

Abstract
Abundant filamentous tau inclusions in oligodendrocytes (OLGs) are hallmarks of neurodegenerative tauopathies, including sporadic corticobasal degeneration and hereditary frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17). However, mechanisms of neurodegeneration in these tauopathies are unclear in part because of the lack of animal models for experimental analysis. We address this by generating transgenic (Tg) mice expressing human tau exclusively in OLGs using the 2′,3′-cyclic nucleotide 3′-phosphodiesterase promoter. Filamentous OLG tau inclusions developed in these Tg mice as a result of human tau expression in OLGs, especially those expressing the FTDP-17 human P301L mutant tau. Notably, structural disruption of myelin and axons preceded the emergence of thioflavin-S positive tau inclusions in OLGs, but impairments in axonal transport occurred even earlier, whereas motor deficits developed subsequently, especially in Tg mice with the highest tau expression levels. These data suggest that the accumulation of tau in OLG cause neurodegeneration, and we infer they do so by disrupting axonal transport. We suggest that similar defects may also occur in sporadic and hereditary human tauopathies with OLG tau pathologies.
Keywords: tau, transgenic mouse, oligodendrocyte, myelin, neurodegeneration, axonal transport
Introduction
A broad array of evidence implicates CNS oligodendrocytes (OLGs) in regulating the growth and maintenance of axons in the CNS (for review, see Pfeiffer et al., 1993). Axons are ensheathed and myelinated by OLGs, thereby enabling communication between axons and myelin as well as promoting axonal integrity and functions (Lemke, 1988). Thus, abnormal myelination during development and demyelination in adults are linked mechanistically to diverse CNS disorders (for review, see Readhead and Hood, 1990; Bjartmar et al., 1999). In these pathological circumstances, alterations of axonal caliber and axonal transport rates coupled with deficiencies of myelin morphology are observed, whereas axonal disruption without visible disassembly of myelin structure also is seen, and presumably this is related to abnormal myelin components (Griffiths et al., 1998; Brady et al., 1999; Lappe-Siefke et al., 2003). Although the available data imply that several different regulatory signals derived from OLGs affect levels and posttranslational modifications of the axonal cytoskeletal constituents, including microtubules (MTs) and neurofilaments (NFs), the molecular mechanisms by which these signals interact with axonal elements have yet to be clarified.
Disruption of axon-OLG relationships may not be confined to disorders of myelination because it is plausible this could result from CNS degenerative diseases characterized primarily by accumulations of filamentous protein inclusions in OLGs. For example, fibrillary aggregates in OLGs known as glial cytoplasmic inclusions (GCIs) are hallmark lesions of multiple system atrophy (MSA), and GCIs are composed of α-synuclein as well as tau but to a lesser extent (Murayama et al., 1992; Tu et al., 1998) (for review, see Richter-Landsberg, 2000). Other filamentous OLG inclusions known as coiled bodies are primarily composed of hyperphosphorylated tau (Chin and Goldman, 1996; Dickson et al., 1996; Forman et al., 2002), and these inclusions are characteristic of several frontotemporal dementias (FTDs) such as sporadic progressive supranuclear palsy (PSP), Pick's disease, and corticobasal degeneration (CBD), as well as hereditary FTD with parkinsonism linked to chromosome 17 (FTDP-17) (for review, see Komori, 1999; Forman et al., 2000; Richter-Landsberg, 2000). Although GCIs are associated with myelin abnormalities in MSA (Matsuo et al., 1998), altered myelination is not linked to accumulations of coiled bodies in most glial tauopathies despite widespread evidence of neurodegeneration. Hence, it remains unclear how tau inclusions in OLGs lead to neurodegeneration in these tauopathies.
Tau, an MT-associated protein, promotes the stabilization of MTs and polymerization of tubulins into MTs (for review, see Brandt, 1996; Higuchi et al., 2002b). Localization of tau to axons implies roles for tau in the organization of axonal cytoskeletons, whereas tau in cultured OLGs participates in the modulation of MT stability (LoPresti et al., 1995) (for review, see Richter-Landsberg, 2000). Filamentous tau aggregates in neurons are pathological hallmarks of Alzheimer's disease, in addition to most FTDs and other neurodegenerative disorders collectively known as tauopathies. Discovery of pathogenic tau gene mutations in FTDP-17 provided compelling evidence that abnormalities of tau alone are sufficient to cause neurodegenerative disease (for review, see Forman et al., 2000; Lee et al., 2001; Higuchi et al., 2002b). Mechanistic links between formation of neuronal tau inclusions and neurodegeneration are supported by studies of mouse models of neuronal tau pathologies (Ishihara et al., 1999; Lewis et al., 2000; Allen et al., 2002; Tanemura et al., 2002; Zhang et al., 2004). Hence, it is plausible that accumulations of OLG tau inclusions play a mechanistic role in human neurodegenerative diseases such as FTDs with OLG tau pathologies, and this notion is supported by our recent studies of the neurodegenerative phenotype seen in tau transgenic (Tg) mice that expressed human tau in both neurons and glia (Higuchi et al., 2002a). Thus, we tested the hypothesis that OLG tau pathologies contribute to neurodegeneration in FTDs. We reported here on newly generated tau Tg mice engineered to express human mutant and wild-type (WT) tau exclusively in OLGs that developed an FTD-like neurodegenerative phenotype with OLG tau pathologies. Notably, these studies imply that OLG tau inclusions cause neurodegeneration by disrupting axonal transport in our tau Tg mice and in human tauopathies with OLG tau pathologies.
Materials and Methods
Generation of Tg mice overexpressing human tau in OLGs. Human T34 (i.e., the tau isoform with four MT binding repeats and one N-terminal insert) cloned into PRK172 vector was cut out with NdeI and EcoRI and inserted into the EcoRV site of SK+ vector to generate a stop codon just upstream of the T34 sequence. Polyadenylation signal was created downstream of the T34 sequence by mutagenesis (Promega, Madison, WI). The T34/SK+ was then cleaved by EcoRI and ClaI and cloned into the HindIII site in exon 1 of the mouse 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNP) gene (Gravel et al., 1998). Expression of undesirable protein driven by the promoter 2 (P2) was abolished by the stop codon upstream of the T34 sequence. FTDP-17 P301L (PL) mutant tau was then generated in the T34 cDNA (Promega). The DNAs containing the murine CNP promoter, the human WT (hWT) or PL mutant tau cDNA and the polyadenylation signal (Fig. 1 A) were linearized and microinjected into fertilized C57BL/C3H mouse eggs, which were subsequently reimplanted into pseudopregnant females to generate potential founder mice bearing either hWT or PL mutant T34. Samples of genomic DNA were isolated from mouse tails using a Puregene DNA isolation kit (Gentra Systems, Minneapolis, MN) and screened by Southern blot analysis with a 32P-labeled fragment of NotI/EcoRI-cleaved T34/PRK172.
Figure 1.
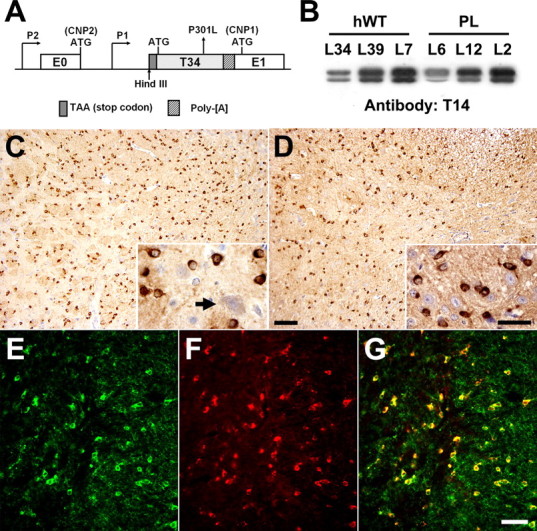
Expression of Tg human tau in OLGs of the murine nervous system. A, Schematic representation of the transgene construct used to generate tau Tg mice, which consists of a stop codon, human T34 with or without the PL mutation, and polyadenylation site (Poly-[A]) inserted into HindIII site between CNP exons 0 and 1 (E0 and E1). Transgene expression in OLGs was directed by the CNP promoter P1, whereas expression from CNP promoter P2 was impeded by a stop codon upstream of the T34 sequence. B, Immunoblots using the human tau-specific anti-tau antibody T14 demonstrate varying levels of transgene expression in the brains of the different tau Tg lines. All protein samples were derived from 3-month-old heterozygous Tg mice. C, D, Sections of the basal ganglia (C) and spinal cord (D) of a 3-month-old line 12 PL Tg mouse immunolabeled with antibody 17026 show intense tau immunoreactivity in glial cells with the morphology of OLGs, whereas neurons show far less intense positivity for endogenous mouse tau (arrow). E-G, Double-immunofluorescence staining with anti-CNP antibody (E) and anti-tau antibody 17026 (F) in spinal cord of a 3-month-old line 12 PL Tg mouse showing robust expression of tau in CNP-positive OLGs. A dualchannel image (G) indicates ∼100% colocalization of CNP and tau positivity in OLGs. Scale bars: C, D, 100 μm; insets in C, D, 20 μm; E-G, 50 μm.
Biochemical assays. Expression of Tg tau in the brain and spinal cord was measured and compared among different lines of non-Tg or WT and Tg mice by an immunoblot analysis as described previously (Higuchi et al., 2002a). Briefly, CNS tissues were dissected after the mice were lethally anesthetized according to protocols approved by the University of Pennsylvania. The samples were homogenized in 2 ml/gm ice-cold high-salt reassembly buffer (HS) [0.1 m MES, 1 mm EGTA, 0.5 mm MgSO4, 0.75 m
NaCl, 0.02 m NaF, 1 mm PMSF, and 0.1% protease inhibitor mixture (100 μg/ml each of pepstatin A, leupeptin, N-tosyl-l-phenylalanyl chloromethyl ketone, N-tosyl-lisine chloromethyl ketone, soybean trypsin inhibitor, and 100 mm EDTA), pH 7.0], and centrifuged at 100,000 × g for 40 min at 4°C in a Beckman Coulter (Fullerton, CA) TL-100 ultracentrifuge. The supernatants were boiled for 5 min, chilled on ice for 5 min, and recentrifuged at 10,000 × g for 20 min at 4°C to eliminate heat-labile proteins. Protein concentration was measured for the final supernatants with a BCA assay kit (Pierce, Rockford, IL). Equal amounts (40 μg) of sample were resolved on 7.5% SDS-PAGE gels and transferred onto nitrocellulose membranes, which were incubated with anti-tau antibodies, followed by 125I-labeled goat anti-mouse IgG or 125I-labled Protein A (PerkinElmer, Boston, MA) for quantitative analysis (Ishihara et al., 1999).
To analyze solubility of CNS tau proteins, tissues were also collected from the non-Tg or WT and Tg mice at the ages of 3, 6, 9, and 12 months as described above, and HS-insoluble pellets were rehomogenized with 1 ml/gm radioimmunoprecipitation assay (RIPA) buffer [50 mm Tris, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40, and 5 mm EDTA, pH 8.0] and centrifuged at 100,000 × g for 40 min at 4°C. Unlike previous studies (Ishihara et al., 1999; Higuchi et al., 2002a), removal of myelin and associated lipids using sucrose buffer was not performed because Tg tau was associated with these components. The resulting pellets were extracted with 70% formic acid (FA) to recover insoluble protein aggregates, and quantitative immunoblotting was performed with anti-tau antibodies and 125I-labeled secondary antibodies. Finally, myelin proteins were analyzed by extracting myelin from homogenized brain samples in 0.32 m sucrose as reported previously (Norton and Poduslo, 1973).
Histochemical and immunohistochemical analyses. Histopathological features of the Tg mice were studied by analyzing 3-, 6-, 9-, 12-, and 15-month-old WT and Tg mice as published previously (Higuchi et al., 2002a). Briefly, mice were deeply anesthetized and transcardially perfused with 15 ml of PBS, followed by 15 ml of either 70% ethanol in isotonic saline or 4% paraformaldehyde in phosphate buffer (PB). Representative 6-μm-thick paraffin sections of the brains and spinal cords were immunostained using standard streptavidin- biotin-peroxidase techniques. Additionally, the sections were histochemically analyzed by Gallyas silver impregnation (Gallyas, 1971) and thioflavin-S staining to visualize mature pathological filaments. Furthermore, myelin was semiquantitatively analyzed by performing Luxol fast blue staining of paraffin sections. To monitor alterations in axons and their myelin sheaths, mice were serially perfused (Li et al., 1997; Higuchi et al., 2002a) with PBS, 3% paraformaldehyde in PB (pH 7.5), 3% paraformaldehyde in sodium borate buffer, pH 9.0, and 10% sucrose in PB, and the CNS tissues were cryoprotected in 25% sucrose/10% glycerol in PB. Double-immunofluorescence staining was then performed on 15-μm-thick frozen sections to localize axonal cytoskeletal and myelin constituents.
Association of OLG tau pathology with myelin degeneration also was investigated in human PSP and CBD brain samples that were serially fixed with 3% paraformaldehyde in PB and 3% paraformaldehyde in sodium borate buffer and then cryoprotected with sucrose/glycerol in PB to generate 15-μm-thick frozen sections for immunofluorescence staining to visualize myelin structures and tau lesions. Furthermore, paraffin sections from FTD patients with abundant OLG tau inclusions were stained with anti-tau antibodies and Luxol fast blue to examine the effects of OLG tau pathology on myelin structures.
Electron microscopic study. Degeneration of axons and myelin sheaths were investigated at an ultrastructural level by transmission EM (TEM) in non-Tg and Tg mice at 6, 12, 15, and 18 months of age as described previously (Ishihara et al., 1999; Higuchi et al., 2002a). After being deeply anesthetized, mice were serially perfused with 10 ml of 0.05% glutaraldehyde/0.5% paraformaldehyde in 0.1 m cacodylate buffer, pH 7.4, and 50 ml of 2% glutaraldehyde/2% paraformaldehyde in 0.1 m cacodylate buffer. The brains and spinal cords were dissected and postfixed in 2% osmium tetroxide for 60 min at 4°C. The tissue samples were dehydrated with graded alcohol and propylene oxide and embedded in Epon-812, which was then polymerized for 72 h at 60°C. Sections, 65-nm-thick, were generated and mounted on 200 mesh copper grids. The sections were subsequently stained with 1% uranyl acetate in 50% ethanol and bismuth subnitrate and examined with a JEM-1010 EM (Jeol, Peabody, MA) at 80 kV.
Additionally, preembedding immuno-EM was performed for the mice to characterize cytoskeletal arrangements/derangements and tau aggregates in OLGs. For perfusing the mice, 10 ml of 0.05% glutaldehyde/0.5% paraformaldehyde in 0.1 m cacodylate buffer and 50 ml of 0.2% glutaldehyde/2% paraformaldehyde in 0.1 m cacodylate buffer were sequentially used. The brain and spinal cord tissues were postfixed in 4% paraformaldehyde/0.2% glutaldehyde/0.2% picric acid in 0.1 m cacodylate buffer overnight. Vibratome sections, 50-μm-thick, were generated, quenched with 0.1% sodium borohydride in Tris-buffered saline for 10 min, and immersed in 20% ethanol for 10 min. The sections were treated with blocking solution (5% donor horse serum/0.1% cold-water fish skin gelatin/1% chicken egg albumin in PBS) for 60 min and incubated with anti-tau or anti-α-tubulin antibody in reaction solution (0.1% bovine serum albumin in PBS) at 4°C overnight. The immunostaining was performed using the streptavidin- biotin-peroxidase method with diaminobenzidine and enhanced by silver-gold intensification (Teclemariam-Mesbah et al., 1997). The sections were then processed and examined as described above for TEM.
Measurement of axonal transport. The impact of OLG tau accumulations on axonal functions was further examined by measuring fast and slow axonal transport in optic nerves using methods similar to those described recently (Zhang et al., 2004) after first subjecting optic nerves of 6-, 9-, and 12-month-old non-Tg and Tg mice to immunohistochemistry, immunoblotting, and TEM as described above. For quantitative immunoblotting, mice were deeply anesthetized, and the optic nerves were dissected, incubated with BUST buffer [0.5% SDS, 8 m urea, 2% b-mercaptoethanol, 0.01% protease inhibitor mixture, and 0.1 m Tris HCl, pH 6.8] at room temperature overnight, homogenized, and centrifuged at 100,000 × g for 40 min at room temperature, followed by the SDS-PAGE using the supernatants, and quantitative immunoblot analysis was performed with anti-tau, anti-α-tubulin, and anti-NF antibodies and 125I-labeled secondary antibodies. Based on data from these studies, we then monitored rates of axonal transport in other cohorts of older Tg mice with myelin and axonal degeneration, younger Tg mice without pronounced optic nerve pathology, and age-matched non-Tg or WT mice that were subjected to bilateral intraocular injections of 0.5 mCi of 35S-methionine (ICN Biochemicals, Costa Mesa, CA) as described previously (Brady, 1985; Kirkpatrick et al., 2001). Groups of mice were killed at 3 h and 14 d after injection to analyze the fast and slow axonal transport rates, respectively. The optic nerves were removed, cut into seven consecutive 1-mm-long segments, incubated with BUST buffer at room temperature overnight, and centrifuged at 100,000 × g for 40 min at room temperature. Radioactively labeled proteins in the supernatants were resolved on SDS-PAGE gels and transferred onto nitrocellulose membranes, and radioactive bands were quantified with a PhosphorImager (Molecular Dynamics/Amersham, Piscataway, NJ).
Phenotypic assessments. Body weights of non-Tg or WT and Tg mice at 3, 6, 9, and 12 months of age were measured and statistically compared, as was motor function using the tail suspension test as described previously (Ishihara et al., 1999; Higuchi et al., 2002a).
Antibodies. The anti-tau antibodies used here included the following: mouse monoclonal antibody (mAb) T14 specific for human tau (1:500 dilution) (Kosik et al., 1988); rabbit polyclonal antibody 17026 raised against the recombinant protein of the longest human tau isoform (1: 2000 dilution) (Ishihara et al., 1999); mAb T46 specific for the C terminus of human tau (1:500 dilution) (Mawal-Dewan et al., 1994); mAb T49 specific for mouse tau (1:500 dilution) (Mawal-Dewan et al., 1994); mAb PHF1 (1:500 dilution) (Greenberg and Davies, 1990), mAb PHF6 (1:500 dilution) (Hoffmann et al., 1997), mAb AT8 (1:2000 dilution) (Goedert et al., 1994), mAb AT270 (1:1000 dilution) (Goedert et al., 1994), mAb 12E8 (1:1000 dilution) (Seubert et al., 1995), and rabbit polyclonal antibody T3P (1:250 dilution) (Lee et al., 1991) specific for phosphorylated tau; and mAb T1 specific for nonphosphorylated tau (1:500 dilution) (Binder et al., 1985). Other antibodies to NF triplet proteins also were used here, including the following: mAb RmdO9 specific for nonphosphorylated heavy NF (NFH) (1:2000 dilution) (Carden et al., 1987); mAb RMO24 to phosphorylated NFH (1:2000 dilution) (Carden et al., 1987); mAb RMO189 specific for a phosphorylation-independent epitope in the mid-size NF (NFM) subunit (1:2000 dilution) (Carden et al., 1987); and rabbit polyclonal antibody specific for the light NF (NFL) subunit (1:2000 dilution) (Tu et al., 1995). Mouse mAb DM1A against α-tubulin was also used (1:1000 dilution; Sigma, St. Louis, MO). The antibodies to myelin components used here included the following: a mouse mAb (1:1000 dilution; Chemicon, Temecula, CA) and rabbit polyclonal antibody (1:1000 dilution; provided by Dr. F. Arthur McMorris, The Wistar Institute, Philadelphia, PA) against CNP; a mouse mAb (1:1000 dilution; Chemicon) and rabbit polyclonal antibody (1:1000 dilution; provided by Dr. F. Arthur McMorris) to myelin basic protein (MBP). Finally, the kinesin heavy chain was detected using a mouse mAb (1:1000 dilution; Chemicon).
Results
Human tau proteins are expressed in mouse CNS OLGs
Three different lines of Tg mice stably expressing hWT (lines 34, 39, and 7) or PL (lines 6, 12, and 2) tau at a low, medium, and high levels, respectively, were established, as shown in Figure 1B. Expression levels of human tau in hWT lines 34, 39, and 7 were comparable with those in PL lines 6, 12, and 2, respectively. Mice that were heterozygous and homozygous for the transgene were generated in each line, and the homozygotes were used for studies described below. Significantly, no overt expression of Tg human tau in the systemic organs, including the heart, lung, liver, and kidney, were detected (data not shown), indicating that the transgene expression driven by the CNP promoter was confined to the nervous system, consistent with previous findings (Gravel et al., 1998).
Expression of hWT and PL T34 in the CNS was visualized by tau immunohistochemistry. All tau Tg lines showed strong tau immunostaining in the cytoplasm and processes of cells with morphological features of OLGs (Fig. 1C,D), although the intensity of tau immunolabeling was approximately proportional to the tau expression levels. Localization of the Tg human tau to OLGs was confirmed by double-immunofluorescence staining with anti-tau and anti-CNP antibodies, and nearly 100% of CNP-immunoreactive cells were tau positive throughout the CNS (Fig. 1E,G). The expression of human T34 in Tg mice became detectable by immunoblotting at 1 week of age and further increased during the following 3 weeks (data not shown), consistent with the developmental regulation of the CNP promoter (Scherer et al., 1994; Gravel et al., 1998).
PL tau in mouse CNS OLGs becomes insoluble and filamentous with aging
Figure 2A illustrates the levels of hWT and PL tau proteins in Tg mice at different ages, and transgenic tau proteins in the HS-soluble fraction remained at a stable level (Fig. 2A). Because the immunoblot analyses for brain and spinal cord were performed under the same conditions (including protein loading and exposure time), these results indicate that the expression levels of transgenic human tau in brain is comparable with those in spinal cord. In contrast to the soluble fraction, there was a progressive increase in the amounts of RIPA- and FA-extractable tau proteins in PL Tg mice, whereas the solubility of human tau in hWT Tg mice remained unchanged with aging (Fig. 2A). Although myelin-associated normal tau in OLGs is recovered primarily in the RIPA-soluble fraction, the age-dependent increase of the RIPA-extractable human tau indicates progressive accumulation of myelin-associated tau. Furthermore, a progressive formation of pathological tau aggregates is suggested by the age-dependent increases in RIPA-extractable and FA-extractable tau in the PL mice in association with increased tau phosphorylation in these fractions (data not shown). Contemporaneously, mice homozygous for PL tau (lines 12 and 2) developed tau-positive OLG inclusions that also were Gallyas sliver and thioflavin-S positive at 15 months of age (Fig. 2B-D). Thus, these tau aggregates exhibit filamentous properties similar to OLG tau pathologies in PSP, CBD, and FTDP-17 (Fig. 2F). In contrast, none of the hWT Tg mice showed Gallyas- and/or thioflavin-S-positive OLG tau inclusions at any age, nor did line 6 PL Tg mice, which expressed the lowest levels of PL tau (data not shown).
Figure 2.

Formation of fibrillary human tau inclusion in OLGs of PL Tg mice as a function of age. A, T14 immunoblots of human tau in HS-, RIPA-, and FA-soluble fractions extracted from the brains (top row) and spinal cords (bottom row) of line 39 hWT and line 12 PL Tg mice at different ages. Levels of HS-soluble tau remained stable during aging, as was the case for RIPA- and FA-extractable tau proteins in the hWT Tg mice, but there was a pronounced increase in these relatively insoluble species of tau in older PL Tg mice. The retarded electrophoretic mobility of the FA-extractable tau in older PL Tg mice is consistent with its increased phosphorylation. B-D, Fibrillary lesions in presumptive OLGs in a Gallyas silver-stained spinal cord section of 15-month-old line 12 PL Tg mouse. High-power photomicrographs of the inclusions in the anterior horn (C) and anterior column (D) reveal that pathological fibrillar tau extends into proximal processes of OLGs. E, Similar OLG inclusion labeled with thioflavin-S [counterstained for nuclei with 4′,6′-diamidino-2-phenylindole (DAPI)] in 15-month-old line 12 PL Tg mouse. F, Gallyas silver-positive coiled bodies in the frontal white matter of a patient with CBD, showing resemblance of fibrillary lesions in the PL Tg mice to those in FTD patients. Scale bars: B, 50 μm; C-F, 20 μm.
OLG alterations emerge at early stages of tau pathology
Luxol fast blue staining revealed progressive loss of myelin in the posterior column of PL tau Tg spinal cord beginning at 9 months of age (Fig. 3A-H), whereas hWT tau Tg and non-Tg or WT mice did not show similar change. Myelin degeneration with similar severity also was observed in the basal ganglia of PL Tg mice (data not shown). Moreover, myelin loss in these CNS areas of the PL Tg mice also was evident by immunohistochemistry using antibodies to CNP and MBP (data not shown).
Figure 3.
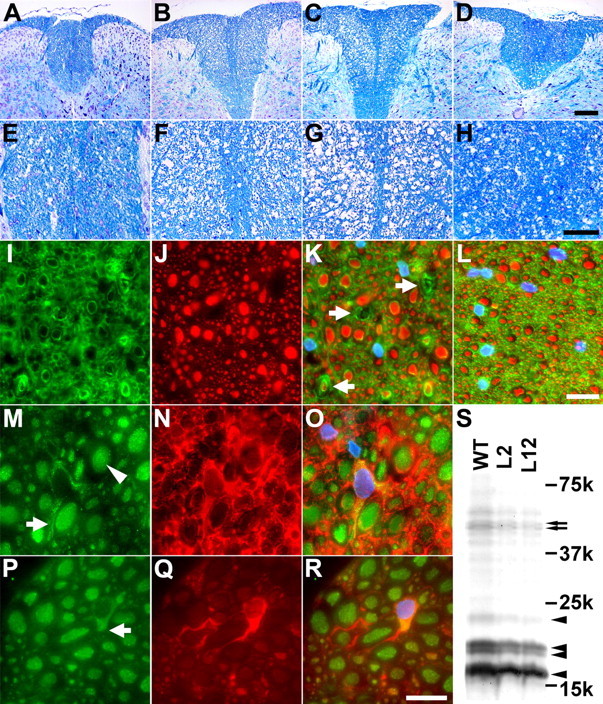
Progressive degeneration of OLGs and axons in the spinal cord of PL Tg mice. A-H, Age-associated loss of myelin in the posterior column of the spinal cord revealed by Luxol fast blue staining on 6- (A, E), 9- (B, F), and 12- (C, G) month-old PL Tg and 12-month-old WT non-Tg mice (D, H). High-power view photomicrographs are displayed in the bottom panels (E-H). I-L, Disrupted myelin accompanies axonal damage in the posterior column of the spinal cord. Double-immunofluorescence staining of a frozen section from 6-month-old PL Tg mouse using anti-CNP (I) and anti-NFH (RMO24) (J) antibodies demonstrates disorganization of myelin and atrophy or loss of axons as indicated by arrows on a merged image (K; counterstained with DAPI), whereas a WT non-Tg mouse (L) at the same age does not show these pathological alterations. M-R, Impaired kinesin transport in spinal cord OLGs of PL Tg mouse at 9 months of age. Photomicrographs of doubly stained OLGs of an age-matched WT mouse with channels for kinesin (M), CNP (N), and merged images (O; counterstained with DAPI) depict normal physiological distribution of kinesin that ranges from the cell body to distal processes (arrow in M) in a CNP-positive OLG. Arrowhead in M indicates kinesin in the axon. Unlike WT mice, PL Tg mouse showed reduced kinesin staining in distal processes of OLGs (arrow in P), which were filled with tau immunoreactivity (Q). Multichannel photomicrograph demonstrates disrupted kinesin staining in tau-positive OLGs (R; counterstained with DAPI). S, Loss of myelin components in the brains of 12-month-old PL Tg mice visualized by Coomassie blue. Myelin samples extracted from line 2 and line 12 mice contained significantly less myelin constituents, including MBP (arrowheads) and CNP (arrows), than age-matched WT mouse-derived sample. Other minor bands are also less intense in the PL Tg mice than those in the WT mouse, implying uniform reduction of myelin proteins as a result of OLG tau pathology. Scale bars: A-D, 200 μm; E-H, 100 μm; I-L, P-R, 20 μm.
Double-immunofluorescence staining of frozen spinal cord sections from PL Tg mice using anti-NFH and anti-CNP antibodies revealed disrupted myelin accompanied by axonal degeneration at 6 months of age (Fig. 3I-L), suggesting that PL tau accumulation in OLGs resulted in the loss of NFs in some myelinated axons. Additionally, impaired vesicular transport in OLGs attributable to the accumulation of PL tau was suggested by abnormal immunolabeling with anti-kinesin antibody (Fig. 3M-R) because kinesin in tau-positive OLGs of the PL Tg mice was restricted primarily to OLG cell bodies even at 9 months of age, which suggests that functional and structural alterations in OLGs precedes the emergence of fibrillary tau lesions.
When the composition of the myelin constituents was quantitatively analyzed by Coomassie blue staining of SDS-PAGE gels of myelin fractions from the whole brain of PL Tg mice (Fig. 3S), it was evident that CNP and MBP in 12-month-old PL Tg mice (line 2 and line 12) were reduced by ∼30% relative to the non-Tg or WT mice, and this suggests that the accumulation of PL tau in OLGs leads to a reduction of these myelin proteins.
Finally, loss of CNP-immunoreactive oligodendrocytes in the spinal cord of PL Tg mice (line 12) was observed in an age-dependent manner. The number of oligodendrocytes was significantly reduced in PL Tg mice at 12 months of age when compared with WT and hWT Tg mice (p < 0.05 by ANOVA; data not shown). Because Gallyas-positive fibrillary tau aggregates emerge in the spinal cord of PL Tg mice at the age of 15 months, this finding supports the view that degenerative changes in OLGs precedes formation of mature tau fibrils.
Ultrastructural alterations in myelin are associated with accumulations of abnormal tau filaments in OLGs
TEM demonstrated myelin degeneration associated with an axonopathy in the spinal cord of PL Tg mice by 12 months of age as exemplified by disruption and fragmentation of myelin lamellae that were devoid of an ensheathed axon (Fig. 4A-C). Furthermore, immuno-EM of older PL Tg mice revealed accumulations of tau-immunoreactive filaments in the processes of OLGs as well as between disrupted myelin lamellae, and these ∼15 nm in diameter filaments were not immunolabeled with anti-α-tubulin antibodies, nor were NFs in adjacent axons labeled by anti-tau antibodies (Fig. 4D-F). These tau-positive filamentous OLG aggregates were observed exclusively in the old PL Tg mice but not in non-Tg or hWT Tg mice, consistent with the other data mentioned above.
Figure 4.
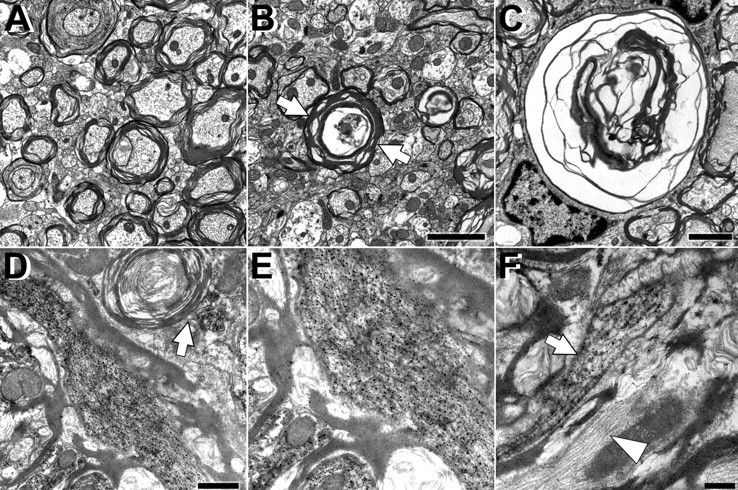
Ultrastructural alterations of myelin and axons associated with accumulation of abnormal tau filaments in the spinal cords of the line 12 PL Tg mice at 12 months of age. A-C, Myelin pathology and axonal degeneration demonstrated by TEM. Although the WT mouse did not develop changes in myelin and axons (A), myelin in the PL mouse is fragmented and disorganized (arrows in B), and the axons within the myelin sheaths are severely atrophic. Disruption of myelin and axon loss are shown at higher magnification (C). D-F, Accumulation of tau-immunoreactive filaments in the PL Tg mouse revealed by immuno-EM with anti-tau antibody 17026. Abnormal filaments were present between detached myelin layers (D) and in areas apposed to degenerating myelin lacking a central axon (arrow in D). High-power view shows intense labeling of well oriented filaments with anti-tau antibody (E). Tau-immunolabeled filaments were also localized to OLG processes (arrow in F), whereas filaments in axons were not tau positive and had side arms characteristic of NFs (arrowhead in F). Scale bars: A, B, 2 μm; C, D, 1 μm; E, F, 500 nm.
Axonal transport is impaired before axonal degeneration in the optic nerves of PL Tg mice
Similar to PL tau Tg mouse brain and spinal cord, by 12 months of age, PL tau Tg mouse optic nerves showed evidence of OLG tau accumulations with attenuation of myelin (Fig. 5A-D), and biochemical analysis demonstrated that CNP and MBP were decreased by ∼10 and ∼35% in line 12 PL Tg mice at 4 and 12 months, respectively, relative to non-Tg or WT mice (data not shown). These mice also exhibited losses of axonal cytoskeletal components, including the NFH, NFM (Fig. 5E,G), and middle molecular weight or so-called “big” tau (Fig. 5F,G) beginning at 4 months of age. There also were less prominent reductions in NFL and α-tubulin in 12-month-old PL Tg (lines 2 and 12) mice compared with non-TG or WT mice (Fig. 5E,G). In contrast, low molecular weight species of endogenous tau isoforms remained at the same levels in the PL Tg and non-Tg or WT mice (Fig. 5F).
Figure 5.
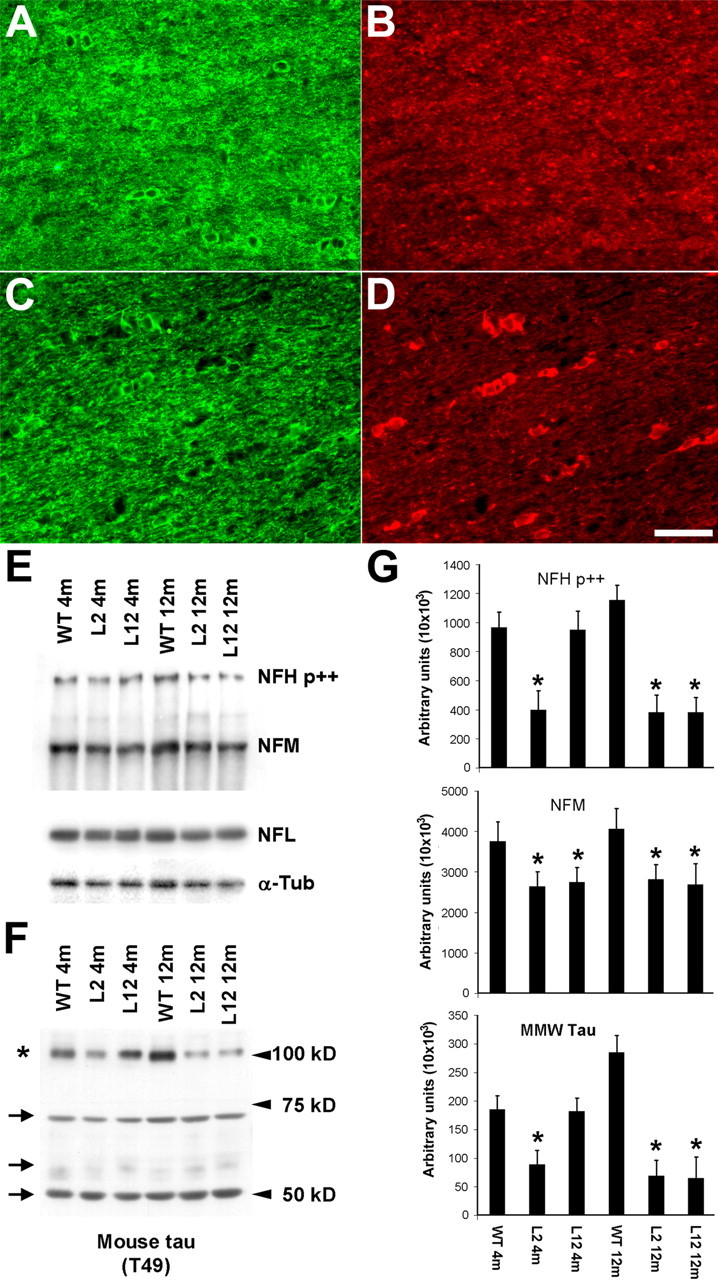
Biochemical impact of OLG tau accumulations on axons in the optic nerve of the PL Tg mice. A, Overexpression of Tg tau in the optic nerve interfascicular OLGs shown by double-immunofluorescence staining with anti-CNP antibody (A, C) and anti-tau antibody 17026 (B, D) for 6-month-old WT non-Tg (A, B) and line 12 PL tau Tg (C, D) mice. In contrast to diffuse tau immunoreactivity in WT OLGs, strong tau staining is seen in specific CNP-positive OLGs of the PL Tg mouse. CNP staining in myelin and tau staining in axons were moderately diminished in the PL Tg mouse compared with the WT mouse. E-G, Immunoblotting for optic nerve samples with antibodies against phosphorylated NFH (NFH p++), NFM, NFL, α-tubulin (α-tub) (E), and endogenous mouse tau (F), and quantification of the blots (n = 4 in each group) (G). Substantial reduction of NFH and NFM occurred in the line 2 and line 12 PL Tg mice at 4 months of age (E and top and middle panels in G). The 12-month-old PL Tg mice also showed slight decrease in the amounts of NFL and α-tubulin. There were no remarkable differences in the levels of low-molecular-weight tau isoforms ranging from 50 to 70 kDa (indicated by arrows) between the WT and PL Tg mice, whereas middle-molecular-weight tau (∼100 kDa, indicated by an asterisk) in the PL Tg mice was pronouncedly reduced relative to the WT mice (F and bottom panel in G). *p < 0.05 by multiple comparison by ANOVA. Scale bar: A-D, 50 μm.
In the PL tau Tg mice, no prominent pathological changes were found by TEM at 6 months of age (Fig. 6A), whereas disruption of myelin layers, axonal atrophy and loss, and expansion of interstitial space were notable at 9 months (Fig. 6B,C). In contrast to the PL Tg mice, the non-Tg or WT and hWT tau Tg mice developed neither marked myelin degeneration nor axonopathy at 6 and 9 months of age (Fig. 6D,F). When the rates of fast and slow axonal transport were measured in the optic nerves of 6- and 9-month-old-non-TG or WT and PL tau Tg mice, 6-month-old PL tau Tg mice exhibited significant reductions of fast axonal transport relative to age-matched non-Tg WT mice (Fig. 6G,I), and this fast axonal transport deficit became more prominent in 9-month-old PL tau Tg mice (Fig. 6J). Additionally, there also was evidence of a slow axonal transport deficit in 6-month-old PL tau Tg mice as exemplified by retarded translocation of tubulin and NFs relative to age-matched WT non-TG mice (Fig. 6H,K,L). Together with the ultrastructural data, these findings indicate that impaired axonal function precedes the onset of overt axonal degeneration in optic nerves after the accumulation of OLG tau aggregates.
Figure 6.
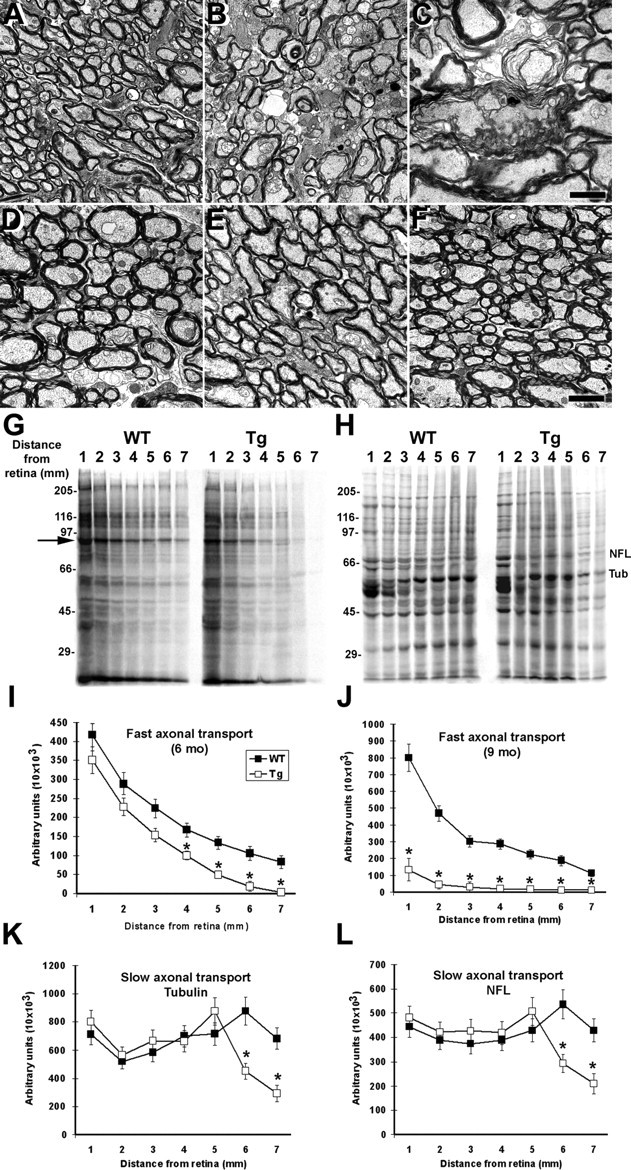
Impaired axonal transport in the optic nerves of the PL Tg mice before ultrastructural disruption of myelin and axons. A-F, TEM of the optic nerve sections of line 12 PL mice at 6 (A) and 9 (B, C) months of age, WT mice at 6 (D) and 9 (E) months, and line 39 h WT tau Tg mouse at 9 months (F). The 6-month-old PL tau Tg mouse exhibited relatively normal myelin sheaths and axons morphologically (A), whereas there were marked losses of these structures and expansion of interstitial space in the 9-month-old PL Tg mice (B). Severe disorganization of myelin and axonal atrophy are also shown in a high-power view (C). The WT and hWT Tg mice did not develop overt pathologies until 9 months of age (D-F). G-L, Quantitative assays of fast (G, I, J) and slow (H, K, L) axonal transport in the optic nerve. SDS-PAGE of protein samples from optic nerve segments indicates significant retardation of fast axonal transport in 6-month-old line 12 PL Tg mice compared with age-matched WT mice (G, I). This deficit became greater at 9 months of age (J). The arrow in G identifies a major protein band undergoing fast axonal transport and is used here as a marker for the quantification of fast transport. Slow axonal transport of proteins such as tubulin and NFL (corresponding bands are indicated in H) were also significantly slower in the 6-month-old PL Tg mice relative to the WT mice (K, L). Error bars in I-L represent SE (n = 3 in each group). *p < 0.05 by t test. Scale bars: A, B, D-F, 2 μm; C, 1 μm.
Weight loss and progressive motor impairments are augmented in PL tau Tg relative to hWT tau Tg mice
Tg mice expressing PL tau at medium and high levels (lines 12 and 2) exhibited progressive weight loss relative to WT non-Tg and hWT tau Tg mice at 12 months of age (Fig. 7A). Presumably, this weight loss is a consequence of neurodegeneration and neurogenic muscle atrophy, which was observed by histochemical examination in hindlimb muscles of 12-month-old PL tau Tg mice (data not shown), which has be observed in other tau Tg mice with a prominent axonopathy (Probst et al., 2000). These findings also are consistent with the emergence of motor weakness in the PL tau Tg mice as evidenced by a high frequency of limb twitching in these mice relative to the hWT tau Tg mice (Fig. 7B,C) as well as the non-TG or WT mice (data not shown).
Figure 7.

Weight loss and progressive motor deficits in the PL Tg mice. A, Weights of different lines of PL and hWT Tg mice at 12 months of age. Values are normalized by the mean body weight of the WT mice at the same age. Homozygotes of the line 7 PL Tg mice are not included, because most of them did not survive until this age. n = 6 in each group. B, Frequency of limb twitching in tail suspension test. Although the hWT Tg mice (left) showed this pathologic motor phenotype at a frequency that was higher than that of WT mice reported previously (Higuchi et al., 2002a), the frequency was further augmented in the PL mice (right). The emergence of limb twitching was approximately proportional to the dosage of the transgenes in both of the hWT and PL Tg mice.
Myelin is altered in association with OLG tau inclusions in human FTD brains similar to PL tau Tg mice
To assess the verisimilitude of the neurodegenerative phenotype in the PL tau Tg mice with authentic human tauopathies, we compared the myelin disruption caused by OLG tau pathology in the Tg mice with that seen in several PSP and CBD brains by histochemistry and immunohistochemistry. These studies revealed focal alterations in myelin sheaths adjacent to OLGs with fibrillary tau inclusions in PSP and CBD brains, but disruption of myelin was not prominent (Fig. 8A) except when there were numerous OLG tau inclusions (Fig. 8B), which was associated with extensive myelin loss (Fig. 8C). Thus, the myelin abnormalities in the PL tau Tg mice recapitulate similar pathologies in authentic human FTD tauopathies.
Figure 8.

Oligodendrocytic tau accumulation and disruption of myelin sheaths in patients with FTD. A, Frozen section of the frontal white matter from a PSP patient immunolabeled with antibodies against CNP (green) and tau (PHF1, red) and counterstained with DAPI (blue). Myelin sheaths that were connected to a phospho-tau-immunoreactive OLG showed marked disorganization (arrows), although a large portion of myelin appeared intact (arrowhead). B, C, Paraffin sections of the frontal white matter from an FTD patient stained with PHF1 (B) and Luxol fast blue (C). Tau-positive inclusions were frequently found in putative OLGs (B). Round, well bordered deposits were localized to the cytoplasm of OLG as revealed in high-power photomicrograph (inset in B). Severe loss of myelin was also observed in the same region (C). Scale bars: A, inset in B, 15 μm; B, 25 μm; C, 100 μm.
Discussion
Our studies of CNP tau Tg mice demonstrate for the first time that OLG tau accumulations lead to a primary oligodendrocytic as well as a secondary neuronal degenerative phenotype. Specifically, we demonstrated that impairments in axonal transport precede overt axonal degeneration, and this appears to correlate with early stages in the accumulation of OLG tau aggregates. At later time points, myelin disruption, formation of Gallyas-positive tau inclusions, overt axonal degeneration, and motor impairments occur in this mouse model in an age-dependent manner. Thus, our data support a significant contribution of oligodendrocytic degeneration to causing neurodegeneration in human FTD tauopathies.
Although many Tg mouse models of tauopathies have been reported (including the Tα-1 tau Tg mice that produce both neuronal and glial tau pathologies), these mice did not allow a dissection of the independent contributions of oligodendrocytic versus astrocytic and neuronal tau pathology to neuronal dysfunction and degeneration (Higuchi et al., 2002a). This is attributable to the use of heterologous promoters to drive tau overexpression simultaneously in neurons and glia of mouse models of tauopathies, resulting in the inability to attribute the underlying degeneration to neuronal, astrocytic, or OLG tau lesions (Higuchi et al., 2002a; Lin et al., 2003). Significantly, the exclusive overexpression of tau in OLGs driven by the CNP promoter here allowed us to dissect out the primary role of OLG tau accumulations in the onset of functional deficits and morphological alterations in OLGs versus neurons in the CNS.
Our study also indicates that tau aggregation into thioflavin-S-positive amyloids leads to degeneration of OLGs and axons followed by neuromuscular impairments, and we believe that FTDP-17 tau gene mutations and the expression levels of Tg tau are the major determinants responsible for this. Indeed, the non-mutant hWT Tg mice exhibited no Gallyas-positive tau inclusions, very mild myelin disruption, and mild motor weakness relative to the PL Tg mice when mice with similar tau expression levels were compared. Thus, tau gene mutations cause a robust acceleration of pathological changes as reported here and in previous studies (Lewis et al., 2000; Tanemura el al., 2002; Zhang et al., 2004). However, the amount of Tg tau expressed in OLGs is conceivably a major determinant of the CNS degeneration, at least in these tau Tg mice, because the formation of tau fibrils and the associated degeneration occurred most prominently in Tg mice with the highest expression levels of PL tau. Line 6 PL tau Tg mice, which expressed Tg tau at the lowest level, did not develop OLG tau inclusions, although they did so when they were crossbred with OLG-directed α-synuclein Tg mice (Giasson et al., 2003), thereby implying that cofactors such as α-synuclein, when expressed in OLG, can lower the threshold at which tau pathologies form in OLGs.
Tau gene mutations, including the P301L mutation, have been shown to reduce the ability of tau to bind and stabilize MTs in the polymerized state in vitro, as well as augment the self-aggregation of tau into fibrils (for review, see Lee et al., 2001; Higuchi et al., 2002b). Diminished binding of PL tau to MTs increases free tau in the cytosol that is not bound to MTs, and high concentrations of cytosolic tau can enhance tau aggregation. Increased phosphotau can be observed in the PL Tg mice, but it remains to be determined whether tau phosphorylation occurs before aggregation and thereby enhances the polymerization of tau, or whether this occurs after tau has already assembled into fibrils (Goedert et al., 1996; Schneider et al., 1999; Geschwind, 2003). However, because tau phosphorylation negatively regulates the binding of tau to MTs (Drechsel et al., 1992; Biernat et al., 1993; Bramblett et al., 1993), this increase in the levels of free tau not bound to MTs may also facilitate tau fibrillization.
Indeed, loss of stable MTs with attendant impairments of intracellular transport in OLGs may explain the altered distribution of kinesin that we observed in OLGs of PL tau Tg mice because kinesin binds to stable detyrosinated MTs with higher affinity than to tyrosinated MTs (Liao and Gundersen, 1998). However, it also has been speculated that excess binding of tau to MTs might block the association of kinesin with MTs and its translocation (Ebneth et al., 1998; Trinczek et al., 1999; Stamer et al., 2002). Other evidence to support the redistribution of kinesin comes from recent studies of tau-transfected cultured astrocytes wherein there was a tight association of elevated tau level and decreased detyrosinated MTs with an altered distribution of kinesin (Yoshiyama et al., 2003). Thus, it is likely that free tau that is increased by the PL mutation reduces detyrosinated MTs and consequently induces decline in the attachment of kinesin to MTs.
The present study has also provided insights into the temporal relationship between tau aggregate formation, glial and neuronal degeneration, and defects in axonal transport. Whereas the formation of thioflavin-S-positive tau amyloid in OLGs can only be observed in late stages (> 12 months of age), impairments in axonal transport and a reduction in NFs can be detected in 4- to 6-month-old PL mice. Concomitant with the latter changes, insoluble PL tau that can be extracted by formic acid is already present at this stage. Therefore, the accumulation of this pool of insoluble tau might be responsible for the earlier changes observed here. However, the nature of the insoluble tau is unclear, although EM analyses identified tau fibrils in OLGs that are not stained by Gallyas or thioflavin-S at 6-9 months. Other explanations include the presence of tau oligomers, but this is highly unlikely in view of the very insoluble nature of this pool of tau.
The analyses of our CNP-tau mice also provided insights into the molecular machinery governing alterations of axon-OLG interrelationships in age-associated neurodegenerative disorders. Although several different mouse models of myelin-related disorders have been established, most of these animal models develop early onset degeneration. Thus, it is unclear whether the phenotypes reflect aberrant neurodevelopment rather than neurodegenerative processes (Griffiths et al., 1998; Brady et al., 1999; Lappe-Siefke et al., 2003). The Tg PL tau in the mice investigated here had no overt abnormalities in myelination and axonal growth during development. Hence, the onset of disease in fully mature PL tau Tg mice undoubtedly reflects neurodegenerative processes induced by the formation of potentially cytotoxic tau aggregates and filaments with aging. Therefore, these Tg mice are highly suitable in vivo models of OLG degeneration with verisimilitude to authentic human FTD tauopathies. Interestingly, the myelin and axonal pathologies observed in the PL Tg mice are distinct from those observed in previously reported mouse models of primary myelin diseases (Griffiths et al., 1998; Brady et al., 1999; Kirkpatrick et al., 2001; Lappe-Siefke et al., 2003), thereby suggesting that there are multiple molecular pathways leading to glial induced neuronal degeneration.
Thus, the PL Tg mice define a novel category of pathological alterations in axon-OLG interactions leading to disease because these mice develop axonal transport deficits before substantial myelin loss. Because abnormal tau filaments are formed in the OLG processes, these filaments may perturb the functions of CNP (Braun et al., 1988; Trapp et al., 1988), and this could impair axonal transport rates without visible demyelination in young PL tau Tg mice. Subsequently, disorganization of compact myelin sheaths also occurs conceivably as a consequence of global OLG degeneration induced by filamentous tau inclusions, and this could give rise to axonal dysfunction and loss. Although the mechanisms linking myelin-axon communication and axonal transport deficits remain to be elucidated, comparison of axonal cytoskeletal alterations in the PL mice and shiverer mice, which show increased slow axonal transport rates (Brady et al., 1999; Kirkpatrick et al., 2001), may facilitate the elucidation of mechanisms underlying axonal transport deficits in neurodegenerative disease. Furthermore, our observation on perturbed axonal transport before substantial structural alterations of myelin is consistent with previous investigations using the myelin proteolipid protein null mice (Edgar et al., 2004). Analyses of these and other mouse models could shed light on molecular signals transmitted from OLG to the axons for regulating axonal functions. These speculations notwithstanding, the PL tau Tg mouse reported here provide experimental systems for elucidating glial-induced neurodegeneration in human FTD tauopathies characterized by prominent OLG tau pathologies, and this may lead to better therapies to treat these disorders.
Footnotes
This work was supported by grants from the National Institute on Aging. V.M.-Y.L. is the John H. Ware III Professor for Alzheimer's Research, and J.Q.T. is a William Maul Measey-Truman G. Schnabel Jr, M.D. Professor of Geriatric Medicine and Gerontology. We thank Dr. F. Arthur McMorris for providing antibodies against CNP and MBP, David Wilson and James Soper for technical support, Dr. Christiane Richter-Lansberg (University of Oldenburg, Oldenburg, Germany) for helpful suggestions, and the Biochemical Imaging Core Facility of the University of Pennsylvania for assistance in the EM studies.
Correspondence should be addressed to Dr. Virginia M.-Y. Lee, Maloney 3, Hospital of the University of Pennsylvania, 3600 Spruce Street, Philadelphia, PA 19104-4283. E-mail: vmylee@mail.med.upenn.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259434-10$15.00/0
References
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M (2002) Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci 22: 9340-9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E (1993) Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron 11: 153-163. [DOI] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI (1985) The distribution of tau in the mammalian central nervous system. J Cell Biol 101: 1371-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjartmar C, Yin X, Trapp BD (1999) Axonal pathology in myelin disorders. J Neurocytol 28: 383-395. [DOI] [PubMed] [Google Scholar]
- Brady S (1985) Axonal transport methods and applications. In: Neuromethods, general neurochemical techniques (Boulton A, Baker G, eds), pp 419-476. Clifton, NJ: Humana.
- Brady ST, Witt AS, Kirkpatrick LL, de Waegh SM, Readhead C, Tu PH, Lee VM-Y (1999) Formation of compact myelin is required for maturation of the axonal cytoskeleton. J Neurosci 19: 7278-7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramblett G, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM-Y (1993) Abnormal tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding. Neuron 10: 1089-1099. [DOI] [PubMed] [Google Scholar]
- Brandt R (1996) The tau proteins in neuronal growth and development. Front Biosci 1: d118-d130. [DOI] [PubMed] [Google Scholar]
- Braun PE, Sandillon F, Edwards A, Matthieu JM, Privat A (1988) Immunocytochemical localization by electron microscopy of 2′3′-cyclic nucleotide 3′-phosphodiesterase in developing oligodendrocytes of normal and mutant brain. J Neurosci 8: 3057-3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carden MJ, Trojanowski JQ, Schlaepfer WW, Lee VM-Y (1987) Two-stage expression of neurofilament polypeptides during rat neurogenesis with early establishment of adult phosphorylation patterns. J Neurosci 7: 3489-3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin SSM, Goldman JE (1996) Glial inclusions in CNS degenerative diseases. J Neuropathol Exp Neurol 55: 499-508. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Feany MB, Yen SH, Mattiace LA, Davies P (1996) Cytoskeletal pathology in non-Alzheimer degenerative dementia: new lesions in diffuse Lewy body disease, Pick's disease, and corticobasal degeneration. J Neural Transm Suppl 47: 31-46. [DOI] [PubMed] [Google Scholar]
- Drechsel DN, Hyman AA, Cobb MH, Kirschner MW (1992) Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell 3: 1141-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E (1998) Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol 143: 777-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Yool D, Zhang SC, Fowler JH, Montague P, Barrie JA, McCulloch MC, Duncan ID, Garbern J, Nave KA, Griffiths IR (2004) Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol 166: 121-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman MS, Lee VM-Y, Trojanowski JQ (2000) New insights into genetic and molecular mechanisms of brain degeneration in tauopathies. J Chem Neuroanat 20: 225-244. [DOI] [PubMed] [Google Scholar]
- Forman MS, Zhukareva V, Bergeron C, Chin SS, Grossman M, Clark C, Lee VM-Y, Trojanowski JQ (2002) Signature tau neuropathology in gray and white matter of corticobasal degeneration. Am J Pathol 160: 2045-2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallyas F (1971) Silver staining of Alzheimer's neurofibrillary changes by means of physical development. Acta Morphol Acad Sci Hung 19: 1-8. [PubMed] [Google Scholar]
- Geschwind DH (2003) Tau phosphorylation, tangles, and neurodegeneration: the chicken or the egg? Neuron 40: 457-460. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM-Y (2003) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300: 636-640. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Crowther RA, Cohen P, Vanmechelen E, Vandermeeren M, Cras P (1994) Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer's disease: identification of phosphorylation sites in tau protein. Biochem J 301: 871-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA (1996) Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383: 550-553. [DOI] [PubMed] [Google Scholar]
- Gravel M, Di Polo A, Valera PB, Braun PE (1998) Four-kilobase sequence of the mouse CNP gene directs spatial and temporal expression of lacZ in transgenic mice. J Neurosci Res 53: 393-404. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P (1990) A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA 87: 5827-5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths IR, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA (1998) Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280: 1610-1613. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Ishihara T, Zhang B, Hong M, Andreadis A, Trojanowski J, Lee VMY (2002a) Transgenic mouse model of tauopathies with glial pathology and nervous system degeneration. Neuron 35: 433-446. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Lee VM-Y, Trojanowski JQ (2002b) Tau and axonopathy in neurodegenerative disorders. Neuromolecular Med 2: 131-150. [DOI] [PubMed] [Google Scholar]
- Hoffmann R, Lee VM-Y, Leight S, Varga I, Otvos Jr L (1997) Unique Alzheimer's disease paired helical filament specific epitopes involve double phosphorylation at specific sites. Biochemistry 36: 8114-8124. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM-Y (1999) Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 24: 751-762. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick LL, Witt AS, Payne HR, Shine HD, Brady ST (2001) Changes in microtubule stability and density in myelin-deficient shiverer mouse CNS axons. J Neurosci 27: 2288-2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T (1999) Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick's disease. Brain Pathol 9: 663-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Orecchio LD, Binder L, Trojanowski JQ, Lee VM-Y, Lee G (1988) Epitopes that span the tau molecule are shared with paired helical filaments. Neuron 1: 817-825. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA (2003) Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33: 366-374. [DOI] [PubMed] [Google Scholar]
- Lee VM-Y, Balin BJ, Otvos Jr L, Trojanowski JQ (1991) A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science 251: 675-678. [DOI] [PubMed] [Google Scholar]
- Lee VM-Y, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24: 1121-1159. [DOI] [PubMed] [Google Scholar]
- Lemke G (1988) Unwrapping the genes of myelin. Neuron 1: 535-543. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25: 402-405. [DOI] [PubMed] [Google Scholar]
- Li J, Hertzberg EL, Nagy JI (1997) Connexin32 in oligodendrocytes and association with myelinated fibers in mouse and rat brain. J Comp Neurol 379: 517-591. [DOI] [PubMed] [Google Scholar]
- Liao G, Gundersen GG (1998) Kinesin is a candidate for cross-bridging microtubules and intermediate filaments. Selective binding of kinesin to detyrosinated tubulin and vimentin. J Biol Chem 273: 9797-9803. [DOI] [PubMed] [Google Scholar]
- Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW (2003) Filamentous tau in oligodendrocytes and astrocytes of transgenic mice expressing the human tau isoform with the P301L mutation. Am J Pathol 162: 213-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI (1995) Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci USA 92: 10369-10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo A, Akiguchi I, Lee GC, McGeer EG, McGeer PL, Kimura J (1998) Myelin degeneration in multiple system atrophy detected by unique antibodies. Am J Pathol 153: 735-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawal-Dewan M, Henley J, Van de Voorde A, Trojanowski JQ, Lee VM-Y (1994) The phosphorylation state of tau in the developing rat brain is regulated by phosphoprotein phosphatases. J Biol Chem 269: 30981-30987. [PubMed] [Google Scholar]
- Murayama S, Arima K, Nakazato Y, Satoh J, Oda M, Inose T (1992) Immunocytochemical and ultrastructural studies of neuronal and oligodendroglial cytoplasmic inclusions in multiple system atrophy. 2. Oligodendroglial cytoplasmic inclusions. Acta Neuropathol (Berl) 84: 32-38. [DOI] [PubMed] [Google Scholar]
- Norton WT, Poduslo SE (1973) Myelination in rat brain: method of myelin isolation. J Neurochem 21: 749-757. [DOI] [PubMed] [Google Scholar]
- Pfeiffer SE, Warrington AE, Bansal RB (1993) The oligodendrocyte and its many cellular processes. Trends Cell Biol 3: 191-197. [DOI] [PubMed] [Google Scholar]
- Probst A, Götz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, Hong M, Ishihara T, Lee VMY, Trojanowski JQ, Jakes R, Crowther RA, Spillantini MG, Burki K, Goedert M (2000) Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol (Berl) 99: 469-481. [DOI] [PubMed] [Google Scholar]
- Readhead C, Hood L (1990) The dysmyelinating mouse mutants shiverer (shi) and myelin deficient (shimld). Behav Genet 20: 213-234. [DOI] [PubMed] [Google Scholar]
- Richter-Landsberg C (2000) The oligodendroglia cytoskeleton in health and disease. J Neurosci Res 59: 11-18. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Braun PE, Grinspan J, Collarini E, Wang DY, Kamholz J (1994) Differential regulation of the 2′,3′-cyclic nucleotide 3′-phosphodiesterase gene during oligodendrocyte development. Neuron 12: 1363-1375. [DOI] [PubMed] [Google Scholar]
- Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM (1999) Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 38: 3549-3558. [DOI] [PubMed] [Google Scholar]
- Seubert P, Mawal-Dewan M, Barbour R, Jakes R, Goedert M, Johnson GV, Litersky JM, Schenk D, Lieberburg I, Trojanowski JQ, Lee VM-Y (1995) Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J Biol Chem 270: 18917-18922. [DOI] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM (2002) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 156: 1051-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanemura K, Murayama M, Akagi T, Hashikawa T, Tominaga T, Ichikawa M, Yamaguchi H, Takashima A (2002) Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J Neurosci 22: 133-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teclemariam-Mesbah R, Wortel J, Romijin HJ, Buijs RM (1997) A simple silver-gold intensification procedure for double DAB labeling studies in electron microscopy. J Histochem Cytochem 45: 619-621. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Bernier L, Andrews SB, Colman DR (1988) Cellular and subcellular distribution of 2′,3′-cyclic nucleotide 3′-phosphodiesterase and its mRNA in the rat central nervous system. J Neurochem 51: 859-868. [DOI] [PubMed] [Google Scholar]
- Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E (1999) Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J Cell Sci 112: 2355-2367. [DOI] [PubMed] [Google Scholar]
- Tu PH, Elder G, Lazzarini RA, Nelson D, Trojanowski JQ, Lee VM-Y (1995) Overexpression of the human NFM subunit in transgenic mice modifies the level of endogenous NFL and the phosphorylation state of NFH subunits. J Cell Biol 129: 1629-1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu PH, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, Iwatsubo T, Trojanowski JQ, Lee VM-Y (1998) Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble α-synuclein. Ann Neurol 44: 415-422. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Zhang B, Bruce J, Trojanowski JQ, Lee VM-Y (2003) Reduction of detyrosinated microtubules and Golgi fragmentation are linked to tau-induced degeneration in astrocytes. J Neurosci 23: 10662-10671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Higuchi M, Yoshiyama Y, Ishihara T, Forman MS, Martinez D, Joyce S, Trojanowski JQ, Lee VM-Y (2004) Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. J Neurosci 24: 4657-4667. [DOI] [PMC free article] [PubMed] [Google Scholar]