expression at neuromuscular synapses is modulated by synaptic activity and neurotrophic factors. J Neurosci 22: 2206-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longart M, Liu Y, Karavanova I, Buonanno A (2004) Neuregulin-2 is developmentally regulated and targeted to dendrites of central neurons. J Comp Neurol 472: 156-172. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT (2001) Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29: 243-254. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44: 5-21. [DOI] [PubMed] [Google Scholar]
- Montgomery JM, Madison DV (2004) Discrete synaptic states define a major mechanism of synapse plasticity. Trends Neurosci 27: 744-750. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC (1999) Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann NY Acad Sci 868: 515-525. [DOI] [PubMed] [Google Scholar]
- Petryshen TL, Middleton FA, Kirby A, Aldinger KA, Purcell S, Tahl AR, Morley CP, McGann L, Gentile KL, Rockwell GN, Medeiros HM, Carvalho C, Macedo A, Dourado A, Valente J, Ferreira CP, Patterson NJ, Azevedo MH, Daly MJ, Pato CN, et al. (2005) Support for involvement of neuregulin 1 in schizophrenia pathophysiology. Mol Psychiatry 10: 366-374, 328. [DOI] [PubMed] [Google Scholar]
- Pickard L, Noel J, Duckworth JK, Fitzjohn SM, Henley JM, Collingridge GL, Molnar E (2001) Transient synaptic activation of NMDA receptors leads to the insertion of native AMPA receptors at hippocampal neuronal plasma membranes. Neuropharmacology 41: 700-713. [DOI] [PubMed] [Google Scholar]
- Staubli U, Chun D (1996) Factors regulating the reversibility of long-term potentiation. J Neurosci 16: 853-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Steinthorsdottir V, Thorgeirsson TE, Gulcher JR, Stefansson K (2004) Neuregulin 1 and schizophrenia. Ann Med 36: 62-71. [DOI] [PubMed] [Google Scholar]
- Tsai G, Coyle JT (2002) Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol 42: 165-179. [DOI] [PubMed] [Google Scholar]
- Wagner JJ, Alger BE (1995) GABAergic and developmental influences on homosynaptic LTD and depotentiation in rat hippocampus. J Neurosci 15: 1577-1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JJ, Alger BE (1996) Homosynaptic LTD and depotentiation: do they differ in name only? Hippocampus 6: 24-29. [DOI] [PubMed] [Google Scholar]