

Abstract
Rad, Gem/Kir, Rem, and Rem2 are members of the Ras-related RGK (Rad, Gem, and Kir) family of small GTP-binding proteins. Heterologous expression of RGK proteins interferes with de novo calcium channel assembly/trafficking and dramatically decreases the amplitude of currents arising from preexisting high-voltage-activated calcium channels. These effects probably result from the direct interaction of RGK proteins with calcium channel β subunits. Among the RGK family, Rem2 is the only member abundantly expressed in neuronal tissues. Here, we examined the ability of Rem2 to modulate endogenous voltage-activated calcium channels in rat sympathetic and dorsal root ganglion neurons. Heterologous expression of Rem2 nearly abolished calcium currents arising from preexisting high-voltage-activated calcium channels without affecting low-voltage-activated calcium channels. Rem2 inhibition of N-type calcium channels required both the Ras homology (core) domain and the polybasic C terminus. Mutation of a putative GTP/Mg2+ binding motif in Rem2 did not affect suppression of calcium currents. Loading neurons with GDP-β-S via the patch pipette did not reverse Rem2-mediated calcium channel inhibition. Finally, [125I]Tyr22-ω-conotoxin GVIA cell surface binding in tsA201 cells stably expressing N-type calcium channels was not altered by Rem2 expression at a time when calcium current was totally abolished. Together, our results support a model in which Rem2 localizes to the plasma membrane via a C-terminal polybasic motif and interacts with calcium channel β subunits in the preassembled N-type channel, thereby forming a nonconducting species.
Keywords: channel, sympathetic, dorsal root ganglion, Rem2, small GTPase, conotoxin
Introduction
High-voltage-activated (HVA) Ca2+ channels are heterooligomeric complexes consisting of a pore-forming α subunit and auxiliary β and α2δ subunits. At present, seven Cavα, four β, and four α2δ genes have been cloned. The CaVα11.x family encodes L-type Ca2+ channels, whereas CaVα12.1, CaVα12.2, and CaVα12.3 encode P/Q-, N-, and R-type Ca2+ channels, respectively. The Cavα subunit contains four homologous domains, with each domain consisting of six transmembrane segments. Cavα subunits determine the ion selectivity and single-channel conductance and contain gating charges within the S4 segments and high-affinity binding sites for peptide neurotoxins. Coexpression of Cavβ and/or α2δ facilitates the cell surface trafficking of Cavα1, increases Ca2+ current (ICa) amplitude, and alters the channel gating properties (Dolphin, 2003).
Using Cavβ3 as bait, a yeast two-hybrid screen identified Kir/Gem (Béguin et al., 2001) as an interacting protein. Overexpression of Kir/Gem prevents de novo L-type ICa expression and decreases endogenous HVA-ICa density (Béguin et al., 2001; Murata et al., 2004; Ward et al., 2004). Kir/Gem, originally cloned from lymphocytes (Cohen et al., 1994; Maguire et al., 1994), is a member of the recently defined RGK (Rad, Gem, and Kir) family of Ras-related small GTP-binding proteins. The distinct structural features of the RGK family include the following: (1) substitution of critical amino acid residues within the central Rashomologous domain involved in GTP/GDP binding and GTP hydrolysis; (2) a unique G2 domain putatively involved with effector interactions; (3) long N and C termini relative to other Ras-like molecules; and (4) a polybasic C terminus lacking the typical prenylation site present in virtually all other Ras-like GTPases (Reynet and Kahn, 1993; Finlin and Andres, 1997; Finlin et al., 2000). Like Kir/Gem, the other RGK family members, including Rad, Rem, and Rem2, also interact with Cavβ subunits and prevent de novo expression of L-type ICa (Finlin et al., 2003, 2005). Accordingly, it might be presumed that members of the RGK family use a unified mechanism to inhibit L-type Ca2+ channels. However, examination of L-type Ca2+ channel surface expression has produced conflicting results. Immunostaining of hemagglutinin-tagged CaVα11.2/β3 expressed in HEK293 cells indicates that both Kir/Gem and Rem2 reduce Ca2+ channel expression at the cell surface (Béguin et al., 2001, 2005). Additionally, overexpression of Kir/Gem in cardiomyocytes resulted in marked reduction of the gating currents arising from L-type Ca2+ channels, indicating a reduced expression of functional channels at the cell surface (Murata et al., 2004). In contrast, Rem2 overexpression had little effect on the cell surface density of endogenous L-type Ca2+ channels in insulin-secreting cells as determined by surface biotinylation (Finlin et al., 2005). Thus, the mechanism(s) of Rem2-mediated downregulation of L-type Ca2+ channels remains controversial.
Among the RGK family, Rem2 is the only member abundantly expressed in neuronal tissues (Finlin et al., 2000). Although recent studies have examined the effects of Rem2 on L-type Ca2+ channels in clonal cells lines, the effects of Rem2 on N-type Ca2+ channels natively expressed in mammalian neurons is unknown. We have thus examined modulation by Rem2 of endogenous voltage-dependent Ca2+ channels in rat sympathetic and sensory neurons and tsA201 cells stably expressing an N-type Ca2+ channel of defined composition (Cavα12.2/β3/α2δ).
Materials and Methods
All experiments were approved by the Institutional Animal Care and Use Committee.
cDNA plasmid constructs and recombinant adenoviruses. Most of our experiments were based on the originally deposited rat Rem2 sequence (GenBank accession number AF084464) (Finlin et al., 2000). During the course of these experiments, examination of expressed sequence tags and genomic DNA sequences across several species (while searching for alternative splicing variants) revealed two in-frame ATGs upstream of the start codon assigned in AF084464. The longest predicted open reading frame (deposited as GenBank accession number AY916790) encodes an additional 69 residues at the N terminus. The open reading frame assigned in AF084464 likely arose from a sequencing error rather than representing a genuine alternative splice variant. Recently, this discrepancy was noted by two other groups (Béguin et al., 2005; Finlin et al., 2005). To maintain consistency with the nomenclature used in recent publications, we term the shorter construct Rem2t (truncated; equivalent to Rem2NΔ69 by Finlin et al., 2005) and the longer sequence Rem2.
The coding sequences of Rem2t and Rem2 were amplified from mRNA of rat dorsal root ganglia (DRGs) by reverse transcription-PCR, and the resulting PCR products were ligated into the multiple cloning region of mammalian expression vectors, including pCI (Promega, Madison, WI), pEGFP-C1, and pEGFP-N1 (BD Biosciences Clontech, Mountain View, CA). The forward primer was designed such that the start codon was under the optimal context for translational initiation (Kozak, 1999). All truncation, green fluorescent protein (GFP)-fusion, and point mutant constructs were prepared using PCR-based methods. Fidelity of the PCR products was confirmed by automated DNA sequencing. Adenoviruses expressing GFP (Ad-GFP) or GFP-Rem2t were generated using the AdEasy system (He et al., 1998; Chen et al., 2004a).
Cell cultures. Single superior cervical ganglion (SCG) neurons were isolated from 10- to 14-week-old male Wistar rats as described previously (Ikeda, 2004). Briefly, after dissection, the ganglia were desheathed, cut into small pieces, and incubated with 0.7 mg/ml collagenase type D (Roche Molecular Biochemicals, Indianapolis, IN), 0.4 mg/ml trypsin (Worthington, Lakewood, NJ), and 0.1 mg/ml DNase 1, type IV, (Sigma, St. Louis, MO) in minimal essential medium (MEM) saturated with 5% CO2/95% O2 at 35°C for 1 h. Thereafter, cells were dissociated by vigorous shaking of the flask. After centrifugation twice at 50 × g, the dispersed neurons were resuspended in MEM containing 10% fetal bovine serum, 1% glutamine, and 1% penicillin-streptomycin solution (all from Invitrogen, Carlsbad, CA). Neurons were then plated into polystyrene tissue culture dishes coated with poly-l-lysine and stored in a humidified atmosphere containing 5% CO2 in air at 37°C. Single DRG neurons were isolated from 1- to 2-week-old Sprague Dawley rats using essentially the same procedure except that the trypsin concentration was reduced to 0.1 mg/ml. Hippocampal neurons isolated from newborn Sprague Dawley rats were cultured essentially as described previously (Chen and Lambert, 2000) except that neurons were grown on glass bottom dishes coated with poly-d-lysine (0.1 mg/ml). HEK293 cells and COS-7 cells were cultured in growth media as recommended by the provider (American Type Culture Collection, Manassas, VA). A tsA201 cell line stably expressing Cavα12.2/β3/α2δ (a gift from Dr. Diane Lipscombe, Brown University, Providence, RI) was maintained as described previously (Lin et al., 2004).
Intranuclear injection, transfection, and adenoviral infection. cDNA constructs (50-100 ng/μl in 10 mm Tris, pH 8) were injected into the nucleus of SCG neurons 4-8 h after cell isolation with an Eppendorf (Brinkmann Instruments, Westbury, NY) FemtoJet microinjector and 5171 micromanipulator (Ikeda, 2004). When necessary, neurons were coinjected with pEGFP-N1 (5 ng/μl) to facilitate later identification of successfully injected cells. SCG neurons expressing GFP were used for whole-cell patch-clamp recordings within 21-28 h after injection. Transfection of mammalian cell lines and hippocampal neurons (1 week in culture) was mediated by Lipofectamine 2000 following the instructions of the manufacturer (Invitrogen). DRG neurons were infected 6-12 h after isolation by adding 1-3 × 108 infectious units (diluted in MEM) of recombinant adenoviruses to each 35 mm dish, followed by replacement of the culture medium after 12 h. Recording was made 36-48 h after infection. SCG neurons were infected 2 h after isolation by adding 4 × 108 infectious units of Ad-GFP.
Electrophysiology. Whole-cell currents were recorded with a patch-clamp amplifier (Axopatch 200B; Axon Instruments, Union City, CA) at room temperature (21-24°C) using the conventional whole-cell variant of the patch-clamp technique (Hamill et al., 1981). For recording Ca2+ current (ICa), patch electrodes were pulled from borosilicate glass capillaries (Corning 7052; Garner Glass, Claremont, CA) on a P-97 Flaming-Brown micropipette puller (Sutter Instruments, Novato, CA), fire polished on a microforge, and filled with a solution containing the following (in mm): 120 N-methyl-d-glucamine, 20 tetraethylammonium-OH, 10 HEPES, 11 EGTA, 1 CaCl2, 14 Tris-creatine phosphate, 4 MgATP, and 0.3 Na2GTP, pH adjusted to 7.24 with methanesulfonic acid and HCl (20 mm final [Cl-]). In some experiments, as specified, GTP was replaced with 2 mm GDP-β-S. Pipette resistances ranged from 1 to 3 MΩ, yielding uncompensated series resistances of 2-6 MΩ. Electronic series resistance compensation of 80% was used in all recordings. For recording ICa from SCG neurons and tsA201 cells stably expressing Cavα12.2/β3/α2δ, the external solution consisted of 145 mm tetraethylammonium methanesulfonate, 10 mm HEPES, 15 mm glucose, 10 mm CaCl2, and 300 nm tetrodotoxin, pH 7.35, 320 mOsm/kg. Unless indicated otherwise, ICa recorded from SCG neurons and tsA201 cells was evoked by a double-pulse voltage protocol (Elmslie et al., 1990) consisting of an initial 20 ms test pulse (“prepulse”) stepped from the holding potential of -80 mV, a 50 ms depolarizing conditioning pulse to +80 mV, and a subsequent 20 ms test pulse to +10 mV (“postpulse”). ICa density was determined from the amplitude of ICa evoked at the midpoint of the prepulse normalized to the cell capacitance. For recording ICa from DRG neurons, the bathing solution CaCl2 was reduced to 5 mm, and the HEPES was increased by an identical amount. Low-voltage-activated (LVA) ICa from DRG neurons was evoked at -40 mV from a holding potential of -80 mV. High-voltage-activated ICa from DRG neurons was evoked at +10 mV. Voltage protocol generation and data acquisition were performed using custom data-acquisition software on a Macintosh G4 computer (Apple Computers, Cupertino, CA) with an ITC-18 interface board (InstruTech, Port Washington, NY). ICa traces were low-pass filtered at 2-5 kHz and digitized at 10 kHz.
[125I]Tyr22-ω-conotoxin GVIA binding assay. Unless indicated otherwise, 2 × 105 (per well) tsA201 cells stably expressing a high density of Cavα12.2/β3/α2δ (N-type) Ca2+ channels (Lin et al., 2004) were cultured overnight in a 24-well plate (Visiplate-24 TC; PerkinElmer, Wellesley, MA) precoated with rat tail collagen. Plasmid constructs expressing GFP, GFP-Rem2t, and GFP-Rem2 were then transfected for ∼26 h using Lipofectamine 2000 following the instructions of the manufacturer (Invitrogen). The culture medium was removed from each well, and the cells were rinsed once with PBS buffer containing 0.3% bovine serum albumin (supplemented with 1 mm Ca2+ and 1 mm Mg2+ to prevent cell detachment). Cells were then incubated with [125I]Tyr22-ω-conotoxin GVIA (1 nm, 300 μl per well, 2200 Ci/mmol; PerkinElmer), a specific N-type Ca2+ channel antagonist (Terlau and Olivera, 2004) for 45 min at room temperature. To remove the unbound radioactive ligand, cells were washed gently with the above-modified PBS buffer (eight times for 5 min). One milliliter of Optiphase Supermix (1200-439; PerkinElmer) was added to each well, and the bound ligand was measured from the radioactivity detected in a microplate scintillation counter (MicroBeta Trilux LSC; PerkinElmer). Nonspecific binding was <5% as calculated from the radioactivity in control wells containing a comparable number of HEK293 cells.
Statistical analysis. Numerical values are expressed as mean ± SEM. Statistical comparisons were made by using Student's t test or ANOVA, followed by Dunnett's test as appropriate. The differences were considered significant if p < 0.05.
Results
Heterologous expression of Rem2 nearly abolishes ICa in adult rat sympathetic neurons within ∼24 h
To examine the effect of Rem2 on endogenous neuronal Ca2+ channels, we expressed Rem2 cDNA constructs in adult rat sympathetic neurons derived from the SCG. The composition of ICa in these neurons is relatively homogenous arising primarily from N-type (Cav2.2) Ca2+ channels (Ikeda, 1991). Representative traces of ICa recorded from a control (uninjected) neuron and a neuron injected with Rem2 cDNA are depicted in Figure 1A. Ca2+ currents were evoked from a holding potential of -80 mV with 70 ms test pulses to the indicated voltages. Recordings made 21-26 h after cDNA injection revealed a dramatic decrease in ICa amplitude across the entire voltage range (approximately -30 to +60 mV) without apparent alteration in the shape of the evoked currents. To facilitate comparisons from neurons of different size, ICa amplitude (measured isochronally at 10 ms after initiation of the test pulse) was normalized to membrane capacitance (Cm) and expressed as ICa density (in picoamperes per picofarad). Cm is related to cell surface area and was determined by integrating the capacitance transient produced by a 5 mV depolarizing pulse. Mean ± SEM current density-voltage (I-V) relationships from control neurons and neurons expressing Rem2 or Rem2t are shown in Figure 1B. Both Rem2 constructs produced nearly identical results, with ICa density being severely depressed across the entire voltage range. The effects of Rem2, Rem2t, GFP-Rem2t, and Rem2t-GFP on peak ICa density (determined for a test pulse to +10 mV) are summarized in Figure 1C. Mean peak ICa density in neurons injected with Rem2 or Rem2t cDNA was greatly decreased when compared with control neurons (4.4 ± 0.7 and 2.4 ± 0.8, respectively, versus 24.2 ± 2.1 pA/pF; p < 0.001, ANOVA). Whereas expression of GFP alone had no effect on ICa density (27.3 ± 3.7 pA/pF; p = 0.997), expression of the fusion constructs GFP-Rem2t and Rem2t-GFP significantly (p < 0.001) reduced ICa density to 4.4 ± 1.7 and 4.7 ± 2.0 pA/pF, respectively, indicating that fusion of GFP did not disrupt the effects of Rem2t and that a free N or C terminus was not required for inhibition of ICa.
Figure 1.
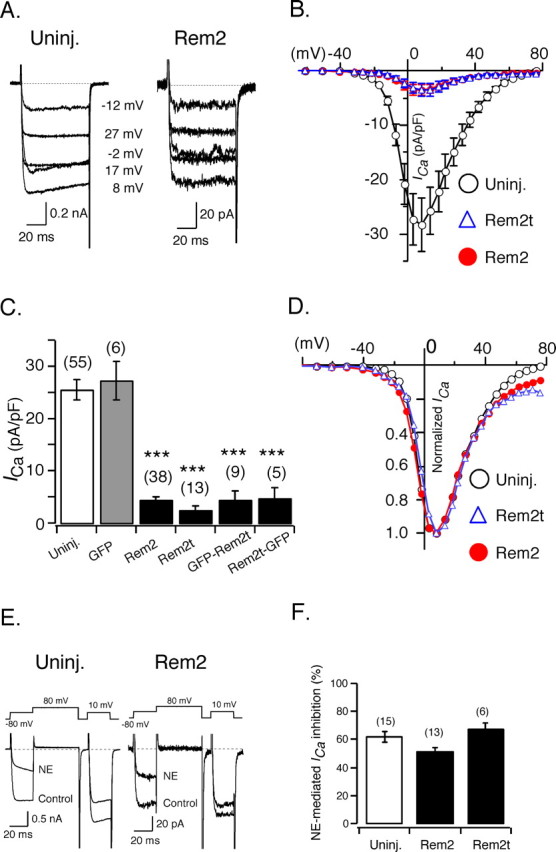
Effects of heterologous expression of Rem2 on N-type Ca2+ channels of sympathetic neurons. A, Superimposed representative ICa traces recorded from a control (uninjected) neuron (left) and a neuron expressing Rem2 (right). Expression was accomplished with nuclear microinjection (see Materials and Methods). ICa was evoked by a 70 ms step to the indicated voltages from a holding potential of -80 mV. B, Average ± SEM ICa I-V relationships for ICa recorded from control neurons (open circles) and neurons expressing Rem2 (filled circles) or Rem2t (open triangles). ICa density was calculated from ICa amplitude at a series of test pulses normalized to cell membrane capacitance. C, Average ± SEM ICa density in control neurons (open bar) and neurons expressing GFP, Rem2, Rem2t, GFP-Rem2t, or Rem2t-GFP as indicated (filled bars). ICa density was calculated from ICa amplitude at a test pulse of +10 mV normalized to cell membrane capacitance. D, The mean values from B were scaled to facilitate comparison of the current density-voltage relationship. E, Superimposed ICa traces recorded in the absence or presence of NE (10 μm) from a control neuron (left) and a neuron expressing Rem2 (right). ICa was evoked at 0.1 Hz with the voltage protocol illustrated. F, Average ICa inhibition mediated by NE in control neurons and neurons expressing Rem2 or Rem2t. The number of neurons tested is shown in parentheses. ***p < 0.001 versus controls, ANOVA followed by Dunnett's test. Uninj., Uninjected.
In 13 of 65 neurons expressing Rem2, Rem2t, Rem2t-GFP, or GFP-Rem2t, ICa was reduced below detectable levels (<1 pA/pF). However, because a measurable residual ICa was seen in the majority of neurons, the question arose as to whether the residual ICa differed substantially from the unmodified ICa. As shown in Figure 1D, scaling of the mean ICa density data (from Fig. 1A) reveals a nearly identical relationship between ICa density and test pulse potential. The apparent deviation seen at very positive potentials (more than +40 mV) likely results from errors introduced during leak subtraction of the very small currents in the Rem2-expressing neurons. The response of the residual ICa to heterotrimeric G-protein-mediated modulation was also examined (Fig. 1E,F). N-type Ca2+ channels in sympathetic neurons are inhibited after activation of endogenous α2A adrenergic receptors with norepinephrine (NE) (Schofield, 1991). The modulation is mediated by the Gβγ subunit and defined as voltage dependent based on several distinct characteristics (Ikeda and Dunlap, 1999) as illustrated for the control ICa in Figure 1E (left). Here, currents were evoked with a double-pulse voltage protocol (Elmslie et al., 1990) composed of two identical test pulses separated by a depolarizing conditioning pulse. In the presence of NE (10 μm), the current evoked by the first pulse was reduced in amplitude and the rising phase of the current slowed (a phenomenon termed kinetic slowing). After the conditioning pulse, the current inhibition was markedly relieved and the kinetic slowing reversed. Application of NE to an Rem2-expressing neuron produced similar effects despite the large reduction in ICa amplitude (Fig. 1E, right). Comparison of the mean ICa inhibition (determined from the first test pulse) produced by NE for the control, Rem2-modified, and Rem2t-modified ICa (Fig. 1F) revealed a similar magnitude of inhibition (∼50-60%).
Together, these results show that expression of Rem2 nearly abolished N-type ICa in adult rat sympathetic neurons within ∼24 h. Expression of Rem2t and constructs encoding GFP-fusions to either terminus of the molecule produced nearly identical results. The small component of ICa remaining after ∼24 h has macroscopic properties similar to the total ICa. Hence, this component likely arises from a population of unmodified channels.
Blockade of protein translation did not replicate the effects of Rem2 expression
To determine whether the ICa reduction after Rem2 expression resulted from actions on preexisting channels or interference with the synthesis of new channels, we tested the effect of blocking protein translation on ICa in rat SCG neurons. We reasoned that, if Rem2 produced a reduction in ICa by inhibiting channel synthesis, then pretreatment of the neurons for 20-29 h with cycloheximide (CHX) (0.35 mm), an established protein translation blocker (Lee and Hollenbeck, 2003), should produce similar results. As shown in Figure 2, A and B, mean ICa amplitude was unaltered (2.12 ± 0.30 vs 1.98 ± 0.22 nA in control and treated neurons, respectively) by CHX pretreatment. When the data were normalized to Cm, CHX treatment actually produced an increase, rather than a decrease, in mean ICa density (Fig. 2B, right). Although these results might suggest an increase in the population of Ca2+ channels at the plasma membrane, the increased ICa density resulted from a decrease in Cm (47.2 ± 4.3 vs 78.3 ± 3.7 pF for CHX-treated and control neurons, respectively; p < 0.001, unpaired t test) rather than an increase in ICa amplitude. Consistent with this finding, CHX appeared to decrease the diameter of neuron somata (Fig. 2C, phase contrast images), although this was not examined in detail. As a positive control for CHX treatment, adenovirus-mediated GFP expression in neurons was monitored by epifluorescence microscopy. As shown in Figure 2C, 22 h after infection with Ad-GFP (4 × 108 infectious units/ml), GFP fluorescence was detected in both neurons and glia in control but not CTX-treated cells. To ensure that adenoviral entry into cells was not encumbered, Ad-GFP lysate was added to the neuronal culture for 3 h before initiating CHX treatment.
Figure 2.

Treatment of sympathetic neurons with cycloheximide, a protein translation blocker, did not alter ICa amplitude. A, Representative whole-cell ICa traces from a control neuron and a neuron pretreated with cycloheximide (0.35 mm) for ∼24 h. The voltage protocol above the current traces was repeated at 0.1 Hz. B, Average ± SEM ICa amplitude and density in control neurons (open bar) and neurons pretreated with cycloheximide (filled bar) for 22-29 h. C, Phase-contrast (left column) and fluorescent (right column) micrographs illustrating adenovirus-mediated GFP expression in control neurons (top row) and neurons treated with cycloheximide (bottom row).
Because the amplitude of ICa in SCG neurons was not altered after block of protein translation for ∼24 h, we conclude that Ca2+ channel turnover is relatively slow within the time course of these experiments. Therefore, the ability of Rem2 to decrease ICa density likely arises from effects on preexisting N-type Ca2+ channels rather than alterations in channel synthesis as has been seen for other molecules (e.g., Cavγ7) (Moss et al., 2002).
Domains of Rem2 important for cellular localization and Ca2+ channel downregulation
Rem2 directly interacts with Cavβ subunits as shown by coimmunoprecipitation and GST pull-down assays (Béguin et al., 2005; Finlin et al., 2005). If this association is required for the downregulation of preexisting functional Ca2+ channels, we anticipated that Rem2 would be enriched at the plasma membrane. To examine this notion, GFP fusion constructs were heterologously expressed in clonal cell lines and primary neuronal cultures. Expression of GFP-Rem2t and Rem2t-GFP in cultured rat hippocampal neurons resulted in a “rim-type” fluorescence pattern consistent with enrichment at the plasma membrane (Fig. 3, top row). However, a similar fluorescence pattern was seen in COS-7 cells (Fig. 3, bottom row), a cell line shown to be devoid of Cav subunits (Berrow et al., 1997). Therefore, trafficking of Rem2t to the plasma membrane was not dependent on Ca2+ channel subunits and likely arose from an intrinsic affinity of Rem2t for the plasma membrane.
Figure 3.

Heterologously expressed Rem2 is enriched at the plasma membrane in hippocampal neurons and COS-7 cells. Fluorescent micrographs were taken from hippocampal neurons (top row) and COS-7 cells (bottom row) ∼24 h after transfection of cDNA constructs encoding GFP-Rem2t (left column) or Rem2t-GFP (right column). To facilitate the visualization of GFP fluorescence at plasma membrane areas, COS-7 cells were briefly treated with PBS containing trypsin-EDTA to cause cell rounding.
To define the role of various Rem2 domains in plasma membrane targeting and downregulation of Ca2+ channels, a series of truncation constructs, based around the central Ras homology domain, were prepared and expressed in SCG neurons by intranuclear injection (Fig. 4A, left). Deletion of the N-terminal domain (Rem2ΔN) altered neither the fluorescence pattern of GFP-Rem2t in HEK293 cells and SCG neurons nor the effect of Rem2 on ICa density. As shown in Figure 4, both GFP-Rem2t and GFP-Rem2ΔN were enriched at the plasma membrane when heterologously expressed in HEK293 cells and SCG neurons. In addition, ICa density was dramatically reduced in SCG neurons expressing Rem2ΔN (1.65 ± 0.79 pA/pF) or GFP-Rem2ΔN (2.46 ± 0.65 pA/pF) when compared with control neurons (Fig. 4A, open bar). In contrast, deletion of the C-terminal domain (GFP-Rem2tΔC) not only confined the truncated protein to the cytoplasm, consistent with a previous report (Finlin et al., 2000), but also rendered both Rem2tΔC and GFP-Rem2tΔC ineffective in modulating ICa density in SCG neurons (30.4 ± 6.2 and 26.7 ± 3.1 pA/pF, respectively). Like GFP-Rem2tΔC, the Ras-homology core domain alone (GFP-Rem2core) displayed a cytosolic fluorescence pattern and was ineffective at altering ICa density (25.7 ± 2.6 pA/pF). The results of these experiments implicate the C terminus in targeting Rem2 to the plasma membrane. To substantiate this idea, soluble GFP was fused to the C-terminal domain of Rem2 (GFP-Rem2ct). Expression of GFP-Rem2ct resulted in a rim-type fluorescence pattern consistent with the idea that the Rem2ct possesses intrinsic affinity for the plasma membrane. However, ICa density in neurons expressing GFP-Rem2ct was comparable with control neurons. Thus, the core domain may be responsible for Ca2+ channel downregulation but is unable to function in the absence of appropriate plasma membrane targeting.
Figure 4.
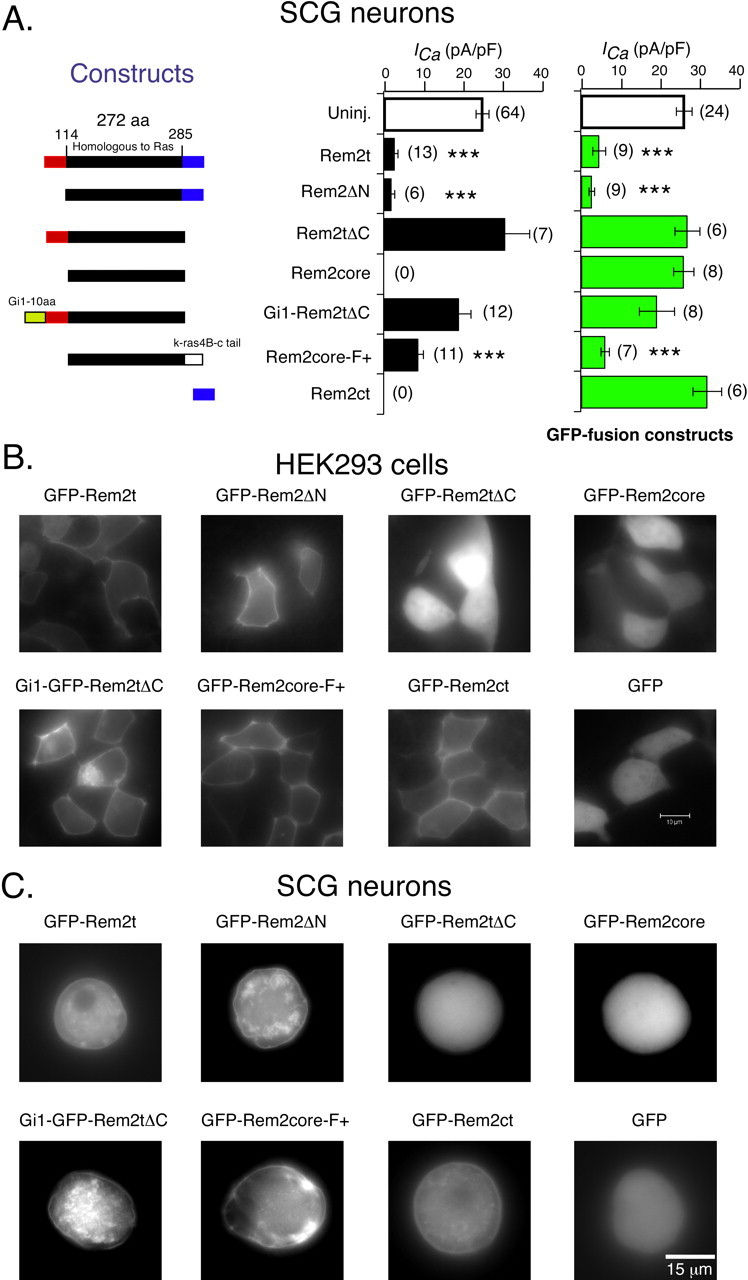
The structure-function relationship of Rem2. A, Average ± SEM ICa density in uninjected neurons (open bar) and neurons injected (filled bars) with cDNA constructs encoding one of the truncated or recombinant Rem2 (middle panel) as illustrated in the schematic diagram (left panel). Average ± SEM ICa density in uninjected neurons (open bar) and neurons injected (filled bars) with the corresponding GFP-fusion cDNA constructs were summarized in the right panel. ICa density was calculated from ICa amplitude at a test pulse of +10 mV normalized to cell membrane capacitance. ***p < 0.001 versus control neurons, ANOVA. B, Fluorescent micrographs were taken from HEK293 cells ∼24 h after transfection with the cDNA construct as indicated above each graph. C, Fluorescent micrographs were taken from sympathetic neurons ∼20 h after intranuclear injection with the cDNA construct as indicated above each graph. Gi1-10aa, The first 10 residues of Gαi1; K-ras4B-c-tail, the last 23 residues at the C-end of K-ras4B; Uninj., uninjected.
To further examine this hypothesis, the Rem2 core domain was fused to the C terminus (23 residues) of K-Ras4B, an extensively studied membrane-targeting domain consisting of a polybasic region and a farnesylation motif (Prior and Hancock, 2001). The resulting construct and GFP adduct were termed Rem2core-F+ and GFP-Rem2core-F+, respectively. Expression of the Rem2core-F+ constructs in SCG neurons resulted in a reduction of ICa density (8.46 ± 1.2 and 5.85 ± 1.1 pA/pF, respectively; p < 0.001 when compared with ICa density from control neurons). Consistent with the rescue of function, the fluorescence pattern of GFP-Rem2core-F+ expressed in HEK293 cells and SCG neurons was similar to that of GFP-Rem2t (i.e., plasma membrane enriched). A complementary experiment was undertaken in which the first 10 residues of the heterotrimeric G-protein α subunit, Gαi1, was fused to the N terminus of Rem2tΔC or GFP-Rem2tΔC (termed Gi1-Rem2tΔC and Gi1-GFP-Rem2tΔC, respectively). The N terminus of Gαi1 contains both myristoylation and palmitoylation motifs (Fivaz and Meyer, 2003) that efficiently targets fusion proteins to the plasma membrane. As expected, the fluorescence pattern of Gi1-GFP-Rem2tΔC expressed in HEK293 cells and SCG neurons revealed apparent plasma membrane enrichment. However, ICa density in neurons expressing Gi1-Rem2tΔC was comparable with control neurons (18.7 ± 3.0 vs 24.6 ± 1.67 pA/pF; p = 0.42, ANOVA), demonstrating that plasma membrane targeting, although necessary, was not the sole factor in conferring function activity to the Rem2 core domain. Similarly, ICa density in neurons expressing Gi1-GFP-Rem2tΔC was also not significantly different from control neurons (19.0 ± 4.4 vs 25.9 ± 2.0 pA/pF; p = 0.23, ANOVA).
From these results, we propose that the Ras homology core domain of Rem2 contains the elements necessary for the observed decrease in N-type ICa density. However, to be effective, the Rem2 core domain must be targeted to the plasma membrane by motifs in the C, but not the N, terminus.
Rem2 effects may be independent of GTP/Mg2+ binding
Small GTP binding proteins are inactive in the GDP-bound form, become active after release of GDP and binding of GTP [a process stimulated by guanine nucleotide exchange factors (GEFs)], and return to the inactive state when GTP is hydrolyzed to GDP by the intrinsic GTPase activity of small GTP binding proteins, a process accelerated by GAPs (Takai et al., 2001). However, because Rem2 (as well as other RGK family proteins) has several nonconservative amino acid substitutions within regions involved in GTP binding and hydrolysis (Finlin et al., 2000), we designed a series of experiments to examine whether the activity of Rem2 conforms to the canonical small G-protein cycle.
The S17N mutation interferes with GTP and Mg2+ binding, thereby rendering the Ras protein ineffective (Feig, 1999). Serine 17 is located within the so-called G1 domain (Bourne et al., 1991), a region perfectly conserved in Rem2 (Finlin et al., 2000), thus allowing unambiguous assignment of the cognate residue (S129). Unexpectedly, the mean ICa density in SCG neurons expressing Rem2t-S129N or GFP-Rem2t-S129N (Fig. 5A) was markedly reduced (6.74 ± 1.4 and 8.88 ± 0.91 pA/pF, respectively) compared with control neurons (26.9 ± 2.9 pA/pF; p < 0.001, ANOVA). These results suggest that GTP/Mg2+ binding to Rem2 may not be required for the Rem2-mediated decrease in ICa density observed in SCG neurons. We also observed that GFP-Rem2t-S129N, when expressed in COS-7 cells, was excluded from the nucleus, whereas GFP-Rem2t was distributed throughout the cell (Fig. 5B). It should be noted that, with flat cells like COS-7, conventional wide-field fluorescence microscopy is not well suited to distinguish fluorescence originating from the plasma membrane area versus the cytoplasm. Because Ras S17N binds Ras-specific GEFs with higher affinity than wild-type Ras (Feig, 1999), we speculate that Rem2t S129N might form a tight complex with a unique GEF and thus be excluded from the nucleus as a consequence of an increased effective mass. If this is the case, then the Rem2t-S129N/GEF complex was apparently still effective in reducing ICa density.
Figure 5.
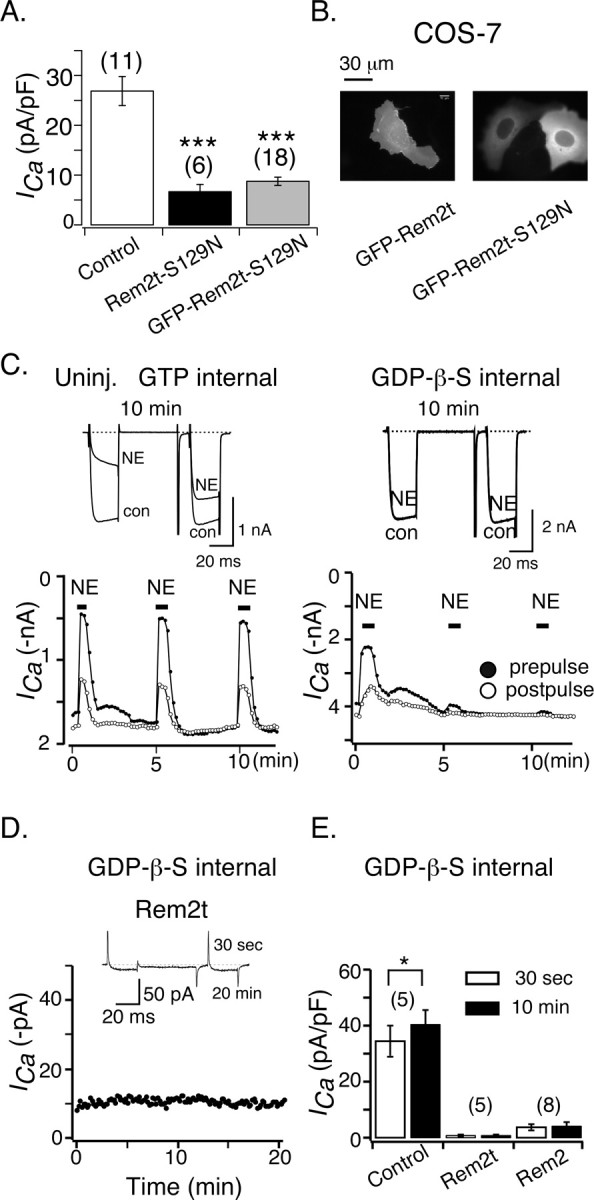
GTP binding may not be required for Rem2-mediated downregulation of ICa in sympathetic neurons. A, Average ± SEM ICa density in control neurons (open bar) and neurons expressing (filled bars) Rem2t-S129N or GFP-Rem2t-S129N. ICa density was calculated as in Figure 1C. ***p < 0.001 versus control neurons, ANOVA. B, Fluorescent micrographs were taken from COS-7 cells ∼24 h after transfection with GFP-Rem2t or GFP-Rem2t-S129N cDNA constructs as indicated. C, Comparison of NE-mediated inhibition of ICa recorded with pipette solutions containing either GTP (left) or GDP-β-S (right). ICa was evoked as in Figure 2 A. D, Time course of ICa recorded from a neuron expressing Rem2t. GDP-β-S (2 mm) was included in the patch-pipette solution. Superimposed ICa traces recorded at 30s and 20 min after patch rupture. E, Mean ± SEM ICa density at 30 s and 10 min after the establishment of whole-cell recording (2 mm GDP-β-S was included in the pipette solution) in control neurons and neurons expressing Rem2t or Rem2. *p < 0.05, paired t test. Uninj., Uninjected; con, control.
Because the function of the Rem2t-S129N mutation relies on extrapolation from a cognate mutation in Ras, we performed two additional experiments to explore a potential role for GTP-binding in the actions of Rem2. As shown in Figure 5C, when GTP (0.3 mm) was included in the patch pipette solution, application of NE (10 μm) to SCG neurons repeatedly inhibited ICa in a voltage-dependent manner (Ikeda and Dunlap, 1999). Replacement of GTP with the hydrolysis-resistant GDP analog GDP-β-S (2 mm) resulted in an attenuation of NE-mediated inhibition within 5 min and abolition within 10 min (Schofield, 1991). These results suggest that GDP-β-S replaces the cellular guanine nucleotide, thereby locking heterotrimeric G-proteins in an inactive state. Dialysis of neurons with GDP-β-S for 10-20 min, however, was unable to acutely rescue the ICa amplitude from the attenuated state produced by heterologously expressing Rem2t or Rem2 (Fig. 5D,E). If we assume that dialysis with GDP-β-S produces effects on Rem2 similar to those produced on heterotrimeric G-protein α subunits and Kir/Gem (Béguin et al., 2001), then these data provide additional evidence that GTP binding is not a requisite for the functional effects of Rem2.
Finally, we tested whether Rem2 might be inactive when cells were cultured in serum-free medium. The rationale for the experiment derived from the observation that Ras family proteins are often activated by extracellular factors that couple to receptor tyrosine kinases (Takai et al., 2001). ICa density determined from SCG neurons expressing Rem2t and deprived of serum for at least 7 h before the initiation of whole-cell recording was still dramatically reduced when compared with the ICa density of control neurons cultured under the same condition (1.73 ± 0.78 vs 29.0 ± 9.32 pA/pF; n = 5; p < 0.05, unpaired t test).
Specificity of Rem2 for high-voltage-activated Ca2+ channels
The interaction of RGK proteins with the CaVβ subunit is believed to underlie the functional effects of these proteins on Ca2+ channel function. To examine this hypothesis, the ability of Rem2 to modulate endogenous voltage-activated Ca2+ channels in primary sensory neurons of DRGs was examined. Unlike SCG neurons, DRG neurons express both LVA and HVA Ca2+ channels (Fedulova et al., 1985; Kostyuk et al., 1988; Regan et al., 1991; Mintz et al., 1992; Yusaf et al., 2001). Because large functional T-type currents are recorded from cells expressing only the CaVa13.x subunit, it is assumed that endogenous LVA Ca2+ channels neither require nor associate with CaVβ subunits (Perez-Reyes, 2002). Given these assumptions, one would predict differential modulation of HVA versus LVA Ca2+ currents after Rem2 expression.
To separate ICa components arising from LVA and HVA Ca2+ channels, a voltage protocol (Chen and Ikeda, 2004) consisting of 30 ms test pulses to -40 and +10 mV evoked from a holding potential of -80 mV was used (Fig. 6A, top). The test pulses were separated by a 50 ms pulse to -60 mV, which served to inactivate the majority of LVA Ca2+ channels. With this voltage protocol, ICa evoked at -40 mV should arise primarily from LVA Ca2+ channels because few HVA Ca2+ channels are activated near this voltage. Conversely, ICa recorded during the +10 mV test pulse is highly enriched in the HVA current component, although a small fraction may arise from a population of LVA Ca2+ channels that have not undergone inactivation. As shown in Figure 6A, the composition of ICa in the DRG neuron population was heterogeneous (Fig. 6A, left vs right). ICa recorded from DRG neurons 36-48 h after infection with recombinant Ad-GFP consistently showed a high density of HVA Ca2+ current, whereas the LVA current component was highly variable. To facilitate analysis, DRG neurons were divided into two groups: one having a large (>15 pA/pF) LVA ICa component and the other displaying a small LVA ICa component (<3.5 pA/pF). As shown in Figure 6, HVA ICa recorded from DRG neurons 36-48 h after infection with adenovirus expressing GFP-Rem2t was dramatically reduced, whereas the amplitudes of LVA-type current varied from cell to cell. Within the first group, although the amplitudes of LVA-type ICa were comparable in neurons expressing GFP and GFP-Rem2t (29.0 ± 3.8 and 32.2 ± 5.3 pA/pF, respectively; p = 0.54, unpaired t test), HVA ICa was markedly reduced (27.6 ± 2.8 and 6.89 ± 1.0 pA/pF, respectively; p < 0.001, unpaired t test). Within the second group, HVA ICa was nearly abolished in neurons expressing GFP-Rem2t compared with neurons expressing GFP (110 ± 19 and 4.87 ± 1.1 pA/pF, respectively; p = 0.003, unpaired t test). Therefore, we conclude that expression of GFP-Rem2t in DRG neurons selectively suppressed current arising from all HVA Ca2+ channel types, although having little or no effect on endogenous LVA-type Ca2+ channels. In addition to providing insight into the mechanism of Rem2 action, the results of these experiments show that LVA-type Ca2+ channels of DRG neurons can be effectively isolated without using a mixture of HVA-type Ca2+ channel blockers.
Figure 6.

Expression of GFP-Rem2t in DRG neurons attenuated HVA but spared LVA ICa. A, ICa traces recorded from two separate DRG neurons infected with Ad-GFP. The left panel illustrates a neuron with a large LVA ICa component. The right panel illustrates a neuron with negligible LVA ICa. The voltage protocol used to evoke ICa is illustrated above the traces. The current evoked at -40 mV arises from LVA Ca2+ channels, whereas the current evoked at +10 mV arises primarily from HVA Ca2+ channels (see Materials and Methods). To facilitate analysis, DRG neurons were divided into a group displaying a high density of LVA ICa (>15 pA/pF; left) and a group displaying virtually no LVA ICa (< 3pA/pF; right). Bar graphs summarizing the average ICa density arising from LVA (black) and HVA (gray) channels 36-48 h after infection with Ad-GFP. B, The same analysis as shown in A for neurons infected with adenoviruses expressing GFP-Rem2t. LVA ICa amplitude was measured 15 ms after initiation of the voltage step to -40 mV, whereas HVA ICa amplitude was measured 10 ms from the start of the voltage to +10 mV. ICa density was calculated from ICa amplitude normalized to cell membrane capacitance. Number of neurons tested is shown in parentheses.
Expression of Rem2 abolished ICa recorded from tsA201 cells stably expressing N-type channels without affecting surface expression
The preceding data have demonstrated that heterologous expression of Rem2 dramatically reduced the amplitude of ICa derived from the endogenous N-type Ca2+ channels in sympathetic neurons and HVA Ca2+ channels in primary sensory neurons. Mechanistically, a key question is whether the Ca2+ channels were removed from or remained at the cell surface after Rem2 expression. To address this issue, we opted to use tsA201 cells stably expressing molecularly defined N-type Ca2+ channels composed of Cavα12.2/β3/α2δ (Lin et al., 2004). This cell line expresses functional Ca2+ channels at high density (Lin et al., 2004) and supports a high transfection efficiency (at least 60% as shown in Fig. 7D). The data in Figure 7A show that N-type ICa was abolished in cells transfected with GFP-Rem2t or GFP-Rem2 cDNA for 19-53 h (0.16 ± 0.10 and 0.71 ± 0.60 pA/pF, respectively) when compared with cells expressing GFP only (66.3 ± 13.1 pA/pF), thereby recapitulating the results obtained in the neuronal systems. To examine whether Rem2 alters Ca2+ channel density at the cell surface, a live-cell surface ligand-binding assay was performed with cells cultured in 24-well plates. As summarized in Figure 7B, application of [125I]Tyr22-ω-conotoxin GVIA (1 nm, 300 μl), an antagonist specific to N-type Ca2+ channels (Terlau and Olivera, 2004), to wells containing the overnight culture of the stable cell line expressing Cavα12.2/β3/α2δ resulted in ligand binding correlated with the number of cells (22.7 ± 1.1 × 103 cpm for 0.5 × 105 cells; 32.5 ± 1. 7 × 103 cpm for 1 × 105 cells; and 48.2 ± 1.4 × 103 cpm for 2 × 105 cells). Note that the number of the cells in each well was measured ∼24 h before the ligand binding assay. Nonspecific ligand binding was <5%, as estimated from binding to control HEK293 cells devoid of N-type Ca2+ channels. As shown in Figure 7C, 26 h after transfection, cells expressing either GFP-Rem2t or GFP-Rem2 bound the same amount of the ligand as cells expressing GFP alone (43.8 ± 1.9 × 103; 43.3 ± 2.6 × 103, and 44.2 ± 1.7 × 103 cpm, respectively; p = 0.96, ANOVA). Similar results were obtained when the concentration of [125I]Tyr22-ω-conotoxin GVIA was doubled in the binding assay (data not shown). Therefore, we conclude that Rem2 inhibited the activity of N-type Ca2+ channels located at the plasma membrane without affecting surface expression of the Cavα12.2 subunit. This conclusion was based on assumptions that the expression of Rem2 does not alter the radioligand equilibrium dissociation constant (Kd) and that the nonspecific binding measured from HEK293 cells is reflective of tsA201 cells.
Figure 7.

Heterologous expression of Rem2 in tsA201 cells stably expressing Cavα12.2/β3/α2δ abolished ICa without affectingω-conotoxin GVIA binding. A, Bar graphs summarizing the average ICa density of cells transfected 19-53 h earlier with cDNA constructs encoding GFP, GFP-Rem2t, or GFP-Rem2. ICa density was calculated as in Figure 1C. The number of cells tested is shown in parentheses. B, Comparison of [125I]Tyr22-ω-conotoxin GVIA binding to different numbers of tsA201 cells expressing Ca2+ channels. To compensate for potential nonspecific binding, HEK293 cells were added to maintain a total of 2 × 105 cells per well. After overnight culture, cells were incubated with [125I]Tyr22-ω-conotoxin GVIA (1 nm) for 40 min and washed thoroughly, and the bound ligand was quantified with a scintillation counter. C, Mean [125I]Tyr22-ω-conotoxin GVIA bound to tsA201 cells 26 h after transfection with cDNA constructs encoding GFP, GFP-Rem2t, or GFP-Rem2. D, Transfection efficiency (measured as the percentage of cells displaying GFP fluorescence) was determined from parallel experiments. A total of 500 cells were examined for each construct by phase contrast and epifluorescence microscopy (40× objective, 0.60 numerical aperture).
Discussion
Kir/Gem, Rad, Rem, and Rem2 comprise a recently defined family of small Ras-related GTP-binding proteins termed RGK proteins. Unique features of this family are the ability to directly interact with Cavβ and greatly reduce ICa arising from HVA Ca2+ channels in both heterologous and native expression systems (Béguin et al., 2001; Finlin et al., 2003, 2005; Murata et al., 2004; Ward et al., 2004). Among the RGK family, Rem2 stands out as the sole member displaying a high level of expression in the CNS (Finlin et al., 2000). Given the importance of HVA Ca2+ channels in regulating neuronal excitability and synaptic function, we examined the effects of expressing Rem2 on endogenous neuronal Ca2+ channels in mammalian neurons.
Mechanisms of Rem2-mediated decrease in ICa density
Several distinct mechanisms could underlie the decrease in ICa density after Rem2 expression. First, Rem2 might accelerate the degradation and/or internalization of Ca2+ channels, leading to a decrease in the surface density of channels. Our data seem to firmly rule out this possibility because binding of [125I]Tyr22-ω-conotoxin GVIA, a specific high-affinity antagonist of N-type Ca2+ channels (Terlau and Olivera, 2004), was unaltered after Rem2 expression for 26 h (Fig. 7). At this time, whole-cell patch-clamp recordings demonstrated a virtual elimination of ICa. Because ω-conotoxin GVIA binds to Cavα12.2, the subunit conferring ion permeation and voltage-sensitive gating, we cannot rule out the degradation or removal of CaVβ (or perhaps other auxiliary subunits), a possibility that will be considered later. Second, an inhibition of Ca2+ channel synthesis (without compensatory alterations in channel removal) would eventually lead to a decrease in ICa density. In this regard, expression of truncated Cavα12.2 inhibits channels synthesis (Raghib et al., 2001) via activation of an endoplasmic reticulum resident RNA-dependent kinase, PERK (PKR-like endoplasmic reticulum eIF2α kinase) (Page et al., 2004). Similar effects result from the expression of the CACNG7 gene product, CaVγ7, although the mechanism remains to be determined (Moss et al., 2002). The inability of a global protein translation inhibitor cycloheximide to reduce ICa density in sympathetic neurons at a time when Rem2 expression produced significant ICa reduction argues against this mechanism for mediating the short-term effects of Rem2 expression (Figs. 1, 2). In agreement with our cycloheximide data, expression of CaVγ7 for 36-48 h did not affect ICa in sympathetic neurons (Moss et al., 2002), thus implicating a relatively long channel half-life. For this reason, we cannot exclude potential effects of Rem2 on Ca2+ channel synthesis but can eliminate this mechanism as an explanation for the decrease in ICa density seen after ∼24 h of Rem2 expression. Third, Rem2 might interfere with assembly and trafficking of CaV subunits to the plasma membrane surface. In the seminal work of Béguin et al. (2001), expression of Kir/Gem was shown to eliminate functional L-type Ca2+ channels in HEK293 cells after concurrent transfection with both Kir/Gem and CaV subunits. Immunostaining of the epitope-tagged CaVα11.2 demonstrated a disruption of trafficking to the cell surface, suggesting a connection with studies showing that CaVβs are required for appropriate CaVα1 trafficking (Bichet et al., 2000a). Consistent with this notion, prolonged expression of Kir/Gem in guinea pig cardiomyocytes reduced the number of the functional L-type Ca2+ channels, as evidenced by a marked reduction in gating current (Murata et al., 2004). Finally, recent studies performed with Rem2 also demonstrated a block of CaVα11.2 surface expression (Béguin et al., 2005). Thus, it was surprising to find that surface expression of CaVα12.2 in a stably expressing cell line was unaltered by Rem2 expression (Fig. 7). In support of this finding, Finlin et al. (2005) reported that L-type channel surface expression in the MIN6 mouse insulinoma line was not reduced (as determined from surface biotinylation) at a time when channel activity was potently inhibited. Therefore, although Rem2 can alter the trafficking of L-type Ca2+ channels in some cell types, this appears not to be the mechanism by which it inhibits Ca2+ channels in the SCG, DRG neurons, and tsA201 cells in our study. Thus, Rem2 may alter Ca2+ channel function via two distinct pathways. One pathway disrupts CaV subunit trafficking and would require turnover of endogenous channels before functional effects were evident, a mechanism well suited for setting the steady-state level of channels. The other pathway, discussed below, involves modulation of preexisting channels and is thus more suitable for dynamically regulating Ca2+ channel activity on a more rapid timescale.
Modulation of preexisting Ca2+ channels by Rem2
Given the extensive evidence for the direct interaction of RGK proteins with CaVβ, an attractive hypothesis is that binding of Rem2 disrupts the CaVβ interaction with CaVα, thereby creating a population of channels at the cell surface devoid of CaVβ. In support of this notion, there is limited evidence showing that Kir/Gem interferes with the CaVα/β interaction (Béguin et al., 2001, their supplement; Sasaki et al., 2005) and that this interaction may be reversible (Bichet et al., 2000b). The latter finding is controversial because the in vitro determined affinity of CaVβ for the CaVα I-II linker domain is extremely high (for review, see Richards et al., 2004). Although this mechanism could explain defects in the trafficking of newly synthesized (i.e., before subunit assembly) Ca2+ channels, our data do not support such a mechanism for preexisting channels. First, Rem2 expression abolished (below detectable levels) ICa in a subset of both SCG neurons and in cells stably expressing N-type Ca2+ channels. This result is contrary to data obtained from HVA Ca2+ channels expressed without overt CaVβ in which detectable current flowing through the modified channels was recorded (Meir et al., 2000; Yasuda et al., 2004). Second, the loss of CaVβ would be expected to shift the activation of the channel to more depolarized potentials (Dolphin, 2003). In Rem2-expressing SCG neurons with an attenuated but detectable ICa, the voltage dependence of activation and kinetics of ICa appeared unaltered (Fig. 1A,D). In addition, the magnitude and characteristics (i.e., voltage dependent) of ICa block produced by NE was similar for the residual ICa and unmodified channels (Fig. 1E,F). The loss of CaVβ would be expected to render the inhibition much weaker and voltage independent (Meir et al., 2000).
A remaining hypothesis, which we currently favor, is that Rem2 interacts with the CaVβ of assembled HVA Ca2+ channels at the plasma membrane to form a nonconducting species. At this point, the evidence, as outlined above, is circumstantial, and additional experiments are necessary to either exclude or validate the hypothesis. The finding that preexisting LVA Ca2+ channels of DRG neurons were spared by Rem2 expression (Fig. 6) supports a mechanism that requires CaVβ as heterologous expression of the CaVα13.x subunit alone recapitulates native T-type channel properties (Perez-Reyes, 2002). Recent studies of CaVβ structure have confirmed previous predictions (Hanlon et al., 1999) of similarity between CaVβ and the MAGUK (membrane-associated guanylate kinase) class of proteins and further revealed that the interaction between CaVβ and CaVα I-II loop occupies only a small area (Chen et al., 2004b; Opatowsky et al., 2004; Van Petegem et al., 2004). Thus, the majority of the CaVβ surface in the preassembled channels is potentially available to interact with other proteins. One can speculate that Rem2 (and other RGK proteins) interacts with a region of CaVβ, thereby altering the interaction of CaVα/β and/or the interaction between Src homology 3 and guanylate kinase domains within the conserved CaVβ core region.
Regulation of Rem2 function
Regardless of the mechanisms by which Rem2 suppresses preexisting Ca2+ channel function, it was of interest to explore elements of Rem2 that conferred this phenomenon. As a class, Rasrelated proteins are molecular switches controlled by the binding of GTP. As with heterotrimeric G-proteins, the canonical pathway involves the exchange of GDP for GTP, often facilitated by a GEF-type molecule, which promotes transition of the protein from the inactive to active state (Takai et al., 2001). Kir/Gem conforms to this paradigm because a point mutation that disrupts GTP binding rendered the protein impotent in regard to HVA Ca2+ channel suppression (Ward et al., 2004; Sasaki et al., 2005). The cognate mutation (S66N) in Rad also resulted in total loss of GTP binding (Zhu et al., 1995), although the functional consequence has not been tested. Unexpectedly, the cognate mutation (S129N) in Rem2 was without effect (Fig. 5A), thus undermining the role of GTP binding in the Ca2+ channel-suppressing actions of Rem2. At present, there is no direct evidence that this mutation disrupts GTP binding to Rem2. However, the extensive homology of the G1 region (containing the S129 residue) and data from cognate mutations of related proteins Gem and Rad (Zhu et al., 1995; Feig, 1999; Ward et al., 2004) corroborates the interpretation. In addition, Béguin et al. (2005) report that the S129N mutation impairs the ability of Rem2 to interact with CaVβ, thus providing additional evidence for the function of this residue. Although the latter result appears at odds with the findings reported here, it is possible that the S129N mutation decreases, but does not eliminate, the affinity of Rem2 for CaVβ. In addition, dialysis of the neuron with GDP-β-S via the patch pipette did not reverse Rem2-mediated channel inhibition (Fig. 5D), although Ca2+ channel modulation by heterotrimeric G-proteins was abolished (Fig. 5C). Our data indicate that Rem2, in terms of Ca2+ channel suppression, may bypass the canonical G-protein pathway and that different RGK proteins may use distinct activation mechanisms to produce similar functional effects. Future experiments designed to directly measure the affinity of Rem2 S129N to GTP should help clarify this issue.
The domains flanking the Ras homology core domain of Rem2 contain protein interaction motifs that may affect function. The N terminus has a motif for 14-3-3 protein binding, whereas the C terminus contains a polybasic motif, a well conserved calmodulin-binding motif, and a second 14-3-3 binding motif. Béguin et al. (2005) have demonstrated recently that both 14-3-3 binding domains are required to demonstrate in vitro binding of 14-3-3, presumably in a dimerized form. Because expression of Rem2t, which lacks the N terminus 14-3-3 binding site, was as effective as Rem2 (Fig. 1), our data suggest that interaction with 14-3-3 proteins is not required for suppressing Ca2+ channel activity. This result agrees with data obtained from a Rem2 construct containing point mutations (S69A and S334A) that ablated 14-3-3 binding (Béguin et al., 2005). Deletion of the C terminus of Rem2 disrupted Ca2+ channel suppression and targeting of Rem2 to the plasma membrane (Fig. 4) (Finlin et al., 2000). The latter effect presumably arises from polybasic residues in the C terminus. Consistent with this proposal, both plasma membrane targeting and Ca2+ channel inhibition could be restored by fusing the last 23 residues of K-Ras4B to the C terminus of Rem2 core domain (Fig. 4), which contains both a polybasic motif and a farnesylation site (Prior and Hancock, 2001). Interestingly, fusing the initial 10 residues of Gαi1, which has a myristoylation/palmitoylation motif (Fivaz and Meyer, 2003), to the N terminus of the Rem2 core domain did not reconstitute function, although plasma membrane targeting was restored (Fig. 4). Thus, targeting of the Rem2 core domain to the plasma membrane was necessary but not sufficient for attenuating Ca2+ channel function.
Footnotes
This research was supported by the Intramural Research Program of the National Institutes of Health/National Institute on Alcohol Abuse and Alcoholism. We are very grateful to Dr. Diane Lipscombe (Brown University, Providence, RI) for the gift of tsA201 cells stably transfected with Cavα12.2/β3/α2δ. We thank Dr. T.-C. He (Johns Hopkins University, Baltimore, MD) for providing the AdEasy system.
Correspondence should be addressed to Dr. Stephen R. Ikeda, Laboratory of Molecular Physiology, National Institutes of Health/National Institute on Alcohol Abuse and Alcoholism, Fishers Lane 5625, Room TS11, MSC 9411, Bethesda, MD 20892-9411. E-mail: sikeda@mail.nih.gov.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259762-11$15.00/0
References
- Béguin P, Nagashima K, Gonoi T, Shibasaki T, Takahashi K, Kashima Y, Ozaki N, Geering K, Iwanaga T, Seino S (2001) Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature 411: 701-706. [DOI] [PubMed] [Google Scholar]
- Béguin P, Mahalakshmi RN, Nagashima K, Cher DH, Kuwamura N, Yamada Y, Seino Y, Hunziker W (2005) Roles of 14-3-3 and calmodulin binding in subcellular localization and function of the small G protein Rem2. Biochem J 390: 67-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrow NS, Brice NL, Tedder I, Page KM, Dolphin AC (1997) Properties of cloned rat α1A calcium channels transiently expressed in the COS-7 cell line. Eur J Neurosci 9: 739-748. [DOI] [PubMed] [Google Scholar]
- Bichet D, Cornet V, Geib S, Carlier E, Volsen S, Hoshi T, Mori Y, De Waard M (2000a) The I-II loop of the Ca2+ channel α1 subunit contains an endoplasmic reticulum retention signal antagonized by the β subunit. Neuron 25: 177-190. [DOI] [PubMed] [Google Scholar]
- Bichet D, Lecomte C, Sabatier JM, Felix R, De Waard M (2000b) Reversibility of the Ca2+ channel α1-β subunit interaction. Biochem Biophys Res Commun 277: 729-735. [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F (1991) The GTPase superfamily: conserved structure and molecular mechanism. Nature 349: 117-127. [DOI] [PubMed] [Google Scholar]
- Chen H, Ikeda SR (2004) Modulation of ion channels and synaptic transmission by a human sensory neuron-specific G-protein-coupled receptor, SNSR4/mrgX1, heterologously expressed in cultured rat neurons. J Neurosci 24: 5044-5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lambert NA (2000) Endogenous regulators of G protein signaling proteins regulate presynaptic inhibition at rat hippocampal synapses. Proc Natl Acad Sci USA 97: 12810-12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Honse Y, Ikeda SR (2004a) Alternative modalities of adenovirus-mediated gene expression in hippocampal neurons cultured on microisland substrate. Neurosci Lett 368: 221-225. [DOI] [PubMed] [Google Scholar]
- Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L, Yang J (2004b) Structural basis of the α1-β subunit interaction of voltage-gated Ca2+ channels. Nature 429: 675-680. [DOI] [PubMed] [Google Scholar]
- Cohen L, Mohr R, Chen YY, Huang M, Kato R, Dorin D, Tamanoi F, Goga A, Afar D, Rosenberg N, Witte O (1994) Transcriptional activation of a ras-like gene (kir) by oncogenic tyrosine kinases. Proc Natl Acad Sci USA 91: 12448-12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC (2003) β subunits of voltage-gated calcium channels. J Bioenerg Biomembr 35: 599-620. [DOI] [PubMed] [Google Scholar]
- Elmslie KS, Zhou W, Jones SW (1990) LHRH and GTP-γ-S modify calcium current activation in bullfrog sympathetic neurons. Neuron 5: 75-80. [DOI] [PubMed] [Google Scholar]
- Fedulova SA, Kostyuk PG, Veselovsky NS (1985) Two types of calcium channels in the somatic membrane of new-born rat dorsal root ganglion neurones. J Physiol (Lond) 359: 431-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig LA (1999) Tools of the trade: use of dominant-inhibitory mutants of Ras-family GTPases. Nat Cell Biol 1: E25-E27. [DOI] [PubMed] [Google Scholar]
- Finlin BS, Andres DA (1997) Rem is a new member of the Rad- and Gem/Kir Ras-related GTP-binding protein family repressed by lipopolysaccharide stimulation. J Biol Chem 272: 21982-21988. [DOI] [PubMed] [Google Scholar]
- Finlin BS, Shao H, Kadono-Okuda K, Guo N, Andres DA (2000) Rem2, a new member of the Rem/Rad/Gem/Kir family of Ras-related GTPases. Biochem J 347: 223-231. [PMC free article] [PubMed] [Google Scholar]
- Finlin BS, Crump SM, Satin J, Andres DA (2003) Regulation of voltagegated calcium channel activity by the Rem and Rad GTPases. Proc Natl Acad Sci USA 100: 14469-14474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlin BS, Mosley AL, Crump SM, Correll RN, Ozcan S, Satin J, Andres DA (2005) Regulation of L-type Ca2+ channel activity and insulin secretion by the Rem2 GTPase. J Biol Chem, in press. [DOI] [PubMed]
- Fivaz M, Meyer T (2003) Specific localization and timing in neuronal signal transduction mediated by protein-lipid interactions. Neuron 40: 319-330. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch 391: 85-100. [DOI] [PubMed] [Google Scholar]
- Hanlon MR, Berrow NS, Dolphin AC, Wallace BA (1999) Modelling of a voltage-dependent Ca2+ channel β subunit as a basis for understanding its functional properties. FEBS Lett 445: 366-370. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95: 2509-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR (1991) Double-pulse calcium channel current facilitation in adult rat sympathetic neurones. J Physiol (Lond) 439: 181-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR (2004) Expression of G-protein signaling components in adult mammalian neurons by microinjection. Methods Mol Biol 259: 167-182. [DOI] [PubMed] [Google Scholar]
- Ikeda SR, Dunlap K (1999) Voltage-dependent modulation of N-type calcium channels: role of G protein subunits. Adv Second Messenger Phosphoprotein Res 33: 131-151. [DOI] [PubMed] [Google Scholar]
- Kostyuk PG, Shuba YM, Savchenko AN (1988) Three types of calcium channels in the membrane of mouse sensory neurons. Pflügers Arch 411: 661-669. [DOI] [PubMed] [Google Scholar]
- Kozak M (1999) Initiation of translation in prokaryotes and eukaryotes. Gene 234: 187-208. [DOI] [PubMed] [Google Scholar]
- Lee SK, Hollenbeck PJ (2003) Organization and translation of mRNA in sympathetic axons. J Cell Sci 116: 4467-4478. [DOI] [PubMed] [Google Scholar]
- Lin Y, McDonough SI, Lipscombe D (2004) Alternative splicing in the voltage-sensing region of N-type CaV2.2 channels modulates channel kinetics. J Neurophysiol 92: 2820-2830. [DOI] [PubMed] [Google Scholar]
- Maguire J, Santoro T, Jensen P, Siebenlist U, Yewdell J, Kelly K (1994) Gem: an induced, immediate early protein belonging to the Ras family. Science 265: 241-244. [DOI] [PubMed] [Google Scholar]
- Meir A, Bell DC, Stephens GJ, Page KM, Dolphin AC (2000) Calcium channel β subunit promotes voltage-dependent modulation of α1B by Gβγ Biophys J 79: 731-746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Adams ME, Bean BP (1992) P-type calcium channels in rat central and peripheral neurons. Neuron 9: 85-95. [DOI] [PubMed] [Google Scholar]
- Moss FJ, Viard P, Davies A, Bertaso F, Page KM, Graham A, Canti C, Plumpton M, Plumpton C, Clare JJ, Dolphin AC (2002) The novel product of a five-exon stargazin-related gene abolishes Cav2.2 calcium channel expression. EMBO J 21: 1514-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M, Cingolani E, McDonald AD, Donahue JK, Marban E (2004) Creation of a genetic calcium channel blocker by targeted gem gene transfer in the heart. Circ Res 95: 398-405. [DOI] [PubMed] [Google Scholar]
- Opatowsky Y, Chen CC, Campbell KP, Hirsch JA (2004) Structural analysis of the voltage-dependent calcium channel β subunit functional core and its complex with the α1 interaction domain. Neuron 42: 387-399. [DOI] [PubMed] [Google Scholar]
- Page KM, Heblich F, Davies A, Butcher AJ, Leroy J, Bertaso F, Pratt WS, Dolphin AC (2004) Dominant-negative calcium channel suppression by truncated constructs involves a kinase implicated in the unfolded protein response. J Neurosci 24: 5400-5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E (2002) Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev 83: 117-161. [DOI] [PubMed] [Google Scholar]
- Prior IA, Hancock JF (2001) Compartmentalization of Ras proteins. J Cell Sci 114: 1603-1608. [DOI] [PubMed] [Google Scholar]
- Raghib A, Bertaso F, Davies A, Page KM, Meir A, Bogdanov Y, Dolphin AC (2001) Dominant-negative synthesis suppression of voltage-gated calcium channel CaV2.2 induced by truncated constructs. J Neurosci 21: 8495-8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan LJ, Sah DWY, Bean BP (1991) Ca2+ channels in rat central and peripheral neurons: high-threshold current resistant to dihydropyridine blockers and ω-conotoxin. Neuron 6: 269-280. [DOI] [PubMed] [Google Scholar]
- Reynet C, Kahn CR (1993) Rad: a member of the Ras family overexpressed in muscle of type II diabetic humans. Science 262: 1441-1444. [DOI] [PubMed] [Google Scholar]
- Richards MW, Butcher AJ, Dolphin AC (2004) Ca2+ channel β-subunits: structural insights AID our understanding. Trends Pharmacol Sci 25: 626-632. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Shibasaki T, Beguin P, Nagashima K, Miyazaki M, Seino S (2005) Direct inhibition of the interaction between alpha-interaction domain and β-interaction domain of voltage-dependent Ca2+ channels by Gem. J Biol Chem 280: 9308-9312. [DOI] [PubMed] [Google Scholar]
- Schofield GG (1991) Norepinephrine inhibits a Ca2+ current in rat sympathetic neurons via a G-protein. Eur J Pharmacol 207: 195-207. [DOI] [PubMed] [Google Scholar]
- Takai Y, Sasaki T, Matozaki T (2001) Small GTP-binding proteins. Physiol Rev 81: 153-208. [DOI] [PubMed] [Google Scholar]
- Terlau H, Olivera BM (2004) Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol Rev 84: 41-68. [DOI] [PubMed] [Google Scholar]
- Van Petegem F, Clark KA, Chatelain FC, Minor DL Jr (2004) Structure of a complex between a voltage-gated calcium channel β-subunit and an α-subunit domain. Nature 429: 671-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward Y, Spinelli B, Quon MJ, Chen H, Ikeda SR, Kelly K (2004) Phosphorylation of critical serine residues in Gem separates cytoskeletal reorganization from down-regulation of calcium channel activity. Mol Cell Biol 24: 651-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Chen L, Barr W, McRory JE, Lewis RJ, Adams DJ, Zamponi GW (2004) Auxiliary subunit regulation of high-voltage activated calcium channels expressed in mammalian cells. Eur J Neurosci 20: 1-13. [DOI] [PubMed] [Google Scholar]
- Yusaf SP, Goodman J, Pinnock RD, Dixon AK, Lee K (2001) Expression of voltage-gated calcium channel subunits in rat dorsal root ganglion neurons. Neurosci Lett 311: 137-141. [DOI] [PubMed] [Google Scholar]
- Zhu J, Reynet C, Caldwell JS, Kahn CR (1995) Characterization of Rad, a new member of Ras/GTPase superfamily, and its regulation by a unique GTPase-activating protein (GAP)-like activity. J Biol Chem 270: 4805-4812. [DOI] [PubMed] [Google Scholar]