

Abstract
In many principal brain neurons, the fast, all-or-none Na+ spike initiated at the proximal axon is followed by a slow, graded afterdepolarization (ADP). The spike ADP is critically important in determining the firing mode of many neurons; large ADPs cause neurons to fire bursts of spikes rather than solitary spikes. Nonetheless, not much is known about how and where spike ADPs are initiated. We addressed these questions in adult CA1 pyramidal cells, which manifest conspicuous somatic spike ADPs and an associated propensity for bursting, using sharp and patch microelectrode recordings in acutely isolated hippocampal slices and single neurons. Voltage-clamp commands mimicking spike waveforms evoked transient Na+ spike currents that declined quickly after the spike but were followed by substantial sustained Na+ spike aftercurrents. Drugs that blocked the persistent Na+ current (INaP), markedly suppressed the sustained Na+ spike aftercurrents, as well as spike ADPs and associated bursting. Ca2+ spike aftercurrents were much smaller, and reducing them had no noticeable effect on the spike ADPs. Truncating the apical dendrites affected neither spike ADPs nor the firing modes of these neurons. Application of INaP blockers to truncated neurons, or their focal application to the somatic region of intact neurons, suppressed spike ADPs and associated bursting, whereas their focal application to distal dendrites did not. We conclude that the somatic spike ADPs are generated predominantly by persistent Na+ channels located at or near the soma. Through this action, proximal INaP critically determines the firing mode and spike output of adult CA1 pyramidal cells.
Keywords: burst, hippocampus, phenytoin, persistent sodium current, afterdepolarization, M current, PKC, CA1 pyramidal cell, riluzole
Introduction
In many types of central and peripheral neurons, the rapid repolarization of the somatic spike is incomplete, and the spike is followed by a slow afterdepolarization (ADP) lasting tens or hundreds of milliseconds. The spike ADP comprises a brief, exponentially declining component that reflects passive recharging of the membrane capacitor by outward current flowing mainly through leak channels. This passive ADP component, however, may be augmented in size and duration by a slow inward current that activates during or immediately after the spike (Jensen et al., 1996). The classes of ion channels responsible for this spike aftercurrent vary across different types of neurons and include voltage-gated Ca2+ (Wong and Prince, 1981; Jung et al., 2001; Su et al., 2002) and Na+ channels (Azouz et al., 1996; Su et al., 2001), as well as Ca2+-activated nonspecific cationic channels (Hasuo et al., 1990; Haj-Dahmane and Andrade, 1997). Depending on the density ratio of the inward spike aftercurrent and opposing outward currents, such as the M-type K+ current (IM) (Yue and Yaari, 2004), the spike ADP may be subthreshold or may surpass spike threshold. In the latter case, it may lead to the formation of a high-frequency spike burst. Thus, the size of the inward spike aftercurrent is a critical determinant of neuronal discharge behavior (Jensen et al., 1996; Jung et al., 2001).
Both in vivo (Kandel and Spencer, 1961; Fujita, 1975) and in vitro (Schwartzkroin, 1975), adult hippocampal CA1 pyramidal cells display a conspicuous active somatic spike ADP. The ADP begins at ∼10 mV above resting potential and lasts, on average, ∼40 ms (Jensen et al., 1996). During the ADP, the neuron may re-depolarize by a few millivolts, but in ordinary in vitro conditions the ADP rarely reaches the spike threshold. Consequently, CA1 pyramidal cells normally fire one spike in response to brief depolarizing stimuli. It has been suggested that the inward spike aftercurrent in these neurons is predominantly a persistent Na+ current (INaP) (Azouz et al., 1996; Su et al., 2001), but direct proof of this contention is still lacking. It is also not known whether this spike aftercurrent is generated in the proximal portion of the neuron (i.e., at or near the axonal site of spike initiation) (Spruston et al., 1995; Turner et al., 1991; Colbert and Johnston, 1996). Alternatively, or in addition to that, spike aftercurrents activated at the apical dendrites after their invasion by the backpropagating spike may contribute to spike ADPs. The resultant dendritic depolarization would augment the somatic spike ADP in the same way that dendritic EPSPs depolarize the soma.
In this study, we used a variety of preparations and electrophysiological techniques to further characterize the currents generating the spike ADP in adult CA1 pyramidal cells. Our results support the notion that activation of persistent Na+ channels residing in the proximal portion of these neurons, but not of Ca2+ channels, underlies the generation of the somatic spike ADP.
Materials and Methods
Hippocampal slices. All animal experiments were conducted in accordance with the guidelines of the Animal Care Committees of the Hebrew and Bonn Universities. Adult male Sabra rats (∼150 g) were decapitated under deep isoflurane anesthesia, and transverse hippocampal slices (400 μm) were prepared with a vibrating microslicer (Leica, Nussloch, Germany) and transferred to a storage chamber perfused with oxygenated (95% O2/5% CO2) artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 3.5 KCl, 2 MgCl2, 1.6 CaCl2, 26 NaHCO3, and 10 d-glucose; pH 7.4; osmolarity, 305 mOsm/L; where they were maintained at room temperature. For sharp microelectrode experiments, slices were placed one at a time in an interface chamber (33.5°C) and perfused with oxygenated ACSF. In some experiments, as indicated, a deep cut was made in the stratum radiatum close to and parallel to the stratum pyramidale (see Fig. 10 A, Ca), using a broken pipette or a razor blade chip propelled by a micromanipulator. The slices were allowed to recover in the chamber for at least 1 h before initiating a recording session. In experiments not involving monitoring of synaptic transmission, 6-cyano-7-nitro-quinoxaline-2,3-dione (15 μm), 2-amino-5-phosphonovaleric acid (50 μm), and picrotoxin (100 μm) were added to the ACSF to block fast excitatory and inhibitory synaptic transmission. Other drugs were added to the ACSF as indicated.
Figure 10.

Spike ADPs in CA1 pyramidal cells lacking most of their apical dendrites. Sharp microelectrode recordings in slices are shown. A, Schematic description of the cut made in the stratum radiatum (parallel to the stratum pyramidale) to truncate the apical dendrites of CA1 pyramidal cells. Also shown are the positions of the stimulating electrodes in distal stratum radiatum (S1) and in alveus (S2) in relation to the recording microelectrode (R). B, Testing the success of dendritic truncation. Orthodromically (S1) and antidromically evoked field potentials (S2) elicited before (control) and 1 h after truncation (truncated). It is apparent that the orthodromically evoked response is abolished by the cut in the stratum radiatum. C, Morphology and spike discharge of truncated CA1 pyramidal cells. The photomicrograph (Ca) shows two neighboring neurons injected with biocytin through the recording microelectrode ∼ 1 h after cutting the slice. The biocytin stains the soma and basilar dendrites of these neurons, but only short stumps of the apical dendrites can be detected. Recordings from one of these neurons (Cb) disclose normally looking spikes and discharge behavior. D, Effects of TTX. Da and Db illustrate the spike before (control) and 15 min after adding 20 nm TTX to the ACSF. Dc shows superposition of the respective ADPs enlarged in size, and Dd shows superposition of the respective spikes expanded in time. As in intact neurons, TTX reduced the active spike ADP before affecting the spike rise time or amplitude. E, Effects of PDB in another neuron. Ea and Eb illustrate the spike before (control) and 25 min after adding 5 μm PDB to the ACSF. Ec shows superposition of the respective ADPs enlarged in size, and Ed shows superposition of the respective spikes expanded in time. Again, PDB reduced the active spike ADP without affecting the spike rise time and amplitude.
Sharp microelectrode recordings. Intracellular recordings were obtained using sharp glass microelectrodes containing 4 m K+-acetate (90-110 MΩ) and an amplifier (Axoclamp 2B; Molecular Devices, Foster City, CA) allowing simultaneous injection of current and measurement of membrane potential. The bridge balance was carefully monitored and adjusted before each measurement. The pyramidal cells included in this series of experiments had stable resting potentials of -60 mV or more, and action potentials ranging from 73.4 to 104.3 mV in amplitude (Table 1). The intracellular signals were filtered on-line at 10 kHz, digitized at a sampling rate of 10 kHz or more, and stored by a personal computer using a data acquisition system (Digidata 1322A) and pClamp software (Molecular Devices).
Table 1.
Passive and active electrophysiological properties of intact versus truncated CA1 pyramidal cells
|
|
Truncated neurons (n = 36) |
Intact neurons (n = 37) |
|---|---|---|
| Resting potential (mV) | −68.2 ± 0.6 | −68.8 ± 0.7 |
| Input resistance (Mω) | 39.2 ± 1.8* | 33.0 ± 1.6* |
| Spike threshold (mV) | −56.9 ± 1.0 | −57.2 ± 0.7 |
| Spike rise time (ms) | 0.15 ± 0.3 | 0.15 ± 0.46 |
| Spike amplitude (mV) | 93.6 ± 6.6 | 93.5 ± 1.4 |
| Spike width (ms) | 0.75 ± 0.17 | 0.77 ± 0.02 |
| Fast AHP (ms) | −59.1 ± 1.3 | −58.6 ± 0.9 |
| ADP size (mV·ms) |
178.8 ± 12.3 |
180.5 ± 7.9 |
Values provided are means ± SEM.
Significant differences between the means of the two groups (unpaired Student's t test;p = 0.017).
In some experiments, bipolar electrodes made of platinum wire (50 μm), connected to a stimulator (Master8; AMPI, Jerusalem, Israel) by an isolation unit, were used for focal stimulation (1-20 V; 50-70 μs) of afferent fibers in stratum radiatum near the CA2/CA3 border (orthodromic stimulation) and CA1 pyramidal cell axons in the alveus near the CA1/subiculum border. Extracellular recordings were made in the stratum pyramidale using sharp glass microelectrodes filled with 1 m NaCl (5-10 MΩ).
The intracellular and extracellular signals were digitized and stored by a personal computer using a data acquisition system (Digidata 1322A) and pClamp software (Molecular Devices).
Drug applications. Drugs were applied to the entire slice by bath application or to discrete locations in the slice using a puff application system. Because the spread of drugs in slices maintained in an interface chamber is predominantly by diffusion, the onset and offset of drug effects are slow (in the order of tens of minutes) (see Fig. 1 B). For all bath-applied drugs used in this study, maximal effects were obtained within 20-30 min of adding them to the ACSF.
Figure 1.

Tetrodotoxin blocks somatic spike ADPs and associated bursting in adult CA1 pyramidal cells. Sharp microelectrode recordings in slices are shown. A, Effects of bath-applied TTX. Stimulation with a brief depolarizing stimulus evoked a solitary somatic spike followed by a conspicuous active ADP(Aa). Bath application of 20 nm TTX suppressed the active ADP component (Ab). The traces in Aa and Ab are enlarged and superimposed in Ac to facilitate comparison of the ADPs. The corresponding spikes are expanded and overlaid in Ad to facilitate comparison of the spike rise times. The same neuron was also stimulated with prolonged depolarizing current pulses of increasing strength (Ae). Each spike was followed by an ADP. At the highest stimulus strength, the early spikes clustered together forming a burst. Bath-applied TTX (Af) dispersed the early burst, concurrent with ADP suppression (compare top traces in Ae and Af). The afterpotentials that follow the spikes indicated by arrows in Ae and Af are enlarged and expanded in Ag. B, Plot of the average time course of the effects of TTX on spike ADP (squares) and spike rise time (triangles) for five similar experiments. C, Scheme of experimental arrangement for focal pressure application of drugs. For somatic applications, the puffing pipette was positioned in the stratum pyramidale ∼25 μm from the recording microelectrode (lodged in the soma of the neuron). To apply drugs to the distal apical dendrites, the puffing pipette was positioned in the stratum radiatum ∼200-300 μm away from and vertical to, the stratum pyramidale. D, Effects of TTX applied focally to apical dendrites on the spike ADP. In another neuron, stimulation with a brief depolarizing stimulus evoked a solitary somatic spike followed by a conspicuous active ADP (Da). Distal TTX application (3 puffs) had no effect during the 10 min observation period (Db). One puff of TTX onto the axo-soma suppressed the ADP within 3 min (Dc). Enlarged portions of the traces in Da, Db, and Dc are superimposed in Dd to facilitate comparison of the ADPs. E, Effects of TTX applied focally to apical dendrites on 4-AP-induced ADP facilitation and bursting. In another neuron, stimulation with a brief depolarizing stimulus evoked a solitary somatic spike followed by a conspicuous active ADP (Ea). Bath application of 3 mm 4-AP caused, within 17 min, progressive ADP facilitation (Eb, solid line) up to the point of bursting (Eb, dotted line). Distal application of TTX (1 puff) completely reversed the 4-AP effects with in 3 min without grossly affecting the somatic spike (Ec). Enlarged portions of the traces in Ea, Eb, and Ec are superimposed in Ed to facilitate comparison of the ADPs. Error bars indicate SEM.
For focal drug applications, we used a puffing system consisting of a pneumatic pump (Picospritzer III; General Valve, Fairfield, NJ) connected to a patch pipette (tip diameter, ∼10 μm) filled with ACSF and the drugs intended for focal application. The tip of the pipette touched the upper surface of the slice. In control experiments, the strength and duration of the pressure pulses were adjusted to produce a drop that covered a circular area ∼50 μm in diameter when ejected onto the surface of the slice (visualized by including fast-green dye in the pipette solution). This yielded pressure pulses of 5 bar for 20 ms, which were used for all applications. For somatic drug applications, the puffing pipette was positioned in the stratum pyramidale ∼25 μm from the recording microelectrode (lodged in the soma of the neuron). To apply drugs to the distal apical dendrites, the puffing pipette was positioned in the stratum radiatum ∼200-300 μm away from, and vertical to, the stratum pyramidale (the experimental arrangement is shown in Fig. 1 C). The effects of focally applied drugs, when present, appeared usually after 3 min of puffing and attained maximum within 8-10 min. These delays are undoubtedly attributable to the slow spread of the drugs from their site of application into the slice.
Dissociated neurons. Acutely dissociated neurons were harvested from adult male Wistar rats (∼150 g). Animals were perfused through the heart under deep anesthesia (ketamine, 100 mg/kg; xylazine, 15 mg/kg) with ice-cold sucrose-based ACSF containing the following (in mm): 60 NaCl, 100 sucrose, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 5 MgCl2, 1 CaCl2, and 25 glucose, and the brain was rapidly removed. Transverse hippocampal slices (400 μm) were prepared with a vibrating microslicer (Leica) and then transferred to a storage chamber perfused with oxygenated (95% O2/5% CO2) ACSF. The chamber was gradually warmed to 36°C in a water bath and maintained at this temperature for 30 min. Finally, the slices were transferred into a chamber filled with ACSF containing the following (in mm): 124 NaCl, 3.5 KCl, 2 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 d-glucose; pH 7.4; osmolarity, 305 mOsm/L; where they were maintained at room temperature for up to 5 h.
For preparation of dissociated CA1 neurons, slices were transferred after an equilibration period of at least 60 min to a tube with 5 ml of saline containing the following (in mm): 145 CH3SO3Na, 3 KCl, 1 MgCl2, 0.5 CaCl2, 10 HEPES, and 15 glucose, pH 7.4 (adjusted with NaOH). Pronase (protease type XIV; 2 mg/ml; Sigma, St. Louis, MO) was added to the oxygenated medium (100% O2). After an incubation period of 15 min at 35°C, the slice was washed in Pronase-free saline of an identical composition. The CA1 region was dissected and triturated with fire-polished glass pipettes of decreasing aperture. The Petri dish containing the cell suspension was then mounted on the stage of an inverted microscope (Telaval; Zeiss, Jena, Germany). Dissociated cells were allowed to settle for 5-10 min before recordings were attempted. Whole-cell recordings were performed only on pyramidal-shaped neurons with a smooth surface and a three-dimensional contour. They all possessed a clearly identifiable apical dendrite and well preserved basal structures, representing basal dendrites and the axon hillock/initial segment.
Experimental ACSFs were applied by hydrostatic pressure via a nearby (50-100 μm) superfusion pipette. Solutions around the neurons were exchanged within <1 min. Therefore, effects of ion channel blockers were obtained in a matter of seconds or tens of seconds, much faster than in slice experiments, and were readily reversible. The standard ACSF composition used to isolate inward (i.e., Na+ and Ca2+) currents (here-after referred to as 2Ca-ACSF) contained the following (in mm): 125 CH3SO3Na, 20 tetraethylammonium, 3 KCl, 2 MgCl2, 10 HEPES, 4 4-aminopyridine (4-AP), 0.3 CdCl2, and 10 glucose; pH 7.4; osmolarity, 310 mOsm/L. In some experiments, as indicated, CaCl2 concentration was decreased to 1.2 mm (hereafter referred to as 1.2Ca-ACSF). To these ACSFs, we added, as necessary, 0.3 mm CdCl2 and/or 0.5 μm tetrodotoxin (TTX) to block voltage-gated Ca2+ and/or Na+ channels, respectively. Additional drugs were added to the ACSFs as indicated.
Whole-cell patch-clamp recordings. Patch pipettes with a resistance of 2.0-3.0 MΩ were pulled from borosilicate glass capillaries (outer diameter, 1.5 mm; inner diameter, 1 mm; Science Products, Hofheim, Germany) on a Narashige PP-830 puller (Narishige, Tokyo, Japan). Pipettes were filled with an intracellular solution containing the following (in mm): 110 CsF, 2 MgCl2, 1 CaCl2, 10 HEPES, 10 1,2-bis(2-aminophenoxy)ethane-N, N,N′,N′-tetra-acetic acid (BAPTA), 10 ATP, and 0.5 GTP, pH adjusted to 7.4 with CsOH. The osmolarity was adjusted with mannitol to 295 ± 5 mOsm/L. For these solutions, the liquid junction potential was calculated (5.1 mV) and the command voltages were corrected accordingly. Tight-seal whole-cell recordings were obtained at room temperature. Seal resistance was >1 GΩ in all recordings. The series resistance was 6 ± 2 MΩ measured by the dial settings for series resistance compensation on the Axopatch 200A amplifier (Molecular Devices). To improve the voltage control, series resistance compensation between 70 and 90% was used. The maximal residual voltage error caused by the series resistance errors was negligible (<0.5 mV) for measurements of small currents, such as INaP (<150 pA). For larger currents, such as Na+ spike currents (∼15 nA), the maximal residual voltage error was considerably higher (∼17 mV). Voltage commands were delivered via the Axopatch 200A amplifier, and the resulting current was recorded on-line with the pClamp 8.0 acquisition and analysis program. Current signals were filtered at 10 kHz (mock AP protocols) or 1 kHz (ramp protocols), sampled at 20 kHz or more, and stored by a personal computer using a data acquisition system (Digidata 1322A) and pClamp software (Molecular Devices).
Drugs. Stock solutions of 4β-phorbol 12,13-dibutyrate (PDB) (10 mm), 4α-phorbol 12,13-didecanoate (PDD) (10 mm), riluzole (10 mm), and phenytoin (100 mm) were prepared in dimethylsulfoxide (DMSO) and stored at -20°C. They were usually diluted 1:1000 when added to the ACSFs. Control ACSFs contained equal amounts of DMSO (0.001%), which by itself had no effects on the measured parameters. All other drugs were added to the ACSFs from aqueous stock solutions. Chemicals and drugs were obtained from Sigma, apart from apamin and iberiotoxin (IBTX) (Alomone Labs, Jerusalem, Israel).
Data analysis. To measure passive membrane properties, the pyramidal cells were injected with small (0.1-0.5 nA) 200 ms negative current pulses. The apparent input resistance was provided by the slope of the linear regression line fitted through the linear portion of the steady-state voltage versus current amplitude plot. The fast spike afterhyperpolarization (fAHP) (Storm, 1987) was measured as the potential attained at the end of the spike downstroke. Spike width was measured at 50% of spike amplitude. Spike rise time was measured as the time in which the spike rose from 10 to 90% of its maximal amplitude. The size of the spike ADP was measured as the integrated “area under the curve” between the fAHP and the point at which membrane voltage returned to resting potential.
The voltage dependence of INaP was determined using standard protocols (see Fig. 2). The ramp current was converted to conductance G(V) according to the following equation:
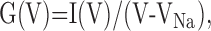 |
(1) |
Figure 2.

Properties of INaP and Na+ spike aftercurrents in CA1 pyramidal cells. Whole-cell patch-clamp recordings in acutely dissociated neurons are shown. A, Isolation of INaP. The neuron was superfused with 2Ca-ACSF containing 0.3 mm CdCl2 (to block Ca2+ channels) and subjected to slow voltage ramps (50 mV/s) (inset). The current responses shown were obtained before (Aa) and after application of 0.5 μm TTX (Ab). INaP was isolated by the subtraction of responses obtained under TTX from those in control conditions (Ac). The subtracted current traces were converted to conductance (Eq.1) and plotted versus the ramp voltage. The resultant persistent Na+ conductance-voltage relationship is depicted in Ad. B, Isolation of Na+ spike currents and aftercurrents. Representative currents elicited by mock action potentials are shown (upstroke 1 ms, from -70 to + 30 mV; downstroke 2 ms to potentials ranging from -70 to -30 mV) (inset). The current responses shown were obtained before (Ba) and after application of 0.5 μm TTX (Bb). The Na+ spike currents and aftercurrents were isolated by subtraction of responses obtained under TTX from those in control conditions (Bc). The amplitudes of Na+ spike aftercurrents were measured at two time points, namely, 10 and 100 ms after repolarization of the mock spike (Bd, filled and open circles, respectively).
where VNa is the Na+ reversal potential, V the ramp potential, and I(V) the peak current amplitude. G(V) was then fitted with the following Boltzmann function:
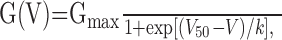 |
(2) |
where Gmax is the maximal Na+ conductance, V50 is the membrane voltage (V) at which G(V) is 50% of Gmax, and k is the slope of the G(V)/V relationship at V50.
In some cases, a compound ramp current was measured comprising both high threshold-activated Ca2+ current (ICa) and INaP (see Fig. 3). In these cases, the ramp current was fitted directly with the sum of two Boltzmann functions of the following form:
 |
(3) |
Figure 3.

The Ca2+ and Cd2+ content of the ACSF modulates INaP-voltage relationship. Whole-cell patch-clamp recordings in acutely dissociated neurons are shown. A, Recordings in 2Ca-ACSF without CdCl2 resulted in the appearance of a dual-component current response, consisting of a high-threshold component that corresponds to ICa, in addition to the low-threshold INaP (ramp current, ICa plus INaP; gray line). The current response was fitted with a function comprised of the sum of two modified Boltzmann functions, one for INaP and one for ICa (Eq. 3). The resultant fitted function (labeled ICa + INaP; solid line) is superimposed on the current trace. The parameter values of the fitted function were used to derive separately the INaP-voltage and ICa-voltage relationships, which are also shown (ICa,fit and INaP,fit, respectively). B, In the same neuron, ICa was isolated by subtracting traces obtained in Cd2+- and TTX-containing ACSF from those obtained after washout of Cd2+ (yielding the Cd2+-sensitive current component corresponding to ICa without contamination by INaP; gray line). Overlaying ICa,fit (black line) on the current response shows that ICa,fit accurately reflects the ICa-voltage relationship. C, Voltage dependence of INaP-voltage relationships in three different ACSF compositions. The INaP component was isolated from the dual-component current responses evoked by voltage ramp commands by subtracting ICa,fit. It was then converted to persistent Na+ conductance-voltage relationship (gray lines) and fitted with the Boltzmann function (superimposed black lines). D, Bar diagram showing V50 values of persistent Na+ conductance in the different ACSF compositions. The V50 in 1.2Ca-ACSF was 8.9 ± 2.0 mV more negative than in Cd2+-containing 2Ca-ACSF. *Statistically significant differences between compared groups. Error bars indicate SEM.
with VCa standing for the apparent Ca2+ reversal potential, and GNa and GCa being described by two Boltzmann functions of the form given in Equation 2. A nonlinear Levenberg-Marquardt algorithm was used for all fits.
All results are presented as the mean ± SEM. The significance of the differences between the measured parameters was evaluated using, as required, Student's unpaired or paired t test. For multiple comparisons, ANOVA was used. The significance level in all tests was set to p < 0.05.
Results
Spike ADPs in intact CA1 pyramidal cells
When stimulated with brief (4 ms) suprathreshold depolarizing current pulses, all but one of 97 intact neurons fired a solitary spike followed by a distinct ADP (Fig. 1Aa). The membrane voltage at which the spike downstroke ended and the ADP began (i.e., the fAHP) was -58.6 ± 0.9 mV (n = 37), which is ∼10 mV more depolarized than the resting potential of these neurons (-68.8 ± 0.7 mV). The spike ADPs varied in size across the neurons (from 107.0 to 350.3), averaging 180.5 ± 7.9 mV · ms (n = 37). They were evidently larger in neurons having a tendency to generate bursts in response to prolonged depolarizing current pulses (see Fig. 11Ba). Indeed, in the atypical neuron, the ADP was so large that it triggered two additional spikes, causing the neuron to fire bursts of three spikes even in response to brief stimuli. The wave-forms of the spike ADPs were stable over time (1-3 h), provided resting membrane potentials did not change by more than ±1 mV. In a series of six control experiments, the spike ADPs measured 159.3 ± 13.3 mV · ms at the beginning of the recording session and 159.0 ± 12.5 mV · ms 1 h later.
Figure 11.

Spike ADPs and firing modes in intact and truncated CA1 pyramidal cells. Sharp microelectrode recordings in slices are shown. A, Pie chart diagrams depicting the incidence of nonbursters (NBs), high-threshold bursters (HTBs) and low-threshold bursters (LTBs) in intact (Aa) and in truncated CA1 pyramidal cells (Ab). B, Spike ADPs in intact (Ba) and intruncated neurons (Bb). In each panel, the spike ADPs recorded in a nonburster, a high-threshold burster, and a low-threshold burster are superimposed to facilitate comparison of ADP size. C, Bar diagrams comparing sizes of spike ADPs in nonbursters, high-threshold bursters, and low-threshold bursters in both intact (Ca) and truncated neurons (Cb). In both intact and truncated neurons, the spike ADPs in bursters are significantly larger than in nonbursters. *Statistically significant differences between compared groups. Error bars indicate SEM.
Effects of tetrodotoxin on spike ADPs
Previous studies have suggested that the depolarizing drive for the active spike ADP in adult CA1 pyramidal cells is furnished by an Na+ spike aftercurrent (Azouz et al., 1996; Su et al., 2001). However, in those experiments, the slices were bathed in ACSFs containing elevated K+ concentrations ([K+]o) or reduced Ca2+ concentrations ([Ca2+]o). These conditions augment INaP (Li and Hatton, 1996; Somjen and Muller, 2000) and thereby could artificially amplify its contribution to the generation of the spike ADP. Therefore, in the present series of experiments, we bathed the slices in ACSFs containing 3.5 mm K+ and 1.6 mm Ca2+. The rationale for using 1.6 mm Ca2+ is that it yields a free Ca2+ ion concentration of ∼1.2 mm in bicarbonate-based ACSF (U. Heinemann, personal communication), thus resembling physiological [Ca2+]o in the adult brain (Heinemann et al., 1977).
To evaluate the role of an Na+ current in generating the spike ADP, we first tested the effects of the classical Na+ channel blocker TTX. Because TTX blocks both transient Na+ current (INaT) and INaP (French et al., 1990), we carefully monitored spike threshold and amplitude, as well as its rate of rise (which reflects the maximal transient Na+ conductance) (Hodgkin and Katz, 1949). Representative results from one neuron are shown in Figure 1A. Bath application of 20 nm TTX reduced the spike ADP (measured as area under the curve; see Materials and Methods) from 170.6 to 84.4 mV · ms within 30 min (Fig. 1Aa;Ab;Ac, superimposed traces). Despite the 50.5% reduction in ADP size, TTX did not notably affect the properties of the fast spike (Fig. 1Aa;Ab;Ad, superimposed traces). Thus, the spike rise time was 160.5 μs before TTX application and 160.0 μs after 30 min of exposure to the drug (Fig. 1B). In nine similar experiments, quantification of ADP size (measured before any changes in spike morphology became apparent) revealed a significant reduction of the ADP by 28.5 ± 9.1% (from 167.6 ± 13.4 to 119.7 ± 19.3 mV · ms; p = 0.024). The time course of this TTX effect in five experiments is depicted in Figure 1B. Reduction of spike ADPs by TTX was evident also in spikes elicited by prolonged (180 ms) positive current pulses (Fig. 1Ae-Ag). At the same time, TTX caused dispersion of the early spike cluster but did not modify the total number of evoked spikes (Fig. 1Ae,Af, top traces).
The above results are congruent with our original contention that the spike ADP is generated predominantly by an Na+ spike aftercurrent (Azouz et al., 1996; Su et al., 2001). However, we also considered an alternative hypothesis, namely, that the somatic spike ADP is generated by activation of Ca2+ channels in the apical dendrites after their invasion by the backpropagating somatic spike. In such a situation, low TTX concentrations may suppress the somatic spike ADP secondary to a block of spike backpropagation into the apical dendrites. To evaluate the validity of the latter hypothesis, we examined how TTX applied focally to apical dendrites affects the somatic spike ADP. The experimental arrangement is described in Figure 1C (see also Materials and Methods). The puffing pipette contained 50 nm TTX dissolved in ACSF. The pressure pulses used for puffing were of 1 bar, 10 ms duration. Representative results are illustrated in Figure 1D. Distal repetitive applications of TTX (three puffs) had no significant effect on the spike ADP during the 10 min observation period (from 199.4 ± 28.2 to 185.8 ± 23.4 mV · ms; n = 3) (Fig. 1Da;Db;Dd, superimposed traces). When the puffing pipette was moved near the soma and TTX was applied (one puff), it suppressed the spike ADP by 27.9 ± 2.2% within 3 min (144.5 ± 22.9 mV · ms) (Fig. 1Db;Dc;Dd, superimposed traces). In the latter experiments, however, spike depression also appeared early, so it was difficult to document selective modulation of the spike ADP.
To ensure that distally applied TTX effectively blocks spike backpropagation into the apical dendrites, we examined how this procedure affects bursting induced by 3 mm 4-AP in the ACSF. It is known that exposing CA1 pyramidal cells to millimolar 4-AP facilitates spike backpropagation and activation of dendritic Ca2+ channels by blocking A-type K+ currents (IA) (Hoffman et al., 1997; Magee and Carruth, 1999). The resultant dendritic Ca2+ spike depolarizes the soma, thereby triggering two to three additional spikes (Fig. 1Ea,Eb). Accordingly, 4-AP-induced bursts are blocked by millimolar Ni2+ but not by drugs that inhibit INaP (Y. Yue and Y. Yaari, unpublished observations). Likewise, puffing TTX onto the apical dendrites suppresses 4-AP-induced bursts simply by blocking Na+ spike backpropagation (Magee and Carruth, 1999). This effect was readily replicated in our experiments (n = 5). A single puff of TTX onto the apical dendrites suppressed bursting and reversed the augmentation of the ADP within 2-3 min in all cases (Fig. 1Ec;Ed, superimposed traces), indicating that spike backpropagation into this compartment is effectively blocked. At the same time, the rise time and amplitude of the first spike were not affected, indicating that distally applied TTX did not reach the soma in a blocking concentration.
Isolation of Na+ spike aftercurrents
Together, the above data suggest that recruitment of a TTX-sensitive Na+ spike aftercurrent in the proximal region of the neuron plays an essential role in ADP generation. Because the spike ADPs last tens of milliseconds (Fig. 1Aa), this spike aftercurrent likely results from activation of INaP (French et al., 1990). To characterize the contribution of INaP to the Na+ spike aftercurrent, we obtained tight-seal whole-cell recordings from dissociated adult CA1 pyramidal cells. The neurons were superfused with 2Ca-ACSF to which 0.3 mm CdCl2 was added (see Materials and Methods). When slow (50 mV/s) depolarizing voltage ramps from -85 to 25 mV (Fig. 2A, inset at top) were applied to these neurons (causing inactivation of transient Na+ channels), a net inward current was elicited in all cases at potentials between -60 and 5 mV. At more positive potentials, an outward current dominated the response to the voltage command (n = 28 neurons) (Fig. 2Aa). The inward current was completely abolished by adding 0.5 μm TTX to the ACSF (Fig. 2Ab), identifying it as INaP (French et al., 1990). Subtraction of the current traces obtained before and after TTX application yielded the pure INaP component of the current response to the voltage command (Fig. 2Ac). The activation threshold of INaP was about -55 mV, and it increased steeply with depolarization, attaining maximal amplitude at -37.1 ± 1.0 mV. These INaP-voltage relationships were converted to conductance-voltage plots (Eq. 1, Fig. 2Ad). Fitting them with the Boltzmann function (Eq. 2) yielded Gmax, V50, and k values of 1.1 ± 0.1 nS, -48.6 ± 1.1 mV, and 3.6 ± 0.2 mV, respectively (n = 28).
The contribution of INaP to Na+ spike aftercurrent after a spike would depend not only on steady-state conductance values but also on its activation and deactivation kinetics, which are very fast (τ values, <1 ms) (Kay et al., 1998). Therefore, to further assess whether this conductance can generate significant spike aftercurrents, we examined the currents evoked by voltage commands that replicate native spikes. The neurons were maintained at holding potentials of -70 mV, approximating their average native resting potential. The duration of the rising phase (from -70 to +30 mV) of the mock spikes was 1 ms, whereas repolarization lasted 2 ms. In different trials, the mock spikes repolarized completely (i.e., to -70 mV) or to more positive values of -60, -50, -40, or -30 mV for 100 ms (Fig. 2B, inset at top). The mock spikes evoked both transient and sustained inward currents (Fig. 2Ba), which were blocked almost completely by 0.5 μm TTX (Fig. 2Bb), indicating that they are carried by transient and persistent Na+ channels.
The Na+ currents evoked during the mock spike were very large (up to 20 nA) (Fig. 2Ba,Bc; the current traces are truncated), but declined rapidly with the downstroke of the mock spike. The size and waveform of the Na+ spike aftercurrents depended on the membrane voltage after the mock spike. When the mock spikes repolarized to -70 or -60 mV, the Na+ spike aftercurrents were very small, ranging from 0 to 22 pA, and declined to zero within 10-20 ms. However, when the mock spikes repolarized to more positive potentials (-50 to -30 mV), the Na+ spike aftercurrents declined slowly (over the time course of tens of milliseconds) and only partially, so that a substantial Na+ current remained throughout the duration of the 100 ms depolarizing command (Fig. 2Bc). Both transient and sustained components of the Na+ spike aftercurrents were larger at more depolarized potentials (Fig. 2Bc). For quantitative comparison between these two components, we plotted the size of the spike aftercurrents flowing at 10 and 100 ms after the end of the mock spike (Fig. 2Bd).
Interestingly, when the mock spike was repolarized to -50 mV, the Na+ spike aftercurrent declined to a minimum during the first 5 ms but then increased slowly in amplitude over the time course of tens of milliseconds (Fig. 2Bc, arrow). This peculiar small increase in Na+ spike aftercurrent within a narrow voltage range (-50 to -40 mV) was observed in most, if not all, CA1 pyramidal neurons (see Figs. 4Da,5Ca,6Ea).
Figure 4.

Phenytoin inhibits INaP and Na+ spike aftercurrents. Whole-cell patch-clamp recordings in acutely dissociated neurons are shown. The representative neuron was superfused with 2Ca-ACSF containing 0.3 mm Cd2+ to which 100 μm phenytoin and 0.5 mm TTX were sequentially added. Current responses were obtained in the three conditions. The Na+ currents were isolated by subtracting current responses remaining after adding TTX from previous responses. A, In a representative neuron, INaP responses to voltage ramp commands (inset) were obtained before (control) and during exposure to 100 μm phenytoin (phenytoin). B, Pooled results from nine neurons showing INaP evoked at different ramp potentials (ranging from -60 to 0 mV) before (control, filled circles) and during exposure to phenytoin (phenytoin, open circles). All amplitudes are normalized to the peak INaP amplitude in control conditions. The block of peak INaP amounted to 58 ± 4% (n = 9). C, Block of INaP by phenytoin is voltage dependent. The bars of the diagram depict the fractional inhibition of INaP at various potentials. Note that the efficacy of block increases with depolarization, indicating a voltage-dependent blocking action. D, In the same neuron shown in A, Na+ aftercurrents evoked by mock spikes repolarizing to different membrane potentials (from -70 to -30 mV; insets) were obtained before (Da, control) and during exposure to 100 μm phenytoin (Db, phenytoin). E, Pooled results from the same nine neurons showing early (10 ms) (Ea) and late (100 ms) (Eb) components of Na+ spike aftercurrents, obtained as shown in (D) before (control, filled circles) and during exposure to phenytoin (phenytoin, open circles). Error bars indicate SEM.
Figure 5.

Riluzole inhibits INaP and Na+ spike aftercurrents. Whole-cell patch-clamp recordings in acutely dissociated neurons are shown. The representative neuron was superfused with 2Ca-ACSF containing 0.3 mm Cd2+ to which 10 μm riluzole and 0.5 mm TTX were sequentially added. Current responses were obtained in the three conditions. The Na+ currents were isolated by subtracting current responses remaining after adding TTX from previous responses. A, In a representative neuron, INaP responses to voltage ramp commands (inset) were obtained before (control) and during exposure to 10 μm riluzole (riluzole). B, Pooled results from nine neurons showing INaP evoked at different ramp potentials (ranging from -60 to 0 mV) before (control, filled circles) and during exposure to riluzole (riluzole, open circles). All amplitudes are normalized to the peak INaP amplitude in control conditions. The block of peak INaP amounted to 90 ± 2% (n = 9). C, In the same neuron shown in A, Na+ aftercurrents evoked by mock spikes repolarizing to different membrane potentials (from -70 to -30 mV; insets) were obtained before (Ca, control) and during exposure to 100 μm riluzole (Cb, riluzole). D, Pooled results from the same nine neurons showing early (10 ms) (Da) and late (100 ms) (Db) components of Na+ spike aftercurrents, obtained as shown in D before (control, filled circles) and during exposure to riluzole (riluzole, open circles). Error bars indicate SEM.
Figure 6.

The protein kinase C activator PDB inhibits INaP and Na+ spike aftercurrents. Whole-cell patch-clamp recordings in acutely dissociated neurons are shown. The representative neuron was superfused with 2Ca-ACSF containing 0.3 mm Cd2+ to which 10 μm PDB and 0.5 mm TTX were sequentially added. Current responses were obtained in the three conditions. The Na+ currents were isolated by subtracting current responses remaining after adding TTX from previous responses. A, In a representative neuron, INaP responses to voltage ramp commands (inset) were obtained before (control) and at different times (1, 3, 5, and 10 min) after adding 5 μm PDB to the ACSF (PDB). PDB caused a progressive reduction in INaP amplitude together with a hyperpolarizing shift in INaP-voltage relationship. Eventually, PDB blocked INaP completely. B, Example of the hyperpolarizing shift of the INaP-voltage relationship induced by 5 min exposure to PDB. The INaP values were converted to persistent Na+ conductance values (gray lines) and fitted with the Boltzmann function, which is superimposed on the original data (black lines). C, Pooled results from 13 neurons showing the PDB-induced hyperpolarizing shift in V50 of INaP activation 1, 3, and 5 min after adding PDB to the ACSF. The V50 values were obtained by fitting the Boltzmann function to the conductance-voltage relationships (as shown B). D, Pooled results from seven neurons showing INaP amplitudes evoked at different ramp potentials (ranging from -60 to 0 mV) before (control, filled circles) and during 1 (squares), 3 (triangles), 5 (diamonds), and 10 min (open circles) of exposure to PDB. All amplitudes are normalized to the peak INaP amplitude in control conditions. The block of peak INaP amounted to 90 ± 2% (n = 7). During more prolonged exposures to PDB (>10 min), INaP was invariably blocked completely at all ramp potentials (open circles). E, In the same neuron shown in A, Na+ aftercurrents evoked by mock spikes repolarizing to different membrane potentials (from -70 to -30 mV; insets) were obtained before (Ea) and after 5 min of exposure to PDB (Eb). F, Pooled results from the same seven neurons showing early (10 ms) (Fa) and late (100 ms) (Fb) components of Na+ spike aftercurrents, obtained as shown in E before (control, filled circles) and during exposure to PDB (PDB, open circles). Error bars indicate SEM.
Effects of Ca2+ and Cd2+ on INaP
Together, these data suggest that that the spike would be followed by a significant Na+ spike aftercurrent, provided it does not repolarize beyond -50 mV. Given that the fAHPs in this study averaged -58.6 mV, it first seemed unlikely that an Na+ conductance could generate a significant inward spike aftercurrent that would generate the ADP. However, fAHP measurements were made in slices bathed in ACSF containing 1.6 mm Ca2+ (in bicarbonate-based buffer, yielding [Ca2+]o of 1.2 mm), whereas Na+ currents were measured in ACSF containing 2 mm Ca2+ (in HEPES-based buffer in which [Ca2+]o would remain 2 mm) and 0.3 mm Cd2+. It is well known that divalent cations, by reducing the surface potential of the membrane, shift the Na+ current-voltage relationship to more hyperpolarized potentials (McLaughlin et al., 1971; Hille et al., 1975). Hence, it is conceivable that Na+ spike aftercurrents in the voltage range of the spike ADP (between -60 and -50 mV) are larger in Cd2+-free ACSF containing a more physiological Ca2+ concentration.
To test whether the divalent ion content in the ACSF used for isolating Na+ currents indeed displaces activation of these currents to more positive potentials, we exposed neurons sequentially to 1.2Ca-ACSF, 2Ca-ACSF, 2Ca-ACSF containing only TTX and 2Ca-ACSF containing both 0.3 mm CdCl2 and 50 μm TTX. In each of these four conditions, we obtained current responses to slow voltage ramps, as described above. Subtracting the current responses obtained in the Cd2+- and TTX-containing ACSF from all other responses yielded composite current-voltage relationships of INaP and ICa in 1.2Ca-ACSF and 2Ca-ACSF. A representative INaP-plus-ICa-voltage relationship obtained in 2Ca-ACSF is illustrated in Figure 3A (gray line). Because of the clear separation between the INaP and ICa components, the INaP-plus-ICa-voltage relationship could be well fitted with a composite function comprised of the sum of two modified Boltzmann functions (Eqs. 2, 3; Fig. 3A, black line superimposed on ICa plus INaP). The parameter values of the fitted functions were used to derive separately the INaP-voltage and ICa-voltage relationships (ICa,fit and INaP,fit, respectively) (Fig. 3A).
To validate this technique of separating INaP from ICa, we also derived the ICa-voltage relationship independently by subtracting traces obtained in Cd2+- and TTX-containing ACSF from those obtained after washout of Cd2+ (yielding the Cd2+-sensitive current component corresponding to ICa without contamination by INaP). The current-voltage relationship of ICa is depicted in Figure 3B (gray line), superimposed by ICa,fit (derived from the INaP-plus-ICa-voltage relationship) (Fig. 3A). Thus, because ICa,fit accurately reflects ICa, we were able to subtract ICa,fit from compound ramp current responses obtained in 1.2Ca-ACSF and 2Ca-ACSF to yield INaP in isolation.
The isolated INaP-voltage relationships were converted to normalized conductance-voltage plots and fitted with the Boltzmann function (Eq. 2). Representative results from one experiment are shown in Figure 3C. It is evident that increasing [Ca2+]o from 1.2 to 2.0 mm or adding 0.3 mm Cd2+ to the ACSF shifted the voltage dependence of activation of INaP to more positive potentials. In five experiments, V50 values shifted by 4.0 ± 1.0 mV (from -56.2 ± 0.9 to -52.3 ± 1.4; p = 0.015) after increasing [Ca2+]o, and by 4.9 ± 1.6 mV (from -52.3 ± 1.4 to -47.3 ± 2.0 mV; p = 0.038) after adding Cd2+ (Fig. 3D). At the same time, the slope parameter k did not change (7.4 ± 2.0, 6.8 ± 1.3, and 6.2 ± 1.6 mV, respectively). Together, the V50 in 1.2Ca-ACSF was 8.9 ± 2.0 mV more negative than in Cd2+-containing 2Ca-ACSF. Thus, these data show that the contribution of INaP to spike aftercurrents strongly depends on the Ca2+ and Cd2+ content of the ACSF. In physiological [Ca2+]o (1.2 mm) and the absence of Cd2+, INaP starts activating near resting membrane potential (approximately -70 mV). In this case, a significant INaP will flow at the time of the fAHP (i.e., at voltages around -58.6 mV), contributing to the generation of the spike ADP.
Effects of phenytoin, riluzole, and PDB on INaP and Na+ spike aftercurrents
The use of TTX in characterizing the contribution of INaP to the spike ADP and discharge behavior in current-clamp experiments is confounded by its concurrent potent block of INaT. To over-come this drawback of using TTX, we evaluated three drugs shown previously to block INaP preferentially over INaT in a variety of cell types. These include the anticonvulsant phenytoin (Chao and Alzheimer, 1995; Segal and Douglas, 1997; Niespodziany et al., 2004), the neuroprotective drug riluzole (Urbani et al., 2000; Spadoni et al., 2002), and the protein kinase C (PKC) activator PDB (Cantrell et al., 1996). We examined the effects of these three drugs on INaP and on Na+ spike and afterspike currents. All of these experiments were performed in 2Ca-ACSF containing 0.3 mm Cd2+ to block ICa.
Phenytoin
Application of 100 μm phenytoin caused a marked reduction in INaP elicited by the voltage-ramp commands without an apparent shift in the INaP-voltage relationship (Fig. 4A). This effect appeared rapidly and attained its maximum within seconds of drug application. The peak INaP was inhibited by 58 ± 4% (from -121 ± 16 to -51 ± 9 pA; n = 9; p = 0.0001). A summary of phenytoin block at different ramp potentials is provided in Figure 4B (all values were normalized to the peak INaP in control conditions). Interestingly, the blocking effect of phenytoin progressively increased with depolarization from 42 ± 6% at -50 mV to 72 ± 5% at 0 mV (n = 9) (Fig. 4C). This trend, suggesting voltage-dependent block of INaP by phenytoin, was statistically significant (p = 0.0005). Na+ spike aftercurrents after mock spikes were also profoundly reduced by phenytoin throughout the voltage range examined (Fig. 4Da,Db). Both early (10 ms) (Fig. 4Ea) and late (100 ms) (Fig. 4Eb) (n = 7) components of these spike aftercurrents were similarly blocked by phenytoin. At the same time, peak Na+ spike currents decreased by 27 ± 7% (from 16.1 ± 0.7 to 12.0 ± 1.5 nA; n = 7; p = 0.0075).
Riluzole
Similar experiments were conducted with riluzole. INaP elicited by voltage-ramp commands was rapidly (within a few seconds) and almost completely blocked by 10 μm riluzole (Fig. 5A). The peak INaP was inhibited by 90 ± 2% (from -109 ± 14 to 12 ± 3 pA; n = 9; p = 0.00003). The potent blocking effects of riluzole are summarized for different ramp potentials in Figure 5B (all values were normalized to the peak INaP in control conditions). At membrane potentials of -50, -40, and -30 mV, the block of INaP amounted to 86 ± 3, 92 ± 3, and 93 ± 2%, respectively (n = 9). No voltage dependence of the blocking effect was noted. Both early and late components of Na+ spike aftercurrents were also profoundly reduced by riluzole throughout the voltage range examined (Fig. 5Ca,Cb,Da,Db). At the same time, peak Na+ spike current decreased by only 32 ± 7% (from 12.8 ± 0.9 to 8.6 ± 1.0 nA; n = 9; p = 0.0084). This is in agreement with previous findings showing that riluzole affects INaP at far lower concentrations than INaT (e.g., IC50 of 2 vs 50 μm, respectively) (Urbani and Belluzzi, 2000).
PDB
Finally, we examined the effects of PDB, an activator of PKC (Castagna et al., 1982), on INaP and Na+ spike aftercurrents. In neocortical pyramidal cells, PKC activation was shown to reduce maximal persistent Na+ conductance, but at the same time to shift the INaP-voltage relationship to more negative potentials (Astman et al., 1998; Franceschetti et al. 2000; Curia et al., 2004). The combination of these two effects led to an increase in INaP at near-threshold potentials that caused increased excitability (Astman et al., 1998). Because of this multiplicity of effects, we examined the action of PDB in more detail than the former drugs. Application of 5 μm PDB led to a progressive block of INaP and a concomitant negative shift of the INaP-voltage relationship over a period of 5-10 min (n = 13) (Fig. 6A). This slow time course is consistent with an effect mediated via a second messenger cascade. To quantify the shifts in INaP-voltage relationships, we converted them to conductance-voltage curves and fitted them with the Boltzmann function. During the time in which persistent Na+ conductance-voltage curves could still be measured accurately (up to 5 min after onset of PDB application), PDB caused a significant hyperpolarizing shift of V50 (13.6 ± 1.3 mV at 5 min after PDB application; n = 6) (Fig. 6B). The mean V50 values in control ACSF and at 1 min, 3 min (n = 13), and 5 min (n = 6) after adding PDB, are presented in Figure 6C. The negative shift of the conductance-voltage relationship caused the blocking effect of PDB to be voltage dependent; that is, the block increased with depolarization. Indeed, at negative membrane potential near spike threshold (approximately -60 mV), INaP transiently increased, albeit very slightly (Fig. 6D). However, longer (>10 min) exposures to PDB caused complete block of INaP at all ramp potentials. To confirm that the effects of PDB are mediated by PKC activation, we also tested the effects of the inactive phorbol ester PDD (Castagna et al., 1982). In a series of six experiments, superfusing cells with ACSF containing 5 μm PDD exerted no effects on INaP elicited by slow voltage ramps (data not shown).
As expected, application of PDB also progressively caused a potent reduction of Na+ spike aftercurrents (Fig. 6Ea,Eb). This effect was more pronounced for late (Fig. 6Fb) versus early (Fig. 6Fa) components of these currents (n = 7), similar to the effects of riluzole. In contrast, peak Na+ spike current decreased by only 39 ± 12% (from 14.8 ± 1.6 to 9.4 ± 2.4 nA; n = 7; p = 0.025). This is in good agreement with previous results describing the effects of PDB on INaT (Colbert and Johnston, 1998).
Together, these data show that, at the doses used, phenytoin, riluzole, and PDB potently reduce Na+ spike aftercurrents consequent to their effective block of INaP. Although the blocking effects of Na+ spike currents, we determined, are likely contaminated by series resistance errors (see Materials and Methods), it is clear that all three blockers affect INaT far less than INaP.
Isolation of Ca2+ spike aftercurrents
In several types of neurons, including CA1 pyramidal cells in a chronic model of epilepsy (Su et al., 2002), Ca2+ currents were implicated in the generation of the spike ADP (Wong and Prince, 1981; Jung et al. 2001). We therefore further examined the potential contribution of these currents to spike ADP generation in CA1 pyramidal cells. To this end, we isolated ICa in two complementary ways. First, we recorded the current responses to mock spikes in 2Ca-ACSF before and after adding 0.3 mm CdCl2. The Ca2+ spike and afterspike currents were obtained by subtracting the two sets of recordings from each other. A representative set of such currents is shown in Figure 7Aa. Second, we recorded the current responses to mock spikes in 2Ca-ACSF containing 0.5 μm TTX. A representative set of such currents is shown in Figure 7Ca. Both procedures yielded similar results. Activation of ICa began only at the peak of the mock spike. During the repolarization phase, it increased to maximal amplitude and then declined (Fig. 7Ab). After repolarization of the mock spike to different holding potentials (between -70 and -30 mV), the residual ICa deactivated rapidly and almost completely within 1-2 ms. Only small (<30 pA) persistent Ca2+ spike aftercurrents remained thereafter at the more positive potentials examined (Fig. 7Ab). Figure 7B depicts the mean amplitudes of early (Fig. 7Ba) and late Ca2+ spike aftercurrents (Bb) (n = 11 for both) as a function of holding membrane potential after the mock spike, and compares them with those of Na+ spike aftercurrents shown in Figure 2B. It is evident that Ca2+ spike aftercurrents are about one order of magnitude smaller than the respective Na+ spike aftercurrents. These data suggest that, in normal conditions, spike aftercurrents are mediated predominantly by Na+ channels.
Figure 7.

Properties of Ca2+ spike aftercurrents in CA1 pyramidal cells. Whole-cell patch-clamp recordings in acutely dissociated neurons are shown. A, The representative neuron was superfused with 2Ca-ACSF to which 0.3 mm CdCl2 was subsequently added. Currents evoked by mock spikes repolarizing to different membrane potentials (from -70 to -30 mV; insets) were elicited before and after adding CdCl2. The ICa components isolated by subtraction of currents recorded in the two conditions are shown in Aa and Ab. The vertical dashed lines in Ab demarcate the beginning, peak, and end of the mock spike. B, Comparison between Ca2+ (filled symbols) and Na+ spike aftercurrents (open symbols) at early (10 ms) (Ba) and late (100 ms) (Bb) time points after the mock spikes. Note that Na+ spike aftercurrents are considerably larger than Ca2+ spike aftercurrents. C, The effects of 100 μm Ni2+ on Ca2+ spike aftercurrents. The aftercurrents were recorded in 2Ca-ACSF containing 0.5 μm TTX (Ca), during exposure to Ni2+ (Cb), and after washout of Ni2+ (Cc). D, Summary of the effects of 100 μm Ni2+ on Ca2+ spike aftercurrents. The plots in Da depict the amplitudes of Ca2+ spike aftercurrents (measured 10 ms after the end of the mock spike) at different membrane potentials before (control, filled circles) and during exposure to Ni2+ (100 μm Ni2+, open circles). All amplitudes were normalized to the peak ICa amplitude in control conditions (n = 15). The bar diagram in Db depicts the fractional block of Ca2+ spike aftercurrents by Ni2+ at three membrane potentials. The degree of block was the same in the three potentials, indicating that it is voltage independent. Error bars indicate SEM.
Like many CNS neurons, CA1 pyramidal cells express multiple types of voltage-gated Ca2+ channels, but only a subset of these have slow deactivation kinetics by which they could contribute to sustained spike aftercurrents underlying ADPs. A substantial portion of Ca2+ spike aftercurrents is most probably mediated by slowly deactivating T-type voltage-gated Ca2+ channels, most probably corresponding to Ni2+-sensitive CaV3.2 channels (Sochivko et al., 2003; Su et al., 2002; Perez-Reyes, 2003). Therefore, we examined the effects of 100 μm Ni2+ on Ca2+ spike currents and aftercurrents isolated by superfusing the neurons with TTX-containing 2Ca-ACSF. As shown in Figure 7C, adding Ni2+ to this ACSF reversibly inhibited both of these currents. The peak Ca2+ spike current was reduced by 37.5 ± 2.7% (from 402.4 ± 81.4 to 246.9 ± 48.4 pA; n = 5; p = 0.012) (Fig. 7C). This effect is most probably caused by block of CaV3.2 channels, as well as by block of CaV2.3 (R-type) channels, which are also highly sensitive to Ni2+ (Ellinor et al., 1993; Sochivko et al., 2003). Likewise, Ca2+ spike aftercurrents measured 10 ms after the end of the mock spike at membrane potentials of -50, -40, and -30 mV were blocked by 43 ± 15% (p = 0.011), 44 ± 9% (p = 0.016), and 54 ± 8% (p = 0.002), respectively (n = 5) (Fig. 7Da,Db). There was no obvious voltage dependence in the blocking action of Ni2+.
Effects of phenytoin, riluzole, and PDB on the spike ADPs
Having established that phenytoin, riluzole, and PDB potently block INaP and Na+ spike aftercurrents in CA1 pyramidal cells, we used these drugs to further examine the role of INaP in spike ADP electrogenesis. We first tested how they affect the spike ADPs when applied in the ACSF. Representative results are shown in Figure 8A-C. Applications of phenytoin (100 μm) (Fig. 8Aa,Ab), riluzole (10 μm) (Ba,Bb), or PDB (5 μm) (Da,Db) for 20-30 min consistently suppressed the spike ADP (Fig. 8Ac,Bc,Dc, superimposed traces). Thus, phenytoin reduced the overall size of the spike ADP by 33.4% (from 185.1 ± 20.9 to 130.6 ± 26.5 mV · ms; n = 8; p = 0.006), riluzole by 34.9 ± 7.3% (from 163.8 ± 15.5 to 104.7 ± 13.7 mV · ms; n = 6; p = 0.016), and PDB by 41.7 ± 8.3% (from 208.7 ± 18.1 to 123.0 ± 21.2 mV · ms; n = 8; p = 0.003). The effects of the three drugs on the spike ADPs are summarized in Figure 8F (black bars). Suppression of the spike ADP by the three drugs was achieved without noticeable effects on the rate of rise and amplitude of the spike (Fig. 8Ad,Bd,Dd, superimposed traces), indicating that they are not secondary to inhibition of INaT. In another four experiments, we also tested the effects of the inactive phorbol ester PDD. Bath application of 5 μm PDD had no effect on the spike ADP (data not shown), consistent with the notion that PDB suppresses the spike ADP via a PKC-mediated inhibition of INaP (Alroy et al., 1999).
Figure 8.

Active spike ADPs are suppressed by INaP blockers. Sharp microelectrode recordings in slices are shown. A-D, Effects of bath-applied phenytoin, riluzole, and PDB on spike ADPs (evoked by brief depolarizing stimuli) in four representative experiments. In each panel, a and b illustrate the spike before (control) and 20-30 min after drug application (phenytoin, riluzole, PDB), respectively. c shows superposition of the respective ADPs enlarged in size, and d shows superposition of the respective spikes expanded in time. A, Effects of phenytoin (100 μm). B, Effects of riluzole (10 μm) applied to a neuron bathed in normal ACSF. C, Effects of riluzole (10 μm) applied to a neuron bathed in ACSF containing IBTX (100 nm) and apamin(100 nm). D, Effects of PDB (5 μm). The three drugs markedly reduced the active spike ADPs without affecting the rise times or amplitudes of the corresponding spikes, indicating the essential involvement of INaP in ADP generation. E, Effects of 100 μm Ni2+ on spike ADPs and EPSPs. Spikes and EPSPs were evoked intermittently in control ACSF (Ea1, Ea2) and 30 min after adding 100 Ni2+ to the bathing ACSF (Eb1, Eb2). The traces in Ea1 and Eb1 are enlarged and overlaid in Ec1 to facilitate comparison of the ADPs. The traces in Ea2 and Eb2 are enlarged and overlaid in Ec2 to facilitate comparison of the EPSPs. It is evident that Ni2+ inhibits the EPSPs while sparing the spike ADPs. F, Bar diagram summarizing the effects of the INaP blockers (black bars) and Ni2+ (gray bars) on the spike ADPs. *Statistically significant differences between compared groups. Error bars indicate SEM.
Riluzole was shown to stimulate the IBTX-sensitive big-conductance (BK) Ca2+-activated K+ channels in neuroendocrine cells (EC50 = 5 μm) (Wu and Li, 1999). Riluzole was also shown to enhance the activity of heterologously expressed small-conductance (SK) Ca2+-activated K+ channels, especially the apamin-sensitive SK2 and SK3 channels (EC50, 43 μm). However, in the dose used in our experiments (10 μm), it did not affect the apamin-sensitive AHP in hippocampal neurons (Cao et al., 2002). Although blocking BK and SK Ca2+-activated channels (with iberiotoxin and apamin, respectively) does not enhance the spike ADP (Yue and Yaari, 2004), activating these channels may reduce it. Thus, it may be argued that riluzole suppresses the spike ADP by activating Ca2+-activated K+ channels. To explore this possibility, we pretreated slices with 100 nm IBTX (n = 2), 100 nm apamin (n = 3), or both (n = 3) for at least 1 h before adding riluzole (10 μm) to the toxin-containing ACSF. A representative experiment in a neuron pretreated with both toxins is shown in Figure 8C. In this, as in all other seven experiments, riluzole effectively suppressed the active spike ADP (Fig. 8Ca;Cb;Cc, superimposed traces) without noticeably affecting the rate of rise and amplitude of the spike (Fig. 8Cd, superimposed traces). These experiments exclude the possibility that riluzole suppresses the spike ADP by enhancing the activity of BK and SK Ca2+-activated K+ channels.
Effects of Ni2+ on the spike ADPs
Although it seemed unlikely that the small Ca2+ spike aftercurrents could contribute significantly to the generation of the spike ADP, we further tested this possibility. We reasoned that, in such a case, a >40% block of Ca2+ spike aftercurrent by 100 μm Ni2+ (similar in magnitude to the block of Na+ spike aftercurrents by phenytoin) (Fig. 4) should cause a noticeable suppression of the spike ADP. In five experiments, bathing the slices with ACSF containing 100 μm Ni2+ for up to 45 min did not modify the spike ADP (Fig. 8Ea1-Ec1;F, gray bars). To assure ourselves that Ni2+ indeed blocks Ca2+ channels in the slices, we monitored in parallel, in three experiments, the orthodromically evoked EPSPs. In all three cases, Ni2+ substantially reduced EPSP amplitudes (Fig. 8Ea2-Ec2), an effect presumably attributable to blockage of R-type Ca2+ channels in the presynaptic terminals (Wu and Saggau, 1995) and T-type Ca2+ channels in the respective postsynaptic dendrites (Gillessen and Alzheimer, 1997). Together, these results indicate that Ca2+ spike aftercurrents are normally too small to contribute significantly to the generation of the active spike ADP in adult CA1 pyramidal neurons.
Interestingly, we recently showed that 100 μm Ni2+ effectively suppresses the active spike ADP and associated bursting in CA1 pyramidal cells from rats that have experienced convulsant-induced status epilepticus (Su et al., 2002). However, in those neurons, the densities of Ni2+-sensitive T-type ICa and Ca2+ spike aftercurrent were found to be markedly up-regulated compared with their densities in neurons from normal rats.
Focal dendritic and somatic applications of INaP blockers
Our data thus far showed that four potent blockers of INaP (TTX, phenytoin, riluzole, and PDB) suppress the active spike ADP at concentrations that markedly reduce INaP and abolish Na+ spike aftercurrents in the same neurons. These effects were achieved without conspicuous changes in spike waveform and, therefore, cannot be secondary to depression of INaT. Although phenytoin, riluzole, and PDB were shown also to reduce voltage-gated ICa currents in some neurons (Yaari et al., 1987; Doerner et al., 1990; Huang et al., 1997; Stefani et al., 1997), we found that Ca2+ spike aftercurrents are normally too small to play a major role in spike ADP electrogenesis. Therefore, suppression of the spike ADP by these drugs unlikely results from block of ICa currents. Enhancement of IM will suppress the spike ADP (Yue and Yaari, 2004), but there is no evidence that TTX, phenytoin, riluzole, or PDB enhance IM.If anything, PDB suppresses IM (Marrion, 1994). Thus, the most parsimonious explanation for our results is that INaP drives the spike ADP in CA1 pyramidal cells.
As discussed above, the spike ADP may be generated proximally (i.e., at or near the soma) or may arise distally at the apical dendrites after their invasion by the backpropagating spike and spread secondarily to the soma. To distinguish between these two possibilities, we compared how INaP blockers, applied focally at proximal or distal locations, affect the spike ADP. The puffing pipette contained either 100 μm riluzole or 50 μm PDB dissolved in standard ACSF. Representative results obtained with riluzole and PDB are illustrated in Figure 9A,B. Application of riluzole to the apical dendrites (three puffs) had no significant effect on the spike ADP (213.0 ± 30.4 and 193.7 ± 24.9 mV · ms before and after riluzole, respectively; n = 4) (Fig. 9Aa;Ab;Ad, superimposed traces), but suppressed it by 35.6 ± 6.3% when puffed only once at the somatic region (137.3 ± 25.0 mV · ms; p = 0.038) (Fig. 9Ab;Ac;Ad, superimposed traces). Likewise, PDB had no effect on the spike ADP when applied to the apical dendrites (three puffs; 171.4 ± 8.7 and 163.7 ± 5.0 mV · ms before and after PDB, respectively; n = 6; p = 0.009) (Fig. 9Ba1;Bb1;Bd1, superimposed traces), but suppressed it by 49.4 ± 14.6% when applied to the somatic region (82.9 ± 17.7 mV · ms) (Fig. 9Bb1;Bc1;Bd1, superimposed traces). The results of these experiments are summarized in Figure 9D.
Figure 9.

Blocking proximal, but not distal, persistent Na+ channels suppresses the active somatic spike ADP. Sharp microelectrode recordings in slices are shown. This experimental arrangement is the same as in Figure 1C. A, Effects of locally applied riluzole on the spike ADP. The puffing pipette contained 100 μm riluzole in ACSF. Multiple ejections (3 puffs) of riluzole onto the distal dendrites had no detectable effects the spike ADP (Aa, Ab) but substantially reduced it within 3 min when applied (1 puff) to the somatic region without affecting the spike itself (Ab, Ac). The traces in Aa, Ab, and Ac are enlarged in size and overlaid in Ad to facilitate comparison of the spike ADPs. The traces in Aa and Ac are expanded in time and overlaid in Ae to facilitate comparison of the spike rise times. B, Effects of locally applied PDB on the spike ADP and on the EPSP. The puffing pipette contained 100 μm PDB in ACSF. Spikes and EPSPs were evoked intermittently in control ACSF (Ba1, Ba2) and after puffing PDB onto the dendrites (3 puffs) (Bb1, Bb2) and onto the soma (1 puff) (Bc1, Bc2). The traces in Ba1, Bb1, and Bc1 are enlarged in size and overlaid in Bd1 to facilitate comparison of the ADPs. The traces in Ba1 and Bc1 are expanded in time and overlaid in Be to facilitate comparison of the spike rise times. The traces in Ba2, Bb2, and Bc2 are enlarged in size and overlaid in Bd2 to facilitate comparison of the EPSPs. All traces were obtained 10 min after puffing PDB to the respective region. It is evident that distally applied PDB facilitated the EPSPs without affecting the spike ADPs. When applied to the soma, PDB suppressed both the ADP and the EPSP. C, Time course plots of the effects of focally applied PDB on spike ADP (squares), spike rise time (triangles), and EPSP amplitude (circles) in the experiment illustrated in B. D, Bar diagram summarizing the effects of riluzole and PDB applied first to the dendrite and then to the soma on the spike ADP. *Statistically significant differences between compared groups. Error bars indicate SEM.
It may be argued that distal applications of riluzole and PDB were ineffective because the drugs did not reach their target dendrites. This is unlikely because we established previously that distal applications of TTX were effective in reversing 4-AP-induced ADP enhancement and associated bursting (Fig. 1Eb,Ec). Nevertheless, we searched for independent confirmation that these particular drugs indeed reach the target dendrites. It was shown before that PDB facilitates EPSP amplitudes in CA1 pyramidal cells (Malenka et al., 1986). Therefore, in three of the above experiments, we also monitored the size of EPSPs evoked by stimulating afferent fibers in the upper one-third of the stratum radiatum. Distally applied PDB, although having no effects on the spike ADP, caused a marked increase (>300%) in EPSP amplitude in all three experiments (Fig. 9Ba2;Bb2;Bd2, superimposed traces). When subsequently applied to the somatic region, however, PDB reduced and curtailed both the ADPs and the EPSPs (Fig. 9Bb2;Bc2;Bd2, superimposed traces). The latter effect of PDB on the EPSPs may also be attributable to suppression of INaP, given that EPSPs in pyramidal cells are amplified by somatic INaP (Stuart and Sakmann, 1995; Andreasen and Lambert, 1999). The time courses of the effects of focally applied PDB on spike ADP, spike rise time, and EPSP amplitude are depicted in Figure 9C. These results indicate that our focal application technique delivers sufficient amounts of PDB to apical dendrites of CA1 pyramidal neurons. Together, our data support the contention that proximal persistent Na+ channels generate the somatic spike ADPs.
Spike ADPs after truncation of apical dendrites
In a complementary approach to assess the role of apical dendrites in spike ADP generation and associated bursting, we investigated, in 36 CA1 pyramidal cells, the consequences of truncating the apical dendrites close (∼50-70 μm) to the soma (see Materials and Methods) (Fig. 10A). We confirmed the success of truncation in all slices by comparing orthodromically and antidromically evoked field potentials elicited before and after truncation. In intact slices, stimulating orthodromically or antidromically (via S1 or S2, respectively) (Fig. 10A) evoked archetypal field potentials comprised of single population spikes (Fig. 10B, control). After truncation and recuperation of the slice for 1 h, the orthodromic response was entirely abolished (Fig. 10B, Truncated, S1). In contrast, the antidromic field potential was reduced by only ∼30% (Fig. 10B, Truncated, S2), indicating that the majority of neurons remains viable and excitable despite the surgical damage. To estimate the length of the residual dendrite stump, we injected biocytin into 10 neurons in six cut slices during microelectrode impalements. Staining these neurons confirmed that no more than a 50-70 μm segment of the apical dendrite remained attached to the soma (Fig. 10Ca). When the truncated neurons were compared with their intact counterparts, with respect to passive (resting potential and apparent input resistance) and active (spike threshold, rise time, amplitude, and half width; fAHP and ADP) membrane properties, they were surprisingly similar (Table 1). They differed significantly only in apparent input resistance, which was 19% higher in truncated neurons. Likewise, the discharge behavior of truncated pyramidal neurons also appeared entirely normal (Fig. 10Cb) (see below).
Like in intact slices, spike ADPs in truncated neurons varied markedly in size across the neurons (from 57.9 to 360.6 mV · ms, averaging 178.8 ± 12.3 mV · ms; n = 36) and were conspicuously larger in bursting neurons (Fig. 11Bb). They were also highly sensitive to block by TTX, as illustrated in Figure 10D. Bath application of 20 nm TTX substantially reduced the spike ADP (Fig. 10Da;Db;Dc, superimposed traces) before notably affecting the properties of the fast spike (Fig. 10Da;Db; Dd, superimposed traces). In four such experiments, TTX reduced the spike ADP by 51.5 ± 3.9% (from 145.7 ± 14.1 to 71.4 ± 10.6 mV · ms; p = 0.007). Likewise, the INaP blockers used in the above experiments also strongly and consistently suppressed the active spike ADPs in truncated neurons. The decreases in ADP size brought about by phenytoin (100 μm), riluzole (10 μm), and PDB (5 μm) were 24.0 ± 10.1% (n = 3), 50.4 ± 8.4% (n = 3), and 36.3 ± 2.1% (n = 4), respectively. Representative results showing the effects of PDB are shown in Figure 10E. Again, the strong suppression of the spike ADP (Fig. 10Ea;Eb;Ec, superimposed traces) commenced without noticeable changes in the preceding spike (Fig. 10Ea;Eb;Ed, superimposed traces).
Cumulatively, our findings plainly show that proximal persistent Na+ channels furnish the depolarizing drive for active spike ADPs in normal adult CA1 pyramidal cells. In addition, our data also suggest that Ca2+ channels do not play a major role in the genesis of these potentials.
Role of proximal INaP in bursting
It is well established that CA1 pyramidal cells manifest variant firing patterns that can be loosely classified into two main groups, namely, regular firing cells (or nonbursters) and bursters (Schwartzkroin, 1975; Jensen et al., 1994, 1996; Su et al., 2001). When injected with prolonged (e.g., 200 ms) depolarizing current pulses, nonbursters generate a single spike as their minimal response and a train of temporally dispersed spikes in response to all suprathreshold stimuli, whereas bursters fire an early tight cluster of two to five spikes, or a distinct burst, in response to strong depolarizations (high-threshold bursters) or, in rare cases, even to threshold-straddling depolarizations (low-threshold bursters). In the present new series of 97 intact adult CA1 pyramidal cells, 56 neurons (57.7%) were nonbursters, 35 neurons (36.1%) were high-threshold bursters, and 6 neurons (6.2%) were low-threshold bursters (Fig. 11Aa). The overall proportion of bursters (42.3%) is much larger than in previous studies [17.4% (Jensen et al., 1996); 17.0% (Su et al., 2001)], most probably because of the use in the present study of a lower, more physiological Ca2+ concentration in the ACSF [1.6 mm instead of 2 mm (Su et al., 2001)]. Expectedly, we found that the spike ADPs were larger in bursters than in nonbursters, as illustrated in Figure 11Ba, where the spike ADPs of nonburster, high-threshold burster, and low-threshold burster neurons are superimposed. Because ADP size depends critically on resting potential (Schwartzkroin, 1975; Jensen et al., 1996), we compared nonbursters to bursters with respect to this variable, but found no significant difference between the two groups (-67.6 ± 0.6 and -68.3 ± 0.6 mV for nonbursters and bursters, respectively) (Fig. 11C).
The proportions of nonbursters and bursters in truncated neurons were identical to those found in intact neurons. Thus, of 36 truncated neurons, 20 (55.6%) were nonbursters, 13 (36.1%) were high-threshold bursters, and 3 (8.3%) were low-threshold bursters (Fig. 11Ab). Again, the spike ADPs in high- (192.0 ± 12.6 mV · ms; n = 16) and low-threshold bursters (318.5 ± 19.7 mV · ms; n = 4) were significantly larger than in nonbursters (130.6 ± 11.0 mV · ms; n = 16; p = 0.00002) (Fig. 11Bb,Cb).
Given that bursts are the product of large spike ADPs (Jensen et al., 1996), we hypothesized that bursting in adult CA1 pyramidal cells also depend on activation of proximal persistent Na+ channels. We tested this hypothesis first by monitoring the effects of bath-applied phenytoin, riluzole, and PDB on the bursting behavior of intact neurons. Representative results are shown in Figure 12A-C. Bursts were evoked by 2-3× threshold-long depolarizing current pulses. They were consistently dispersed by phenytoin (100 μm; n = 4) (Fig. 12A), riluzole (10 μm; n = 7) (Fig. 12B), or PDB (5 μm; n = 7) (Fig. 12C), concurrent with the suppression of their underlying spike ADPs. In a second series of experiments, we applied the INaP blockers focally. Puffing (three puffs) riluzole (n = 3) (Fig. 12Da,Db) or PDB (n = 3) (Fig. 12Ea,Eb) onto the distal dendrites had no detectable effects on bursting, whereas somatic applications (one puff) (Fig. 12Db,D-c,Eb,Ec) converted this firing mode to regular firing in all cases. Finally, we examined how bursting in truncated CA1 pyramidal cells is affected by the INaP blockers. Bath application of phenytoin (100 μm; n = 3), riluzole (10 μm; n = 3), and PDB (5 μm; n = 4) invariably converted bursting to regular firing (Fig. 12F-H). Cumulatively, our findings suggest that native bursting in adult CA1 pyramidal cells is underlain by recruitment of persistent Na+ channels located at or near the soma.
Figure 12.

Blocking persistent Na+ channels suppresses bursting in CA1 pyramidal cells. Sharp microelectrode recordings in slices are shown. All neurons included in this series of experiments were high-threshold bursters, generating an early high-frequency burst in response to large (2-3× threshold) and prolonged (180 ms) positive current pulses. A-C, Effects of bath applied drugs on intact neurons. The three representative experiments illustrate the effects of phenytoin (100 μm) (A), riluzole (10 μm) (B), and PDB (5 μm) (C). In each panel, a and b illustrate the firing of the neuron before (control) and 20-30 min after drug application, respectively. In all three experiments, the applied drugs converted the initial spike burst to a regular firing pattern. D, E, Effects of focally applied drugs on intact neurons. The experimental arrangement and the drug concentrations in the puffing pipettes are the same as in Figure 9. D, Effects of focally applied riluzole. Multiple ejections (3 puffs) of riluzole onto the distal dendrites had no detectable effects on the firing pattern of the neuron (Da, Db). A single puff of riluzole at the somatic region converted the initial spike burst evoked by the strong stimulus to a regular firing pattern (Db, Dc). E, Effects of focally applied PDB. Like riluzole, PDB applied to the distal dendrites had no effect on high-threshold bursting (Ea, Eb) but suppressed it when applied to the somatic region (Eb, Ec). F-H, Effects of bath-applied drugs on truncated neurons. The three representative experiments illustrate the effects of phenytoin (100 μm) (F), riluzole (10 μm) (G), and PDB (5 μm) (H). In each panel, a and b illustrate the firing of the neuron before (control) and 20-30 min after drug application, respectively. As in intact neurons, the applied drugs converted the initial spike burst to a regular firing pattern in all cases.
Discussion
In this study, we investigated the ionic mechanisms underlying the somatic spike ADP in a widely studied principal cortical neuron, the CA1 pyramidal cell. We show that a large component of this ADP is generated via activation of persistent Na+ channels localized in the proximal portions of the neuron. In fact, Na+ channels and other types of membrane channels in apical dendrites are not ordinarily involved in generation of the somatic spike ADP, as indicated by its resistance to almost complete truncation of the apical dendritic tree. Although Ca2+ channels are also coactivated during and after the spike, they do not normally contribute to spike ADP electrogenesis.
The somatic spike ADP is generated by persistent Na+ channels
Here, we present several new findings regarding the mechanisms generating the spike ADP in adult CA1 pyramidal cells. First, we show that the inward spike aftercurrents flowing at voltages within the range of the spike ADP are predominantly Na+ currents. When the divalent cation content of the ACSF approximates physiological levels, substantial persistent Na+ currents (∼50 pA at room temperature) are observed at membrane voltages attained at the end of the spike downstroke. Given the high Q10 value of INaP (2.7) (Kay et al., 1998), the magnitude of these currents are expected to be approximately threefold larger at 33-34°C (the temperature at which recordings in slices were performed). A 150 pA current would depolarize a 33 MΩ neuron (Table 1) by ∼5 mV, which is within the range of the depolarization that occurs after the fAHP (Fig. 11B). Second, we demonstrate that spike ADPs are readily suppressed by TTX, as well as by a battery of structurally diverse drugs that potently block INaP and Na+ spike aftercurrents in CA1 pyramidal neurons. In contrast, substantial reduction of Ca2+ spike aftercurrents with Ni2+ does not significantly affect the spike ADP. Together, these data strongly suggest that persistent Na+ channels, rather than Ca2+ channels, provide the major depolarizing drive for the spike ADP. They also render unlikely a major contribution of Ca2+-activated nonspecific cationic channels to ADP electrogenesis, consistent with our previous finding that chelating cytoplasmic Ca2+ with BAPTA does not reduce the spike ADPs in CA1 pyramidal cells (Su et al., 2001).
The amplitude and duration of the spike ADP are governed not only by INaP but also by the density of opposing repolarizing currents. Notably, it has been shown that blocking IM (but not other slow K+ currents) in CA1 pyramidal neurons strongly augments active spike ADPs and converts most regular firing neurons to a bursting mode (Yue and Yaari, 2004; Gu et al., 2005). This suggests that, under ordinary conditions, the depolarizing action of INaP is restrained by coactivation of IM, normally preventing the spike ADP from attaining threshold potential. Blocking IM unleashes the depolarizing action of INaP, allowing for a regenerative buildup of the spike ADP that leads to bursting (Yue and Yaari, 2004). Indeed, bursting induced by blocking IM is readily suppressed by phenytoin, riluzole, and PDB, but not by blocking ICa currents (C. Yue and Y. Yaari, unpublished observations).
The somatic spike ADP is generated in the proximal portion of the neuron
It is firmly established that spike initiation in CA1 pyramidal cells normally occurs at the axon hillock or proximal nodes of Ranvier (Turner et al., 1991; Spruston et al., 1995), although patterned synaptic excitation may also initiate Na+ spikes in the dendrites (Golding and Spruston, 1998). In contrast, the subcellular compartments at which the various spike afterpotentials (particularly the spike ADP) arise, are ill defined. Once a spike is initiated in the axonal compartment, it backpropagates into the soma and apical dendrites and causes widespread depolarization of the neuron. Both soma and apical dendrites of CA1 pyramidal cells reportedly contain persistent Na+ channels (French et al., 1990; Lipowsky et al., 1996; Andreasen and Lambert, 1999), although it appears that they aggregate in the proximal portion of the neuron (French et al., 1990). Therefore, the somatic spike ADP could arise in the proximal part of the neuron or, alternatively, originate at a remote compartment of the neuron that expresses persistent Na+ channels and spread secondarily to the soma. Two lines of evidence suggest that the somatic spike ADP arises locally. First, applying INaP blockers to the distal apical dendrites had no effect on the somatic spike ADP, whereas puffing these drugs onto the proximal part of the neuron readily abolished this afterpotential. Second, somatic ADPs in neurons lacking apical dendrites were indistinguishable from those in intact neurons. Thus, the density of persistent Na+ channels in distal dendrites recruited by the backpropagating spike is insufficient to mediate a local depolarizing response that would enhance the somatic spike ADP.
Although we could not resolve differences between axon initial segment, soma, and proximal dendrites with respect to the site of ADP generation, indirect evidence suggests that such differences may exist. Of all of the α-subunit isoforms of voltage-activated Na+ channels expressed in the brain (NaV1.1, NaV1.2, NaV1.3, NaV1.5, and NaV1.6) (Goldin, 1999; Hartmann et al. 1999), NaV1.6 is the main subunit underlying INaP (Smith et al., 1998; Maurice et al., 2001; Burbidge et al., 2002). The axon initial segment and nodal membranes in many neurons express a high density of NaV1.6 channels (Jenkins and Bennett, 2001; Boiko et al., 2003). Given the small diameter (and hence, high input resistance) of these structures, even a relatively small INaP will strongly depolarize them. If that is the case in CA1 pyramidal cells, then it is likely that the active spike ADP commences at the site where the spike threshold is lowest (i.e., the axon initial segment). Interestingly, recent data suggest that KCNQ2 channels that coassemble with KCNQ3 subunits to yield the neuronal M-type K+ channel responsible for IM (Wang et al., 1998; Shah et al., 2002) also aggregate in the axon initial segment and nodes (Devaux et al., 2004). This raises the intriguing possibility that the two opposing low-threshold currents that govern active spike ADPs are closely colocalized. The presence of these conductances at the axon initial segment will likely render it a strategic site, not only for spike initiation but also for generating the active spike ADPs and associated bursting.
Persistent Na+ channels, spike ADP, and bursting
A substantial fraction of adult CA1 pyramidal cells (42.3% in 1.6 mm Ca2+) fire in burst mode when strongly depolarized, and a few of them burst even in response to threshold-straddling stimuli. We found that a high propensity for bursting is associated with large-spike ADPs. Here, we show that bursting is readily suppressed by the same battery of drugs that block INaP and suppress the spike ADPs, suggesting that the depolarizing envelope underlying native bursts in adult CA1 pyramidal cells is also generated by INaP. Whether the large ADP and associated bursting are attributable to a large INaP density in the proximal portion of the neuron, to a smaller density of the opposing IM, or to both, is not yet known.
In the principal trans-hippocampal pathway, CA1 pyramidal cells receive their main excitatory input from CA3 pyramidal cells and project predominantly to subicular neurons. Intrinsic bursting, reportedly, is more abundant in the latter types of neurons than in CA1 pyramidal cells (Wong et al., 1979; Mattia et al., 1997; Staff et al., 2000). In both adult CA3 and subicular pyramidal cells, the spike ADPs and associated bursting have been attributed to a Ca2+ spike aftercurrent (Wong and Prince, 1981; Jung et al., 2001), although a contribution of an Na+ spike aftercurrent to these potentials has not been excluded (Mattia et al., 1997). Testing the effects of phenytoin, riluzole, and PDB on bursting in these neurons may help to resolve this issue. Here, we show that the ADP and associated bursting in adult CA1 pyramidal cells are mediated predominantly by INaP, as hypothesized previously (Azouz et al., 1996; Su et al., 2001). A similar role for INaP was suggested also in studies of other types of intrinsically bursting cortical and subcortical neurons (Mantegazza et al., 1998; Brumberg et al., 2000; Wu et al., 2005). It is interesting to note, however, that Ni2+-sensitive (i.e., T- and/or R-type) Ca2+ channels contribute significantly to the generation of the enlarged spike ADPs found in many immature CA1 pyramidal cells (Chen et al., 2005; Metz et al., 2005), as well as in adult neurons that have experienced repetitive seizures (Sanabria et al., 2001; Su et al., 2002). Thus, normal and pathological developmental programs regulate the size of the spike ADP and the associated propensity for bursting by modifying the expression and/or function of Na+ and Ca2+ channels.
The firing mode of CA1 pyramidal cells in vivo alternates between regular firing and bursting (Ranck, 1973). Both INaP (Mittmann and Alzheimer, 1998; Alroy et al., 1999; Carr et al., 2002) and IM (for review, see Brown and Yu, 2000) are strongly modulated by many neurotransmitters and second messenger cascades. By altering the INaP-IM density ratio, these neurotransmitters may modify the size of the spike ADP and thereby control the ongoing firing mode of CA1 pyramidal cells.
Footnotes
This work was supported by the German-Israel collaborative research program of the Bundesministerium für Bildung und Forschung and the Ministry of Science, the Deutsche Forschungsgemeinschaft (DFG) SFB TR3 (H.B., Y.Y.), the Henri J. and Erna D. Leir Chair for Research in Neurodegenerative Diseases, and the Mercator Program of the DFG (Y.Y.).
Correspondence should be addressed to Dr. Yoel Yaari, Department of Physiology, Hebrew University School of Medicine, P.O. Box 12272, Jerusalem 91121, Israel. E-mail: yaari@md.huji.ac.il.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259704-17$15.00/0
C.Y. and S.R. contributed equally to this work.
References
- Alroy G, Su H, Yaari Y (1999) Protein kinase C mediates muscarinic block of intrinsic bursting in rat hippocampal neurons. J Physiol (Lond) 518: 71-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen M, Lambert JD (1999) Somatic amplification of distally generated subthreshold EPSPs in rat hippocampal pyramidal neurones. J Physiol (Lond) 519: 85-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astman N, Gutnick MJ, Fleidervish IA (1998) Activation of protein kinase C increases neuronal excitability by regulating persistent Na+ current in mouse neocortical slices. J Neurophysiol 80: 1547-1551. [DOI] [PubMed] [Google Scholar]
- Azouz R, Jensen MS, Yaari Y (1996) Ionic basis of spike afterdepolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol (Lond) 492: 211-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiko T, Van Wart A, Caldwell JH, Levinson SR, Trimmer JS, Matthews G (2003) Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci 23: 2306-2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BS, Yu SP (2000) Modulation and genetic identification of the M channel. Prog Biophys Mol Biol 73: 135-166. [DOI] [PubMed] [Google Scholar]
- Brumberg JC, Nowak LG, McCormick DA (2000) Ionic mechanisms underlying repetitive high-frequency burst firing in supragranular cortical neurons. J Neurosci 20: 4829-4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbidge SA, Dale TJ, Powell AJ, Whitaker WR, Xie XM, Romanos MA, Clare JJ (2002) Molecular cloning, distribution and functional analysis of the NaV1.6 voltage-gated sodium channel from human brain. Brain Res Mol Brain Res 103: 80-90. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Ma JY, Scheuer T, Catterall WA (1996) Muscarinic modulation of sodium current by activation of protein kinase C in rat hippocampal neurons. Neuron 16: 1019-1026. [DOI] [PubMed] [Google Scholar]
- Cao YJ, Dreixler JC, Couey JJ, Houamed KM (2002) Modulation of recombinant and native neuronal SK channels by the neuroprotective drug riluzole. Eur J Pharmacol 449: 47-54. [DOI] [PubMed] [Google Scholar]
- Carr DB, Cooper DC, Ulrich SL, Spruston N, Surmeier DJ (2002) Serotonin receptor activation inhibits sodium current and dendritic excitability in prefrontal cortex via a protein kinase C-dependent mechanism. J Neurosci 22: 6846-6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y (1982) Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem 257: 7847-7851. [PubMed] [Google Scholar]
- Chao TI, Alzheimer C (1995) Effects of phenytoin on the persistent Na+ current of mammalian CNS neurones. NeuroReport 6: 1778-1780. [DOI] [PubMed] [Google Scholar]
- Chen S, Yue C, Yaari Y (2005) A transitional period of Ca2+-dependent bursting triggered by spike backpropagation into apical dendrites in developing rat CA1 neurons. J Physiol (Lond) 567: 79-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert CM, Johnston D (1996) Axonal action-potential initiation and Na+ channel densities in the soma and axon initial segment of subicular pyramidal neurons. J Neurosci 16: 6676-6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert CM, Johnston D (1998) Protein kinase C activation decreases activity-dependent attenuation of dendritic Na+ current in hippocampal CA1 pyramidal neurons. J Neurophysiol 79: 491-495. [DOI] [PubMed] [Google Scholar]
- Curia G, Aracri P, Sancini G, Mantegazza M, Avanzini G, Franceschetti S (2004) Protein-kinase C-dependent phosphorylation inhibits the effect of the antiepileptic drug topiramate on the persistent fraction of sodium currents. Neuroscience 127: 63-68. [DOI] [PubMed] [Google Scholar]
- Devaux JJ, Kleopa KA, Cooper EC, Scherer SS (2004) KCNQ2 is a nodal K+ channel. J Neurosci 24: 1236-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerner D, Abdel-Latif M, Rogers TB, Alger BE (1990) Protein kinase C-dependent and -independent effects of phorbol esters on hippocampal calcium channel current. J Neurosci 10: 1699-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinor PT, Zhang JF, Randall AD, Zhou M, Schwarz TL, Tsien RW, Horne WA (1993) Functional expression of a rapidly inactivating neuronal calcium channel. Nature 363: 455-458. [DOI] [PubMed] [Google Scholar]
- Franceschetti S, Taverna S, Sancini G, Panzica F, Lombardi R, Avanzini G (2000) Protein kinase C-dependent modulation of Na+ currents increases the excitability of rat neocortical pyramidal neurones. J Physiol (Lond) 528: 291-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CR, Sah P, Buckett KJ, Gage PW (1990) A voltage-dependent persistent sodium current in mammalian hippocampal neurons. J Gen Physiol 95: 1139-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y (1975) Two types of depolarizing after-potentials in hippocampal pyramidal cells of rabbits. Brain Res 94: 435-446. [DOI] [PubMed] [Google Scholar]
- Gillessen T, Alzheimer C (1997) Amplification of EPSPs by low Ni2+- and amiloride-sensitive Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Neurophysiol 77: 1639-1643. [DOI] [PubMed] [Google Scholar]
- Goldin AL (1999) Diversity of mammalian voltage-gated sodium channels. Ann NY Acad Sci 868: 38-50. [DOI] [PubMed] [Google Scholar]
- Golding NL, Spruston N (1998) Dendritic sodium spikes are variable triggers of axonal action potentials in hippocampal CA1 pyramidal neurons. Neuron 21: 1189-1200. [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF (2005) Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium afterhyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol (Lond) 566: 689-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R (1997) Calcium-activated cation nonselective current contributes to the fast afterdepolarization in rat prefrontal cortex neurons. J Neurophysiol 78: 1983-1989. [DOI] [PubMed] [Google Scholar]
- Hartmann HA, Colom LV, Sutherland ML, Noebels JL (1999) Selective localization of cardiac SCN5A sodium channels in limbic regions of rat brain. Nat Neurosci 2: 593-595. [DOI] [PubMed] [Google Scholar]
- Hasuo H, Phelan KD, Twery MJ, Gallagher JP (1990) A calcium-dependent slow afterdepolarization recorded in rat dorsolateral septal nucleus neurons in vitro. J Neurophysiol 64: 1838-1846. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Lux HD, Gutnick MJ (1977) Extracellular free calcium and potassium during paroxysmal activity in the cerebral cortex of the cat. Exp Brain Res 27: 237-243. [DOI] [PubMed] [Google Scholar]
- Hille B, Woodhull AM, Shapiro BI (1975) Negative surface charge near sodium channels of nerve: divalent ions, monovalent ions, and pH. Philos Trans R Soc Lond B Biol Sci 270: 301-318. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Katz B (1949) The effect of sodium ions on the electrical activity of the giant axon of the squid. J Physiol (Lond) 108: 37-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D (1997) K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387: 869-875. [DOI] [PubMed] [Google Scholar]
- Huang CS, Song JH, Nagata K, Yeh JZ, Narahashi T (1997) Effects of the neuroprotective agent riluzole on the high voltage-activated calcium channels of rat dorsal root ganglion neurons. J Pharmacol Exp Ther 282: 1280-1290. [PubMed] [Google Scholar]
- Jenkins SM, Bennett V (2001) Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol 155: 739-746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MS, Azouz R, Yaari Y (1994) Variant firing patterns in rat hippocampal pyramidal cells modulated by extracellular potassium. J Neurophysiol 71: 831-839. [DOI] [PubMed] [Google Scholar]
- Jensen MS, Azouz R, Yaari Y (1996) Spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol (Lond) 492: 199-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HY, Staff NP, Spruston N (2001) Action potential bursting in subicular pyramidal neurons is driven by a calcium spike aftercurrent. J Neurosci 21: 3312-3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER, Spencer WA (1961) Electrophysiology of hippocampal neurons. II. After-potentials and repetitive firing. J Neurophysiol 24: 243-259. [DOI] [PubMed] [Google Scholar]
- Kay AR, Sugimori M, Llinas R (1998) Kinetic and stochastic properties of a persistent sodium current in mature guinea pig cerebellar Purkinje cells. J Neurophysiol 80: 1167-1179. [DOI] [PubMed] [Google Scholar]
- Li Z, Hatton GI (1996) Oscillatory bursting of phasically firing rat supraoptic neurones in low-Ca2+ medium: Na+ influx, cytosolic Ca2+ and gap junctions. J Physiol (Lond) 496: 379-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipowsky R, Gillessen T, Alzheimer C (1996) Dendritic Na+ channels amplify EPSPs in hippocampal CA1 pyramidal cells. J Neurophysiol 76: 2181-2191. [DOI] [PubMed] [Google Scholar]
- Magee JC, Carruth M (1999) Dendritic voltage-gated ion channels regulate the action potential firing mode of hippocampal CA1 pyramidal neurons. J Neurophysiol 82: 1895-1901. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Madison DV, Nicoll RA (1986) Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature 321: 175-177. [DOI] [PubMed] [Google Scholar]
- Mantegazza M, Franceschetti S, Avanzini G (1998) Anemone toxin (ATX II)-induced increase in persistent sodium current: effects on the firing properties of rat neocortical pyramidal neurones. J Physiol (Lond) 507: 105-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV (1994) M-current suppression by agonist and phorbol ester in bullfrog sympathetic neurons. Pflügers Arch 426: 296-303. [DOI] [PubMed] [Google Scholar]
- Mattia D, Kawasaki H, Avoli M (1997) In vitro electrophysiology of rat subicular bursting neurons. Hippocampus 7: 48-57. [DOI] [PubMed] [Google Scholar]
- Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ (2001) D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci 21: 2268-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin SG, Szabo G, Eisenman G (1971) Divalent ions and the surface potential of charged phospholipid membranes. J Gen Physiol 58: 667-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz AE, Jarsky T, Martina M, Spruston N (2005) R-type calcium channels contribute to afterdepolarization and bursting in hippocampal CA1 pyramidal neurons. J Neurosci 25: 5763-5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittmann T, Alzheimer C (1998) Muscarinic inhibition of persistent Na+ current in rat neocortical pyramidal neurons. J Neurophysiol 79: 1579-1582. [DOI] [PubMed] [Google Scholar]
- Niespodziany I, Klitgaard H, Georg Margineanu D (2004) Is the persistent sodium current a specific target of anti-absence drugs? NeuroReport 15: 1049-1052. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E (2003) Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev 83: 117-161. [DOI] [PubMed] [Google Scholar]
- Ranck Jr JB (1973) Studies on single neurons in dorsal hippocampal formation and septum in unrestrained rats. I. Behavioral correlates and firing repertoires. Exp Neurol 41: 461-531. [DOI] [PubMed] [Google Scholar]
- Sanabria ER, Su H, Yaari Y (2001) Initiation of network bursts by Ca2+-dependent intrinsic bursting in the rat pilocarpine model of temporal lobe epilepsy. J Physiol (Lond) 532: 205-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzkroin PA (1975) Characteristics of CA1 neurons recorded intracellularly in the hippocampal in vitro slice preparation. Brain Res 85: 423-436. [DOI] [PubMed] [Google Scholar]
- Segal MM, Douglas AF (1997) Late sodium channel openings underlying epileptiform activity are preferentially diminished by the anticonvulsant phenytoin. J Neurophysiol 77: 3021-3034. [DOI] [PubMed] [Google Scholar]
- Shah M, Mistry M, Marsh SJ, Brown DA, Delmas P (2002) Molecular correlates of the M-current in cultured rat hippocampal neurons. J Physiol (Lond) 544: 29-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Smith RD, Plummer NW, Meisler MH, Goldin AL (1998) Functional analysis of the mouse Scn8a sodium channel. J Neurosci 18: 6093-6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sochivko D, Chen J, Becker A, Beck H (2003) Blocker-resistant Ca2+ currents in rat CA1 hippocampal pyramidal neurons. Neuroscience 116: 629-638. [DOI] [PubMed] [Google Scholar]
- Somjen GG, Muller M (2000) Potassium-induced enhancement of persistent inward current in hippocampal neurons in isolation and in tissue slices. Brain Res 885: 102-110. [DOI] [PubMed] [Google Scholar]
- Spadoni F, Hainsworth AH, Mercuri NB, Caputi L, Martella G, Lavaroni F, Bernardi G, Stefani A (2002) Lamotrigine derivatives and riluzole inhibit INa,P in cortical neurons. NeuroReport 13: 1167-1170. [DOI] [PubMed] [Google Scholar]
- Spruston N, Schiller Y, Stuart G, Sakmann B (1995) Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science 268: 297-300. [DOI] [PubMed] [Google Scholar]
- Staff NP, Jung HY, Thiagarajan T, Yao M, Spruston N (2000) Resting and active properties of pyramidal neurons in subiculum and CA1 of rat hippocampus. J Neurophysiol 84: 2398-2408. [DOI] [PubMed] [Google Scholar]
- Stefani A, Spadoni F, Bernardi G (1997) Differential inhibition by riluzole, lamotrigine, and phenytoin of sodium and calcium currents in cortical neurons: implications for neuroprotective strategies. Exp Neurol 147: 115-122. [DOI] [PubMed] [Google Scholar]
- Storm JF (1987) Action potential repolarization and a fast afterhyperpolarization in rat hippocampal pyramidal cells. J Physiol (Lond) 385: 733-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart G, Sakmann B (1995) Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron 15: 1065-1076. [DOI] [PubMed] [Google Scholar]
- Su H, Alroy G, Kirson ED, Yaari Y (2001) Extracellular calcium modulates persistent sodium current-dependent intrinsic bursting in rat hippocampal neurons. J Neurosci 27: 4173-4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Sochivko D, Becker A, Chen J, Jiang Y, Yaari Y, Beck H (2002) Upregulation of a T-type Ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus. J Neurosci 22: 3645-3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RW, Meyers DE, Richardson TL, Barker JL (1991) The site for initiation of action potential discharge over the somatodendritic axis of rat hippocampal CA1 pyramidal neurons. J Neurosci 11: 2270-2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbani A, Belluzzi O (2000) Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci 12: 3567-3574. [DOI] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282: 1890-1893. [DOI] [PubMed] [Google Scholar]
- Wong RK, Prince DA (1981) Afterpotential generation in hippocampal pyramidal cells. J Neurophysiol 45: 86-97. [DOI] [PubMed] [Google Scholar]
- Wong RK, Prince DA, Basbaum AI (1979) Intradendritic recordings from hippocampal neurons. Proc Natl Acad Sci USA 76: 986-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Saggau P (1995) Block of multiple presynaptic Ca2+ channel types by omega-conotoxin-MVIIC at hippocampal CA3 to CA1 synapses. J Neurophysiol 73: 1965-1972. [DOI] [PubMed] [Google Scholar]
- Wu N, Enomoto A, Tanaka S, Hsiao CF, Nykamp DQ, Izhikevich E, Chandler SH (2005) Persistent sodium currents in mesencephalic v neurons participate in burst generation and control of membrane excitability. J Neurophysiol 93: 2710-2722. [DOI] [PubMed] [Google Scholar]
- Wu SN, Li HF (1999) Characterization of riluzole-induced stimulation of large-conductance calcium-activated potassium channels in rat pituitary GH3 cells. J Investig Med 47: 484-495. [PubMed] [Google Scholar]
- Yaari Y, Hamon B, Lux HD (1987) Development of two types of calcium channels in cultured mammalian hippocampal neurons. Science 235: 680-682. [DOI] [PubMed] [Google Scholar]
- Yue C, Yaari Y (2004) KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci 24: 4614-4624. [DOI] [PMC free article] [PubMed] [Google Scholar]