

Abstract
Endocannabinoid release from a single neuron has been shown to cause presynaptic inhibition of transmitter release at many different sites. Here, we demonstrate that hypothalamic proopiomelanocortin (POMC) neurons release endocannabinoids continuously under basal conditions, unlike other release sites at which endocannabinoid production must be stimulated. The basal endocannabinoid release selectively inhibited GABA release onto POMC neurons, although exogenous administration of cannabinoid agonists also inhibited glutamate release. The CB1 cannabinoid receptor antagonist AM 251 [N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] blocked endocannabinoid-mediated inhibition of GABA release without affecting excitatory synaptic currents, whereas the CB1 receptor agonist WIN 55,212-2 [R-(+)-(2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrol [1,2,3-de]-1,4-benzoxazin-6-yl)(1-naphthalenyl) methanone monomethanesulfonate] inhibited both inhibitory and excitatory synaptic currents in POMC neurons. These data demonstrate that endogenously released cannabinoids and exogenously applied CB1 receptor agonists can have markedly different effects on synaptic inputs. Furthermore, the data suggest a novel form of endocannabinoid-mediated retrograde inhibition, whereby the regulation of a subset of inputs requires either the removal of tonic presynaptic inhibition caused by endocannabinoids or the engagement of a mechanism that actively inhibits endocannabinoid production.
Keywords: POMC, hypothalamus, GABA, presynaptic, retrograde, food intake
Introduction
Endocannabinoids are produced in and released from many types of neurons, generally in response to specific stimuli, and act via Gi/o-coupled receptors to retrogradely inhibit presynaptic neurotransmitter release (Piomelli, 2003). The production of endocannabinoids can be experimentally driven with high-frequency stimulation, although physiologically relevant stimulation can also be sufficient to drive production ex vivo (Brown et al., 2003). The influx of extracellular calcium is often necessary for the production of endocannabinoids (Di Marzo et al., 1994; Cadas et al., 1997), although calcium-independent production also occurs (Varma et al., 2001; Kim et al., 2002). Endocannabinoid actions are terminated by a combination of reuptake and degradation in both neurons and astrocytes (Beltramo et al., 1997; Hillard et al., 1997; Ligresti et al., 2004).
Hypothalamic arcuate nucleus proopiomelanocortin (POMC) neurons are crucial for maintaining energy balance (Coll et al., 2004) and respond to acute administration of cannabinoids with an increase in Pomc gene transcription (Corchero et al., 1999). In addition, feeding state alters both POMC neuron activity in terms of c-Fos induction (Elias et al., 1999; Shu et al., 2003) and POMC peptide release (Dube et al., 2000; Breen et al., 2005). Feeding state also affects the levels of endocannabinoids in the hypothalamus (Di Marzo et al., 2001; Kirkham et al., 2002). However, the potential synaptic effects of cannabinoids on POMC neurons have not been examined previously, and the identity of the neurons that produce endocannabinoids in the arcuate has remained unknown. Here, we demonstrate that POMC neurons are a source of endocannabinoids in the arcuate nucleus; however, interestingly, endogenous and exogenous cannabinoids differ significantly in their regulation of synaptic inputs to these neurons.
Materials and Methods
Animals. We bred Pomc-egfp (enhanced green fluorescent protein) homozygous mice (Cowley et al., 2001) congenic on the C57BL/6J strain with wild-type C57BL/6J (The Jackson Laboratory, Bar Harbor, ME) mice to produce the heterozygous mice used in these studies. All mice were housed under controlled temperatures (22-24°C) and a constant 12 h light/dark schedule and were given standard chow and tap water ad libitum. All experimental procedures met United States Public Health Service guidelines with the approval of the Institutional Animal Care and Use Committee of Oregon Health and Science University. All brain slices were prepared from 7- to 10-week-old male mice.
Electrophysiology. Whole-cell recordings were performed as described previously (Hentges et al., 2004), except that the internal recording solution contained the following (in mm): 57 KCl, 70 K+ methyl sulfate, 20 NaCl, 1.5 MgCl2, 0.1 EGTA, 10 phosphocreatine, 2 Mg-ATP, and 0.5 GTP, buffered with 5 HEPES, pH 7.3. Sagittal brain slices containing the hypothalamus were prepared from mice expressing enhanced green fluorescent protein under the control of the Pomc promoter to visualize POMC neurons. Slices were continuously perfused with oxygenated Krebs' solution at 37°C, and cells were held at -60 mV. GABAergic inputs were isolated with the addition of glutamate receptor blockers [0.1 μm (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK801) and 10 μm DNQX; Sigma, St. Louis, MO]. Evoked AMPA-mediated EPSCs (eEPSCs) were examined in the presence of MK801 and picrotoxin (100 μm; Sigma). Every 20 s, a stimulating current was applied, and the resulting postsynaptic current was recorded. Once the recording was stable, PSCs were evoked with a bipolar stimulating electrode in pairs (0.5 ms, 10 Hz) once every 20 s. The stimulus for evoking the PSCs varied depending on the preparation and the proximity to the stimulating electrode (between 50 and 200 μm dorsal to the cell). The maximum evoked current was reached, and then the stimulus was decreased to evoke <50% of the peak before beginning the experiment. Stable PSCs were obtained, and then either picrotoxin or DNQX was perfused for 5-10 min before the beginning of the experiment to isolate excitatory or inhibitory currents, respectively. Stimulation artifacts have been reduced or deleted from the presented traces for clarity. In the analysis of the paired-pulse data, the peak amplitudes were measured from the baseline just before the respective stimulus to compensate for incomplete recovery of the current between pulses. Miniature IPSCs were recorded in the presence of tetrodotoxin (TTX; 1 μm; Sigma), and mIP-SCs were detected and analyzed using Axograph software (Molecular Devices, Union City, CA). For stock solutions, all drugs were suspended in DMSO at 10,000 times the final concentration.
Statistics. For statistical analyses, paired Student's t tests were performed comparing the combined data from the last minute in control solution with the last minute in drug or the peak of drug response if it occurred earlier than 10 min.
Results
All results were obtained using brain slices from POMC-EGFP transgenic mice, which allowed the precise identification of fluorescently labeled POMC neurons before performing the whole-cell recording. The CB1 cannabinoid receptor antagonist N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM 251; 1 μm; Tocris Cookson, Ellisville, MO) increased the evoked GABAA-mediated IPSC (eIPSC) in POMC neurons (216 ± 31%) (Fig. 1A), suggesting the presence of local endocannabinoid tone. In the majority of POMC neurons, AM 251 approximately doubled the amplitude of the eIPSC, but four cells displayed an increase of <20%, and, in three cells, there was an increase of more than threefold (30 cells total were tested). The data shown in Figure 1 include all cells tested. Another CB1 receptor antagonist/inverse agonist, AM 281, had a similar effect (203 ± 24% increase; n = 5; data not shown). Conversely, the CB1 receptor agonist R-(+)-(2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl] pyrol[1,2,3-de]-1,4-benzoxazin-6-yl)(1-naphthalenyl) methanone monomethanesulfonate [WIN 55,212-2 (WIN) 1 μm; Tocris Cookson] reduced the eIPSC to 74 ± 7% of control (Fig. 1B). Endocannabinoid production can be driven by activation of metabotropic glutamate receptors (Maejima et al., 2001). However, metabotropic glutamate receptor (mGluR) activation was not necessary for endocannabinoid actions on POMC neurons because the nonselective group I/II mGluR antagonist α-methyl-4-carboxyphenylglycine (1 mm; Sigma) had no effect on the evoked IPSC in POMC neurons (p = 0.13; n = 6; data not shown).
Figure 1.

CB1 receptor activation alters GABAergic tone onto POMC neurons. IPSCs were evoked every 20 s, and three sweeps were combined to give a data point for each minute. The top panels represent the recordings from a single cell. The bottom panels illustrate the compiled normalized data (mean ± SEM) for the experiments. The insets show an average of three eIPSCs in control conditions (last minute of control) and in drug (between 9 and 10 min in drug). AM 251 increased the eIPSC (A; p < 0.01; n = 30) and WIN 55,212-2 inhibited the eIPSC (B; p < 0.01; n = 12). Error bars represent SEM.
Chelation of intracellular calcium with BAPTA (10 mm)inthe recording electrode blocked the increased eIPSC by AM 251 (Fig. 2A) and accentuated the inhibition by the CB1 agonist WIN 55,212-2 (36 ± 4% of control) (Fig. 2B). These data demonstrate that the POMC neurons themselves are the source of calcium-dependent endocannabinoid production. The larger reduction in eIPSCs by WIN 55,212-2 in the presence of BAPTA likely reflects the fact that there was less endocannabinoid present under this condition compared with when the low calcium-buffering internal pipette solution was used. The calcium required for the production of endocannabinoids is likely attributable to extracellular calcium influx because cyclopiazonic acid (10 μm, Sigma), which depletes intracellular calcium stores, had no effect on the eIPSC in POMC neurons (p = 0.69; n = 6; data not shown).
Figure 2.

Endocannabinoids are produced from POMC neurons by a calcium-dependent mechanism. In the presence of 10 mm BAPTA to chelate intracellular calcium, AM251 had no effect on the eIPSC (A;p = 0.15;n = 8). WIN55, 212-2 decreased the eIPSC in the presence of 10 BAPTA (B; p < 0.001; n = 6). The insets show an average of three eIPSCs in control conditions (last minute of control) and in drug (between 9 and 10 min in drug). The top panels represent a single cell; the bottom panels represent the mean ± SEM of the normalized data. Error bars represent SEM.
The constitutive release of endocannabinoids in the arcuate was primarily localized to POMC neurons because no neighboring non-EGFP cells tested responded to AM 251 (zero of eight cells tested) (Fig. 3A). The specificity of endocannabinoid action is consistent with previous reports of low CB1 receptor binding (Herkenham et al., 1991; Fernandez-Ruiz et al., 1997) and localized transcription of CB1 receptor message (Cota et al., 2003) in the hypothalamus. The lack of a response to AM 251 in non-POMC cells implies that these cells do not produce endocannabinoids constitutively, rather than implying an absence of CB1 receptors on inputs to non-POMC neurons because the CB1 agonist inhibited the evoked IPSC in these cells (70 ± 5% of control) (Fig. 3B)
Figure 3.

Cell-selective release of endocannabinoids in the arcuate. Nonlabeled neurons neighboring POMC neurons did not respond to AM 251 (A; p = 0.99; n = 8), but WIN 55,212-2 decreased the IPSC (B; p < 0.001; n = 4). The data represent the mean ± SEM. Three sweeps were averaged so that there is a data point for each min. The insets show an average of three eIPSCs in control conditions (last minute of control) and in drug (between 9 and 10 min in drug). Error bars represent SEM.
Treatments that alter the presynaptic release of transmitter usually result in a change in the paired-pulse ratio of postsynaptic currents. AM 251 increased the eIPSC and reduced the ratio of pulse2/pulse1 (Fig. 4A), consistent with a presynaptic action on the release of GABA. With low calcium buffering, WIN 55,212-2 had a small effect on the size of the first eIPSC and accordingly did not cause a significant change in the paired-pulse ratio (Fig. 4B). However, when calcium was highly buffered by BAPTA, and the effect of WIN 55,212-2 on the first eIPSC was larger, there was a concomitant significant increase in the paired-pulse ratio (Fig. 4C). Together, these results are consistent with a presynaptic effect of endocannabinoids on transmitter release. Additionally, AM 251 increased the frequency, but not the amplitude, of miniature IPSCs (mIPSCs) onto POMC neurons (Fig. 5), further indicating a presynaptic mechanism of action for the tonically released endocannabinoids.
Figure 4.
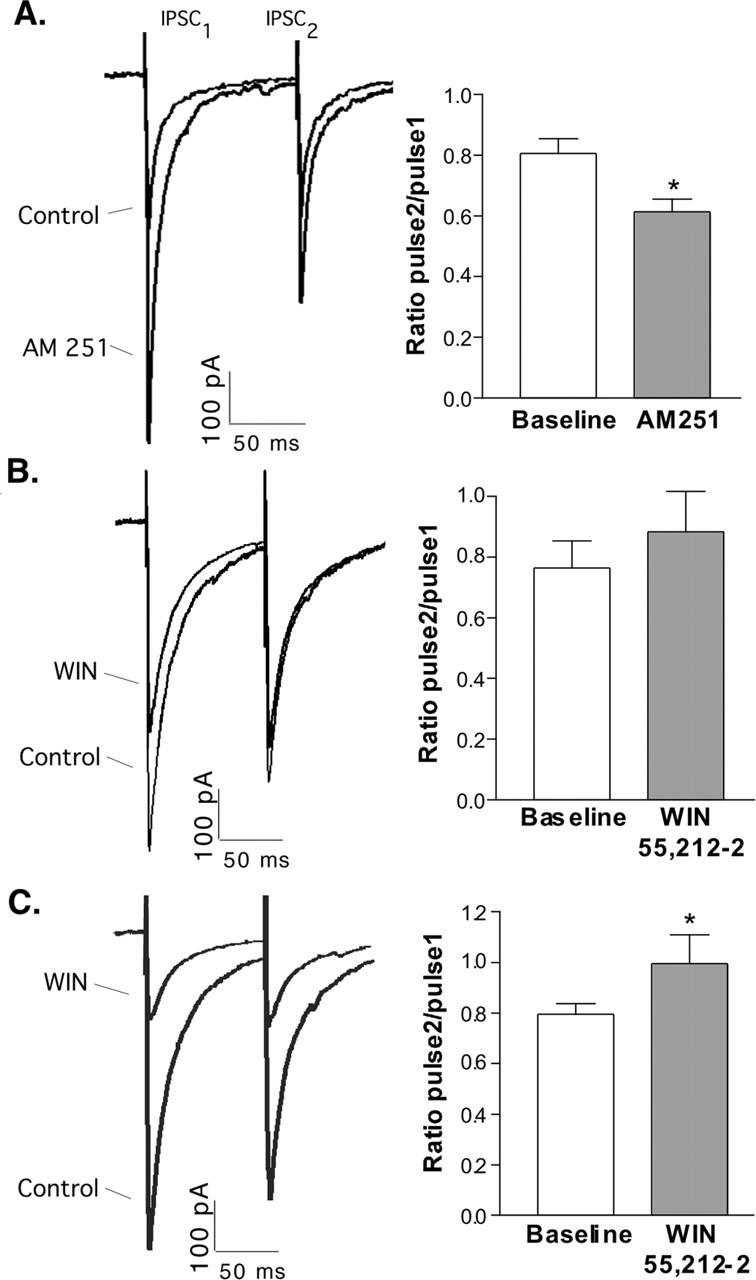
Retrograde inhibition of GABA release onto POMC cells. For each experiment, two stimuli were applied close together, as described in the text. AM 251 decreased the paired-pulse ratio from 0.81 ± 0.05 to 0.61 ± 0.04 (A; p < 0.001; n = 30). WIN 55,212-2 did not have a significant effect under normal calcium-buffering conditions (B; p = 0.09; n = 12) but increased the paired-pulse ratio from 0.80 ± 0.04 to 1.0 ± 0.11 when calcium was tightly buffered (C; p < 0.05; n = 6). The traces on the left show averages of three eIPSCs during the minute just before drug perfusion and three eIPSCs between 9 and 10 min in drug. The graphs on the right represent mean ± SEM. Error bars represent SEM.
Figure 5.

Frequency of miniature IPSCs onto POMC neurons is increased by AM251. Miniature IPSCs were isolated using TTX (1 μm) in addition to DNQX. One or two 30 s recordings were taken every 5 min. A, A representative trace recorded during the last minute in control (top trace) and during the last minute (between 9 and 10 min) in AM251 (1 μm; bottom trace). B, Compiled data from nine cells. Each 30 s recording in control conditions for a given cell displayed a steady number of events (5 and 10 min control; p > 0.01). AM 251 increased the number of events twofold within 10 min after the start of treatment (p < 0.01). The histogram in (C; data from all9 cells; numbers were taken from the 30 s recording made during the last minute in control or AM251) indicates that there was an overall increase in the number of mIPSCs but not the distribution of events induced by AM251. The first bar of the histogram represents all events up to 30 pA, and all other bars represent 10 pA bins. D, The same data as in C, plotted as cumulative frequency, indicate no difference in the size of the events resulting from the AM 251 treatment (p = 0.54). Error bars represent SEM.
To determine whether the effect of endocannabinoids on POMC neurons was selective for inhibitory inputs, evoked AMPA-mediated eEPSCs were examined in the presence of picrotoxin (100 μm; Sigma). AM 251 had no effect on eEPSCs (Fig. 6A). The lack of response to AM 251 could not be attributed to an absence of CB1 receptors on excitatory terminals because the CB1 agonist WIN 55,212-2 inhibited the evoked EPSC to 60 ± 7% of control (Fig. 6B).
Figure 6.

Excitatory terminals onto POMC neurons respond only to exogenous cannabinoids. Evoked excitatory PSCs were not altered by AM 251 in POMC cells (A; p = 0.72, n = 8). WIN 55,212-2 inhibited the evoked excitatory currents in POMC neurons (B; p = 0.02, n = 8). The data represent the mean ± SEM. Three sweeps were averaged so that there is a data point for each minute. The insets show an average of three eIPSCs in control conditions (last minute of control) and in drug (between 9 and 10 min in drug). Error bars represent SEM.
Although AM 251 did not affect the excitatory inputs to POMC neurons, perfusion of the cannabinoid reuptake transport blocker (5Z,8Z,11Z,14Z)-N-(4-hydroxy-2-methylphenyl)-5,8,11,14 eicosatetraenamide (VDM 11) (1 μm; Tocris Cookson) inhibited the eEPSC to 56.4% of control (Fig. 7A,B), similar to WIN 55,212-2 (Fig. 6B). The inhibition caused by VDM 11 was reversed with AM 251 (Fig. 7A,B), indicating that the inhibition of the eEPSC was attributable to an increase in the availability of endogenously released cannabinoids. When BAPTA was added to the internal recording solution to prevent endocannabinoid production (as shown in Fig. 2), there was no effect of VDM 11 on the eEPSC, although WIN 55,212-2 still decreased the eEPSC, and this was reversed with AM 251 (Fig. 7C). In 3 of 10 cells tested, VDM 11 did not change the amplitude of the eEPSC; however, in those cells, WIN 55,212-2 did decrease the eEPSC (Fig. 7D). The selective response to WIN indicated the presence of CB1 receptors and may suggest that excitatory inputs are situated away from areas that are affected by endocannabinoids, even when reuptake is blocked. Furthermore, the lack of an inhibition by VDM 11 in some cells that subsequently responded to WIN demonstrates that there is not a direct action of VDM 11 on the CB1 receptors. It appears that, for POMC-neuron-derived endocannabinoids to reach excitatory inputs, blockade of the transporter is necessary. Attempting to increase endocannabinoid release with prolonged depolarization (5 s to 0 mV) had no effect on the frequency (p = 0.09; n = 5) or amplitude (p = 0.11; n = 5) of spontaneous EPSCs onto POMC neurons or on evoked EPSCs (data not shown).
Figure 7.
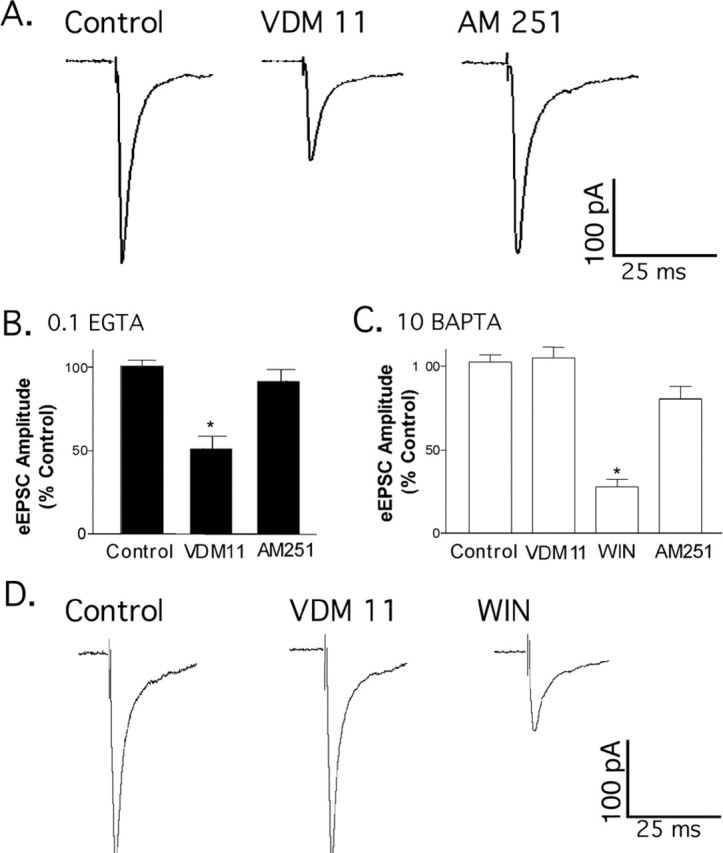
Blocking uptake extends the action of endocannabinoids to excitatory synapses. A, The endocannabinoid membrane transporter inhibitor VDM 11 reduced the amplitude of the evoked EPSC in POMC neurons, and AM251 reversed the effect of VDM 11. B, The compiled data are shown (n = 7; *p < 0.01) as a mean percentage of control ± SEM. C, VDM 11 had no effect on the eEPSC when BAPTA was present in the recording electrode to prevent the production of endocannabinoids (n = 8; p>0.1). WIN55, 212-2 inhibited the eEPSC after the application of VDM 11 in the presence of BAPTA (n = 5; *p < 0.01), indicating the presence of presynaptic CB1 receptors, and this effect was partially reversed by AM 251 (C; n=5). D, Traces from a cell that did not respond to VDM 11, although the low calcium-buffering internal solution was used. CB1 receptors were present because WIN 55,212-2 inhibited the eEPSC (D; right trace). Error bars represent SEM.
Discussion
In summary, POMC neurons in the slice preparation spontaneously released endocannabinoids in a calcium-dependent manner. There was no evidence that non-POMC cells constitutively release endocannabinoids. The endocannabinoids from POMC neurons acted as retrograde transmitters to selectively inhibit presynaptic GABA release. In contrast, exogenous CB1 receptor agonists inhibited both excitatory and inhibitory presynaptic inputs. POMC-derived endocannabinoids only affected excitatory terminals when local endocannabinoid concentrations were increased above baseline levels by the blockade of reuptake transporters.
Release of endocannabinoids from POMC neurons The preparation used in the current studies exhibited constitutive endocannabinoid tone. The consistent presence of endocannabinoids in the hypothalamus has been detected previously in tissue homogenates (Di Marzo et al., 2001), and it has been suggested that several actions of CB1 antagonists in vivo are mediated by the hypothalamus (Jamshidi and Taylor, 2001; Tucci et al., 2004; Verty et al., 2004). In the present study, endocannabinoid release was examined in the arcuate nucleus of the hypothalamus, and constitutive release was detected specifically from POMC neurons. POMC neurons in the slice released endocannabinoid in the absence of electrical stimulation, which has generally been necessary to detect synaptic endocannabinoid release experimentally from many other types of neurons. Unlike previous studies in which local endocannabinoid release was only detectable in the hypothalamus after pharmacological induction (Di et al., 2003), the response to AM 251 alone (Fig. 1A) demonstrates unstimulated, constitutive release of endocannabinoids consistent with the detection of endocannabinoids in hypothalamic tissue extracts (Di Marzo et al., 2001). The constitutive release of endocannabinoids to inhibit GABAergic input observed in the present study is similar to previous findings in the hippocampus (Losonczy et al., 2004) and suggests that basal inhibition of GABA release by endocannabinoids may serve as a tonic regulatory mechanism in the arcuate nucleus as well.
Constitutive activity of CB1 receptors in the absence of agonist binding cannot account for the inhibitory tone that was observed at GABAergic inputs to POMC neurons, because chelating intracellular calcium to halt endocannabinoid production blocked the effect of AM 251. The driving force or intrinsic property of POMC neurons responsible for the production of endocannabinoids remains to be discovered. However, it seems unlikely that endocannabinoid production in the arcuate is unique to the slice preparation because previous studies have also detected measurable endocannabinoid tone in the hypothalamus (Di Marzo et al., 2001; Kirkham et al., 2002). It is also possible that POMC neurons produce endocannabinoids at a rate consistent with other neurons but have an unusually low rate of endocannabinoid degradation and/or uptake.
Specificity of endocannabinoid release and actions
Constitutive endocannabinoid release was only apparent in POMC neurons. Other neurons adjacent to POMC neurons did not respond to AM 251. The functional basis for the selective constitutive endocannabinoid release from POMC neurons remains to be explored. Although the data do not indicate constitutive endocannabinoid release from non-POMC neurons, they do not rule out the possibility that other arcuate neurons may be able to release endocannabinoids after stimulation. Future studies to determine whether depolarization-induced suppression of inhibition or depolarization-induced suppression of excitation (DSE) can be produced in other arcuate neurons will be needed to address this possibility.
The origin of the CB1 receptor-expressing inputs to POMC neurons is not yet known. The arcuate is essentially a GABAergic nucleus, and CB1 receptors have not yet been colocalized with peptide markers in this region. In one study, CB1 receptor RNA message was colocalized with a few peptides expressed in the arcuate (Cota et al., 2003), but there was no CB1 message detected in the subpopulation of neurons that are thought to primarily inhibit POMC neuron function, the neuropeptide Y/AGRP (agouti-related protein)-expressing neurons in the arcuate. However, the GABAergic terminals onto POMC neurons could originate from extra-arcuate/hypothalamic sites. Several peptidergic neurons synapse onto POMC neurons, but there are only a few in which the classical transmitter coreleased has been identified. Therefore, speculating about the identity of CB1-containing inhibitory inputs is difficult. Nonetheless, it appears that nearly all POMC neurons must receive a significant number of endocannabinoid-sensitive GABAergic terminals because AM 251 strongly inhibited IPSCs in almost every cell examined in this study. These inputs could originate from a common source or may reflect origins from many cell types that express CB1 receptors.
Endogenous versus exogenous cannabinoid actions
Under basal conditions, endogenous cannabinoids from POMC neurons only inhibited presynaptic GABA release, although both inhibitory and excitatory inputs expressed functional CB1 receptors (Figs. 2B, 6B). The lack of endocannabinoid-induced inhibition at excitatory synapses could reflect a topographical relationship if these terminals are located farther away from sites of endocannabinoid production than the inhibitory terminals. However, if this is the case, the two types of inputs would have to be separated by a significant distance because endocannabinoids can diffuse distances in the range of 20 μm (Wilson and Nicoll, 2001). Alternatively, there could be a higher density of CB1 receptors at presynaptic inhibitory terminals than excitatory terminals, as reported previously in other brain regions (Matsuda et al., 1993; Marsicano and Lutz, 1999). Another possibility is that uptake and degradation of endocannabinoids selectively occludes their ability to reach the excitatory presynaptic terminals. Consistent with this hypothesis, blocking the anandamide membrane transporter with VDM 11 did allow for POMC-neuron-derived endocannabinoids to reach glutamatergic inputs (Fig. 7A). This raises the possibility that there could be physiologic conditions in which endocannabinoid production is increased to the extent that excitatory sites are affected. Alternatively, uptake or degradation of endocannabinoids could be a regulated step within arcuate circuits such that inhibition of these processes leads to increased endocannabinoid levels sufficient to access these excitatory release sites.
Although it is difficult to determine whether natural states exist to support endocannabinoid actions on excitatory terminals, it is clear that exogenous cannabinoids can inhibit presynaptic glutamate release onto POMC neurons (Fig. 6B). Ohno-Shosaku et al. (2002) demonstrated previously that in the hippocampus presynaptic cannabinoid sensitivity is different between excitatory and inhibitory inputs and determines the extent of depolarization-induced retrograde suppression. This raises the possibility that excitatory inputs to POMC neurons are not sensitive enough to cannabinoids to respond to the constitutive levels of endocannabinoids released from POMC neurons but respond only to higher levels of cannabinoids. Attempts to increase endocannabinoid release from POMC neurons to cause DSE were unsuccessful; however, stimuli >5 s at 0 mV were not tested, and stronger stimuli may be required to cause inhibition at presynaptic excitatory terminals (Ohno-Shosaku et al., 2002). Nonetheless, the data clearly demonstrate that the constitutive endocannabinoid tone from POMC neurons is sufficient only to inhibit presynaptic GABA release and not glutamate release.
Perspective
POMC neurons in the slice produced endocannabinoids that acted in a retrograde manner to inhibit the release of presynaptic GABA but not glutamate. Although there are a few other examples of sites in which endocannabinoids may be produced tonically (Losonczy et al., 2004) or in which exogenous and endogenous cannabinoid actions may differ (Robbe et al., 2003), the current data indicate that POMC neuron regulation involves both tonic endocannabinoid release and synapse-specific actions of the released endocannabinoids. This suggests an interesting level of regulation of POMC neurons whereby increasing the GABAergic tone that is tonically dampened by endocannabinoids would require the active inhibition of endocannabinoid production or action.
The study of POMC neuron activity has received much attention recently because of the prominent role that these neurons play in regulating energy balance. The role of cannabinoids in the regulation of food intake has also been the subject of many recent studies. The discovery that CB1 receptor antagonists can inhibit food intake and body weight has garnered much interest for potential pharmacologic use in weight loss. The mechanisms by which CB1 antagonists reduce food intake/body weight are essentially unknown. Although POMC neurons seem like a reasonable candidate to mediate CB1 receptor antagonist actions on weight loss, the current data may argue against this possibility because CB1 antagonists increase GABA-mediated IPSCs and would therefore be expected to inhibit the activity of POMC neurons, which, in general, inhibit food intake. There are numerous other hypothalamic and extrahypothalamic sites of endocannabinoid actions that may mediate the efficacy of CB1 receptor antagonist-induced weight loss, including areas that mediate aspects of food reward (Harrold and Williams, 2003; Sharkey and Pittman, 2005). The current data are consistent with the possibility that food intake induced by exogenous cannabinoids could result, at least in part, from the inhibition of excitatory inputs to POMC neurons. However, additional studies will be needed to determine whether the cellular regulation of POMC neurons by cannabinoids translates into the regulation of food intake or metabolism.
Footnotes
This work was supported by National Institutes of Health Grants DK062219 (S.T.H.), DK66604 (M.J.L.), and DA08163 (J.T.W.).
Correspondence should be addressed to Shane T. Hentges, Vollum Institute, Oregon Health and Science University, L-474, 3181 Southwest Sam Jackson Park Road, Portland, OR 97239. E-mail: hentgess@ohsu.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259746-07$15.00/0
References
- Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D (1997) Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science 277:1094-1097. [DOI] [PubMed]
- Breen TL, Conwell IM, Wardlaw SL (2005) Effects of fasting, leptin, and insulin on AGRP and POMC peptide release in the hypothalamus. Brain Res 1032:141-148. [DOI] [PubMed]
- Brown SP, Brenowitz SD, Regehr WG (2003) Brief presynaptic bursts evoke synapse-specific retrograde inhibition mediated by endogenous cannabinoids. Nat Neurosci 6:1048-1057. [DOI] [PubMed]
- Cadas H, di Tomaso E, Piomelli D (1997) Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl phosphatidylethanolamine, in rat brain. J Neurosci 17:1226-1242. [DOI] [PMC free article] [PubMed]
- Coll AP, Farooqi IS, Challis BG, Yeo GS, O'Rahilly S (2004) Proopiomelanocortin and energy balance: insights from human and murine genetics. J Clin Endocrinol Metab 89:2557-2562. [DOI] [PubMed]
- Corchero J, Manzanares J, Fuentes JA (1999) Repeated administration of delta9-tetrahydrocannabinol produces a differential time related responsiveness on proenkephalin, proopiomelanocortin and corticotropin releasing factor gene expression in the hypothalamus and pituitary gland of the rat. Neuropharmacology 38:433-439. [DOI] [PubMed]
- Cota D, Marsicano G, Tschop M, Grubler Y, Flachskamm C, Schubert M, Auer D, Yassouridis A, Thone-Reineke C, Ortmann S, Tomassoni F, Cervino C, Nisoli E, Linthorst AC, Pasquali R, Lutz B, Stalla GK, Pagotto U (2003) The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest 112:423-431. [DOI] [PMC free article] [PubMed]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ (2001) Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411:480-484. [DOI] [PubMed]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D (1994) Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372:686-691. [DOI] [PubMed]
- Di Marzo V, Goparaju SK, Wang L, Liu J, Batkai S, Jarai Z, Fezza F, Miura GI, Palmiter RD, Sugiura T, Kunos G (2001) Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 410:822-825. [DOI] [PubMed]
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG (2003) Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci 23:4850-4857. [DOI] [PMC free article] [PubMed]
- Dube MG, Pu S, Kalra SP, Kalra PS (2000) Melanocortin signaling is decreased during neurotoxin-induced transient hyperphagia and increased body-weight gain. Peptides 21:793-801. [DOI] [PubMed]
- Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK (1999) Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron 23:775-786. [DOI] [PubMed]
- Fernandez-Ruiz JJ, Munoz RM, Romero J, Villanua MA, Makriyannis A, Ramos JA (1997) Time course of the effects of different cannabimimetics on prolactin and gonadotrophin secretion: evidence for the presence of CB1 receptors in hypothalamic structures and their involvement in the effects of cannabimimetics. Biochem Pharmacol 53:1919-1927. [DOI] [PubMed]
- Harrold JA, Williams G (2003) The cannabinoid system: a role in both the homeostatic and hedonic control of eating? Br J Nutr 90:729-734. [DOI] [PubMed]
- Hentges ST, Nishiyama M, Overstreet LS, Stenzel-Poore M, Williams JT, Low MJ (2004) GABA release from proopiomelanocortin neurons. J Neurosci 24:1578-1583. [DOI] [PMC free article] [PubMed]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC (1991) Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci 11:563-583. [DOI] [PMC free article] [PubMed]
- Hillard CJ, Edgemond WS, Jarrahian A, Campbell WB (1997) Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J Neurochem 69:631-638. [DOI] [PubMed]
- Jamshidi N, Taylor DA (2001) Anandamide administration into the ventromedial hypothalamus stimulates appetite in rats. Br J Pharmacol 134:1151-1154. [DOI] [PMC free article] [PubMed]
- Kim J, Isokawa M, Ledent C, Alger BE (2002) Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci 22:10182-10191. [DOI] [PMC free article] [PubMed]
- Kirkham TC, Williams CM, Fezza F, Di Marzo V (2002) Endocannabinoid levels in rat limbic forebrain and hypothalamus in relation to fasting, feeding and satiation: stimulation of eating by 2-arachidonoyl glycerol. Br J Pharmacol 136:550-557. [DOI] [PMC free article] [PubMed]
- Ligresti A, Morera E, Van Der Stelt M, Monory K, Lutz B, Ortar G, Di Marzo V (2004) Further evidence for the existence of a specific process for the membrane transport of anandamide. Biochem J 380:265-272. [DOI] [PMC free article] [PubMed]
- Losonczy A, Biro AA, Nusser Z (2004) Persistently active cannabinoid receptors mute a subpopulation of hippocampal interneurons. Proc Natl Acad Sci USA 101:1362-1367. [DOI] [PMC free article] [PubMed]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M (2001) Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31:463-475. [DOI] [PubMed]
- Marsicano G, Lutz B (1999) Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci 11:4213-4225. [DOI] [PubMed]
- Matsuda LA, Bonner TI, Lolait SJ (1993) Localization of cannabinoid receptor mRNA in rat brain. J Comp Neurol 327:535-550. [DOI] [PubMed]
- Ohno-Shosaku T, Tsubokawa H, Mizushima I, Yoneda N, Zimmer A, Kano M (2002) Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J Neurosci 22:3864-3872. [DOI] [PMC free article] [PubMed]
- Piomelli D (2003) The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 4:873-884. [DOI] [PubMed]
- Robbe D, Alonso G, Manzoni OJ (2003) Exogenous and endogenous cannabinoids control synaptic transmission in mice nucleus accumbens. Ann NY Acad Sci 1003:212-225. [DOI] [PubMed]
- Sharkey KA, Pittman QJ (2005) Central and peripheral signaling mechanisms involved in endocannabinoid regulation of feeding: a perspective on the munchies. Sci STKE 2005:pe15. [DOI] [PubMed]
- Shu IW, Lindenberg DL, Mizuno TM, Roberts JL, Mobbs CV (2003) The fatty acid synthase inhibitor cerulenin and feeding, like leptin, activate hypothalamic proopiomelanocortin (POMC) neurons. Brain Res 985:1-12. [DOI] [PubMed]
- Tucci SA, Rogers EK, Korbonits M, Kirkham TC (2004) The cannabinoid CB1 receptor antagonist SR141716 blocks the orexigenic effects of intra-hypothalamic ghrelin. Br J Pharmacol 143:520-523. [DOI] [PMC free article] [PubMed]
- Varma N, Carlson GC, Ledent C, Alger BE (2001) Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci 21:RC188(1-5). [DOI] [PMC free article] [PubMed]
- Verty AN, McFarlane JR, McGregor IS, Mallet PE (2004) Evidence for an interaction between CB1 cannabinoid and oxytocin receptors in food and water intake. Neuropharmacology 47:593-603. [DOI] [PubMed]
- Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410:588-592. [DOI] [PubMed]