

Abstract
Cyclin-dependent kinase 5 (Cdk5) is a serine/threonine kinase with significant homology to cell cycle-related Cdks but is not believed to be active in a typical cell cycle. In Cdk5-deficient embryos and Cdk5 chimeras, migration and survival of postmitotic neurons is compromised in a cell-autonomous manner. In the present study, we show that loss of Cdk5 leads to both failure of neuronal differentiation and loss of cell cycle control. Using specific cytoskeletal proteins as indices of neuronal differentiation, we find that Cdk5-deficient neurons are significantly arrested or delayed in their developmental program both in vivo and in vitro. For example, immunocytochemistry of embryonic day 16 (E16) cortex reveals that the expression of microtubule-associated protein 2c (Map-2c), a marker of mature neurons, is nearly absent in Cdk5-/- cells that have migrated to the cortical plate while these same cells continue to express nestin. Similarly, in vitro, Map-2-positive cells are rare in cultures from E16 Cdk5-/- embryos. Cell cycle control is also deficient in Cdk5-/- cells. In vivo, neurons engaged in cell cycle activities are found in the cortical plate, and, in vitro, class III β-tubulin-positive cells continue to label with bromodeoxyuridine even after 5 d of incubation. Transfection of a wild-type Cdk5 construct reveals that cell cycle control can be regained in Cdk5-/- cells by overexpression of Cdk5. These data indicate that Cdk5 is necessary for both neuronal differentiation and cell cycle inhibition.
Keywords: PCNA, cyclin, nestin, cell death, cerebral cortex, BrdU
Introduction
Cyclin-dependent kinase 5 (Cdk5) is a proline-directed serine/threonine kinase that was discovered by its homology to Cdc2 (Cdk1) (Hellmich et al., 1992). Despite its structural homology with the traditional cyclin-dependent kinases, Cdk5 is not believed to function in a normal cell cycle. Traditional cyclin proteins can bind Cdk5, but they do not activate it (Xiong et al., 1992; Zhang et al., 1993; Miyajima et al., 1995). Activity requires the association of p35 or p39, both of which possess structural but not sequence homologies to typical cyclins. The substrates of Cdk5/p35 include cytoskeletal and cell-signaling proteins (Homayouni and Curran, 2000; Kwon and Tsai, 2000; Dhavan and Tsai, 2001; Maccioni et al., 2001; Smith et al., 2001). The known functions of Cdk5 are diverse; they include neuronal migration (Gilmore et al., 1998), axonal growth (Paglini et al., 1998), neurite extension (Nikolic et al., 1996), stress response (Bibb et al., 2001), and cellular senescence (Alexander et al., 2004).
Cdk5 activity is found almost exclusively in postmitotic neurons. In the rodent brain, Cdk5 protein first appears in young neurons at embryonic day 12 (E12) as they migrate out of the ventricular zone (Tsai et al., 1993). Protein levels peak at postnatal day 7 (P7) and remain high throughout adulthood. Cdk5 activity, in contrast, begins to decline at P14 and remains low thereafter (Wu et al., 2000). The functions of Cdk5 are suggested by the phenotype of homozygous Cdk5-/- mice (Ohshima et al., 1996, 1999; Gilmore et al., 1998; Gilmore and Herrup, 2001). Homozygous mutants die before birth with neuronal migration deficits throughout the brain. Neocortical Cdk5-/- neurons destined for layers II to V stall below the subplate (SP) (Gilmore et al., 1998), whereas cerebellar Purkinje cells are almost completely blocked in their migration (Ohshima et al., 1999). The defects, however, are not universal. Some cerebellar granule cells as well as the neurons of neocortical layer VI and the tangentially migrating GABAergic neurons are unaffected in Cdk5-/- brains (Gilmore and Herrup, 2001).
CNS neurons are permanently postmitotic cells that will not re-enter a cell cycle once they leave the ventricular zone and the subventricular zone (SVZ). The retinoblastoma (pRb) and p27 proteins are important players in this blockage (Lee et al., 1992, 1994; Ferguson et al., 2000). Based on its developmental pattern of expression, we wondered whether Cdk5 might play an unappreciated role in this cell cycle suppression. In support of this idea, Cdk5 is able to bind to cyclins D1, D3, and E, although it is not activated by these associations (Xiong et al., 1992; Zhang et al., 1993; Miyajima et al., 1995). Moreover, Cdk5 can also phosphorylate retinoblastoma both in vitro and in vivo (Lee et al., 1997; Hamdane et al., 2005).
In the current study, we examined the effect of Cdk5 deficiency on the processes of cell cycle control and neuronal differentiation. The combined in vivo and in vitro data indicate that Cdk5 is necessary and sufficient for neuronal cell cycle arrest and subsequent differentiation.
Materials and Methods
Animals. A colony of Cdk5+/- mice was interbred to maintain them on a mixed (C57BL/6J × 129SvjS1) background (Gilmore et al., 1998). Cdk5-/- mice are not viable, so a breeding colony of Cdk5+/- mice was established for Cdk5+/- × Cdk5+/- matings. Vaginal plugs were considered E0.5. Embryos were taken at E16.5 for either cortical cultures or histology. Genotyping of embryos was performed by PCR of tail DNA as described previously (http://www.hms.harvard.edu/pathol/tsai/ProtocolsPage.html). Primers were Cdk5F1 (ATTGTGGCTCTGAAGCGTGTC), Cdk5R1 (CTTGTCACTATGCAGGACATC), and phosphoglycerate kinase 1 (TCCATCTGCACGAGACTAGT). All procedures with mice were performed in accordance with the local Institutional Animal Care and Use Committee as well as Association for Assessment and Accreditation of Laboratory Animal Care standards.
Primary neuronal cultures. Embryonic cortical neurons were isolated by standard procedures. Briefly, embryos were collected in ice-cold PBS glucose, and the cortical lobes were dissected out. Meninges were removed, and the cortices were placed in 1× trypsin-EDTA for 20 min at 37°C. The tissue was removed from the trypsin solution and placed in DMEM with 10% FBS to inactivate the trypsin, followed by transfer to Neurobasal media supplemented with B-27, penicillin-streptomycin (1×), and l-glutamine (2 mm). Tissue was triturated 10 times through a 5 ml pipette and allowed to settle to the bottom of a 15 ml conical tube. Cells in the solution above the 1 × g pellet were removed. Surviving cells were counted with Trypan Blue to identify dead cells and were plated on poly-l-lysine-coated (0.05 mg/ml) and laminin-coated (5 μg/ml) plates or glass coverslips. Cells were plated in 24-well plates at 50,000 cells/well. For Cdk5-deficient cultures, all embryos were treated separately, and the tail DNA of each embryo was isolated for genotyping. All cultures were grown for a minimum of 5 d in vitro (DIV) before any treatment. To assess cell cycle activity after 5 DIV, media was changed to new media containing 10 μm bromodeoxyuridine (BrdU). Cells were left in BrdU-containing media for 48 h and then fixed with 4% paraformaldehyde in phosphate buffer at room temperature for 30 min and then washed and stored in PBS. All experiments were performed on a minimum of three litters in triplicate.
Histology. Pregnant dams were killed by cervical dislocation, and embryos were removed from the uterine horns. The tails of each embryo were removed for genotyping. The entire head of each embryo was immersion fixed in 4% paraformaldehyde in phosphate buffer for 24 h. Brains were removed and cryoprotected in 30% sucrose for a minimum of 24 h. Brains were then oriented and frozen in OCT for cryosectioning. Ten micrometer serial sections were taken of the entire cortex of each animal and kept frozen until immunohistochemistry was completed. For all experiments, a minimum of three litters was used, and littermates were compared. To analyze cell cycle activity in the embryos, pregnant dams were injected with BrdU (100 mg/kg body weight) at E16.5. After 5 h, the mouse was killed and processed as above.
Immunohistochemistry. Brain sections were blocked and permeabilized in 5% normal goat serum in PBS with 0.1% Triton X-100 for 1 h. For proliferating cell nuclear antigen (PCNA), BrdU, Ki67, chondroitin sulfate proteoglycan (CS56), RC2, activated caspase-3, and phosphoH2AX staining, antigen retrieval was performed by treatment for 5 min at 100°C in citrate buffer. For BrdU visualization, sections were treated with 2N HCl for 15 min, neutralized in 0.1 m sodium borate, pH 8.6, for 15 min, and then washed extensively in PBS (three times) for 45 min before blocking. For micro-tubule-associated protein 2 (Map-2), class III β-tubulin (Tuj1), and nestin labeling, no antigen retrieval was performed. Immunohistochemistry of cell cultures was done without antigen retrieval. For BrdU labeling, DNA was hydrolyzed by 2N HCl for 10 min and then neutralized in 0.1 m sodium borate, pH 8.6, for 10 min and then washed extensively in PBS (three times) for 45 min before blocking. Cells were blocked in 5% normal goat serum with 0.1% Triton X-100 for 1 h before application of the primary antibody.
Antibody concentrations used for mouse brain sections were as follows: mouse anti-PCNA (1:10,000; Santa Cruz Biotechnology, Santa Cruz, CA), rat anti-BrdU (1:100; Abcam, Cambridge, UK), mouse anti-Map-2 (1:1000; Sigma, St. Louis, MO), mouse anti-Tuj1 (1:1000; Covance, Princeton, NJ), mouse anti-nestin (1:500; Chemicon, Temecula, CA), rabbit anti-caspase-3 (1:100; Cell Signaling Technology, Beverly, MA), rabbit anti-H2AX (1:100; Trevigen, Gaithersburg, MD), mouse anti-RC2 (1:40; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), mouse anti-CS56 (1:200; Abcam), and rabbit anti-Ki67 (1:50). Secondary antibodies used were as follows: goat anti-rat rhodamine (1:300; The Jackson Laboratory, Bar Harbor, ME) and goat anti-mouse Alexa 488 (1:1000; Invitrogen, Eugene, OR). Sections were counterstained with green or red NeuroTrace (1:300; Invitrogen) or 1 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI).
Immunoblotting. Pregnant dams were killed by cervical dislocation. Embryos were removed from the uterine horns in ice-cold calcium- and magnesium-free PBS. Tails were collected for genotyping. Embryo brains were rapidly dissected and placed in lysis buffer (20 mm Tris, 1% Triton X-100, 100 mm NaCl, 40 mm NaF, 1 mm EDTA, 1 mm EGTA, 1 mMNa3VO4, aprotinin, leupeptin, and PMSF) on ice and homogenized. Lysates were then sonicated for 15 s and centrifuged at 13,000 rpm for 15 min at 4°C. Supernatant was collected and protein was measured by the Bradford method. Equal amounts of protein (10 or 15 μg) in Laemmli buffer were boiled for 7 min. The samples were run on SDS-PAGE gels and transferred onto polyvinylidene difluoride membranes as described previously (Combs et al., 2001). Antibodies used for immunoblotting were as follows: mouse anti-Map-2a and 2b (1:2000; Sigma), mouse anti-PCNA (1:500; Chemicon), polyclonal rabbit anti-Map-2 (1:2000; Chemicon), mouse anti-Tuj1 (1:2000; Covance), rabbit anti-Erk1/2 (1: 1000; Santa Cruz Biotechnology), rabbit anti-actin (1:4000; Abcam), sheep anti-mouse HRP (1:1000; Amersham Biosciences, Arlington Heights, IL), donkey anti-mouse HRP (1:20,000; Jackson Immuno-Research, West Grove, PA), and donkey anti-rabbit HRP (1:1000; Amersham Biosciences). Blots were developed with ECL (Amersham Biosciences) according to the instructions of the manufacturer.
Cdk5 construct. Mouse Cdk5 cDNA was amplified by reverse transcription (RT)-PCR from the mRNA of an adult brain. Briefly, 4 μg of total RNA was primed with Oligo dT(14) and reverse transcribed with SSII according to the instructions of the manufacturer. To amplify Cdk5 cDNA, 80 ng of total RNA from the RT reaction was subjected to PCR with murine-specific Cdk5 primers. Primers used were 5′ to 3′ GCCGCTGCAATGCAGAAA and 5′ to 3′ GTTCAGAGAGCCTATGGG. These primers contain the entire coding sequence of Cdk5 and start 12nt upstream of the first start site. The RT-PCR product was then subcloned into pCR.II TOPO vector. Sequences and direction of transformants were confirmed by automated sequencing (Northwoods DNA, Solway, MN), and the construct was removed from the TOPO vector by BamH1 and XhoI restriction digest. This fragment was ligated into phosphorylated internal ribosomal entry site 2 (pIRES2)-enhanced green fluorescent protein (EGFP) vector for mammalian expression. The Cdk5-pIRES2-EGFP construct was transformed into D5α chemically competent cells, which were grown to confluence. Plasmid DNA was isolated and subjected to cesium chloride gradient twice and resuspended in TE buffer for use in transient transfections.
Transient transfections. Cesium chloride-prepared constructs were transfected into neuronal cultures by the calcium phosphate precipitation method or were performed using Lipofectamine 2000, according to the directions of the manufacturer. All transfections were performed on primary cultures of cortical neurons after a minimum of 5 DIV. Transfection experiments were performed in triplicate on whole litters of animals in at least seven separate experiments. Cell counts were completed by counting all transfected cells in three separate wells of two separate experiments.
Results
Cdk5-/- and Cdk5+/- neurons continue to cycle in the cortical plate
Previous studies of the cerebral cortex of Cdk5-deficient mice have indicated that the neurons of layers II-V of the neocortex are stalled in their migration (Gilmore et al., 1998). Although the earlier migrating layer VI neurons split the preplate and migrate to their normal position, the neurons of other layers appear arrested below the subplate in the intermediate zone (IZ). To determine whether the migrating and settled neuroblasts had efficiently exited the cell cycle, we examined Cdk5-/-, Cdk5+/-, and wild-type embryos 5 h after BrdU injection of the pregnant dams at E16.5. Apart from the expected levels of cell cycle activity in the ventricular zone (data not shown), a few BrdU-positive cells were found in the SVZ of all three genotypes. In wild-type embryos (Fig. 1A), no additional labeling was observed. In contrast, substantial BrdU labeling was found in the cortical plate (CP), SP, and IZ of the Cdk5-/- mutants (Fig. 1C, arrows). The phenotype of the heterozygous animals was intermediate, with a few BrdU-positive cells found in the subplate and intermediate zone (Fig. 1B). We confirmed these findings by immunostaining sections with antibodies against PCNA, an S-phase marker. High levels of PCNA-positive neurons were found throughout the Cdk5-/- neocortex in the same regions identified by the BrdU labeling (Fig. 1F). Western blot analysis of PCNA protein levels (Fig. 1J) also revealed an overall increase in PCNA protein levels. Additional confirmation of this persistent cell cycle activity was found after immunostaining for the Ki67 antigen (Fig. 1G-I). The nuclear transcription factor Ki67 is expressed from the S phase to the M phase of the cell cycle. In the wild-type cortex, Ki67-positive cells were restricted to proliferative areas of the subventricular and ventricular zones, with a few positive cells scattered in the intermediate (Fig. 1G). In contrast, cells in all areas of the Cdk5-/- cortex are labeled with Ki67 (Fig. 1I). Double labeling with an antibody against CS56, a subplate marker (Fig. 1G-I, green), revealed that many cells in the cortical plate were positive for proliferation markers. As with the BrdU labeling, the Cdk5+/- heterozygote appeared intermediate, with some Ki67-positive cells in the subplate (Fig. 1, arrows) as well as many positive cells in the intermediate zone (Fig. 1H). Additional cell cycle markers were also used. Cyclin D is a regulatory subunit of Cdk4 and Cdk6 that is expressed as cells leave the restriction point and complete the G1 phase of the cell cycle. Its pattern of expression in the three Cdk5 genotypes we examined closely follows that described above for the other cell cycle markers (Fig. 2A-C). Cyclin A is a Cdk2 regulator that is predominantly found during S-phase. This cell cycle marker is also found in regions of the Cdk5-/- embryonic cortex, where only postmitotic cells should be located (Fig. 2F, arrows; wild-type and heterozygote cortices are shown in D and E, respectively). Cyclin B is a regulatory subunit of Cdc2 (Cdk1) and is normally found only during the G2 phase of the cell cycle. No immunostaining was found for this marker outside of the ventricular zone in any of the three genotypes (data not shown). Together, the cell cycle markers we examined indicate that cells in the Cdk5-/- cortex remain actively engaged in the cell cycle even after leaving the ventricular zone. Based on the clear presence of multiple markers of G1 and S-phase markers and the absence of cyclin B, it appears that the cells enter a cell cycle and proceed through most or all of the S-phase but stop or die before entering G2. It is unknown whether the cells of the Cdk5-/- cortex are stalled in a particular phase of the cell cycle yet still emigrate from the ventricular zone, whether the cell cycle is exceedingly protracted, or whether these cells have migrated and then reentered the cell cycle.
Figure 1.
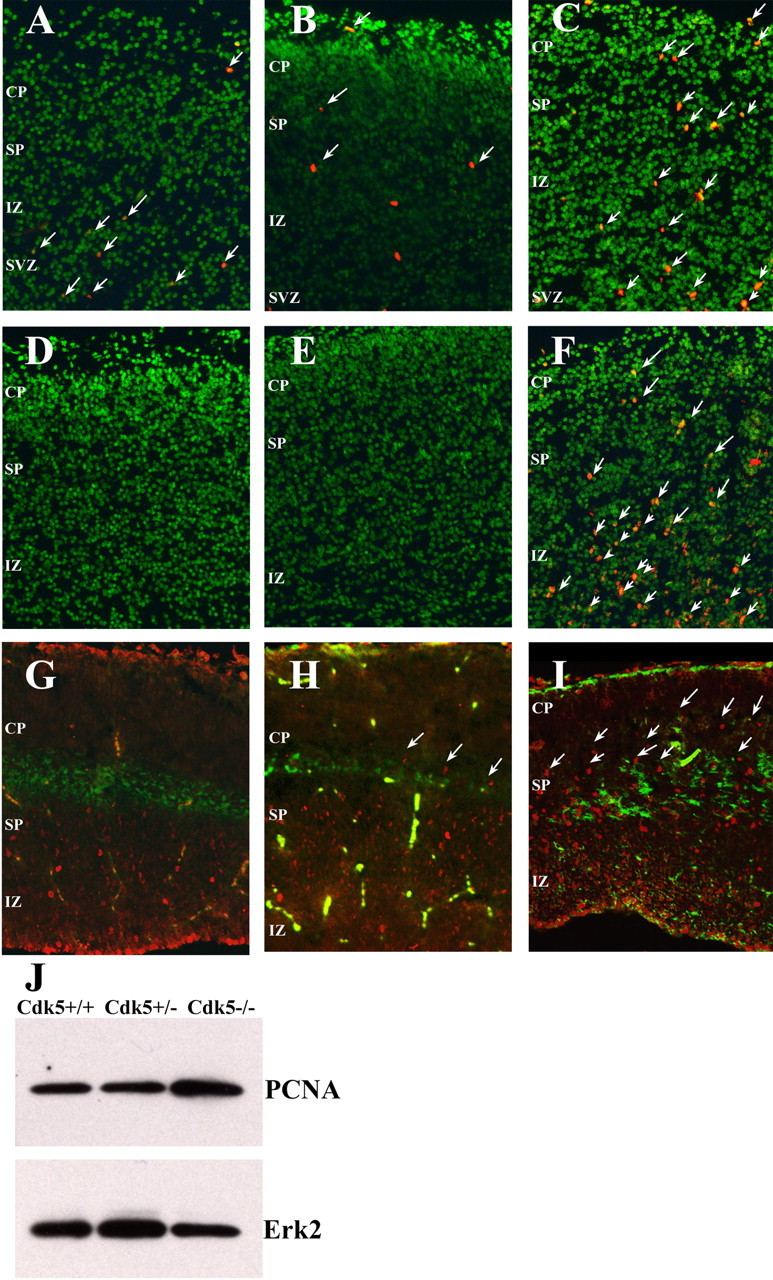
Loss of Cdk5 leads to loss of cell cycle control in E16.5 neocortex. A-C, Coronal sections of Nissl-stained neocortex (green) shows the location and number of BrdU-positive neurons (red) in Cdk5+/+ (A), Cdk5+/- (B), and Cdk5-/- (C)embryos. Arrows denote BrdU-positive cells. D-F, Coronal sections labeled with anti-PCNA (red) in Cdk5+/+ (D), Cdk5+/- (E), and Cdk5-/- (F) cortex. Labeled cells are apparent in the subventricular zone (SVZ), intermediate zone (IZ), subplate (SP), and cortical plate (CP) of the Cdk5-/- brain. Arrows denote examples of PCNA-positive cells. G-I, Sagittal sections labeled with anti-chondritin sulfate proteoglycan (green) and anti-Ki67, a proliferation marker (red). The Cdk5-/- cortex (I) has many Ki67-positive cells in both SP and CP. In the wild-type cortex (G), there are no positive cells in or above the subplate, and, in the heterozygote cortex, there is an intermediate phenotype with some Ki67-positive cells in the subplate (H). J, Western blot of an E16.5 neocortical lysate shows an increase in the PCNA protein content in the Cdk5-/- brain (lane three).
Figure 2.
Cell cycle markers are expressed in the Cdk5-/- E16.5 neocortex. A-C, Sagittal sections immunostained with Map-2 and cyclin D (red) in Cdk5+/+ (A), Cdk5+/- (B), and Cdk5-/- (C). In the Cdk5-/- cortex, cells that are Map-2 positive in the SP and CP are also cyclin D positive (arrows). D-F, Sagittal sections labeled with cyclin A (red) in Cdk5+/+ (D), Cdk5+/- (E), and Cdk5-/- (F) show neurons in cell cycle throughout SVZ, IZ, SP, and CP in Cdk5-/- brain. Arrows denote examples of cyclin A-positive cells.
Cdk5-deficient neurons fail to differentiate in vivo
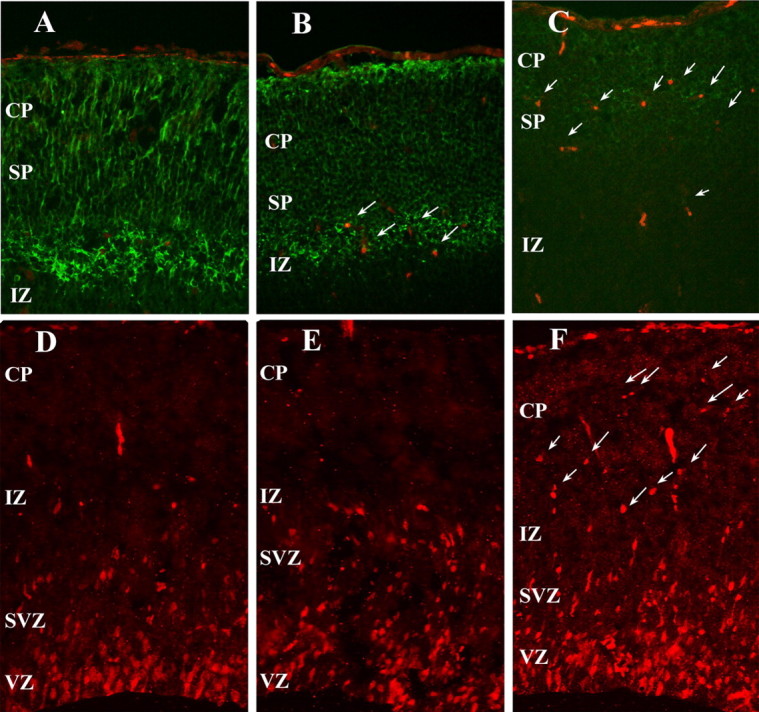
In the normal mouse embryo, cell division is restricted to the ventricular and subventricular zones. During this period, the dividing precursors express the intermediate filament protein, nestin. As neurogenesis ends and migration begins, cells lose their immunoreactivity to nestin and begin expression of the neuron-specific class III β-tubulin protein (revealed by the TuJ1 antibody). Later, during migration across the intermediate zone, immature forms of Map-2 appear. As development proceeds, the early Map-2 isoform (Map-2c) is replaced by the more mature Map-2a and Map-2b isoforms. In the wild-type embryos we examined, the neurons of the neocortex faithfully recapitulate this progression. By E16.5, cells in and above the intermediate zone have lost their nestin immunoreactivity in wild-type (Fig. 3A) and heterozygous animals (Fig. 3B). In contrast, cells in the Cdk5-/- cortex (Fig. 3C) continue to express significant levels of nestin, long after they have migrated out of the ventricular zone. Thus, the postmigratory Cdk5-/- neurons continue to express cytoskeletal components that are more typical of mitotic progenitor cells than of mature cortical neurons. The qualitative results seen with immunocytochemistry apply throughout the cortex as shown by Western blots of E16.5 cortical lysates of the three genotypes (Fig. 3J,K). There is a 10-fold increase in the amount of nestin protein present in the Cdk5-/-cortex compared with the wild-type cortex (Fig. 3K). Curiously, the Cdk5 heterozygote has levels of nestin protein that are intermediate to the mutant and wild-type values.
Figure 3.
Cdk5 is required for neuronal differentiation. A-C, Intermediate filament protein, nestin, is found throughout the E16.5 Cdk5-/- cortex (C), although it is completely lost in both the Cdk5+/+ (A) and Cdk5+/- (B) embryos. D-F, TuJ1 labeling of sagittal sections of the E16.5 cortex shows a decrease in class IIIβ-tubulin in both CP and IZ of Cdk5-/- brains (F), compared with either Cdk5+/+ (D) or Cdk5+/- (E) samples. G-I, Radial glia are present and extend processes to the pia in a radial mode in Cdk5+/+ (G), Cdk5+/- (H), and Cdk5-/- (I). J, Representative Western blot analysis of nestin expression in littermates. Note the increased nestin in lane 3 (Cdk5-/-). Erk2 is used as a loading control. K, Quantification of the Western blots shows a 10× increase in nestin expression in the Cdk5-/- cortex compared with wild-type littermates (n = 3; **p < 0.001, ANOVA). Error bars represent SEM.
As expected, in most regions outside of the ventricular zone, the wild-type and heterozygous cortices show strong TuJ1 immunostaining for the immature neuron marker β-III tubulin (Fig. 3D,E), with particularly robust staining in subplate and intermediate zone. In the Cdk5-/- neocortex, however, TuJ1 immunoreactivity is noticeably reduced (Fig. 3F). Tuj1 immunoreactivity in Cdk5-deficient cortex also revealed an abnormal pattern of neurite growth. In the wild type, the apical dendrites of most neurons extend in a predominantly radial direction, perpendicular to the ventricular and pial surfaces (Fig. 3D). In the mutant animals, however, these apical processes appear disoriented (Fig. 3F), an observation that is further supported by the pattern of Map-2 immunolabeling (Fig. 4C). The reasons for this loss of radial orientation are unclear, because the system of radial migratory guides, revealed by labeling with the RC2 antibody, is mostly intact (Fig. 2G-I, insets). As was observed with nestin immunostaining, there are subtle changes in the heterozygote cortex in the apparent levels of TuJ1 staining and the orientation of the neurites (Fig. 3E).
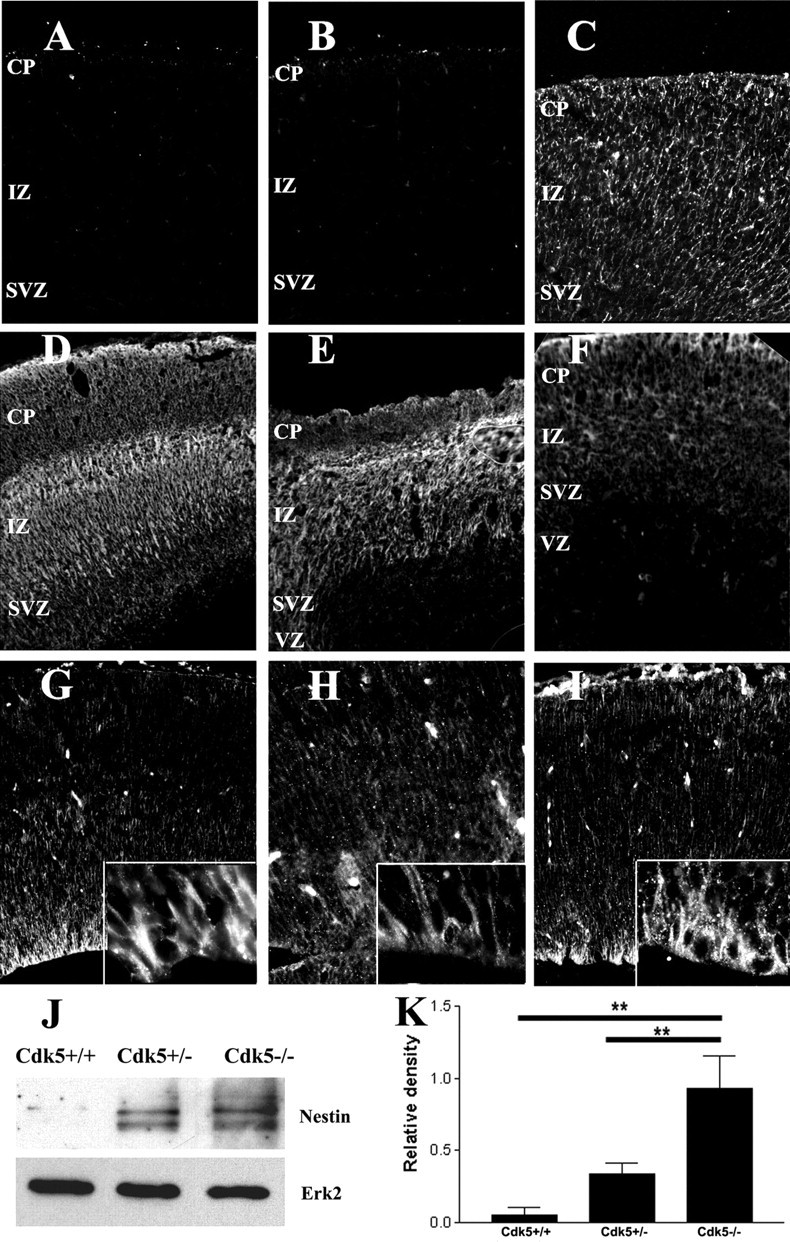
Figure 4.

Mature and immature isoforms of Map-2 fail to develop in the E16.5 Cdk5-/- neocortex. Map-2 is present in the CP and SP in Cdk5+/+ (A) and Cdk5+/- (B) neocortices, whereas the levels of staining Cdk5-/- neocortex (C) are reduced. D, Representative Western blot of three separate E16.5 neocortices of each genotype showing a decrease in the expression of Map-2c. E, Representative Western blot of littermates showing a decrease in Map-2a and Map2b of three separate embryos. β-Actin is used as a loading control. F, Quantification of Western blots shows a relative decrease of Map-2c protein. Compared with Cdk5+/+, protein levels were reduced in both Cdk5+/- (n = 4; p < 0.001, ANOVA) and Cdk5-/- cortices (n = 3; p < 0.001, ANOVA). G, Quantification of Map-2a/b Western blots shows a loss of protein in the Cdk5-/- cortex compared with wild type (n = 6; p < 0.001, ANOVA). Map-2a/b levels in the Cdk5+/-, however, do not change significantly with respect to Cdk5+/+. Error bars represent SEM.
The spatial pattern of Map-2a/b expression is also disrupted in the mutant cortex. The mature isoforms Map-2a and Map-2b are located predominantly in the cells of the cortical plate and subplate in wild-type and heterozygous neocortices (Fig. 4A,B). In contrast, there is a significant decrease in the levels of both Map-2a and Map-2b immunolabeling in the Cdk5-/- neocortex (Fig. 4C). To determine the levels of the particular isoforms of Map-2 expressed in the knock-out cortex, we performed Western blot analysis with two different antibodies, one that recognizes Map-2c (Fig. 4D) and a second that recognizes both Map-2a and Map-2b (Fig. 4E). In our analysis of three separate embryos from each genotype, we found that the Map-2c isoform is decreased by >50% in both Cdk5+/- (20 ± 5 relative band intensity units) and Cdk5-/- (3.5 ± 0.5 U) cortices compared with the wild-type cortex (42 ± 5 U) (Fig. 4F). We also wished to determine the levels of expression of the more mature Map-2 isoforms. We considered the doublet we found at ∼280 kDa as the total of Map-2a plus Map-2b protein. We analyzed lysates from six separate embryos of each genotype. A representative Western blot of three separate embryos is shown in Figure 4E. In keeping with the findings from the immunocytochemistry, there was a 15-fold decrease in the levels of Map-2a/b in the Cdk5-/- cortex compared with wild type (0.3 ± 0.13 and 4.6 ± 1.9 U, respectively). The levels in the heterozygote cortex (3.4 ± 1.0 U) were modestly, but not significantly, reduced (Fig. 4G).
Cell death in the Cdk5-/- cortex
Previous work from our laboratory has indicated that Cdk5 deficiency is not cytotoxic to all neurons. Nonetheless, fewer than the expected numbers of Cdk5-/- cortical neurons are found to survive in Cdk5 chimeric mice (Gilmore and Herrup, 2001). This finding suggests a Cdk5-dependent neuronal survival function. We extended these findings using activated caspase-3 as an index of cell death. We found dying neurons in all zones of the Cdk5-/- neocortex, especially in the subplate and intermediate zone (Fig. 5C). As expected, there was little cell loss in either wild-type or heterozygote cortex (Fig. 5A,B). We found a similar picture when anti-phospho-histone2AX was used as a marker of apoptotic cells (Fig. 5D-F). Although neuron specific markers tend to be lost during the cell death process precluding their positive identification, many of the caspase-3-positive cells had an unmistakably neuronal morphology (arrows and inset). We were, however, able to show the coincidence of cell cycling and cell death. Using PCNA/terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) (Fig. 5G-I) or PCNA/activated caspase-3 (Fig. 5J-L) as cell cycle and cell death markers, respectively, we were able to find cells that were double labeled for both cell cycle and cell death proteins. In addition, we previously found cyclin D-positive cells in the area of the Cdk5-/- cortex where there is some Map-2a/b expression (Fig. 2A-C). We believe that these data extend our previous findings and suggest that Cdk5 is required for the survival of many neuronal cell types in vivo. They further suggest that the death of the Cdk5-deficient cells is accompanied by a re-expression of cell cycle proteins.
Figure 5.
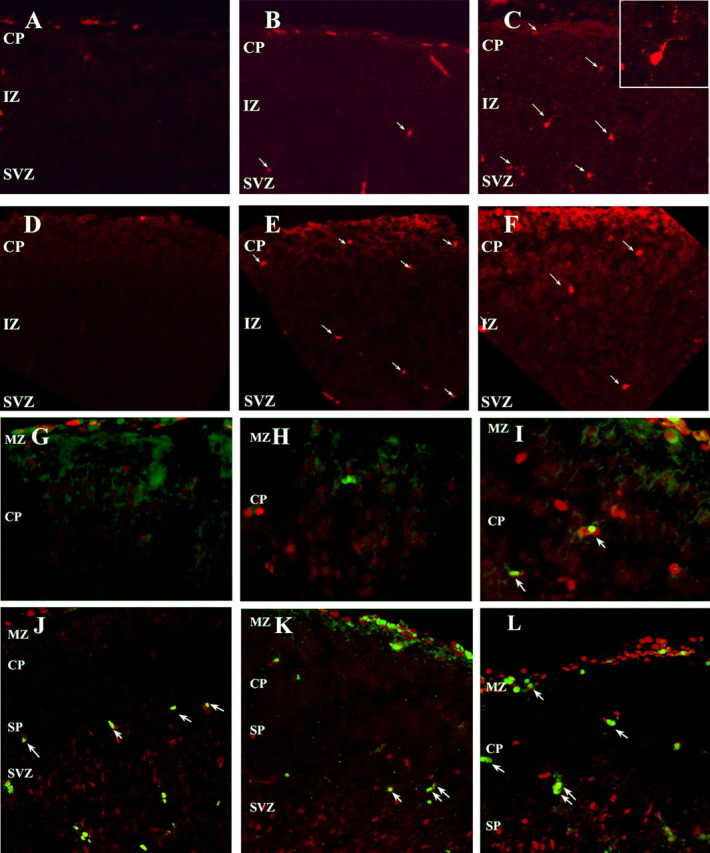
Increased cell death in the E16.5 Cdk5-/- neocortex is correlated with continued cell cycling. Sagittal sections of Cdk5+/+ (A), Cdk5+/- (B), and Cdk5-/- (C) neocortex labeled with anti-activated caspase 3 indicates a greater number of labeled neurons (arrows). Inset, Distinct morphology marks this caspase-positive Cdk5-/- as neuronal. Phospho-histone H2AX labeling, a hallmark of apoptosis independently demonstrates the increased cell loss in mutant cortex. Cdk5+/+ (D), Cdk5+/- (E), and Cdk5-/- (F) are shown. Cells in the Cdk5-/- cortex are cell cycle positive and apoptotic. TUNEL-positive cells (green) in the Cdk5-/- cortex (I) are also PCNA positive (red). There are few to no TUNEL-positive cells in the Cdk5+/+ (G) or Cdk5+/- (H) neocortices and no double-labeled cells. A similar picture is found using caspase-3. Cdk5+/+ (J), Cdk5+/- (K), and Cdk5-/- (L) neocortices labeled with activated caspase-3 (green) and PCNA (red) revealed double-labeled cells (arrows) in the CP only in the mutant (L). Double-labeled cells in Cdk5+/+ (J) and Cdk5+/- (K) cortices are found below the subplate and in the SVZ.
Cdk5-/- cortical neurons continue to cycle in vitro and do not differentiate
The deficits seen in the in vivo preparation represent a phenotype that is the result of the interactions of many different cell types. We used dissociated cultures to isolate those features that are intrinsic to the neurons themselves. Cultures of dissociated E16.5 neurons from Cdk5+/+, Cdk5+/-, and Cdk5-/- brains were grown in serum-free NeuroBasal medium for a minimum of 5 d before analysis. As shown in Figure 6A-C, there were distinct differences in the TuJ1-stained neurite out-growth in mutant cultures (Fig. 6C) compared with cells in wild-type (Fig. 6A) and heterozygote (Fig. 6B) cultures. The Cdk5-/- dendrites were sparsely branched and thin, even near the cell soma. Note that, once again, the properties of the heterozygote were intermediate between those of the mutant and wild type. The neurites in the Cdk5+/- cultures were consistently abnormal. They appeared thinner and more disorganized than wild-type dendrites but not as severely atrophic as the Cdk5-/- cells (Figs. 6 B, 7B). Although there are phenotypic differences in their processes, the actual numbers of TuJ1-positive neuronal cell bodies in Cdk5-/-, Cdk5+/-, and Cdk5+/+ cultures were equal (Fig. 6G).
Figure 6.

Neurons from Cdk5-/- cortex fail to differentiate in culture. A-C, E16.5 cortical neurons cultured for 5 d were fixed and labeled with anti-βIII tubulin (Tuj1, green) and counterstained with the nuclear marker DAPI. Note that neurite extension and complexity are both decreased in Cdk5-/- (C) cortical cultures compared with Cdk5+/+ (A) and Cdk5+/- (B) cultures. D-F, E16.5 cortical neurons cultured for 5 d were labeled for all Map-2 isoforms with a polyclonal anti-Map-2 antibody (red) and counterstained with DAPI. In Cdk5-/- cultures, there was a significant loss of Map-2-positive neurons (F) with the few cells that were stained showing fluorescence near the soma only. G, Counts of TuJ1-positive neurons indicate no loss of cells (n = 3; ANOVA). H, Quantification of Map-2-positive cells reveals a significant reduction of neurons that express Map-2 in mutant cultures (n = 3; **p < 0.01, ANOVA). There was, however, no decrease in total neuron number as shown in G. Error bars represent SEM.
Figure 7.

Cdk5-/- neurons continue to incorporate BrdU in vitro. A-C, E16.5 cultured cortical neurons grown for 5 DIV were given a 24-48 h pulse of BrdU. TuJ1-positive cells (green) were labeled with BrdU (red) only in the Cdk5-/- cultures. Separate cultures were set up from each embryo and genotype determined retrospectively. A, Cdk5+/+; B, Cdk5+/-; C, Cdk5-/-. Inset, Cdk5-/- BrdU-positive and β-III tubulin-positive neuron. D, Percentage of BrdU-positive Tuj1-positive cells in culture. n = 3. **p < 0.05, ANOVA. Error bars represent SEM.
The differences among the cultured neurons of the three genotypes are even more dramatic when viewed with a polyclonal antibody raised against both embryonic and mature Map-2 isoforms (Fig. 6D-F). Although the cultures contained equal numbers of Map-2-positive neurons in Cdk5 heterozygote and wild-type cultures (Fig. 6H), we consistently observed a decrease in the intensity of Map-2 staining in Cdk5+/- cultures (Fig. 6E) compared with wild-type cultures (Fig. 6D). The number of Map-2-positive neurons in the mutant Cdk5-/- cultures were decreased by 75% overall (Fig. 6H), and the few Map-2-positive cells that were present had weak staining that was restricted to the cell soma (Fig. 6F). Virtually no Map-2 immunoreactivity was found in the TuJ1-positive processes (data not shown). Cdk5-/- cultures stained with a mouse monoclonal specific for Map-2a/b (the mature isoforms) contained no detectable immunopositive cells (data not shown).
After 5 d in culture, wild-type neurons are mitotically inactive. Thus, in wild-type and heterozygote cultures, only a small number of Tuj1-positive cells incorporate BrdU (Fig. 7A,B,D). Many Cdk5-/- TuJ1-positive neurons, in contrast, continue to incorporate BrdU in culture (Fig. 7C). We are confident that these dividing cells were neuronal because we could find many examples of cells (Fig. 7C, inset) that were immunopositive for both TuJ1 (green) and BrdU (red). Cell counts show that 12% of all TuJ1-positive Cdk5-/- cells are also BrdU positive. Less than 1% of the neurons in Cdk5+/- and Cdk5+/+ cultures were double positive (Fig. 7D). These data suggest that the loss of Cdk5 either releases the immature neurons so that they are free to re-enter another cell cycle or prevents them from fully leaving the cell cycle to differentiate.
Overexpression of Cdk5 blocks cell cycle events in Cdk5-/- neurons in vitro
We determined whether the loss of Cdk5 directly causes changes in cell cycle regulation. We transfected cultured cortical neurons from E16.5 embryos with a bicistronic expression vector that contained a full-length wild-type Cdk5 cDNA as well as the EGFP gene, the protein of which was translated from a separate IRES. Cells successfully transfected with the Cdk5/EGFP vector were easily identified by their fluorescence under UV illumination. Twenty-four hours after transfection with control vector, ∼13-17% of all cells were fluorescent, indicating a successful transfection. Cells of all three genotypes showed comparable transfection efficiencies when an empty EGFP vector (without Cdk5) was used. Transfection efficiency decreased to ∼6% when the full bicistronic vector (with Cdk5) was used, although again there were no differences in efficiency among the three genotypes. To test expression of the Cdk5 protein, we used a polyclonal antibody directed against Cdk5 in Cdk5-/- cultures transfected with wild-type Cdk5. In these cultures, EGFP-positive cells were also immunopositive for Cdk5 (data not shown). Twenty-four hours after transfection, BrdU-containing media was added to the cultures, which were then left for an additional 24-48 h. After fixation, cells were stained for BrdU (red) and visualized in green for either EGFP or Cdk5.
Cultures of all three genotypes transfected with the GFP-only vector contained cells that were both GFP and BrdU positive. In wild-type cultures, ∼17% of all transfected cells incorporated BrdU. As expected, in both wild-type and heterozygote cultures, most of these cells were non-neuronal (by morphological criteria) (Fig. 8A,B). In Cdk5-/- cultures, BrdU-positive neurons were also readily found (Fig. 8C). On average, 33% of the transfected cells in Cdk5-/- cultures incorporated BrdU. When the Cdk5 expression vector was used, however, BrdU incorporation decreased dramatically (Fig. 8D-F). Of the total transfected Cdk5-/- cells, only 11% continued BrdU incorporation during the 24-48 exposure (a threefold decrease). Unexpectedly, this was true for non-neuronal as well as the Tuj1-positive cells (Fig. 8D-F). Indeed, even in wild-type cultures, overexpression of Cdk5 decreased the percentage of transfected cells that are BrdU positive to ∼9%. The heterozygote cultures behaved in a similar manner. These data show that expression of Cdk5 is sufficient to suppress the cell cycle in cultured wild-type cells and “rescue” the dysregulation of the cell cycle in cells from the Cdk5-/-mutant. Overexpression of Cdk5 in genetically wild-type cells can also apparently override their normal cell cycle program, slowing or stopping their cell division altogether.
Figure 8.

Cdk5 is sufficient for cell cycle suppression. A-C, Cells transfected with pIRES2-EGFP incorporate BrdU (red) and express EGFP (green) in Cdk5+/+ (A), Cdk5+/- (B), and Cdk5-/- (C) cultures. Arrows indicate cells that are transfected and still incorporate BrdU. Very few neuronal cells incorporate BrdU in Cdk5+/+ and Cdk5+/- cultures. D-F, Transfected cells labeled with exogenous Cdk5 (green) do not incorporate BrdU (red) in any of the three genotypes. Arrows indicate BrdU-positive cells, whereas asterisks indicate Cdk5-transfected cells. G, Cell counts indicate a significant decrease in a percentage of total cells that are BrdU positive with overexpression of Cdk5. Black bars indicate a percentage of total transfected cells in three wells that were BrdU positive (average of two independent experiments). Striped bars indicate a percentage of total cells that are BrdU positive and Cdk5-GFP positive. Total cells were determined by DAPI and GFP staining. H, Graphical representation of a decrease in BrdU incorporation with Cdk5 overexpression. There is an approximate three-times decrease in BrdU-positive transfected cells in Cdk5+/- and Cdk5-/- cultures. There is also a significant decrease in BrdU-positive cells in wild-type cultures when transfected with Cdk5.
Cdk5 activity plays a role in differentiation but not in cell cycle regulation
The data thus far suggest that the presence of Cdk5 is necessary for both normal differentiation as well as for the cessation of cell division. We determined whether these results were based merely on the presence of the Cdk5 protein or whether they also required enzyme activity. We used 10 μm roscovitine as a pharmacological inhibitor of Cdk5 activity in vitro. Six-day-old cultures were fixed and stained for Map-2a/b to determine the state of their differentiation. Roscovitine was added to replicate cultures for different times before fixation. The goal of this treatment was to block enzyme activity after varying amounts of differentiation. As shown in Figure 9, the addition of the drug after 1 or 2 d in vitro (5 or 4 d total activity block, respectively) led to dramatic decreases in both the length and complexity of the neurites (Fig. 9B,C) and the presence of Map-2 (Fig. 9G). The average number of Map-2-positive cells decreases early in differentiation in vitro, whereas after the cells have differentiated (after 3 DIV), there is no change in total numbers of Map-2-positive cells. There were decreases in total cell numbers on days 1 and 2 in vitro, as visualized by DAPI, but these decreases were not statistically significant. When normalized to total cell numbers, the number of Map-2-positive cells is decreased by 50% on days 1 and 2 in vitro and is near control levels by days 4 and 5. Surprisingly, there were also no differences in the number of cells demonstrating cell cycle activity. Examples of this are shown in Figure 9, H (no treatment) and I (10 μm roscovitine after 2 DIV). Given the results from the Cdk5-/- cultures, one might expect that blocking kinase activity would release a significant resurgence of cell cycling, but this was not the case. Together, these pharmacological data point to a situation in which the role of Cdk5 in promoting morphological differentiation is a function that requires full kinase activity, whereas the role of Cdk5 in cell cycle inhibition involves the protein but may not need an active kinase to work.
Figure 9.

Inhibition of Cdk5 kinase activity blocks neuronal differentiation in culture. A-F, Wild-type E16.5 cells were either left untreated (A, H) or were treated with 10 μm roscovitine, as elective Cdk5 inhibitor, after 1 (B), 2 (C, I), 3 (D), 4 (E), or 5 (F) DIV. On the fifth day, all cells were exposed to 10 μm BrdU for 24 h. Cells were stained for BrdU (red) and Map-2a/b (green). G, Counts of surviving Map-2-positive cells at the end of the experiment. Labels on the x-axis indicate the day of culture on which roscovitine was added. H, I, Inhibition of kinase activity by roscovitine does not induce cell cycle re-entry. Untreated cells (H) and cells treated with 10 μm roscovitine at 2 DIV (I) were fixed and stained for BrdU and Map-2. There was no increase in the number of cells incorporating BrdU at any treatment stage. Two independent experiments were counted in triplicate (**p < 0.01; Student's t test). Error bars represent SEM.
Discussion
Neurons lacking Cdk5 fail to complete their differentiation program
The differentiation of most CNS neurons is marked by the orderly expression of a series of cytoskeletal proteins. We focused on three: nestin, βIII tubulin, and Map-2. During normal development, the earliest expressed marker is nestin, an intermediate filament protein found in both glial and neural progenitors that is downregulated after differentiation begins. In the Cdk5-deficient cortex, however, nestin expression continues, even in neurons that have entered the cortical plate. Nestin is a potential target of the Cdk5 kinase, and phosphorylation may target it for subsequent ubiquitination and degradation (Sahlgren et al., 2003). Thus, in Cdk5-/- cells, nestin protein may be abnormally stable. This may account for some aspects of the retarded differentiation program we observed in the Cdk5-/- mutant.
The picture of slowed or blocked development in the absence of Cdk5 is enhanced by the patterns of both TuJ1 (βIII tubulin) and Map-2 immunostaining in the three genotypes of mice we examined. In wild-type animals, βIII tubulin is first expressed in neurons as they exit their final mitosis. In the Cdk5-/- cortex, however, neurons are not reliably TuJ1-positive until they reach the cortical plate. In wild-type cortex, Map-2 is expressed in the cortical plate during the final stages of differentiation of neurons. In the Cdk5-/- mutant, however, there is virtually no Map-2 in either the intermediate zone or cortical plate. Cdk5 is known to be involved in the posttranslational modification of microtubule-associated proteins such as tau (Patrick et al., 1999) and Map1b (Pigino et al., 1997). We have not verified that Cdk5 acts directly on Map-2, but we note that serines 313, 1539, and 1783 are potential consensus sites for Cdk phosphorylation. In the aggregate, these data suggest that in the absence of Cdk5, neuronal cytoskeletal maturation is either blocked or severely retarded. Significantly, our studies of dissociated cortical cultures leads to an identical conclusion.
Cdk5-deficient neurons fail to exit the cell cycle
In the developing wild-type cortex, neurons in the ventricular and subventricular zones complete a terminal mitosis, migrate away from the ventricular zone, and begin to differentiate. Once they emigrate, they will never divide again. In the Cdk5-deficient cortex, we found not only a lack of advanced differentiation markers but also the continued neuronal incorporation of BrdU and expression of PCNA, Ki67, and cyclin D in the cortical plate and intermediate zone. We believe that these cells have lost their ability to fully suppress the cell cycle and may either die (Lee et al., 1994; Herrup and Busser, 1995; Busser et al., 1998; Herrup et al., 2004; Kuan et al., 2004) or remain in a less differentiated mitotically active state. The persistence of nestin is more consistent with the latter interpretation, but the presence of cells dying in the process of division (Fig. 5I,L) tends to support the former. In either case, the immature precursor-like state is found in vitro as well as in vivo. TuJ1-positive cells in Cdk5-/- cortical cultures continue to incorporate BrdU. As in vivo, this cell cycle activity is coupled with the failure of the cells to express Map-2a/b and little Map-2c.
In both the developing mouse and rat cortex, the Cdk5 protein is expressed at the beginning of the postmitotic migratory stage of development (Tsai et al., 1993; Wu et al., 2000). We propose that Cdk5 functions at these stages to suppress the cell cycle and promote the differentiation program of these early cells. The sequence of events underlying these effects, however, is unknown. The neurofilament proteins are well known targets of Cdk5, and as mentioned above, their phosphorylation may be a key step in their developmental program. Cdk5 is also known to interact with components of the cell cycle machinery such as cyclin D, PCNA, and cyclin E (Xiong et al., 1992; Zhang et al., 1993; Miyajima et al., 1995). Although Cdk5 will form complexes with other cyclins, none of these has been shown to have kinase activity. It is possible that the binding of other cyclins by Cdk5 buffers their access to “true” cell cycle kinases such as Cdk4 and Cdk6. This model is supported by our roscovitine treatment data (Fig. 9), which suggests that Cdk5 protein but not activity is required for the Cdk5-dependent cell cycle suppression. In contrast, kinase activity does appear to be required for the protein to promote differentiation. Alternatively, Cdk5 might catalyze a specific phosphorylation of pRB that tightens its binding of E2F family members and/or promotes its differentiation functions (Tsai et al., 1993; Lee et al., 1994, 1997). Indeed, in the developing brain, there is an inverse relationship between Cdk5 expression and Cdk2, cdc2, and cyclin B expression (Tsai et al., 1993). A third possible mechanism by which Cdk5 could suppress cell cycle re-entrance would be through the phosphorylation of RNA polymerase (Sausville, 2002) or transcription factors such as mSds3 (Li et al., 2004). If a specific population of messages were affected, cell cycling might be forced to stop.
The role of Cdk5 in neuronal differentiation proposed here finds analogy in the requirement of Cdk5 activity for some cells to advance to the senescent state. Neurons are considered to be quiescent; they do not divide after becoming differentiated. Many cells reach a senescent state in which they are fully differentiated and do not divide again. This failure to divide can be overridden in specific situations such as DNA damage or mutations leading to cancer. In cell lines, the introduction of either Cdk5 short-interfering RNA or the Cdk5 inhibitor roscovitine results in a loss of the senescent phenotype (Alexander et al., 2004). The implication is that Cdk5 activity is required to maintain the nondividing senescent state. The means by which the kinase achieves this are unknown; however, it may be noteworthy that changes in the levels of Cdk5 activity are linked to cytoskeletal changes such as actin polymerization via Rac1 activity. In a neuron, we might speculate that because of its elaborate morphological state, coupled with the life-long cessation of cell division, the cell biology is analogous to the senescent state in fibroblasts and other cell lines. Perhaps, just as in cell lines (Alexander et al., 2004), Cdk5 may promote differentiation through its interaction with these components of the neuronal cytoskeleton, and that loss of Cdk5 can lead to loss of a differentiated phenotype and return to a state in which cell division is possible.
Although Cdk5-/- neurons fail to exit cell cycle, we have no evidence that suggests they are capable of completing mitosis. There are no increases in cell number either in vivo or in vitro, and we found only one instance (among thousands of cells examined) of a mitotic figure in a neuron-like (TuJ1-positive) cell. We propose that the “cycling” neurons we observed in the Cdk5-/- cortical plate (and intermediate zone) are in the process of cell death. In this process, they are similar to the target-deprived neurons of the olivocerebellar system (Herrup and Busser, 1995) and the neurons lost in the Levine stroke model (Kuan et al., 2004). The findings are also consistent with work reported by Li et al. (2002) who showed that cultured Cdk5-/- neurons were more susceptible to apoptotic stimuli.
Transfection of a Cdk5 expression construct into cultured Cdk5-/- neurons blocks their continued re-entrance into the cell cycle. Nearly identical results were observed in the non-neuronal cells in culture (of any genotype). This is consistent with the view that Cdk5 is both necessary and sufficient to block the cell cycle. In addition, failure to exit the cell cycle is concurrent with a decrease in neuronal differentiation as shown with the pattern of nestin, β-III tubulin, and Map-2 immunostaining. Finally, we note that Cdk5 is linked to the machinery of neuronal migration via Ndel1 (Niethammer et al., 2000; Shu et al., 2004; Tanaka et al., 2004), and that the same cellular processes control mitotic spindle formation and movement (Yan et al., 2003; Bond et al., 2005). Like migration, the mitotic process may be altered in Cdk5-deficient neurons. Together, our data provide evidence that Cdk5 has a dual role in development: to help maintain the state of mitotic arrest and to enable the normal process of neuronal differentiation.
Footnotes
This work was supported by grants from the National Institute of Neurological Disorders and Stroke (R01-NS20591), the National Institute on Aging (P50-AG08012), and the Blanchette Hooker Rockefeller Fund.
Correspondence should be addressed to Karl Herrup, Departments of Neurosciences and Neurology, Case Western Reserve University, 10900 Euclid Avenue, School of Medicine E504, Cleveland, OH 44106. E-mail: kxh26@case.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259658-11$15.00/0
References
- Alexander K, Yang HS, Hinds PW (2004) Cellular senescence requires CDK5 repression of Rac1 activity. Mol Cell Biol 24: 2808-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibb JA, Chen J, Taylor JR, Svenningsson P, Nishi A, Snyder GL, Yan Z, Sagawa ZK, Ouimet CC, Nairn AC, Nestler EJ, Greengard P (2001) Effects of chronic exposure to cocaine are regulated by the neuronal protein Cdk5. Nature 410: 376-380. [DOI] [PubMed] [Google Scholar]
- Bond JR, E Springell K, Lizarraga S, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO, Costa SMR, Baralle D, Raponi M, Karbani G, Rashid Y, Jafri H, Bennet C, Corry P, Walsh CA, Woods CG (2005) A centrosomal mechanism involving Cdk5Rap2 and CenpJ controls brain size. Nat Gen 37: 353-355. [DOI] [PubMed] [Google Scholar]
- Busser J, Geldmacher DS, Herrup K (1998) Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci 18: 2801-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CK, Karlo JC, Kao SC, Landreth GE (2001) β-Amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci 21: 1179-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH (2001) A decade of CDK5. Nat Rev Mol Cell Biol 2: 749-759. [DOI] [PubMed] [Google Scholar]
- Ferguson KL, Callaghan SM, O'Hare MJ, Park DS, Slack RS (2000) The Rb-CDK4/6 signaling pathway is critical in neural precursor cell cycle regulation. J Biol Chem 275: 33593-33600. [DOI] [PubMed] [Google Scholar]
- Gilmore EC, Herrup K (2001) Neocortical cell migration: GABAergic neurons and cells in layers I and VI move in a cyclin-dependent kinase 5-independent manner. J Neurosci 21: 9690-9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore EC, Ohshima T, Goffinet AM, Kulkarni AB, Herrup K (1998) Cyclin-dependent kinase 5-deficient mice demonstrate novel developmental arrest in cerebral cortex. J Neurosci 18: 6370-6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdane M, Bretteville A, Sambo AV, Schindowski K, Begard S, Delacourte A, Bertrand P, Buee L (2005) p25/Cdk5-mediated retinoblastoma phosphorylation is an early event in neuronal cell death. J Cell Sci 118: 1291-1298. [DOI] [PubMed] [Google Scholar]
- Hellmich MR, Pant HC, Wada E, Battey JF (1992) Neuronal cdc2-like kinase: a cdc2-related protein kinase with predominantly neuronal expression. Proc Natl Acad Sci USA 89: 10867-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, Busser JC (1995) The induction of multiple cell cycle events precedes target-related neuronal death. Development 121: 2385-2395. [DOI] [PubMed] [Google Scholar]
- Herrup K, Neve R, Ackerman SL, Copani A (2004) Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci 24: 9232-9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayouni R, Curran T (2000) Cortical development: Cdk5 gets into sticky situations. Curr Biol 10: R331-R334. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Schloemer AJ, Lu A, Burns KA, Weng WL, Williams MT, Strauss KI, Vorhees CV, Flavell RA, Davis RJ, Sharp FR, Rakic P (2004) Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci 24: 10763-10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, Tsai LH (2000) The role of the p35/cdk5 kinase in cortical development. Results Probl Cell Differ 30: 241-253. [DOI] [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A (1992) Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359: 288-294. [DOI] [PubMed] [Google Scholar]
- Lee EY, Hu N, Yuan SS, Cox LA, Bradley A, Lee WH, Herrup K (1994) Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes Dev 8: 2008-2021. [DOI] [PubMed] [Google Scholar]
- Lee KY, Helbing CC, Choi KS, Johnston RN, Wang JH (1997) Neuronal Cdc2-like kinase (Nclk) binds and phosphorylates the retinoblastoma protein. J Biol Chem 272: 5622-5626. [DOI] [PubMed] [Google Scholar]
- Li BS, Zhang L, Takahashi S, Ma W, Jaffe H, Kulkarni AB, Pant HC (2002) Cyclin-dependent kinase 5 prevents neuronal apoptosis by negative regulation of c-Jun N-terminal kinase 3. EMBO J 21: 324-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, David G, Hung KW, Depinho RA, Fu AK, Ip NY (2004) Cdk5/p35 phosphorylates mSds3 and regulates mSds3-mediated repression of transcription. J Biol Chem 279: 54438-54444. [DOI] [PubMed] [Google Scholar]
- Maccioni RB, Otth C, Concha II, Munoz JP (2001) The protein kinase Cdk5. Structural aspects, roles in neurogenesis and involvement in Alzheimer's pathology. Eur J Biochem 268: 1518-1527. [DOI] [PubMed] [Google Scholar]
- Miyajima M, Nornes HO, Neuman T (1995) Cyclin E is expressed in neurons and forms complexes with cdk5. NeuroReport 6: 1130-1132. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee MS, Morabito M, Tsai LH (2000) NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 28: 697-711. [DOI] [PubMed] [Google Scholar]
- Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH (1996) The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev 10: 816-825. [DOI] [PubMed] [Google Scholar]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB (1996) Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA 93: 11173-11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima T, Gilmore EC, Longenecker G, Jacobowitz DM, Brady RO, Herrup K, Kulkarni AB (1999) Migration defects of cdk5(-/-) neurons in the developing cerebellum is cell autonomous. J Neurosci 19: 6017-6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paglini G, Pigino G, Kunda P, Morfini G, Maccioni R, Quiroga S, Ferreira A, Caceres A (1998) Evidence for the participation of the neuron-specific CDK5 activator P35 during laminin-enhanced axonal growth. J Neurosci 18: 9858-9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402: 615-622. [DOI] [PubMed] [Google Scholar]
- Pigino G, Paglini G, Ulloa L, Avila J, Caceres A (1997) Analysis of the expression, distribution and function of cyclin dependent kinase 5 (cdk5) in developing cerebellar macroneurons. J Cell Sci 110: 257-270. [DOI] [PubMed] [Google Scholar]
- Sahlgren CMMA, Vaittinen S, Pallari H-M, Kalimo H, Pant HC, Eriksson JE (2003) Cdk5 regulates the organization of nestin and its association with p35. Mol Cell Biol 23: 5090-5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausville EA (2002) Complexities in the development of cyclin-dependent kinase inhibitor drugs. Trends Mol Med 8: S32-S37. [DOI] [PubMed] [Google Scholar]
- Shu TA, Ayala R, Nguyen M-D, Xie Z, Gleeson JG, Tsai L-H (2004) Ndel1 operates in a common pathway with Lis1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron 44: 263-277. [DOI] [PubMed] [Google Scholar]
- Smith DS, Greer PL, Tsai LH (2001) Cdk5 on the brain. Cell Growth Differ 12: 277-283. [PubMed] [Google Scholar]
- Tanaka TS, Serneo FF, Higgins C, Gambello MJ, Wynshaw-Boris A, Gleeson JG (2004) Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J Cell Biol 165: 709-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LH, Takahashi T, Caviness Jr VS, Harlow E (1993) Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development 119: 1029-1040. [DOI] [PubMed] [Google Scholar]
- Wu DC, Yu YP, Lee NT, Yu AC, Wang JH, Han YF (2000) The expression of Cdk5, p35, p39, and Cdk5 kinase activity in developing, adult, and aged rat brains. Neurochem Res 25: 923-929. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Zhang H, Beach D (1992) D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell 71: 505-514. [DOI] [PubMed] [Google Scholar]
- Yan XLF, Liang Y, Shen Y, Zhao X, Huang Q, Zhu X (2003) Human Nudel and NudE as regulators of cytoplasmic dynein in poleward transport along the mitotic spindle. Mol Cell Biol 23: 1239-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Xiong Y, Beach D (1993) Proliferating cell nuclear antigen and p21 are components of multiple cell cycle kinase complexes. Mol Biol Cell 4: 897-906. [DOI] [PMC free article] [PubMed] [Google Scholar]