

Abstract
The opioid-like neuropeptide nociceptin/orphanin FQ (N/OFQ) and its receptor (NOP) are expressed in the substantia nigra (SN), a brain area containing dopamine neurons that degenerate in Parkinson's disease. Endogenous N/OFQ facilitates nigral glutamate release and inhibits nigrostriatal dopamine transmission and motor behavior. Here, we present evidence suggesting that endogenous N/OFQ may contribute to Parkinson's disease. Pharmacological blockade of the SN N/OFQ-NOP receptor system attenuated parkinsonian-like akinesia/hypokinesia in 6-hydroxydopamine hemilesioned or haloperidol-treated rats, whereas deletion of the NOP receptor gene conferred mice partial protection from haloperidol-induced motor depression. The antiparkinsonian action of NOP receptor antagonists was associated with reduction of glutamate release in the SN. In 6-hydroxydopamine hemilesioned rats, enhancement of N/OFQ expression and release was detected in the lesioned compared with the unlesioned SN, indicating that parkinsonism may be associated with overactivation of the N/OFQ-NOP receptor system in the SN. Finally, deletion of the N/OFQ gene conferred mice partial protection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced loss of SN dopamine neurons. Based on these data, we propose that NOP receptor antagonists may represent a novel approach for combined (symptomatic and neuroprotective) therapy of Parkinson's disease.
Keywords: J-113397, microdialysis, neuroprotection, nociceptin, Parkinson's disease, UFP-101
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disorder characterized by motor disturbances such as akinesia, bradykinesia, and tremor, often accompanied by cognitive impairment and depression. Loss of dopamine (DA) neurons projecting from the substantia nigra compacta (SNc) to the striatum, and consequent plastic changes in motor circuits (e.g., overactivation of the subthalamonigral glutamatergic pathway), underlie PD (DeLong, 1990). Pharmacological therapy of PD is presently aimed at symptomatic control because clinically effective neuroprotectants, capable of slowing the progression of nigral DA neuron degeneration, are yet to be identified. Symptomatic therapy is most effectively accomplished by replacing DA with levodopa; however, in a vast majority of PD patients, nonphysiological stimulation of DA receptors generates motor fluctuations (e.g., dyskinesias), which are responsible for a reduction of the clinical response over the years (Obeso et al., 2000). This has prompted the research for novel antiparkinsonian agents, possibly devoid of such disabling side effects and endowed with neuroprotective activity.
Nociceptin/orphanin FQ (N/OFQ) is an opioid-like neuropeptide (Meunier et al., 1995; Reinscheid et al., 1995), which activates the N/OFQ opioid (NOP) receptor (Cox et al., 2000). N/OFQ and its receptor are widely expressed in cortical and sub-cortical motor areas (Darland et al., 1998) and, particularly, in the SNc, in which 50% of DA neurons express NOP receptor mRNA, and 50-60% of N/OFQ neurons express glutamic acid decarboxylase mRNA, suggesting N/OFQ release from SNc GABA neurons (Norton et al., 2002). Exogenous N/OFQ inhibits activity of SNc DA neurons in vitro and nigrostriatal DA transmission in vivo (Marti et al., 2004a) and elevates glutamate (GLU) release in the SN reticulata (SNr) in vivo (Marti et al., 2002a). More relevant, endogenous N/OFQ tonically regulates these functions because selective peptide [[Nphe1,Arg14,Lys15]N/OFQ-NH2 (UFP-101)] (Calò et al., 2002) and nonpeptide [1-[3R,4R)-1-cyclooctylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3-dihydro-2H benzimidazol-2-one (J-113397)] (Kawamoto et al., 1999) NOP receptor antagonists facilitate nigrostriatal DA transmission and motor behavior (Marti et al., 2004a) and inhibit SNr GLU release (Marti et al., 2002a, 2004b). Based on preliminary evidence that an NOP receptor antagonist improves motor performance in haloperidol-treated rats, we recently suggested that endogenous N/OFQ contributes to PD symptoms (Marti et al., 2004b). To prove this concept, we tested whether (1) UFP-101 (experiment 1) and J-113397 (experiment 2) relieved parkinsonian-like symptoms in rats made akinetic/hypokinetic by unilateral lesion of SNc DA neurons with 6-hydroxydopamine (6-OHDA) (hemiparkinsonian rats) and (2) NOP receptor knock-out (NOP-/-) mice (Nishi et al., 1997) were resistant to haloperidol-induced akinesia (experiment 3). The mechanism underlying the antiakinetic action of NOP receptor antagonists was also investigated by measuring SNr GLU release in haloperidol-treated (experiment 4) or hemiparkinsonian (experiment 5) rats. Moreover, to test whether parkinsonism was associated with plasticity of the N/OFQ-NOP receptor system, preproN/OFQ (ppN/OFQ) and NOP receptor mRNA expression (experiment 6) as well as extracellular N/OFQ levels (experiment 7) were measured in the SN of hemiparkinsonian rats. Finally, to investigate whether endogenous N/OFQ plays a role in degeneration of SNc DA neurons, ppN/OFQ knock-out (ppN/OFQ-/-) mice (Koster et al., 1999) were challenged with toxic doses of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (experiment 8).
Materials and Methods
Animals used in the study (see below) were kept under regular lighting conditions (12 h light/dark cycle) and were given food and water ad libitum. The experimental protocols performed in the present study were approved by the Italian Ministry of Health (license number 71-2004-B) and by the Ethic Committee of the University of Ferrara or by the Institutional Animal Care and Use Committee of the Uniformed Services University of the Health Sciences.
Measurement of antiakinetic effects of UFP-101 (experiment 1) and J-113397 (experiment 2) in hemiparkinsonian rats
6-OHDA lesion. Unilateral lesion of DA neurons (Marti et al., 2002b) was induced in isoflurane-anesthetized male Sprague Dawley rats (150 g; Harlan Italy, S. Pietro al Natisone, Italy). Eight micrograms of 6-OHDA (in 4 μl of saline containing 0.2% ascorbic acid) were stereotaxically injected into the right SN [from bregma: anteroposterior (AP), -4.4 mm; mediolateral (ML), -1.2 mm; ventrodorsal (VD), -7.8 mm below dura (Paxinos and Watson, 1982)]. Sham-operated animals were unilaterally injected with the same saline/ascorbic acid solution (vehicle-injected). The rotational model (Ungerstedt and Arbuthnott, 1970) was used to select the rats that had been successfully lesioned. Two weeks after surgery, rats were injected with amphetamine (5 mg/kg, i.p., dissolved in saline just before use), and only those rats performing more than seven ipsilateral turns per minute were enrolled in the study. This behavior has been associated with >95% loss of striatal extracellular DA levels (Marti et al., 2002b). Experiments were performed 6-8 weeks after lesion.
Microinjection technique. A guide injection cannula (outer diameter, 0.55 mm) was stereotaxically implanted in isoflurane-anesthetized rats 0.5 mm above the right SNr [from bregma: AP, -5.5; ML, -2.2; VD, -7.3 below dura (Paxinos and Watson, 1982)] as described previously (Marti et al., 2004a). After surgery, rats were allowed 7 d to recover, and each rat was opportunely handled before behavioral tests. The day of the experiment, saline or UFP-101 was injected (0.5 μl) through a stainless steel injector (outer diameter, 0.30 mm) protruding 1 mm beyond the cannula tip. At the end of each experiment, the placement of the cannula was verified by microscopic examination.
Behavioral studies in rats. Motor activity in rats was evaluated by means of three behavioral tests specific for different motor abilities: (1) the bar test (Sanberg et al., 1988), which measures rat ability to respond to an externally imposed static posture; (2) the drag test [modification of the postural adjustment test (Lindner et al., 1999)], which measures rat ability to balance body posture using forelimbs in response to an externally imposed dynamic stimulus (dragging); and (3) the rotarod test, which measures rat ability to run on a rotating cylinder (Rozas et al., 1997).
Bar test. Each rat was placed gently on a table, and the contralateral and ipsilateral forepaws were placed alternatively on blocks of increasing heights (3, 6, and 9 cm). Total time (in seconds) spent by each paw on the blocks was recorded (cutoff time, 20 s).
Drag test. Each rat was gently lifted from the tail (allowing forepaws on the table) and dragged backward at a constant speed (∼20 cm/s) for a fixed distance (100 cm). The number of steps made by each paw was counted.
Rotarod test. The fixed-speed rotarod test was used according to a previously described protocol (Marti et al., 2004a). Briefly, rats were trained for 10 d to a specific motor task on the rotarod until their motor performance became reproducible in three consecutive sessions. Rats were tested (t0) at four increasing speeds (10, 15, 20, and 25 rpm for hemiparkinsonian rats; 30, 35, 40, and 45 rpm for vehicle-injected rats; 180 s each), causing a progressive decrement of performance to ∼40% of the maximal response (i.e., the experimental cutoff time). A similar response could be reproduced by applying this protocol in two different sessions performed 50 and 100 min later (t50 and t100). Thus, to quantify drug effect on motor behavior, drugs were administered 10 min before t50, and rotarod performance (total time spent on the rotarod) was calculated at t50 and t100 (i.e., 10 and 60 min after injection) as percentage of control (t0) performance. In some instances, rotational behavior was specifically measured. Rats were allowed to habituate in circular bowls for 20 min before the beginning of the test. Contralateral turns (i.e., turns in direction opposite to the injection side) were counted every 5 min, from 15 min before to 90 min after drug injection.
Measurement of haloperidol-induced akinesia in NOP+/+ and NOP-/- mice (experiment 3)
NOP-/- mice and wild-type (NOP+/+) controls (25-30 g) were generated on a mixed C57BL/6J and 129 genetic background (Nishi et al., 1997) and backcrossed with CD1 mice for nine generations, ensuring that >95% of their genetic background is of the CD1 type (Bertorelli et al., 2002). Akinesia was induced in mice by haloperidol (0.3-3 mg/kg, i.p.) and assessed using the rotarod, the drag, and the bar test every 60 min up to 8 h.
Nonhabituated mice were tested on the rotarod in a wide range of speeds (5-55 rpm) and total time spent on the rod calculated (180 s cutoff time). This protocol was carried out daily for 4 consecutive days (Marti et al., 2004a). The bar test was also performed as described above, using blocks of different heights (1, 3, and 6 cm).
Measurement of SNr GLU levels in haloperidol-treated (experiment 4) and hemiparkinsonian (experiment 5) rats
Microdialysis in haloperidol-treated rats. One probe of concentric design (1 mm dialyzing membrane; AN69; Hospal, Bologna, Italy) was implanted in the SNr (AP, -5.5; ML, ±2.2; VD, -8.3) of isoflurane-anesthetized rats. After surgery, rats were allowed to recover, and experiments were run 48 h after probe implantation.
Recovery and analysis of endogenous glutamate. The microdialysis probes were perfused at a flow rate of 3 μl/min with a modified Ringer's solution (in mm: 1.2 CaCl2, 2.7 KCl, 148 NaCl, and 0.85 MgCl2) and samples were collected every 15 min, starting 6 h after the onset of probe perfusion. Haloperidol was administered systemically (0.8 mg/kg, i.p.) and GLU levels monitored every 15 min up to 3 h using the bar test (see above). To correlate changes of GLU extracellular levels with motor activity, rats undergoing microdialysis were challenged in the bar test every 15 min (Marti et al., 2004b). GLU was measured by HPLC coupled to fluorimetric detection as described previously (Marti et al., 2002b). Fifteen microliters of o-phthaldialdehyde/mercaptoethanol reagent were added to 15 μl aliquots of sample, and 20 μl of the mixture were automatically injected (Triathlon autosampler; Spark Holland, Emmen, The Netherlands) onto a 5-C18 Chromsep analytical column (3 mm inner diameter, 10 cm length; Chrompack, Middelburg, The Netherlands) perfused at a flow rate of 0.75 ml/min (Beckman 125 pump; Beckman Instruments, Fullerton, CA) with a mobile phase containing 0.1 m sodium acetate, 10% methanol, and 2.2% tetrahydrofuran, pH 6.5. GLU was detected by means of a fluorescence spectrophotometer (RF-551; Shimadzu, Kyoto, Japan). The limit of detection for GLU was ∼150 fmol/sample. GLU in vitro recovery was 9.0 ± 0.3%.
Microdialysis in hemiparkinsonian rats. Two microdialysis probes were implanted bilaterally in the lesioned and unlesioned SNr of isoflurane-anesthetized hemiparkinsonsian rats (see above). Forty-eight hours after probe implantation, probes were perfused with a modified Ringer's solution, and sample collection started after a 6 h washout (see above). UFP-101 was perfused in the SNr through the probe, whereas J-113397 was given systemically (intraperitoneally). GLU was measured as detailed above.
Plasticity of the N/OFQ-NOP receptor system in hemiparkinsonian rats (experiments 6 and 7)
In situ hybridization (experiment 6). Rats were killed by decapitation under light diethyl-ether anesthesia. Their brains rapidly were removed, frozen in isopentane cooled in a dry ice/methanol bath, and stored at -70°C until use. Probes were prepared from a full-length cDNA insert cloned in a pBluescript SK(+) plasmid (proN/OFQ, from C. Mollereau, Institute of Pharmacology, Centre National de la Recherche Scientifique, Toulouse, France; NOP, from O. Civelli, Department of Pharmacology, University of California, Irvine, CA). The proN/OFQ plasmid was linearized with BamH1 and transcribed with T7 RNA polymerase to obtain antisense riboprobes or linearized with XhoI and transcribed with T3 RNA polymerase to obtain sense riboprobes. The NOP plasmid was linearized with NdeI and transcribed with T3 RNA polymerase to obtain antisense riboprobes or with T7 RNA polymerase to obtain antisense or sense riboprobes, respectively. All riboprobes were obtained by running the transcription assays in the presence of a[33P]-rUTP and hydrolyzed to fragments of ∼200 bp with sodium carbonate at 60°C (Bregola et al., 2002).
Twenty micrometer coronal sections at the SN level [-5.5 from bregma (Paxinos and Watson, 1982)] were thaw mounted onto polylysine coated slides, fixed in 4% paraformaldehyde, soaked in 3× PBS, rinsed in a graded ethanol series, dried, and stored at -20°C until use. Immediately before in situ hybridization, they were pretreated with proteinase K (1 μg/μl, 10 min, 37°C) and acetic anhydride (0.25% v/v, 10 min, room temperature).
In situ hybridization was performed as described previously (Simonato et al., 1996). Briefly, sections were incubated overnight at 52°C with 40 μl of hybridization mixture (50% deionized formamide, 2 sodium-Tris-EDTA, 5× Denhart's solution, 100 μg/ml single-stranded DNA, 100 μg/ml tRNA, 0.05% sodium pyrophosphate, and 60 ng/ml [33P]-riboprobe). They were then rinsed in 4× SSC, treated with RNase A (20 μg/ml, 30 min, 37°C), washed in 1× SSC for 10 min, 0.1× SSC at 52°C for 30 min, and 0.1× SSC at room temperature for 10 min, and dehydrated. Autoradiograms were generated by apposing these dried sections alongside [33P]-riboprobe standards to Kodak (Rochester, NY) BioMax MR film at -70°C for 30 d.
The mean total optical density within an area of interest was calculated by multiple sampling of that area in two sections taken from each animal using a digital analysis system (RBR Altair, Firenze, Italy). Background optical density in film areas not exposed to sections was subtracted from the total optical density. Finally, data have been expressed as percentage of optical density in the control (noninjected) side.
Recovery and analysis of endogenous N/OFQ (experiment 7). Bilateral implantation of microdialysis probes was performed as detailed above. The microdialysis probes were perfused (3 μl/min) with a modified Ringer's solution (for composition, see above) added with polypep 0.1%, captopril 300 μm, and BSA 0.3% (Aparicio et al., 2004). Samples were collected every 60 min, starting 3 h after the onset of probe perfusion. At least two stable values were obtained before testing the rats on the rotarod. Microdialysis samples were mixed with an equal volume of trifluoroacetic acid (TFA) (1% v/v) and loaded onto C18 cartridges (SEPCOL-1; Bachem, St. Helens, UK). Microcolumns were washed twice with 3 ml of 1% TFA and then eluted with 3 ml of 60% acetonitrile in 1% TFA. Eluates were lyophilized and stored at -70°C until radioimmunoassay. The adopted extraction procedure was validated by measuring the recovery of [3H-leucyl]N/OFQ (Amersham Biosciences Europe, Freiburg, Germany) under identical conditions. More than 95% of the tritiated N/OFQ added to a sample was recovered in the eluate (90-92% in the first milliliter).
N/OFQ-like immunoreactivity (N/OFQ-LI) present in microdialysis fractions was measured by a specific radioimmunoassay (Ploj et al., 2000). Lyophilized samples, reconstituted in 25 μl of methanol in 0.1% HCl (1:1), were mixed with 100 μ l of [125I]N/OFQ (Bachem) and 100 μl of N/OFQ antiserum (antiserum 96:2+, kindly supplied by Dr. I. Nylander, Department of Pharmaceutical Biosciences, Uppsala University, Uppsala, Sweden). The antiserum was used at the appropriate dilution to give 30-34% binding of the [125I]N/OFQ added (4800-5000 cpm). The labeled peptide and the antiserum were diluted in a gelatin buffer containing 0.15 m NaCl, 0.02% sodium azide, 0.1% gelatin, 0.1% Triton X-100, and 0.1% BSA in a 0.05 m sodium phosphate buffer, pH 7.4. RIA tubes were incubated at 4°C for 24 h. To separate free and antibody-bound peptide, 200 μl of a sheep anti-rabbit IgG antiserum (Decanting Suspension 3; Pharmacia SpA Diagnostic Division, Milan, Italy) were added, and the samples were incubated for 1 h at 4°C. After centrifugation for 20 min at 3000 × g, the radioactivity of the pellets was counted on a Beckman 5500 gamma counter. The limit of detection of the assay was 1 fmol. N/OFQ in vitro recovery was 2.0 ± 0.1%.
Measurement of MPTP toxicity in ppN/OFQ+/+ and ppN/OFQ-/- mice (experiment 8)
MPTP treatment and tyrosine hydroxylase immunostaining. Male homozygous wild-type (N/OFQ+/+) and knock-out (N/OFQ-/-) mice (Koster et al., 1999) of 2-3 months of age (∼20 g) were injected with four doses of MPTP, 20 mg/kg, intraperitoneally, at 90 min intervals. Seven days after the MPTP treatment, mice were anesthetized, perfused transcardially with 4% paraformaldehyde, and the brains were removed and processed for immunohistochemistry. Serial coronal sections (20 μm) of the midbrain through the SNc from -3.90 to -2.6 mm caudal to bregma and serial sections (30 μm) of the forebrain through the caudate-putamen (CP) at +1.10 mm anterior to bregma were cut at -22°C and stored at -70°C. Sections containing the SNc were preincubated in an albumen-blocking solution and then incubated overnight at room temperature with rabbit anti-tyrosine hydroxylase (TH) primary antibody (1:1000; Calbiochem, Darmstadt, Germany) and mouse anti-neuronal-specific nuclear protein (NeuN; 1:100; Chemicon, Temecula, CA) in blocking solution. After rinsing, the slides were incubated for 3 h in Alexa Fluor 555-labeled goat anti-rabbit IgG (1:200; Molecular Probes, Eugene OR) and Alexa Fluor 488-labeled goat anti-mouse IgG1 (1:200; Molecular Probes) in blocking solution. After additional rinsing, slides were dried and coverslipped with Vectashield Hard Set mounting medium containing 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) (Vector Laboratories, Burlingame, CA) to permit visualization of nuclei. Sections from mice of each genotype and/or treatment group were processed in parallel at the same time to eliminate variations in labeling efficiency. Similar techniques were used to visualize TH-immunoreactive nerve fibers in the CP, except that anti-NeuN IgG was not used.
Every fifth 20 μm section through SNc from -2.85 to -3.88 mm to bregma (Paxinos and Franklin, 2001) was viewed with a Leica (Nussloch, Germany) DM-RXA fluorescence microscope and scanned at 20× magnification with selective fluorescent filters (Y3 for TH; L5 for NeuN; and A4 for DAPI) to yield 36-46 frames. Montages were created for each scanned section using these frames permitting the entire cross section of the SNc to be viewed at every 100 μm intervals from the anterior to posterior (A-P) extent of the nucleus. The boundaries of the right SNc were outlined on each montage using anatomical structures around the SNc to assist in the definition of the boundary line. There was no significant difference between mice of each genotype and/or treatment in the areas defined as SNc at each level. All TH+/NeuN+-immunoreactive neurons contained within the SNc boundary line, in which the NeuN-stained nucleus was fully present in the plane of the section, were then counted at 1520× magnification. The diameters of SNc TH+ neurons are substantially smaller than the tissue section thickness (20 μm), ensuring that any counted cell (with full nucleus visible) was fully contained within the section under analysis, thus avoiding cell counting bias. Neuron counts at each level from -2.85 to -3.88 mm A-P were plotted graphically to display TH+ neuron distribution through the SNc. Total TH+ neuron counts were estimated from the area under the curve (AUC). Coronal CP sections at +1.10 mm A-P relative to bregma were scanned at 20× magnification using the Y3 filter to yield 36-46 frames from which a montage was created. Region of interest for estimation of fluorescence intensity were within square boxes of 655 × 655 μm superimposed within the dorsolateral or ventral CP area, and fluorescence intensity within this box was measured on an arbitrary unit scale. Background correction was based on fluorescence intensity measurements from an equivalent area of the adjacent cerebral cortex with minimal TH staining.
Metabolism of MPTP. CP or midbrain regions (including the SNc) were dissected from MPTP-treated mice and stored at -80°C. Twelve microliters per milligram of frozen tissue weight of a solution of 15 m 7-nitroindazole (7-NI) as internal standard in 90% H2O (containing 0.6% glacial acetic acid v/v)/10% acetonitrile was added, and the samples were sonicated for 1 min and then centrifuged. Supernatants were stored at -20°C overnight, thawed, centrifuged again, and the filtered supernatants taken for HPLC analysis (Castagnoli et al., 1997) using an Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA) with diode array detector. The Zorbax Eclipse XDB-C8 (Agilent Technologies), 5 μm, 4.6 × 150 mm column was eluted at 1 ml/min with mobile phase (15% acetonitrile: 85% H2O containing 0.6% glacial acetic acid and 1% triethylamine v/v). The eluate was monitored at three wavelengths simultaneously: 244 nm (for MPTP), 285 nm (for MPP+), and 360 nm (for 7-NI). Sample concentrations of MPTP and MPP+ were estimated by comparing the ratios of sample UV absorption peaks at the appropriate retention times for MPTP or MPP+ relative to the UV absorption of the internal standard 7-NI with equivalent ratios for MPTP and MPP+ relative to 7-NI for a series of calibration standards.
Uptake of [3H]MPP+ by synaptosomes from mouse CP. CP were dissected from ppN/OFQ+/+ and ppN/OFQ-/- mice and homogenized in 20 μl/mg tissue weight of cold 0.32 m sucrose to prepare a synaptosomal suspension that was centrifuged, rinsed, recentrifuged and resuspended in 8 ml of ice-cold Krebs'-Ringer's buffer. Aliquots (100 μl) containing 40-70 μg of protein in 1 ml of buffer were incubated in the absence or presence of MPP+ (500 μm final concentration) at 37°C. Uptake was initiated by addition of 5 nm [3H]MPP+I- (85 Ci/mmol; American Radiolabeled Chemicals, St. Louis, MO) and terminated after 5 min by addition of 3 ml of ice-cold buffer and filtration under vacuum through Whatman (Florham Park, NJ) GF/B or GF/C glass fiber filters. Rinsed filters were counted on liquid scintillation counter. Specific uptake of [3H]MPP+ was determined by subtracting retained counts in the presence of 500 μm unlabeled MPP+ from counts in its absence and expressed as femtomoles per milligram of protein, measured by the Lowry method.
Data presentation and statistical analysis
Motor performance in the bar and drag test is expressed as absolute number of seconds and touches, whereas, in the rotarod test, motor performance is expressed as percentage ± SEM of the control session (which represents an internal control for each rat). In microdialysis studies, GLU release has been expressed as percentage ± SEM of basal values (calculated as mean of the two samples before the treatment). In Figures 1, 2, 3, 4, 5, 6, 7, 8, 9 (and in Results), endogenous GLU levels for each group of rats are also given in absolute values (in nanomolar concentration). N/OFQ release is expressed as N/OFQ concentration in a 60 min dialysate fraction (femtomoles per 180 μl) and reported as N/OFQ-LI.
Figure 1.

Characterization of motor activity in vehicle-injected and 6-OHDA-injected (hemiparkinsonian) rats. A-C, Hemiparkinsonian rats displayed overall reduction in motor activity and motor asymmetry compared with vehicle-injected rats, as shown by an increase in the total time spent on the blocks in the bar test (the contralateral forepaw being more severely affected; A), reduction in the number of steps made by the contralateral forepaw in the drag test (B), and reduced rotarod performance (C). C, The rotarod performance of naive rats has also been reported. Data are mean ± SEM of 10-33 determinations obtained from 26 vehicle-injected, 33 hemiparkinsonian, and 18 naive rats. °°p < 0.01 versus the ipsilateral forepaw. ##p < 0.01 versus vehicle-injected and naive rats (ANOVA followed by Newman-Keuls post hoc test). Error bars represent SEM.
Figure 2.
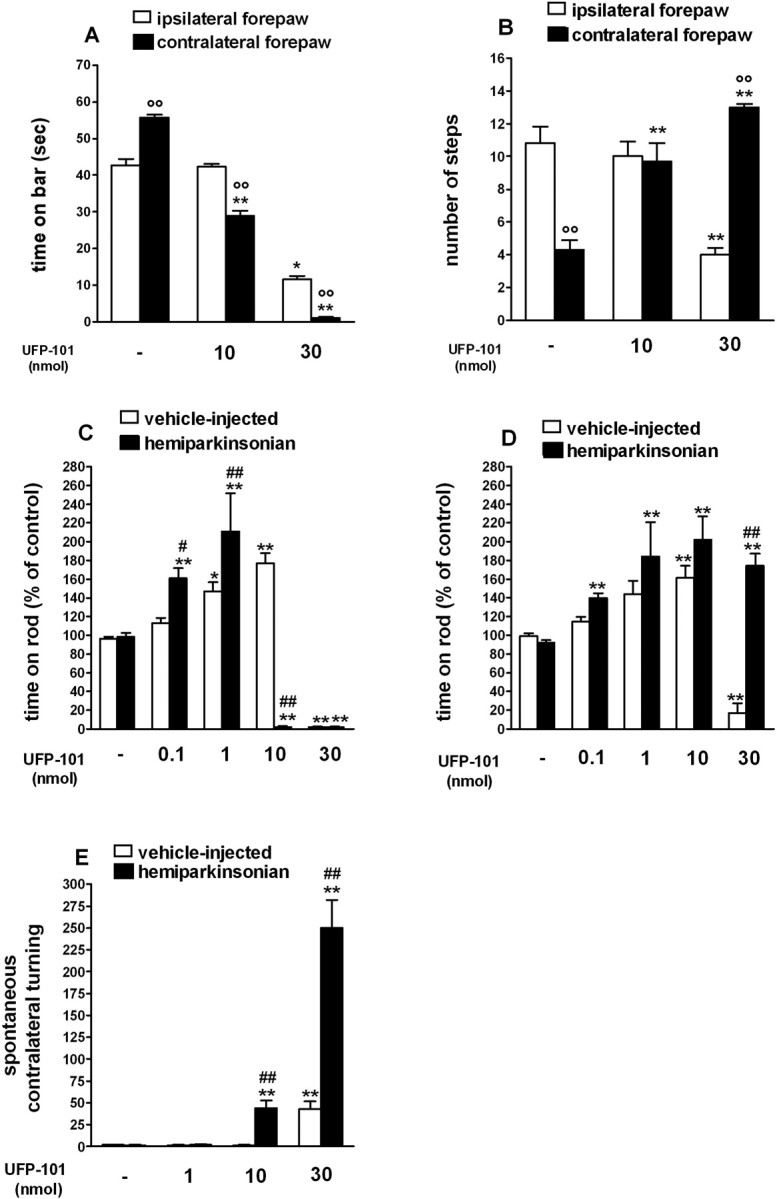
UFP-101 relieved akinesia/hypokinesia in hemiparkinsonian rats. A-E, Injection of UFP-101 (0.1-30 nmol in 0.5 μl) in the SNr reduced the time spent on the blocks in the bar test (in seconds; A), increased the number of steps of the contralateral forepaw in the drag test (B), improved overall motor performance (calculated as percentage of the control session) in the rotarod test (C, D), and induced contralateral rotations (E). A-D, The bar and drag tests (A, B) were performed 10 min after injection; the rotarod test was performed 10 min (C) and 60 min (D) after injection. Motor asymmetry was evaluated by separately measuring activity of the paws ipsilateral and contralateral (parkinsonian) to the lesioned side. E, Turning behavior was assessed by counting the number of rotations in the direction opposite to the injection side (i.e., contralateral) in 90 min. A-E, Data are mean ± SEM of 7-12 determinations obtained from 27 hemiparkinsonian and 30 vehicle-injected rats (A-D) or 36 hemiparkinsonian and 42 vehicle-injected rats (E). *p < 0.05; **p < 0.01 versus saline-treated rats. °p < 0.05; °°p < 0.01 versus the ipsilateral forepaw. #p < 0.05; ##p < 0.01 versus vehicle-injected rats (ANOVA followed by Newman-Keuls post hoc test). Error bars represent SEM.
Figure 3.

J-113397 and levodopa relieved akinesia/hypokinesia and attenuated motor asymmetry in hemiparkinsonian rats. A-D, Systemic (intraperitoneal) injection of J-113397 (0.1-3 mg/kg) or levodopa (1 mg/kg plus 15 mg/kg benserazide) reduced the time spent on the blocks (in seconds) and attenuated motor asymmetry in the bar test (A), increased the number of steps of the contralateral forepaw in the drag test (B), and improved overall motor performance (calculated as percentage of the control session) in the rotarod test (C, D). Motor asymmetry was evaluated by separate measures at the paws ipsilateral and contralateral (parkinsonian) to the lesioned side. The bar and drag tests (A, B) were performed 20 min after injection; the rotarod test was performed 20 min (C) and 70 min (D) after injection. A-D, Data are mean ± SEM of six to eight determinations obtained from 45 hemiparkinsonian and 35 vehicle-injected rats. *p < 0.05; **p < 0.01 versus saline-treated rats. °p < 0.05; °°p < 0.01 versus the ipsilateral paw. #p < 0.05; ##p < 0.01 versus vehicle-injected rats (ANOVA followed by Newman-Keuls post hoc test). Error bars represent SEM.
Figure 4.

Mice lacking the NOP receptor (NOP-/-) are more resistant to haloperidol-induced akinesia than wild-type (NOP+/+) mice. A-F, Mice were tested before (control) and after 0.3 mg/kg (A, C, E) or 0.8 mg/kg (B, D, F) haloperidol (intraperitoneal). A-F, Akinesia was measured for up to 8 h by using the bar (A, B), drag (C, D), and rotarod (E, F) tests. NOP-/- mice having received haloperidol were less akinetic than NOP+/+ mice. Data are expressed as means ± SEM of 7-11 determinations obtained from 22 NOP-/- and 22 NOP+/+ mice. **p < 0.01 versus control. °p < 0.05; °°p < 0.01 versus NOP+/+ mice (ANOVA followed by Newman-Keuls post hoc test). Error bars represent SEM.
Figure 5.

J-113397 normalized GLU release in the rat SNr and reversed haloperidol-induced akinesia. Rats were made akinetic with haloperidol (0.8 mg/kg, i.p.) and treated with J-113397 (1 mg/kg, i.p.) or saline when akinesia was fully instated (i.e., 75 min after haloperidol). A, B, In the same animals, J-113397 reduced SNr GLU release elevated previously by haloperidol (A) and attenuated akinesia, simultaneously evaluated by using the bar test (B). Two-way ANOVA revealed a significant haloperidol by J-113397 interaction. Data are expressed as means ± SEM of eight to nine determinations. Basal GLU (in nanomolar concentration) and AUC (arbitrary units; 75-180 min window) values were, respectively, as follows: 107.6 ± 14.5, 8830 ± 257 (saline n = 8); 110.7 ± 7.1, 8548 ± 252 (J-113397; n = 7); 102.6 ± 14.2, 17349 ± 1038 (haloperidol, n = 9); and 99.1 ± 11.0, 12128 ± 888 (haloperidol plus J-113397; n = 8). A significant correlation (r2 = 0.584; p < 0.0001) was found between reduction of immobility time and nigral GLU release. Error bars represent SEM.
Figure 6.
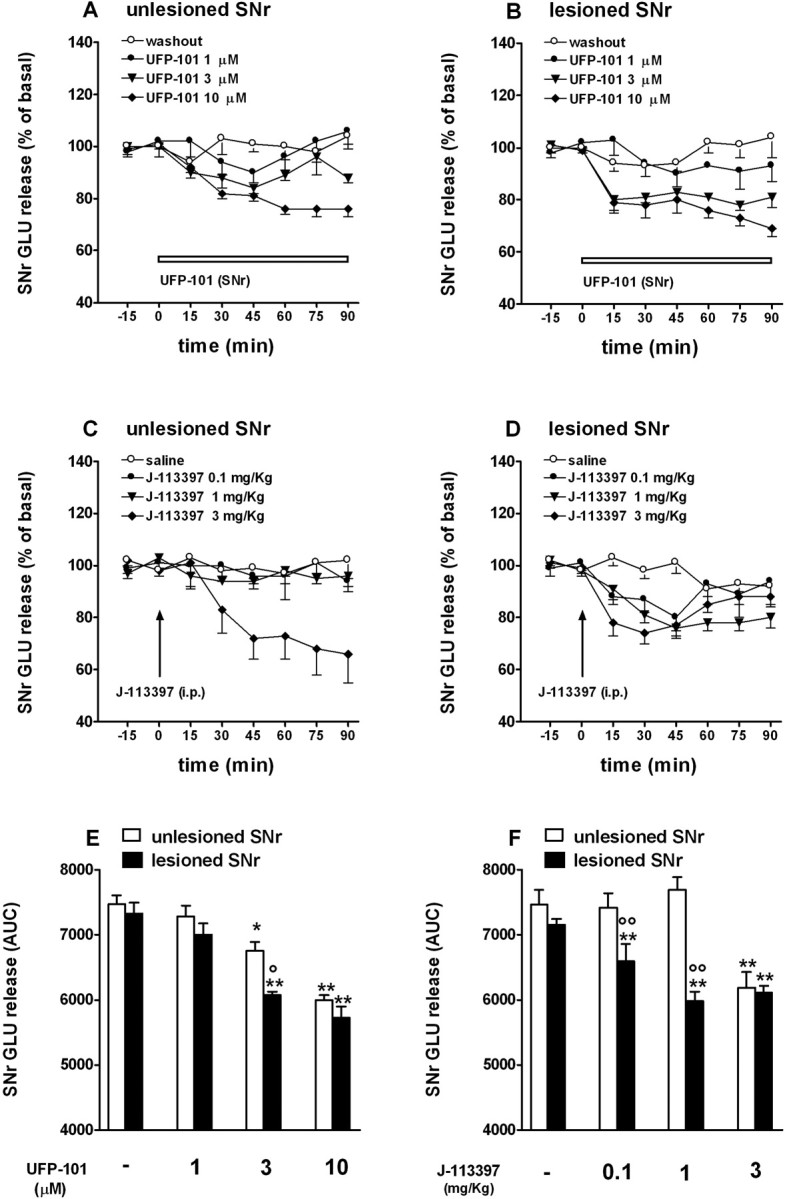
J-113397 and UFP-101 reduce GLU release in the SNr of hemiparkinsonian rats. A-D, Reverse dialysis of UFP-101 (1-10 μm; 60 min; A, B) or systemic administration of J-113397 (0.1-3 mg/kg, i.p.; C, D) reduced GLU extracellular levels in both the lesioned and unlesioned SNr of awake hemiparkinsonian rats. Data (means of 5-12 experiments) are expressed as percentage ± SEM of basal pretreatment levels (calculated as the mean of the two sample before the treatment; A, D) or AUC (arbitrary units in the 0-90 min window; E, F) values. Comparison between effects in the lesioned and unlesioned SNr has been made on AUC values. The probe location is shown in supplemental Fig. 2S (available at www.jneurosci.org as supplemental material). Basal GLU (in nanomolar concentration) values in the unlesioned and lesioned SNr were, respectively, as follows: E, 114.6 ± 16.5 (n = 7) and 107.9 ± 12.9 (n = 10; washout); 94.3 ± 10.1 (n = 8) and 95.3 ± 11.1 (n = 8; 1 μm UFP-101); 114.9 ± 17.6 (n = 5) and 103.6 ± 5.5 (n = 5; 3 μm UFP-101); 118.5 ± 10.7 (n = 12) and 103.2 ± 7.9 (n = 9; 10 μm UFP-101); F, 102.3 ± 7.4 (n = 6) and 89.7 ± 6.5 (n = 5; saline); 87.8 ± 7.6 (n = 6) and 100.6 ± 8.6 (n = 6; 0.1 mg/kg J-113397); 112.4 ± 12.4 (n = 11) and 92.7 ± 7.1 (n = 12; 1 mg/kg J-113397), 95.4 ± 14.0 (n = 6) and 101.4 ± 10.0 (n = 6; 3 mg/kg J-113397). *p < 0.05; **p < 0.01 versus saline-treated rats. °p < 0.05; °°p < 0.01 versus unlesioned SNr (ANOVA followed by Newman-Keuls post hoc test). Error bars represent SEM.
Figure 7.

Plasticity of the N/OFQ-NOP receptor system after 6-OHDA lesion. A, B, Relative optical density values of ppN/OFQ (A) and NOP (B) mRNA hybridization to riboprobes 7 weeks after injection of vehicle (vehicle-injected; open bars; n = 5) or 6-OHDA in the right SN (hemiparkinsonian; solid bars; n = 6) of rats. The results are expressed as mean ± SEM percentage of control signal (in the noninjected side) over SNc and SNr. Representative images of ppN/OFQ mRNA (below A) and NOP receptor mRNA (below B) expression in the SNc (arrow) and SNr (arrowheads) in hemiparkinsonian rats are presented. C, Extracellular levels of N/OFQ were measured by microdialysis in the lesioned and unlesioned SNr of nine hemiparkinsonian rats, 7 weeks after 6-OHDA. Microdialysis was performed at rest and under two 30 min rotarod sessions performed (15 rpm) at the beginning of the third and fourth collection periods. Data (mean ± SEM of 9 determinations) are expressed as N/OFQ-LI in femtomoles. **p < 0.01 versus pretreatment values. °°p < 0.01 versus unlesioned SNr. ##p < 0.01 versus vehicle-injected rats (ANOVA followed by Newman-Keuls post hoc test). Error bars represent SEM.
Figure 8.

Deletion of the ppN/OFQ gene attenuated the MPTP-induced loss of TH/NeuN-immunoreactive neurons in the SN. A, Total number of TH/NeuN-immunoreactive neurons in the SN after MPTP treatment in wild-type (ppN/OFQ+/+) and ppN/OFQ knock-out (ppN/OFQ-/-) mice. MPTP treatment produced a significantly greater decrease in the number of TH-positive cells in the ppN/OFQ+/+ mice (n = 8) than in ppN/OFQ-/- mice (n=7). B, Total TH-positive neurons throughout the rostral- caudal axis of the SN. *p < 0.05 versus saline-treated mice. #p < 0.05 versus ppN/OFQ+/+ mice (ANOVA followed by Newman-Keuls post hoc test). Error bars represent SEM.
Figure 9.

Deletion of the ppN/OFQ gene attenuates the MPTP-induced loss of TH immunoreactivity in the dorsolateral CP. Density of TH immunoreactivity in the rostral (1.10 mm from bregma) CP. No significant difference was detected between saline-treated wild-type (+/+) or knock-out (-/-) mice (2.39 ± 0.17 and 2.75 ± 0.30, respectively). For comparison purposes, all saline-treated animals were combined and termed control. A, B, MPTP produced a significant loss of TH staining in the dorsolateral (A), but not ventral (B), portion of the CP. MPTP significantly decreased TH staining in the dorsolateral CP of ppN/OFQ+/+ (n = 8), but not ppN/OFQ-/- (n = 7), mice. Representative images of the CP sections from saline- and MPTP-treated mice are also presented. *p < 0.05 versus control (ANOVA followed by Newman-Keuls post hoc test). Error bars represent SEM.
Statistical analysis was performed (GraphPad Prism; GraphPad Software, San Diego, CA) by one-way ANOVA on absolute data or AUC values, followed by the Newman-Keuls test for multiple comparisons. p values <0.05 were considered to be statistically significant.
Materials
Haloperidol was purchased from Tocris Neuramin (Bristol, UK), amphetamine, MPTP, and 6-OHDA bromide from Sigma (St. Louis, MO). UFP-101 and J-113397 were synthesized in our laboratories as reported previously (Marti et al., 2004a). All drugs were freshly dissolved in isoosmotic saline solution just before use.
Results
NOP receptor blockade relieves akinesia in hemiparkinsonian rats (experiments 1 and 2)
To study the antiparkinsonian potential of NOP receptor antagonists, UFP-101 and J-113397 were tested for ability to reverse motor disabilities in hemiparkinsonian rats, which most effectively reproduce parkinsonian phenotype in rodents (Gerlach and Riederer, 1996; Schwarting and Huston, 1996). Hemiparkinsonian rats displayed overall marked akinesia/hypokinesia compared with vehicle-injected rats (Fig. 1), as shown by multiple behavioral tests: increase in the total time spent on the blocks in the bar test (96.8 ± 1.6 and 2.0 ± 0.2 s, respectively; p < 0.001), reduction in the total number of steps in the drag test (14.2 ± 0.4 and 23.5 ± 0.4, respectively; p < 0.001), and reduced rotarod performance (406 ± 21 and 1044 ± 50 s in the 5-60 rpm speed range, respectively; p < 0.01). Hemiparkinsonian rats also displayed significant motor asymmetry because the contralateral forepaw spent greater time on the blocks (Fig. 1A) and performed lesser number of steps than the ipsilateral one (Fig. 1B). Vehicle-injected rats performed comparably to naive animals (Fig. 1C).
Effect of UFP-101 (experiment 1)
UFP-101 injection in the lesioned SNr improved motor performance, because it reduced the time spent on the blocks (F(5,48) = 405.4; p < 0.0001) (Fig. 2 A) and increased the number of steps (F(5,48) = 24.5; p < 0.0001) (Fig. 2 B) made by the contralateral paw. This effect was dose dependent, with ∼47% recovery of akinesia (Fig. 2 A) and reinstatement of motor symmetry (Fig. 2 B) observed with the 10 nmol dose at 10 min postinjection time. Thirty nanomoles of UFP-101 induced almost complete recovery of motor activity and better performance of the contralateral compared with the ipsilateral paw in the drag test. At this dose, however, UFP-101 stimulated spontaneous activity and induced marked contralateral rotations (F(7,71) = 28.48; p < 0.0001) (Fig. 2 E). UFP-101 produced biphasic effects on rotarod performance (F(9,58) = 34.29; p < 0.0001) (Fig. 2C), because it relieved akinesia at 0.1 and 1 nmol (∼60 and ∼100%, respectively) but produced motor impairment at 10 and 30 nmol. Turning behavior probably caused the rat to fall off the rod (Rozas et al., 1997). Indeed, when turning subsided (after 60-90 min), rats injected with low and high doses of UFP-101 showed better performances on the rotarod compared with saline (F(9,64) = 15.93; p < 0.0001) (Fig. 2 D). Although less sensitive, vehicle-injected rats also showed a biphasic response to UFP-101, which improved motor performance at 1-10 nmol and impaired it at 30 nmol (Fig. 2C,D), inducing modest contralateral turning (Fig. 2 E).
Effect of J-113397 (experiment 2)
To confirm that NOP receptor blockade reduced akinesia and facilitated motor activity, the nonpeptide NOP receptor antagonist J-113397 was tested, and its effects were compared with those of levodopa (Fig. 3). J-113397 (0.1-3 mg/kg, i.p.) or levodopa (1 mg/kg plus benserazide 15 mg/kg, i.p.) reduced the time spent on the blocks (F(9,81) = 28.81; p < 0.0001) (Fig. 3A) and increased the number of steps made by the contralateral paw (F(9,70) = 34.51; p < 0.0001) (Fig. 3B). Maximal effects were observed with the 1 mg/kg dose of J-113397. Both J-113397 and levodopa improved motor performance on the rotarod (F(9,59) = 29.35; p < 0.0001) (Fig. 3C). These effects were long lasting (F(9,59) = 21.07; p < 0.0001) (Fig. 3D). In hemiparkinsonian rats, improvement was detected early after injection of 1 and 3 mg/kg (Fig. 3C), although a delayed effect was also observed after injection of 0.1 mg/kg J-113397 (Fig. 3D). Again, vehicle-injected rats appeared less sensitive to systemic J-113397, because a significant enhancement of rotarod performance was only observed after the higher dose of J-113397 (3 mg/kg) (Fig. 3C,D). Likewise, vehicle-injected rats were insensitive to such a low dose of levodopa. J-113397 did not induce turning behavior in naive or lesioned rats at doses up to 3 mg/kg.
NOP-/- mice are resistant to haloperidol-induced akinesia (experiment 3)
To investigate the role of N/OFQ in sustaining parkinsonian-like akinesia, a different model of parkinsonism was used, which is based on acute blockade of postsynaptic DA receptors with haloperidol (Sanberg et al., 1988).
NOP-/- mice were challenged with different doses of haloperidol and akinesia/hypokinesia was evaluated in the bar, drag, and rotarod test (Fig. 4). NOP-/- mice did not exhibit differences in the immobility time compared with wild-type controls (1.0 ± 0.1 and 1.3 ± 0.2 s, respectively) (Fig. 4A), although displayed an increased number of steps (16.3 ± 0.1 and 13.8 ± 0.2, respectively; p < 0.01) (Fig. 4C) and improved overall performance on the rod (1063 ± 71 and 729 ± 55 s, respectively; p < 0.01) (see also Marti et al., 2004a). Haloperidol (0.3 mg/kg) significantly increased the immobility time (F(8,118) = 70.91; p < 0.0001) (Fig. 4A), decreased the number of steps (F(8,54) = 47.02) (Fig. 4C) and overall rotarod performance (F(8,54) = 22.9; p < 0.0001) (Fig. 4E) in NOP+/+ mice. Similar, but more intense effects, were evoked by haloperidol 0.8 mg/kg (Fig. 4B,D,F). In all tests, NOP-/- mice appeared more resistant to both doses of haloperidol compared with wild-type controls, as shown by the lower motor impairment and/or latency in onset of haloperidol effects. Rotarod performance of NOP-/- mice was even unaffected by the lower haloperidol dose. Difference between the two genotypes was less marked with 1.5 mg/kg haloperidol and disappeared with 3 mg/kg haloperidol (supplemental Fig. 1S, available at www.jneurosci.org as supplemental material).
NOP receptor antagonists reduce GLU release in the SNr (experiments 4 and 5)
PD is associated with overactivity of the subthalamic nucleus and increased glutamatergic excitatory drive to the SN (Bergman et al., 1990). Therefore, we tested whether UFP-101 and J-113397 reduce GLU release in the SNr of haloperidol-treated or hemiparkinsonian rats.
J-113397 normalizes SNr GLU release and attenuates akinesia in haloperidol-treated rats (experiment 4)
Basal GLU extracellular levels in the SNr (138.9 ± 12.7 nm; n = 26) were progressively elevated by haloperidol injection (Fig. 5A). J-113397 reduced haloperidol-evoked GLU release. Two-way ANOVA revealed main effects of haloperidol (F(1,28) = 56.01; p < 0.001) and J-113397 (F(1,28) = 14.91; p < 0.001) and significant haloperidol by J-113397 interaction (F(1,28) = 12.75; p = 0.0013). These changes of GLU release were associated with antiakinetic effect of J-113397 (Fig. 5B). Haloperidol progressively increased the immobility time during the bar test, whereas saline was without effect. Systemic J-113397 (1 mg/kg, i.p.), ineffective alone, transiently attenuated akinesia, showing maximal efficacy 45-75 min after injection. Statistical analysis revealed main effects of haloperidol (F(1,28) = 504.79; p < 0.001) and J-113397 (F(1,28) = 34.28; p < 0.001) and significant haloperidol by J-113397 interaction (F(1,28) = 33.4; p < 0.001). Statistical analysis on nigral GLU levels and degree of catalepsy, measured in the same animals (n = 32) at different time points (n = 14), revealed a significant correlation (r2 = 0.584; p < 0.0001) between the two variables.
UFP-101 and J-113397 reduce GLU release in the SNr of hemiparkinsonian rats (experiment 5)
GLU extracellular levels were not different in the unlesioned (110.4 ± 6.3 nm; n = 61) compared with lesioned (101.4 ± 4.6 nm; n = 61) SNr. UFP-101 perfusion (1-10 μm) reduced local GLU release in both SNr (F(7,53) = 23.8; p < 0.0001) (Fig. 6A,B). Maximal inhibition was similar but was achieved at lower (3 μm) UFP-101 concentrations in the lesioned side (Fig. 6E). Systemic J-113397 (0.1-3 mg/kg, i.p.) also inhibited GLU levels in the both the unlesioned and lesioned SNr (F(7,50) = 15.14; p < 0.0001) (Fig. 6C,D). However, J-113397 was more potent in the lesioned side, a significant inhibition being observed at 0.1 mg/kg (Fig. 6F).
Plasticity of the N/OFQ-NOP receptor system after 6-OHDA lesion (experiments 6 and 7)
The data described thus far provide strong evidence that blockade of NOP receptors can attenuate the symptoms of PD. One mechanism underlying this effect may be that PD is associated with an increase in N/OFQ release in the SN that would worsen the symptoms. To challenge this hypothesis, we used the 6-OHDA unilateral model, which offers the advantage of using the noninjected side as an internal control, and we analyzed the adaptive changes in ppN/OFQ and NOP gene expression, as well as in N/OFQ release, in the SN.
ppN/OFQ mRNA is increased, whereas NOP mRNA is decreased, in the lesioned SN (experiment 6)
In situ hybridization analysis revealed distribution patterns of ppN/OFQ and NOP mRNA was consistent with those described in previous studies (Neal et al., 1999; Norton et al., 2002). In particular, constitutive ppN/OFQ and NOP receptor gene expression was found in SNc and, at lower levels, in the SNr (data not shown). ppN/OFQ and NOP gene expression was then studied 7 weeks after unilateral injection of vehicle or 6-OHDA. No changes were observed between the injected and noninjected side in vehicle-injected animals. In contrast, 6-OHDA caused a highly significant increase in ppN/OFQ mRNA levels in the lesioned side: ppN/OFQ levels were approximately threefold higher in the SNc [consistent with the study by Norton et al. (2002)] and approximately twofold higher in the SNr (Fig. 7A). NOP mRNA levels decreased in a profound manner (∼50%) in the SNc and, to a lesser level (∼20%), in the SNr (Fig. 7B).
Endogenous N/OFQ levels are increased in the lesioned SNr (experiment 7)
To investigate whether changes of ppN/OFQ mRNA were associated with changes of extracellular endogenous N/OFQ levels and to study the dynamics of N/OFQ release under different motor conditions, N/OFQ levels in the SNr were measured by microdialysis at rest or under motor task (Fig. 7C). N/OFQ levels under resting conditions were approximately threefold higher in the lesioned (64.9 ± 10 fmol) than in the unlesioned (20.1 ± 2.4 fmol) SNr. Two 30 min rotarod sessions (at 15 rpm) consistently elevated N/OFQ extracellular levels both in the lesioned (F(3,8) = 6.251; p = 0.0027) and unlesioned (F(3,8) = 12.81; p < 0.0001) SNr, the increase being approximately fivefold greater in the lesioned than unlesioned SNr (net N/OFQ efflux over 2 h exercise periods, 147.6 ± 29.5 and 28.9 ± 8 fmol, respectively).
ppN/OFQ-/- mice are partially resistant to MPTP (experiment 8)
Finally, we thought to investigate whether N/OFQ may also play a role in DA neuronal loss. To pursue this aim, a different model was used that was thought to replicate those events underlying DA neuron loss in PD and is widely used for the study of neuroprotection in PD: the MPTP-treated mouse (Gerlach and Riederer, 1996; Nicotra and Parvez, 2002). SNc DA neuron toxicity after MPTP treatment was tested in ppN/OFQ-/- mice (Koster et al., 1999) compared with wild-type (ppN/OFQ+/+) mice with the same genetic background.
Numbers of TH-positive cells in SNc differed significantly between genotypes and treatments (F(3,19) = 30.9; p < 0.001) (Fig. 7). In ppN/OFQ+/+ mice, MPTP treatment reduced the number of TH-positive cells in the SNc to ∼34% of the number of TH-positive cells in saline-treated animals. In contrast, in ppN/OFQ-/- mice, the number of TH staining cells in the SNc after MPTP treatment was ∼65% of control levels. The number of TH-positive cells surviving MPTP treatment was significantly higher in ppN/OFQ-/- than ppN/OFQ+/+ mice (p < 0.001) (Fig. 8). The effects of MPTP treatment on the number of TH-positive cells in the SNc of ppN/OFQ-/- and wild-type mice were paralleled by MPTP-induced reductions in the intensity of TH staining in CP (Fig. 9). In wild-type mice, TH staining throughout the CP was substantially reduced at 7 d after MPTP treatment. In contrast, in ppN/OFQ-/- mice, MPTP treatment was associated with a much less substantial loss of TH staining of the terminals of DA neurons in CP (Fig. 9). To show that differences between genotypes in MPP+ production or uptake do not account for the neuroprotective effect of ppN/OFQ gene deletion against MPTP, MPP+ levels, and MPP+ uptake in the brains of mice was analyzed. MPP+ levels in the striatum of mice of each genotype at 2 h after the last MPTP dose were 51 ± 1 and 58 ± 8 pmol/mg tissue (NS; n = 4 per genotype) in wild-type and ppN/OFQ-/- mice, respectively. Moreover, [3H]MPP+ uptake in synaptosomes from the striatum of wild-type mice and ppN/OFQ-/- mice was 1.02 ± 0.17 and 0.96 ± 0.16 pmol/mg protein per 5 min, respectively (NS; n = 6 per genotype).
Discussion
Pharmacological or genetic blockade of NOP receptors in the SN attenuated parkinsonian-like akinesia/hypokinesia, whereas deletion of the ppN/OFQ gene conferred neuroprotection to SN DA neurons. This indicates that endogenous N/OFQ contributes to both symptoms and neurodegeneration associated with experimental parkinsonism, further suggesting that NOP receptors may represent a novel target in PD therapy.
The “gold standard” of PD therapy, levodopa, attenuated selective motor impairment in the limbs contralateral to the DA-depleted side and improved exercise-induced motor performance in hemiparkinsonian rats, suggesting the good predictivity of ethological tests used in the present study (see also Lundblad et al., 2003). UFP-101 and J-113397 reproduced levodopa effects, indicating that blockade of endogenous N/OFQ action on SNr NOP receptors can reverse parkinsonism. The different behavioral profile or UFP-101 and J-113397 may be explained on a pharmacodynamic (presence of splice variants of the NOP receptors) (Marti et al., 2003; Kuzmin et al., 2004) and/or pharmacokinetic (administration route) basis. In particular, J-113397 doses >3 mg/kg were not tested because of the possible loss of selectivity. Indeed, the active enantiomer of J-113397 was reported to increase DA release in NOP-/- mice (i.e., not dependent on NOP receptors) at 10 mg/kg (Koizumi et al., 2004). In the present study, a racemic mixture of J-113397 was used, which makes the possibility of an action beyond NOP receptor fairly remote. To confirm this view, the motor response produced by 10 mg/kg J-113397 racemic mixture in NOP+/+ mice was not observed in NOP-/- mice (M. Marti and M. Morari, unpublished data). Endogenous N/OFQ in the SNr acts as a physiological constraint of motor function (Marti et al., 2004a,b) and reduces DA transmission along the nigrostriatal axis (Marti et al., 2004a). Indeed, NOP receptor antagonists elevated rotarod performance and striatal DA release (Marti et al., 2004a). Thus, stimulation of residual nigrostriatal DA transmission may contribute to the antiakinetic effect of NOP receptor antagonists in hemiparkinsonian animals. Nevertheless, this action is more likely accomplished via DA-independent mechanisms. Indeed, DA loss enhanced the sensitivity toward NOP receptor antagonists, thereby lowering the threshold for motor-stimulating effects. This is suggestive of a functionally negative interaction between endogenous DA and N/OFQ in movement control, leading to increased N/OFQ inhibitory influence under parkinsonian conditions.
Pharmacological and genetic blockade of NOP receptors also attenuated akinesia induced by a DA antagonist (haloperidol), confirming that endogenous N/OFQ sustains hypolocomotion caused by impairment of DA transmission. NOP-/- mice do not behave differently from their control littermates in terms of spontaneous locomotion (Nishi et al., 1997; Murphy et al., 2002; Marti et al., 2004a) or pain threshold (Nishi et al., 1997; Bertorelli et al., 2002; Gavioli et al., 2003), although they display improved spatial learning (Manabe et al., 1998), rotarod performance (Marti et al., 2004a), and behavioral patterns similar to those induced by antidepressants in wild-type mice (Gavioli et al., 2003). The partial resistance of NOP-/- mice to haloperidol cannot be easily ascribed to changes in DA transmission, because striatal DA levels, at rest and after a motor stimulating dose of heroin, were unchanged in NOP-/- mice (Murphy et al., 2002). Conversely, the finding that both UFP-101 (Marti et al., 2004b) and J-113397 attenuated haloperidol-induced akinesia suggests that nigral mechanisms may be involved. It is noteworthy that J-113397 reversed haloperidol- and 6-OHDA-induced akinesia in the same dose range, suggesting that NOP receptor antagonists act on a neurobiological substrate underlying both types of parkinsonism. This may be clinically relevant, because classical antiparkinsonian drugs are scarcely effective in treating neuroleptic-induced parkinsonism.
Although acute DA receptor blockade with haloperidol and DA depletion with 6-OHDA produce parkinsonism through different mechanisms, they share overactivation of the subthalamic nucleus as a common final outcome. According to the classical scheme of basal ganglia functioning (DeLong, 1990), this ensues in increased glutamatergic activation of SNr GABAergic projection neurons, enhanced thalamic inhibition, and impairment of movement initiation. Because N/OFQ increased GLU release in the SNr (Marti et al., 2002a), NOP receptor antagonists likely reverse parkinsonism by depressing an N/OFQergic tone facilitating GLU release in this area. Indeed, both systemic J-113397 and intranigral UFP-101 (Marti et al., 2004b) attenuated akinesia by normalizing haloperidol-induced SNr GLU release. Such a correlation could not be detected in hemiparkinsonian rats, mainly because 6-OHDA lesion did not affect spontaneous GLU release. This was reported previously (Marti et al., 2002b; Galeffi et al., 2003; Robelet et al., 2004) and may be related to methodological reasons, such as the postlesion time at which experiments were performed (Meshul et al., 1999). Nevertheless, J-113397 and UFP-101 depressed SNr GLU levels within the same range of doses effective in facilitating motor performance. Selective blockade of overactive glutamatergic transmission in the SNr has been recognized as an efficient way to restore movement in parkinsonian animals (for review, see Starr, 1995). In parkinsonism models, NR2B-selective NMDA receptor antagonists show greater safety profile than broad-spectrum GLU receptor antagonists, although they only possess mild antiparkinsonian properties (Nash et al., 2004). Blockade of NOP receptor in the SNr may thus represent a novel and indirect way to normalize overactive glutamatergic transmission, with the great advantage that this effect is selectively accomplished in a brain area in which N/OFQ transmission is overactive. Indeed, ppN/OFQ mRNA expression in the SNc and SNr, and N/OFQ extracellular levels in the SNr (both at rest and under exercise), were enhanced after 6-OHDA lesions. Because DA loss resulted in increased N/OFQ tone and N/OFQ-mediated motor constraint (both at rest and under exercise), PD may represent a specific clinical indication for NOP receptor antagonist usage. 6-OHDA lesion was also associated with reduction of NOP receptor expression in the SNc [also reported by Norton et al. (2002)] and, to a lesser extent, in the SNr. Because 75% of NOP receptor expressing neurons in the SNc are TH positive (Norton et al., 2002), reduction of NOP receptor mRNA levels may reflect loss of DA neurons, i.e., structures that may not be involved in the antiparkinsonian action of NOP receptor antagonists. However, additional studies are required to investigate whether adaptive changes involving the residual NOP receptors (located on DA and non-DA structures in the SN) are implicated in the observed enhanced responsiveness of the parkinsonian brain to NOP receptor antagonists. The finding that N/OFQ is released under rotarod performance may also explain why stimulated motor activity is improved by pharmacological or genetic NOP receptor blockade (Marti et al., 2004a), whereas spontaneous locomotion is less affected (Marti et al., 2004a) or unaffected (Noda et al., 1998; Calò et al., 2002). This corroborates the hypothesis that N/OFQ inhibition of motor activity is greater under dynamic conditions (Marti et al., 2004a).
In addition to sustaining parkinsonian-like symptoms, endogenous N/OFQ also contributes to MPTP toxicity. Indeed, MPTP-treated ppN/OFQ-/- mice displayed higher number of surviving TH-positive cells in SN and fibers in the CP. N/OFQ, but not the other products of the ppN/OFQ gene (nocistatin and N/OFQ II), potentiated the excitotoxic white matter lesions induced by ibotenate via NMDA receptor activation (Laudenbach et al., 2001). Glutamatergic mechanisms have been implicated in MPTP toxicity. Indeed, the MPTP active metabolite (MPP+) inhibits mitochondrial complex I, resulting in a loss of intracellular ATP and generation of reactive oxygen species, which contribute to degeneration of DA neurons. Moreover, loss of ATP ultimately causes a fall in neuronal membrane potential, leading to impaired calcium homeostasis and enhanced sensitivity to GLU-mediated excitotoxicity (Nicotra and Parvez, 2002). In keeping with this notion, impairment of nigral GLU transmission via NMDA antagonists (Turski et al., 1991) or ablation of peduncolopontine glutamatergic/cholinergic inputs to the SNc (Takada et al., 2000) protect SNc DA neurons from MPTP toxicity. Because MPTP metabolism or MPP+ uptake were similar in ppN/OFQ-/- and ppN/OFQ+/+ mice it is possible that blockade of the indirect N/OFQ facilitatory drive on nigral GLU release reduces MPTP toxicity on DA neurons. Alternatively, we cannot rule out the possibility that N/OFQ acts postsynaptically on DA neurons to facilitate MPTP toxicity. However, loss of dopaminergic SH-SY5Y cells induced by MPP+ was not enhanced by N/OFQ coapplication (J. M. Brown and B. M. Cox, unpublished observations), supporting the notion of an indirect N/OFQ effect on DA neuronal survival.
Concluding remarks
In summary, we showed that (1) NOP receptor antagonists alleviate parkinsonian-like symptoms in rats; (2) deletion of the NOP receptor gene in mice confers partial resistance to haloperidol; (3) ppN/OFQ mRNA expression and N/OFQ extracellular levels in the SN increase after DA loss; and (4) deletion of the ppN/OFQ gene in mice partially reduces SNc DA neuron loss induced by MPTP. These data suggest that an NOP receptor antagonist endowed with an appropriate pharmacokinetic profile might both reduce the akinesia and retard the progressive loss of SNc DA neurons observed in PD patients. From a clinical perspective, such a drug would offer an antiparkinsonian action presumably accomplished via a novel mechanism (inhibition of nigral GLU release) and independent of direct DA receptor activation. This would provide a rational basis for association with existing therapies. Furthermore, it may offer an advantage over classical antiparkinsonian agents in treating neuroleptic DA-dependent catalepsy and other conditions of subthalamic nucleus overactivation (e.g., dystonia).
Footnotes
This work was supported by grants from the Italian Ministry of the University (Fondo degli Investimenti per la Ricerca di Base 2002; Progetti di Ricerca Scientifica di Rilevante Interesse Nazionale 2004), the University of Bologna (Parkinson's Disease Project “Pluriennale”), the Cassa di Risparmio di Ferrara Foundation, the Michael J. Fox Foundation for Parkinson's research, and the National Institute on Drug Abuse (United States Public Health Service #DA003102 to B.M.C.). We thank Shawn Gouty and John Rosenberger for skilled technical assistance and Hoffman-La Roche for their generous gift of breeding pairs of ppN/OFQ-/- mice. The opinions and assertions contained herein are the private opinions of the authors and are not to be construed as official or as a reflection of the views of the Uniformed Services University of the Health Sciences or the United States Department of Defense.
Correspondence should be addressed to Dr. Michele Morari, Department of Experimental and Clinical Medicine, Section of Pharmacology, and Neuroscience Center, University of Ferrara, via Fossato di Mortara 17-19, 44100 Ferrara, Italy. E-mail: m.morari@unife.it.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259591-11$15.00/0
References
- Aparicio LC, Candeletti S, Binaschi A, Mazzuferi M, Mantovani S, Di Benedetto M, Landuzzi D, Lopetuso G, Romualdi P, Simonato M (2004) Kainate seizures increase nociceptin/orphanin FQ release in the rat hippocampus and thalamus: a microdialysis study. J Neurochem 91: 30-37. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, DeLong MR (1990) Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science 249: 1436-1438. [DOI] [PubMed] [Google Scholar]
- Bertorelli R, Bastia E, Citterio F, Corradini L, Forlani A, Ongini E (2002) Lack of the nociceptin receptor does not affect acute or chronic nociception in mice. Peptides 23: 1589-1596. [DOI] [PubMed] [Google Scholar]
- Bregola G, Zucchini S, Rodi D, Binaschi A, D'Addario C, Landuzzi D, Reinscheid R, Candeletti S, Romualdi P, Simonato M (2002) Involvement of the neuropeptide nociceptin/orphanin FQ in kainate seizures. J Neurosci 22: 10030-10038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calò G, Rizzi A, Rizzi D, Bigoni R, Guerrini R, Marzola G, Marti M, McDonald J, Morari M, Lambert DG, Salvadori S, Regoli D (2002) [Nphe1,Arg14,Lys15]Nociceptin-NH2, a novel potent and selective antagonist of the nociceptin/orphanin FQ receptor. Br J Pharmacol 136: 303-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagnoli K, Palmer S, Anderson A, Bueters T, Castagnoli N (1997) The neuronal nitric oxide synthase inhibitor also inhibits the monoamine oxidase-B-catalized oxidation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Chem Res Toxicol 10: 364-368. [DOI] [PubMed] [Google Scholar]
- Cox BM, Chavkin C, Christie MJ, Civelli O, Evans C, Hamon MD, Hoellt V, Kieffer B, Kitchen I, Mcknight AT, Meunier JC, Portoghese PS (2000) Opioid receptors. In: The IUPHAR compendium of receptor characterization and classification (Girdlestone D, ed), pp 321-333. London: IUPHAR Media.
- Darland T, Heinricher MM, Grandy DK (1998) Orphanin FQ/nociceptin: a role in pain and analgesia, but so much more. Trends Neurosci 21: 215-221. [DOI] [PubMed] [Google Scholar]
- DeLong MR (1990) Primate models of movement disorders of basal ganglia origin. Trends Neurosci 13: 281-285. [DOI] [PubMed] [Google Scholar]
- Galeffi F, Bianchi L, Bolam JP, Della Corte L (2003) The effect of 6-hydroxydopamine lesions on the release of amino acids in the direct and indirect pathways of the basal ganglia: a dual microdialysis probe analysis. Eur J Neurosci 18: 856-868. [DOI] [PubMed] [Google Scholar]
- Gavioli EC, Marzola G, Guerrini R, Bertorelli R, Zucchini S, De Lima TC, Rae GA, Salvadori S, Regoli D, Calo G (2003) Blockade of nociceptin/orphanin FQ-NOP receptor signalling produces antidepressant-like effects: pharmacological and genetic evidences from the mouse forced swimming test. Eur J Neurosci 17: 1987-1990. [DOI] [PubMed] [Google Scholar]
- Gerlach M, Riederer P (1996) Animal models of Parkinson's disease: an empirical comparison with the phenomenology of the disease in man. J Neural Transm 103: 987-1041. [DOI] [PubMed] [Google Scholar]
- Kawamoto H, Ozaki S, Itoh Y, Miyaji M, Arai S, Nakashima H, Kato T, Ohta H, Iwasawa Y (1999) Discovery of the first potent and selective small molecule opioid receptor-like (ORL1) antagonist: 1-[3R,4R)-1-cyclooc-tylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one (J-113397). J Med Chem 42: 5061-5063. [DOI] [PubMed] [Google Scholar]
- Koizumi M, Sakoori K, Midorikawa N, Murphy NP (2004) The NOP (ORL1) receptor antagonist compound B stimulates mesolimbic dopamine release and is rewarding in mice by a non-NOP-receptor-mediated mechanism. Br J Pharmacol 143: 53-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster A, Montkowski A, Schulz S, Stube EM, Knaudt K, Jenck F, Moreau JL, Nothacker HP, Civelli O, Reinscheid RK (1999) Targeted disruption of the orphanin FQ/nociceptin gene increases stress susceptibility and impairs stress adaptation in mice. Proc Natl Acad Sci USA 96: 10444-10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin A, Sandin J, Terenius L, Ogren SO (2004) Evidence in locomotion test for the functional heterogeneity of ORL-1 receptors. Br J Pharmacol 141: 132-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudenbach V, Calo G, Guerrini R, Lamboley G, Benoist JF, Evrard P, Gressens P (2001) Nociceptin/orphanin FQ exacerbates excitotoxic white-matter lesions in the murine neonatal brain. J Clin Invest 107: 457-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner MD, Cain CK, Plone MA, Frydel BR, Blaney TJ, Emerich DF, Hoane MR (1999) Incomplete nigrostriatal dopaminergic cell loss and partial reductions in striatal dopamine produce akinesia, rigidity, tremor and cognitive deficits in middle-aged rats. Behav Brain Res 102: 1-16. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Vaudano E, Cenci MA (2003) Cellular and behavioural effects of the adenosine A2A receptor antagonist KW-6002 in a rat model of L-DOPA-induced dyskinesia J Neurochem 84: 1398-1410. [DOI] [PubMed] [Google Scholar]
- Manabe T, Noda Y, Mamiya T, Katagiri H, Houtani T, Nishi M, Noda T, Takahashi T, Sugimoto T, Nabeshima T, Takeshima H (1998) Facilitation of long-term potentiation and memory in mice lacking nociceptin receptors. Nature 394: 577-581. [DOI] [PubMed] [Google Scholar]
- Marti M, Guerrini R, Beani L, Bianchi C, Morari M (2002a) Nociceptin/orphanin FQ receptors modulate glutamate extracellular levels in the substantia nigra pars reticulata. A microdialysis study in the awake freely moving rat. Neuroscience 112: 153-160. [DOI] [PubMed] [Google Scholar]
- Marti M, Mela F, Bianchi C, Beani L, Morari M (2002b) Striatal dopamine-NMDA receptor interactions in the modulation of glutamate release in the substantia nigra pars reticulata in vivo: opposite role for D1 and D2 receptors. J Neurochem 83: 635-644. [DOI] [PubMed] [Google Scholar]
- Marti M, Stocchi S, Paganini F, Mela F, De Risi C, Calo G, Guerrini R, Barnes TA, Lambert DG, Beani L, Bianchi C, Morari M (2003) Pharmacological profiles of presynaptic nociceptin/orphanin FQ receptors modulating 5-hydroxytryptamine and noradrenaline release in the rat neocortex. Br J Pharmacol 138: 91-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Mela F, Veronesi C, Guerrini R, Salvadori S, Federici M, Mercuri NB, Rizzi A, Franchi G, Beani L, Bianchi C, Morari M (2004a) Blockade of nociceptin/orphanin FQ receptor signaling in rat substantia nigra pars reticulata stimulates nigrostriatal dopaminergic transmission and motor behavior. J Neurosci 24: 6659-6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Mela F, Guerrini R, Calo G, Bianchi C, Morari M (2004b) Blockade of nociceptin/orphanin FQ transmission in rat substantia nigra reverses haloperidol-induced akinesia and normalizes nigral glutamate release. J Neurochem 91: 1501-1504. [DOI] [PubMed] [Google Scholar]
- Meshul CK, Emre N, Nakamura CM, Allen C, Donohue MK, Buckman JF (1999) Time-dependent changes in striatal glutamate synapses following a 6-hydroxydopamine lesion. Neuroscience 88: 1-16. [DOI] [PubMed] [Google Scholar]
- Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, Butour JL, Guillemot JC, Ferrara P, Monsarrat B, Mazarguil H, Vassart G, Parmentier M, Costentin J (1995) Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature 377: 532-535. [DOI] [PubMed] [Google Scholar]
- Murphy NP, Lam HA, Chen Z, Pintar JE, Maidment NT (2002) Heroin-induced locomotion and mesolimbic dopamine release is unchanged in mice lacking the ORL.1 receptor gene. Brain Res 953: 276-280. [DOI] [PubMed] [Google Scholar]
- Nash JE, Ravenscroft P, McGuire S, Crossman AR, Menniti FS, Brotchie JM (2004) The NR2B-selective NMDA receptor antagonist CP-101,606 exacerbates L-DOPA-induced dyskinesia and provides mild potentiation of anti-parkinsonian effects of L-DOPA in the MPTP-lesioned marmoset model of Parkinson's disease. Exp Neurol 188: 471-479. [DOI] [PubMed] [Google Scholar]
- Neal Jr CR, Mansour A, Reinscheid R, Nothacker HP, Civelli O, Watson Jr SJ (1999) Localization of orphanin FQ (nociceptin) peptide and messenger RNA in the central nervous system of the rat. J Comp Neurol 406: 503-547. [PubMed] [Google Scholar]
- Nicotra A, Parvez S (2002) Apoptotic molecules and MPTP-induced cell death. Neurotoxicol Teratol 24: 599-605. [DOI] [PubMed] [Google Scholar]
- Nishi M, Houtani T, Noda Y, Mamiya T, Sato K, Doi T, Kuno J, Takeshima H, Nukada T, Nabeshima T, Yamashita T, Noda T, Sugimoto T (1997) Unrestrained nociceptive response and dysregulation of hearing ability in mice lacking the nociceptin/orphanin FQ receptor. EMBO J 16: 1858-1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda Y, Mamiya T, Nabeshima T, Nishi M, Higashioka M, Takeshima H (1998) Loss of antinociception induced by naloxone benzoylhydrazone in nociceptin receptor-knockout mice. J Biol Chem 273: 18047-18051. [DOI] [PubMed] [Google Scholar]
- Norton CS, Neal CR, Kumar S, Akil H, Watson SJ (2002) Nociceptin/orphanin FQ and opioid receptor-like receptor mRNA expression in dopamine systems. J Comp Neurol 444: 358-368. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Olanow CW, Nutt JG (2000) Levodopa motor complication in Parkinson's disease. Trends Neurosci 23: S2-7. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2001) The mouse brain in stereotaxic coordinates. San Diego: Academic.
- Paxinos G, Watson C (1982) The rat brain in stereotaxic coordinates. Sydney: Academic. [DOI] [PubMed]
- Ploj K, Roman E, Gustavsson L, Nylander I (2000) Basal levels and alcohol-induced changes in nociceptin/orphanin FQ, dynorphin, and enkephalin levels in C57BL/6J mice. Brain Res Bull 53: 219-226. [DOI] [PubMed] [Google Scholar]
- Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma Jr FJ, Civelli O (1995) Orphanin FQ: a neuropeptide that activates an opioid-like G protein-coupled receptor. Science 270: 792-794. [DOI] [PubMed] [Google Scholar]
- Robelet S, Melon C, Guillet B, Salin P, Kerkerian-Le Goff L (2004) Chronic L-DOPA treatment increases extracellular glutamate levels and GLT1 expression in the basal ganglia in a rat model of Parkinson's disease. Eur J Neurosci 20: 1255-1266. [DOI] [PubMed] [Google Scholar]
- Rozas G, Guerra MJ, Labandeira-Garcia JL (1997) An automated rotarod method for quantitative drug-free evaluation of overall motor deficits in rat models of parkinsonism. Brain Res Brain Res Protoc 2: 75-84. [DOI] [PubMed] [Google Scholar]
- Sanberg PR, Bunsey MD, Giordano M, Norman AB (1988) The catalepsy test: its ups and downs. Behav Neurosci 102: 748-759. [DOI] [PubMed] [Google Scholar]
- Schwarting RKW, Huston JP (1996) The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog Neurobiol 50: 275-331. [DOI] [PubMed] [Google Scholar]
- Simonato M, Bregola G, Donatini A, Bianchi C, Beani L, Ferri S, Romualdi P (1996) Kindled seizure-induced c-fos and prodynorphin mRNA expressions are unrelated in the rat brain. Eur J Neurosci 8: 2064-2067. [DOI] [PubMed] [Google Scholar]
- Starr MS (1995) Glutamate/dopamine D1/D2 balance in the basal ganglia and its relevance to Parkinson's disease. Synapse 19: 264-293. [DOI] [PubMed] [Google Scholar]
- Takada M, Matsumura M, Kojima J, Yamaji Y, Inase M, Tokuno H, Nambu A, Imai H (2000) Protection against dopaminergic nigrostriatal cell death by excitatory input ablation. Eur J Neurosci 12: 1771-1780. [DOI] [PubMed] [Google Scholar]
- Turski L, Bressler K, Rettig KJ, Loschmann PA, Wachtel H (1991) Protection of substantia nigra from MPP+ neurotoxicity by N-methyl-d-aspartate antagonists. Nature 349: 414-418. [DOI] [PubMed] [Google Scholar]
- Ungerstedt U, Arbuthnott GW (1970) Quantitative recording of rotational behaviour in rats after 6-hydroxydopamine lesions of the nigrostriatal dopamine system. Brain Res 24: 485-493. [DOI] [PubMed] [Google Scholar]