

Abstract
The primary mechanism responsible for Huntington's disease remains unknown. Postulated early pathogenic events include the following: impaired protein folding, altered protein degradation, mitochondrial dysfunction, and transcriptional dysregulation. Although related therapies can delay disease progression in mouse models, they target downstream and probably indirect effects of mutant-huntingtin expression. Accordingly, in case they prove beneficial in humans, they might only palliate some aspects of disease. Our previous studies in the Tet/HD94 conditional model and the recently reported efficacy of RNA interference against mutant huntingtin in another mouse model support silencing mutant-huntingtin expression as a valid therapeutic approach that has the advantage of targeting toxicity at its root. Here, we address whether gene silencing can still be beneficial in the late stages of disease with detectable striatal neuron loss. Stereological analysis was applied to determine an age at which Tet/HD94 mice show a decrease in the number of striatal neurons. Then, progression of neuropathology and motor phenotype were analyzed in mice that were allowed to continue expressing mutant huntingtin and in mice that no longer expressed it. Neuronal loss did not revert in gene-off mice, but the additional loss that takes place in gene-on mice was prevented. The total number of huntingtin-containing inclusions dramatically reverted, but a small fraction of inclusions positive for the amyloid dye thioflavin-S remained. Interestingly, despite a 20% decrease in striatal neurons and the presence of amyloid-like irreversible inclusions, gene-off mice fully recover from their motor deficit, thus ruling out amyloid-like huntingtin inclusions as the main toxic species and suggesting that gene-silencing therapies might work in late stages of disease.
Keywords: Huntington's disease, reversal, conditional mouse model, amyloid-like inclusions, stereology, neuronal loss
Introduction
Huntington's disease (HD) is an autosomal-dominant neurodegenerative disorder caused by a CAG triplet repeat expansion coding for a poly-glutamine (polyQ) sequence in the N-terminal region of the huntingtin (htt) protein (Huntington's Disease Collaborative Research Group, 1993). Patients suffer from motor dysfunction, cognitive decline, and psychological disturbances over 10-15 years until death, which is caused by brain atrophy mainly in the striatum and cortex (Vonsattel and DiFiglia, 1998).
At least eight additional CAG triplet repeat neurological diseases are caused by a polyQ expansion in their respective proteins (Zoghbi and Orr, 2000). All of these diseases share an interesting commonality: the presence of intraneuronal aggregates containing the expanded polyQ (Ross, 1997; Nakamura et al., 2001). However, the relevance of aggregates to etiology has been controversial. Initially, they were postulated to be the trigger for neurodegeneration, because both aggregation and pathogenicity are caused by the same polyQ length threshold (Scherzinger et al., 1997). Conversely, a recent study clearly establishes that the ability of cultured neurons to develop a htt-contain inclusion protects them from mutant-htt toxicity (Arrasate et al., 2004). However, it is still possible that levels of aggregation previous to inclusion body formation and that more mature and ordered aggregates such as amyloid-like microaggregates or protofibrils (Poirier et al., 2002; Ross and Poirier, 2004) might exert toxicity. In fact, blocking polyQ aggregation by anti-amyloid compounds has been shown to be protective in cell and mouse models of HD (Sanchez et al., 2003).
Independently of the aggregation status of htt, the molecular mechanism leading to HD pathogenesis remains unknown. In addition to impaired protein folding, alterations in protein degradation, mitochondrial dysfunction, and transcriptional dysregulation are among postulated early, but probably indirect, causes of disease (Landles and Bates, 2004). In this context, targeting expression of the pathogenic protein remains as the only approach that targets toxicity at its root. We were pioneers in anticipating the therapeutic potential of silencing mutant-htt expression by demonstrating full recovery of motor symptoms and inclusion neuropathology in the Tet/HD94 conditional mouse model (Yamamoto et al., 2000; Martin-Aparicio et al., 2001). More recently, silencing mutant htt by RNA interference (RNAi) also resulted in improved motor and neuropathological abnormalities in HD-N171-82Q mice (Harper et al., 2005), thus extending to humans the promise of silencing the pathogenic gene.
Interestingly, reversal in Tet/HD94 mice had been analyzed up to the age of 10 months at which stereological analysis still cannot detect neuronal loss despite striatal atrophy (Martin-Aparicio et al., 2001). Similarly, 4-month-old HD-N171-82Q mice like those used in the RNA interference experiments do not show detectable loss of neurons (Schilling et al., 1999). Therefore, previous gene-silencing positive results were achieved in early disease stages at which neuronal atrophy and dysfunction (without neuron loss) account for striatal atrophy and motor impairment. The question therefore remains whether improvement by silencing mutant htt would still be possible in advanced stages of disease characterized by neuronal loss. To test this, we performed stereological analysis in old Tet/HD94 mice. Once a decrease in the number of striatal neurons was found, phenotype progression was compared in mice allowed to continue expressing mutant htt and mice that no longer expressed it.
Materials and Methods
Animals
Tet/HD94 mice were generated as previously described (Yamamoto et al., 2000). Mice were bred at the Centro de Biología Molecular “Severo Ochoa” (Madrid, Spain). Four to five mice were housed per cage with food and water available ad libitum. For reversal experiments, doxycycline was added to the drinking water. Mice were maintained in a temperature-controlled environment on a 12 h light/dark cycle with light onset at 7:00 A.M.
Antibodies
The following antibodies were used: polyclonal anti-N-terminal mutant-huntingtin antibody CAG53b [amino acids 1-118 with a 51 polyQ stretch; a kind gift from Dr. E. Wanker (Berlin, Germany)], monoclonal anti-huntingtin MAB5374 (Chemicon, Temecula, CA), monoclonal anti-α-tubulin (Sigma, St. Louis, MO), polyclonal anti-ubiquitin (Dako-Cytomation, Carpinteria, CA), monoclonal anti-polyubiquitin conjugates (FK-2; Affinity, Golden, CO), polyclonal anti-dopamine- and cAMP-regulated phosphoprotein 32 (DARPP-32) (Chemicon), and monoclonal anti-neuronal nuclei (NeuN) (Chemicon).
Stereological analysis
Histology. Animals were transcardially perfused with 4% paraformaldehyde solution in 0.1 m sodium phosphate, pH 7.2. The brains were postfixed for 2 h in the same solution, cryoprotected in incrementing 10-30% sucrose/PBS, and frozen in dry-ice-cooled isopentane. Serial coronal cryostat sections (30 μm) were processed as free-floating for cresyl violet staining or immunohistochemistry as previously described (Canals et al., 2004). Briefly, to block endogenous peroxidases, sections were incubated for 30 min with PBS containing 10% methanol and 3% H2O2. Sections were then washed three times in PBS and blocked for 1 h with 1-3% normal serum in PBS. Tissue was then incubated with the primary antibodies in PBS containing 2% normal serum for 16 h at 4°C, except for DARPP-32, which was incubated at room temperature. The antibodies used were the following: anti-NeuN (1:100; Chemicon) and anti-DARPP-32 (1:10,000; Chemicon). Sections were washed three times and incubated with a biotinylated secondary antibody (1/200; Vector Laboratories, Burlingame, CA) for 2 h at room temperature in the same buffer as the primary antibody. The immunohistochemical reaction was developed using the Vectastain ABC kit (Vector Laboratories). No signal was detected in controls in which primary antibodies were omitted. DARPP-32-immunostained sections were also counterstained with Nissl dye.
Cell counting. Stereological quantitation was performed using an optical dissector/Cavalieri combination as previously described by Oorschot (1996). All cell counts were performed blind with respect to genotype. Stereological counts were obtained from the entire neostriatum using the Computer Assisted Stereology Toolbox (CAST) software (Olympus, Ballerup, Denmark). The dissector counting method was used to analyze every seventh coronal sections of the brain area containing the striatum or cortex. The counting frames were randomly sampled. The coefficient of variation was between 0.05 and 0.08. In Nissl staining, neurons were identified as cells that have a large- or medium-sized, pale spherical nucleus, which usually contained one prominent nucleolus (Oorschot, 1996).
Immunohistochemistry for inclusion body detection
Sagittal brain sections were pretreated for 1 h with 1% BSA, 5% FBS, and 0.2% Triton X-100, and then incubated with primary antibodies at the following dilutions: anti-ubiquitin (1/500), anti-polyubiquitin FK2 (1/200), or anti-huntingtin MAB5374 (1/200). Then, brain sections were incubated in avidinbiotin complex using the Elite Vectastain kit (Vector Laboratories). Chromogen reactions were performed with 0.05% DAB (Sigma) and 0.01% H2O2 for 10 min. Sections were coverslipped with Fluorosave (Merck, Darmstadt, Germany).
Quantification of immunostained inclusions was performed as follows. For each mouse (four mice per group), a series [21 sections; 60 μm apart; approximately spanning from interaural 1.44 to 1.56 mm lateral plates in Paxinos and Franklin (2001)] of sections (30 μm thick) was immunostained with either the anti-htt Ab or one of the anti-ubiquitin antibodies. Five (three cortical and two striatal) photomicrographs (200× magnification) were taken from each section [captured with a CCD camera (Coolsnap FX Color; Roper Scientific, Tucson, AZ)]. Cortical fields comprised layers III-VI of the primary motor cortex, the primary somatosensory cortex, and the mediomedial area of the secondary visual cortex. Striatal fields corresponded to anterodorsal and anteroventral Cpu. The number of inclusions was then counted for each field on blind-coded slides. Data are presented as mean ± SEM of the number of aggregates per 100 μm2 of either cortex or striatum. Significant differences were determined by ANOVA followed by Bonferroni's test correction.
Thioflavin-S staining of inclusion
For each mouse (four mice per group), a series (21 sections; 60 μm apart) of sections (30 μm thick) was stained with thioflavin-S as follows: sections were treated with 0.1% thioflavin-S in 50% ethanol in the dark for 10 min, and then briefly rinsed with distilled water and coverslipped on aqueous mounting solution.
Fluorescence images were captured with an Axioskop 2 Plus microscope (Zeiss, Oberkochen, Germany) and a CCD camera (Coolsnap FX Color) in the areas, and at the magnification, described for quantification of immunostained inclusions. Signal was considered positive if the mean intensity value was >140 on a 0-255 scale with 0 as white and 255 as black. The cutoff value of 140 was determined from visual analysis of immunolabeling. Data are presented as mean ± SEM of the number of aggregates counted for each brain region (cortex or striatum) per 30 μm section. Significant differences were determined by ANOVA followed by Bonferroni's test correction.
Tissue processing for electron microscopy
For electron microscopy, immunostained vibratome sections were processed as previously described (Lucas et al., 2001). Briefly, the sections were postfixed in 2% OsO4 for 1 h, dehydrated, embedded in Araldite (TABB, Berks, UK) and flat-mounted in Formvar-coated slides (AGAR, Essex, UK), using plastic coverslips. After polymerization, selected areas were photographed, trimmed, reembedded in Araldite, and resectioned at 1 μm. These semithin sections were rephotographed and resectioned in ultrathin sections. The ultrathin sections were observed in a Jeol (Pea-body, MA) electron microscope, without heavy metal staining to avoid artifactual precipitates.
Western blot analysis
Extracts for Western blot analysis were prepared by homogenizing fresh dissected mouse brain regions in ice-cold extraction buffer consisting of 20 mm HEPES, pH 7.4, 100 mm NaCl, 10 mm NaF, 1% Triton X-100, 1 mm sodium orthovanadate, 10 mm EDTA, and protease inhibitors (2 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin). The samples were homogenized at 4°C, and protein content was determined by Bradford. Total protein (20 μg) was electrophoresed on 10% SDS-PAGE gel and transferred to a nitrocellulose membrane (Schleicher and Schuell, Keene, NH). The experiments were performed using the following primary antibodies: polyclonal anti-huntingtin N-terminal CAG53b (1/5000) and monoclonal anti-α-tubulin (1/10000). The filters were incubated with the antibody at 4°C overnight in 5% nonfat dried milk. A secondary goat anti-mouse (monoclonal antibodies) or goat anti-rabbit (polyclonal antibodies) antibody (both 1/5000; Invitrogen, San Diego, CA) was used followed by ECL detection (Amersham Biosciences, Arlington Heights, IL).
Quantification was performed by using a GS-710 calibrated imaging densitometer scanner controller by Quantity One PC software from Bio-Rad (Hercules, CA). In all cases, the average intensity value of the pixels in a background-selected region was calculated and was subtracted from each pixel in the samples. The densitometry values obtained in the linear range of detection with these antibodies were normalized with respect to the values obtained with an anti-α-tubulin antibody to correct for any deviation in loaded amounts of protein. Statistical analysis was performed using one-way ANOVA followed by Bonferroni's test.
Rota-Rod behavioral testing
Twenty control and 21 Tet/HD94 mice were tested for motor coordination at different ages starting at the age of 17 months. More precisely, motor coordination was tested on an accelerating Rota-Rod apparatus (Ugo Basile, Comerio, Italy). Initially, each mouse was pretrained at fixed speed (8 rpm) by being repeatedly placed on the Rota-Rod as many times as necessary until it was able to remain on the apparatus for 60 s. After this pretraining, Rota-Rod was set to accelerate from 4 to 40 rpm over 5 min, and mice were tested four times at 1 h intervals on 2 consecutive days (for a total of eight trials). During accelerating trials, the latency of each mouse to fall from the Rota-Rod was measured.
After the first testing at the age of 17 months, 4 control and 4 Tet/HD94 mice were killed for histological analysis and the remaining 16 control and 17 Tet/HD94 mice were split in two groups (one remaining in plain water and the other receiving 2 mg/ml doxycycline in the drinking water for 4 months and 0.5 mg/ml doxycycline for an additional month).
Data are represented in Figures 1 A and 2 A-D as mean ± SEM of latency to fall from the Rota-Rod for each of the four daily trials, per day. In Figures 1 B and 2 E, data are represented as the mean ± SEM of the latencies to fall in the eight trials per genotype and treatment condition. Significant differences were determined by ANOVA followed by Bonferroni's test correction.
Figure 1.
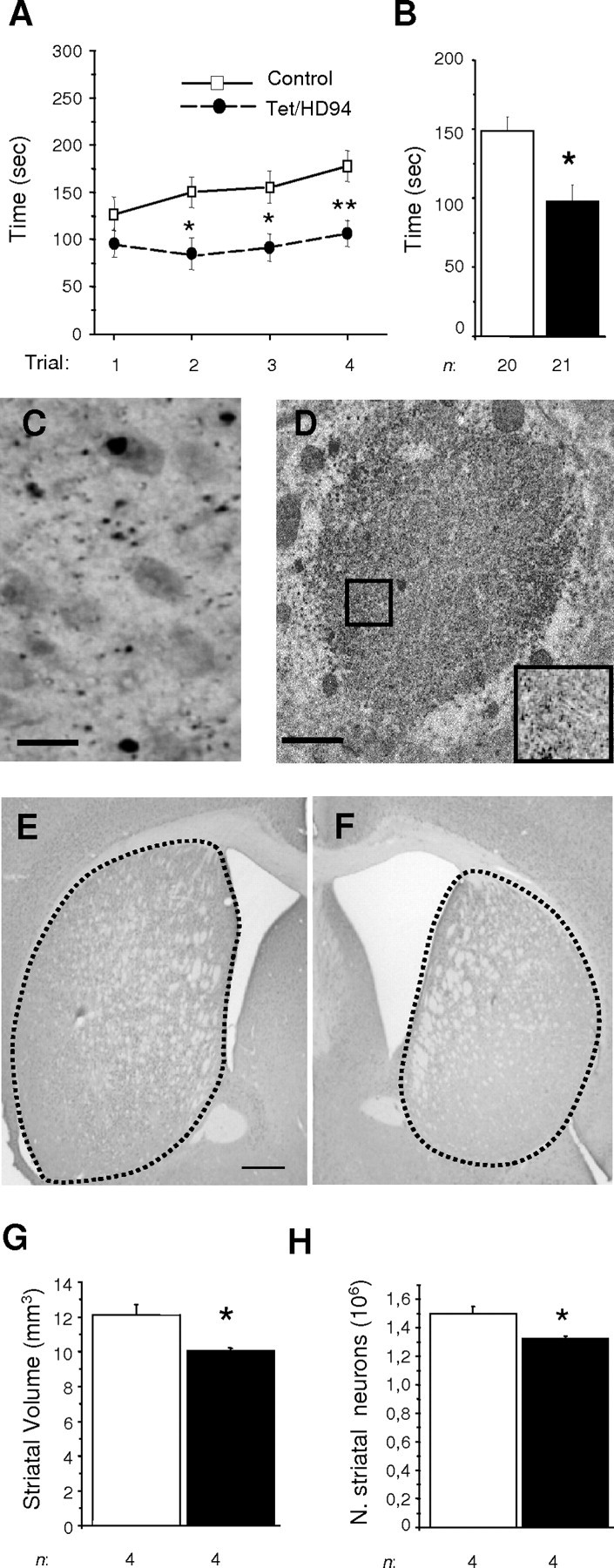
Motor dysfunction and neuropathology (including striatal neuron loss) in 17-month-old Tet/HD94 mice. A, B, Rota-Rod analysis of 17-month-old control (open squares; n = 20) and Tet/HD94 (filled circles; n=21) mice. Values in A represent mean ± SEM time spent on Rota-Rod, per daily trial and genotype (see Material and Methods) (*p < 0.01; **p < 0.005). Values in B represent mean ± SEM of the latencies to fall in the eight trials per genotype (*p < 0.01). C, Immunohistochemistry with anti-N-terminal mutant-htt (CAG53b) antibody revealed the presence of aggregates in the forebrains of Tet/HD94 mice. Scale bar, 20 μm. D, Immunoelectron microscopy analysis of a CAG53b-stained inclusion from a Tet/HD94 mouse. The inset in D corresponds to a 2.0× magnification of the indicated area, showing that inclusions are formed by randomly oriented filamentous structures. Scale bar, 1 μm. E, F, Striatal atrophy can be observed in coronal sections like those stained with anti-Neu-N antibody from control (E) and Tet/HD94 (F) mice. Scale bar: (in E) E, F, 500 μm. G, Stereological measurement of striatal size for control (open bar; n = 4) and Tet/HD94 (filled bar; n = 4) mice. H, Stereological analysis of number (N.) of striatal neurons in control (open bar; n = 4) and Tet/HD94 (filled bar; n = 4) as determined by Neu-N staining. *p < 0.01. Error bars indicate SE.
Figure 2.
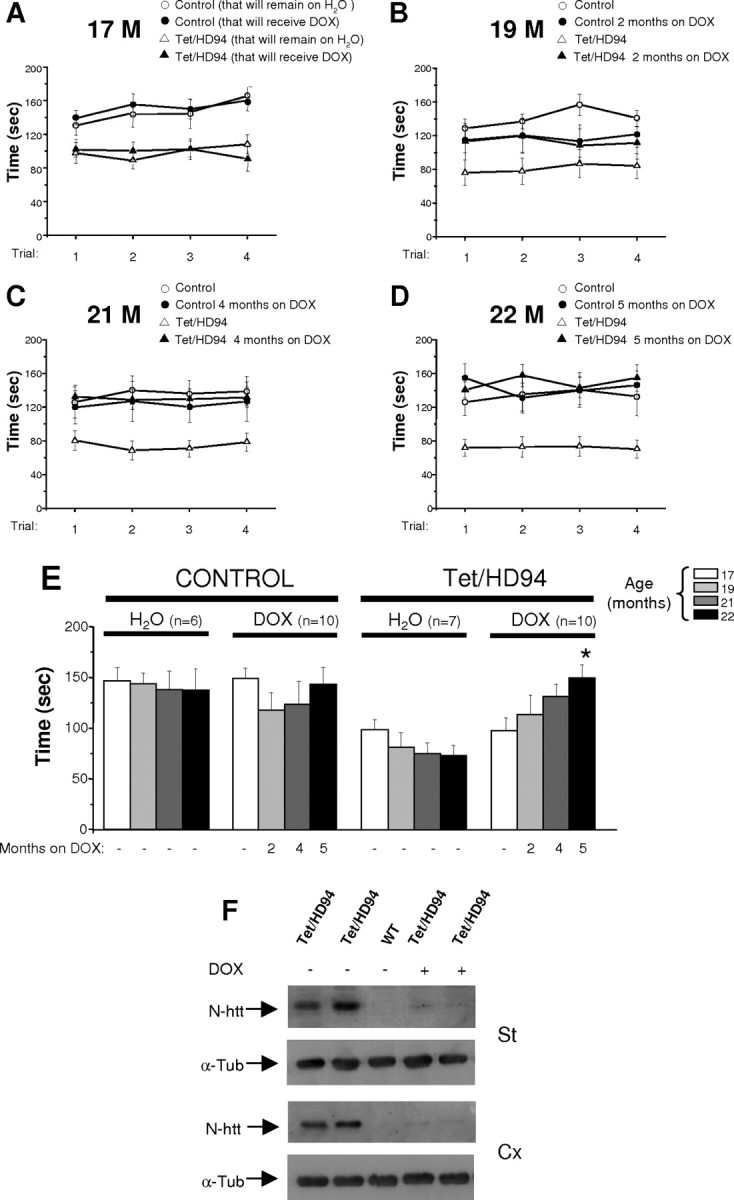
Silencing mutant-htt expression in Tet/HD94 mice with decreased striatal neurons results in full motor recovery. A-E, Rota-Rod performance of control or Tet/HD94 mice either receiving doxycycline to shut down transgene expression (DOX) or kept on water, from the age of 17 months to the age of 22 months. A-D, Values represent mean ± SEM time spent on Rota-Rod at the ages of 17 (A), 19 (B), 21 (C), and 22 (D) months. E, Mean ± SEM of the latencies to fall in the eight trials per genotype, age, and treatment condition. Motor performance of 22-month-old Tet/HD94 mice that have been on DOX for 5 months improve significantly with respect to their performance at the age of 17 months (*p < 0.01). F, Western blot analysis of striatal (St) and cortical (Cx) extracts from 22-month-old Tet/HD94 mice (+/- DOX treatment) and from control littermates probed with antibodies against N-terminal htt (CAG53b). Membranes were probed with anti-tubulin antibody to correct for any possible deviation on protein loading. M, Month; α-Tub, anti-tubulin; WT, wild type.
Results
Loss of striatal neurons in 17-month-old Tet/HD94 mice
Tet/HD94 mice express exon 1 htt with a 94 polyQ repeat in the forebrain (under control of CaMKIIα promoter) in a tetracycline-regulated manner (Yamamoto et al., 2000). As previously reported, up to the age of 10 months, Tet/HD94 mice develop progressive motor decline and striatal atrophy in the absence of striatal neuronal loss (Yamamoto et al., 2000; Martin-Aparicio et al., 2001). It is, however, conceivable that neuronal loss might take place at older ages in Tet/HD94 mice, such as happens in other HD mouse models like the full-length cDNA model (Reddy et al., 1998) and the YAC mice (Hodgson et al., 1999). Even R6/2 mice, despite their short life span and no detectable decrease in the number of striatal neurons, have been reported to experience dark cell degeneration (Turmaine et al., 2000). In this regard, we previously detected dark cell degenerating neurons in 14-month-old Tet/HD94 mice (Diaz-Hernandez et al., 2003). In view of this previous finding, we decided to test whether striatal neuron loss can be detected by stereological analysis in 17-month-old Tet/HD94 mice.
In good agreement with previous findings in younger mice, 17-month-old Tet/HD94 mice show a deficit in the accelerating Rota-Rod test for motor coordination (97.67 ± 11.93 s latency to fall for Tet/HD94 mice with respect to 148.85 ± 10.13 s latency for control littermates; p < 0.01) (Fig. 1A,B). Regarding neuropathology, similarly to what has been described in younger Tet/HD94 mice, N-terminal-htt and ubiquitin-immunopositive inclusions can be detected in the forebrain of 17-month-old Tet/HD94 mice (Fig. 1C). As also previously reported (Diaz-Hernandez et al., 2004b), ultrastructural analysis revealed that they are mostly neuropil inclusions with an even distribution or unordered filamentous structures (Fig. 1D). As can be seen in Figure 1E-G, 17 stereological analysis on Neu-N-immunostained sections revealed that 17-month-old Tet/HD94 show a decrease in striatal volume. More interestingly, and unlike what was described for 10-month-old mice (Yamamoto et al., 2000; Martin-Aparicio et al., 2001), a significant (12.11 ± 1.01%; p < 0.01) decrease in the number of striatal neurons was found in 17-month-old Tet/HD94 with respect to their control littermates (Fig. 1H). Similar results were obtained in stereological analysis of Nissl-stained sections (data not shown).
Full motor recovery is possible despite loss of striatal neurons
In view of the significant neuronal loss in the striatum of Tet/HD94 mice at the age of 17 months, we decided to split these mice and their control littermates into two groups for sequential testing in the Rota-Rod apparatus. One group was allowed to age without any pharmacological intervention, whereas the other was treated with 2 mg/ml doxycycline in the drinking water. We previously showed that this dose of doxycycline completely stops transgene expression in Tet/HD94 mice (Yamamoto et al., 2000; Martin-Aparicio et al., 2001). Each group was then tested for locomotor coordination in the Rota-Rod apparatus every 2 months (Fig. 2A-C). The performance in the accelerating Rota-Rod showed a tendency to improve progressively in Tet/HD94 mice after 2 and 4 months of doxycycline treatment (113.40 ± 19.13 and 130.75 ± 12.34 s at 60 and 120 d of doxycycline treatment, respectively; although this difference did not reach statistical significance) (Fig. 2E). In contrast, Tet/HD94 mice that were allowed to continue expressing mutant htt, worsened their Rota-Rod performance 60 and 120 d later with respect to the age of 17 months, as expected (Fig. 2E). Intriguingly, 2 months of doxycycline treatment worsened the performance of control mice (Fig. 2E) above the age-dependent deterioration that can be seen in control mice on water. We reasoned that this might be attributable to the high dose of doxycycline that was used compared with that used by other groups to achieve shut down of transgene expression in similar conditional-tTA mouse models (Chen et al., 1998). Based on this observation, and to prevent any potential toxicity attributable to an excessive dose of doxycycline, we decided to reduce the dose to 0.5 mg/ml and to retest the mice 1 month later (Fig. 2D,E). Results obtained at 5 months of this doxycycline paradigm showed that control mice that received doxycycline performed the Rota-Rod test in a similar manner as they did at the beginning of treatment (143.18 ± 16.75 s). More interestingly, the improvement in the performance of gene-off Tet/HD94 mice further increased and reached significance after this fifth month of doxycycline treatment in comparison with their performance at the age of 17 months (149.74 ± 12.91 s; p < 0.01), and they were indistinguishable from the control groups regardless of doxycycline administration.
To evaluate whether full shut down of polyQ-htt expression was still taking place despite the decrease in the dose of doxycycline during the last month, Western blot analysis was performed on striatal and cortical extracts from each group. As shown in Figure 2B, virtually total shut down of mutant htt is still achieved following this paradigm.
Silencing mutant-htt expression slows the progression of neuronal loss in the striatum of Tet/HD94 mice
We then analyzed the effect of the 5 month doxycycline treatment on the number of striatal neurons. As expected, Tet/HD94 mice that were not administered doxycycline (and therefore continued expressing mutant htt) presented a clear progression in striatal neuronal loss from 17 to 22 months of age. More precisely, they reached a 44.1 ± 0.82% loss with respect to their control littermates (Fig. 3B). On the contrary, this dramatic additional neuronal loss is decreased in 22-month-old Tet/HD94 mice that received doxycycline. In these mice, the degree of neuronal loss did in fact increase slightly (19.2 ± 2.79% loss; p < 0.02) over the one observed at 17 months (12.11 ± 1.01%). Similar results were obtained regarding progression of striatal atrophy volume (Fig. 3A).
Figure 3.

Silencing mutant-htt expression in Tet/HD94 mice halts additional loss of striatal neurons and boosts DARPP-32 expression in the remaining ones. A, B, Stereological measurement of striatal volume on DARPP-32 immunostained sections (A) or number of striatal neurons on Neu-N-immunostained sections (B). Results are presented as the evolution of percentage of Tet/HD94 values with respect to their control littermates from the age of 17 months (open bar) to the age of 22 months either with continued expression (gene-on; gray bar) or doxycycline-treated (gene-off; black bar). Significant differences between groups were determined by ANOVA: ***p < 0.001; *p < 0.02. Striatal volume values for control mice were as follows: 12.1 ± 0.56 mm3 for 17-month-old, 9.8 ± 0.8 mm3 for untreated 22-month-old, and 10.1 ± 0.6 mm3 for doxycycline-treated 22-month-old mice. Numbers of striatal neurons in control mice were as follows: 1.49 × 106 ± 0.04 neurons in 17-month-old ones, 0.99 × 106 ± 0.01 neurons in untreated 22-month-old ones, and 1.11 × 106 ± 0.02 in doxycycline-treated 22-month-old ones. C, D, Striatal sections from 22-month-old gene-on (C) or gene-off (D) Tet/HD94 mice were immunostained with an antibody against DARPP-32 (brown labeling) and counterstained with Nissl dye (blue labeling). A dramatic increase in the number and intensity of positive DARPP-32 striatal neurons was observed in gene-off compared with gene-on mice. Insets in C and D show a 5× magnification. N., Number; M, month; DOX, doxycycline.
Despite 19.2% loss of striatal neurons in 22-month-old gene-off Tet/HD94 mice, their motor performance is indistinguishable from 22-month-old control mice. Therefore, the remaining 80.8% neurons must be able to compensate for the function of the missing neurons. In good agreement, DARPP-32 immunostaining [a proposed indicator of the functional state of striatal medium-sized spiny neurons (van Dellen et al. 2000)] is dramatically higher in 22-month-old gene-off Tet/HD94 mice than in Tet/HD94 mice that were allowed to continue expressing mutant htt (Fig. 3C,D). More precisely, 22-month-old gene-on Tet/HD94 mice showed a weak staining in only 30.29 ± 7.67% of striatal neurons (Fig. 3C), whereas a strong striatal DARPP-32 immunostaining was found in 67.75 ± 18.55% of striatal neurons of 22-month-old gene-off Tet/HD94 mice (Fig. 3D).
Dramatic but incomplete reversal of inclusions after silencing of mutant-htt expression in Tet/HD94 mice
We previously demonstrated that doxycycline treatment results in complete clearing of inclusions in the striatum of 3- and 9-month-old Tet/HD94 mice (Yamamoto et al., 2000; Martin-Aparicio et al., 2001). To address whether this reversal still happens in more advanced stages of phenotype progression, tissue sections from 22-month-old gene-on and gene-off Tet/HD94 mice were immunostained with anti-N-terminal htt and anti-ubiquitin antibodies. The striatum of 22-month-old gene-on Tet/HD94 mice showed ∼100 inclusions per 100 μm2 (Fig. 4E), regardless of whether they were detected with anti-htt (Fig. 4A) or anti-ubiquitin (Fig. 4C) antibodies. Interestingly, a dramatic but incomplete reduction in the density of aggregates (90%) was observed in the striatum of gene-off Tet/HD94 mice after 5 months of transgene shutdown (Fig. 4B,D). A similar dramatic but incomplete reduction in inclusion density (80%) was seen in the cortex (Fig. 4F-J) of gene-off Tet/HD94 mice respect to gene-on mice.
Figure 4.

Silencing mutant-htt expression in Tet/HD94 mice in advanced stages of disease results in dramatic but incomplete reversal of htt- and ubiquitin-immunopositive inclusions. Immunocytochemistry with anti-N-terminal huntingtin antibody (MAB5374) or anti-polyubiquitin (FK2) revealed the presence of inclusions in the striatum (A-D) and cortex (F-I) of gene-on and gene-off Tet/HD94 22-month-old mice. The number of inclusions was significantly reduced on gene-off with respect to gene-on mice. E, Histogram showing quantification of N-terminal htt- or polyubiquitin-positive inclusions in striatum of gene-on (gray bar) or gene-off (black bar) 22-month-old Tet/HD94 mice. J, Histogram showing quantification of N-terminal htt- or polyubiquitin-positive aggregates in cortex of gene-on (gray bar) or gene-off (black bar) 22-month-old Tet/HD94 mice. Statistical significance was determined by ANOVA: ***p < 0.001. Error bars indicate SE.
Irreversible amyloid-like inclusions in the brain of Tet/HD94 mice
The fact that a fraction of inclusions remains when doxycycline is administered at an old age but not in younger mice, led us to hypothesize that a subset of inclusions might mature with age to a more stable conformation. To test whether amyloid-like inclusions like those found in postmortem brains of HD patients (McGowan et al., 2000) can be detected in the brains of Tet/HD94 mice, we performed thioflavin-S staining on brain sections from Tet/HD94 mice at different ages (initially, 10 and 17 months). Positive thioflavin-S inclusions were detected in cortex, striatum, and hippocampus of 17-month-old Tet/HD94 mice (Fig. 5G) (data not shown) but not in 10-month-old mice (data not shown). To explore progression with age and to test whether amyloid-like inclusions are susceptible to revert, we then performed similar analysis in brain sections of the gene-on and gene-off 22-month-old Tet/HD94 mice as well as control mice (Fig. 5A-G). The number of amyloid-like inclusions increased from 17 to 22 months of age in both gene-on and gene-off Tet/HD94 mice. However, the number of thioflavin-S-positive inclusions was slightly lower in the gene-off mice, but this difference did not reach statistical significance. Because thioflavin-S-positive inclusions do not revert and they are very similar in number to the N-terminal htt- or ubiquitin-positive inclusions remaining in 22-month-old mice gene-off, they probably correspond to the same sub-population of inclusions.
Figure 5.
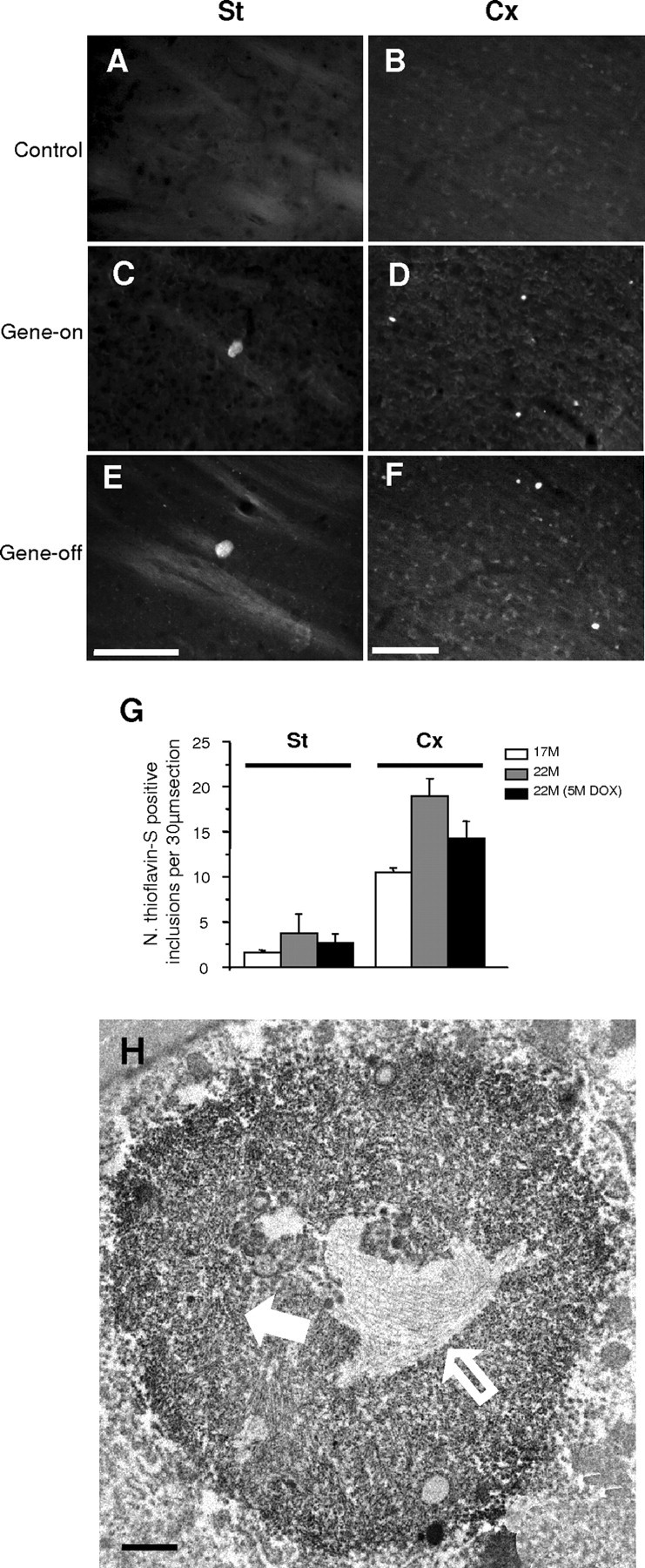
Thioflavin-S amyloid staining and highly ordered core material in the irreversible subset of inclusions. Thioflavin-S staining in the striatum (A, C, E) and cortex (B, D, F) of 22-month-old control or Tet/HD94 mice. Single transgenic BiTetO mice that carry the transgene encoding mutant htt but that do not express it because they lack the tTA transactivator were used as control (A, B). Tet/HD94 mice (C-F) show thioflavin-S-stained inclusions regardless of whether they continued expressing mutant htt (gene-on; C, D) or not (gene-off; E, F). Scale bars: (in E, F) A-F, 50 μm. G, Histogram showing the evolution in number of striatal and cortical thioflavin-S-positive inclusions in Tet/HD94 mice from the age of 17 months (open bar) to the age of 22 months either expressing (gene-on; gray bar) or not expressing mutant htt (gene-off; black bar). Error bars indicate SE. St, Striatal; Cx, cortical. H, Immunoelectron microscopy analysis of N-terminal mutant-htt (CAG53b antibody)-stained sections showing an example of inclusion with two different ultrastructural textures, one consisting of randomly oriented filaments (white filled arrow) and the other consisting of a compact core of highly ordered material (white open arrow). Scale bar, 1 μm.
To gain insight into the ultrastructure that might correspond to the irreversible and amyloid-like inclusions in the brain of 17-month-old Tet/HD94 mice, we performed intensive immunoelectron microscopy analysis of brain sections stained with anti-N-terminal htt antibody. As expected, the most common ultrastructural morphology was like the one previously described for this age (Fig. 1D) and for younger mice (Diaz-Hernandez et al., 2004b) consisting of an even distribution of labeled material that often can be distinguished as randomly oriented filaments. Interestingly, in 17-month-old Tet/HD94 mice, a small fraction of inclusions were found to have a similar external texture but a compact core of highly ordered material (Fig. 5H).
Discussion
The main goal of the present work was to explore whether reversal of motor dysfunction is still possible in advanced stages of disease once neuronal loss has taken place in the brain of a conditional mouse model of HD. We found that, at the age of 17 months, Tet/HD94 mice show significant loss of striatal neurons. Still, these mice were able to fully recover their motor coordination ability when mutant-htt expression was halted for 5 months. Furthermore, suppressing mutant-htt expression also resulted in a reversal of the vast majority of inclusions and in an attenuation of additional neuronal loss. However, we also identified a subset of amyloid-like inclusions that are first detected at the age of 17 months and that do not revert after silencing transgene expression.
Neuronal dysfunction rather than neuronal loss seems to account for HD symptomatology at least in its earliest stages. This notion is supported by previous studies in Tet/HD94 mice and in other mouse models that show motor deficit and/or striatal atrophy without neuronal loss (Hansson et al., 1999; Guidetti et al., 2001; Martin-Aparicio et al., 2001). In this regard, it should be noted that HD mouse models may not mimic the human disease entirely, in that there is more substantial neurological deficit in the mice with less neuronal loss than there is in human HD. However, in good agreement with the notion of neuron dysfunction causing disease at early stages, minimal nonspecific neuropathological features for HD are found in postmortem evaluation of the striatum of some symptomatic patients (Vonsattel's grade 0); and a case of a symptomatic HD patient that died of pneumonia with almost no detectable cell loss in the striatum has also been reported (Mizuno et al., 2000). Nevertheless, significant levels of neuronal death have been demonstrated in postmortem tissue of later stages of HD (Vonsattel et al., 1985; Portera-Cailliau et al., 1995). Hence, HD progression may have an initial period of neuronal dysfunction during which there is initiation and maintenance of the disease, followed by a second period during which neuronal loss would contribute and exacerbate the symptoms. The full reversal of motor symptoms in 9-month-old Tet/HD94 mice (Yamamoto et al., 2000) when neuronal loss is still not detectable (Martin-Aparicio et al., 2001), could then easily be explained by a recovery of the affected but still living neurons when mutant-huntingtin production stops and both aggregated and soluble mutant htt are cleared from the neuron. Here, we now report full motor recovery in mice with a significant loss of striatal neurons.
A possible explanation for motor recovery after onset of neuron loss could have been a repopulation of neurons in the striatum to its normal levels because of increased neurogenesis. In fact, neurogenesis from the subependymal layer has been described in adult human Huntington's disease brain (Curtis et al., 2003), although not from the subgranular layer of the dentate gyrus of the R6/1 mice (Lazic et al., 2004). Additionally, work in mouse models of Huntington's disease has shown that enriching the environment of transgenic animals delays the onset and slows the progression of Huntington's disease-associated motor and cognitive symptoms (Hockly et al., 2002) and environmental enrichment is known to enhance neurogenesis (Kempermann et al., 1997). However, a net repopulation attributable to adult neurogenesis can be excluded, because in gene-off 22-month-old Tet/HD94 mice, the number of striatal neurons had still further decreased from 12% loss at the age of 17 months to 19% loss after the 5 months of doxycycline treatment. Therefore, unless some neurogenesis might have taken place, the most likely explanation for the reversal of motor phenotype is that, on clearing of mutant htt, the 80% remaining striatal neurons are able to recover to a level that allows them to compensate for the function of the missing neurons. This correlates well with the high level of DARPP-32 protein in the striatum of gene-off mice.
Despite intensive research since the identification of the pathogenic mutation (Huntington's Disease Collaborative Research Group, 1993), the primary mechanism responsible for mutant-htt toxicity is still unknown. In the last years, different therapies have been shown to delay disease progression in HD animal models. These include histone deacetylase to increase transcription of neuroprotective genes (Ferrante et al., 2003), caspase inhibitors to prevent apoptosis (Ona et al., 1999), coenzyme Q to enhance energy metabolism (Andreassen et al., 2001; Ferrante et al., 2002), and trehalose or Congo red to inhibit the formation of polyglutamine aggregates (Sanchez et al., 2003; Tanaka et al., 2004). However, these approaches target downstream and indirect effects of mutant-htt expression. Accordingly, in case they prove beneficial in humans, they might only palliate some aspects of disease. Our studies in the conditional mouse model (Yamamoto et al., 2000; Martin-Aparicio et al., 2001) and the recently reported efficacy of RNAi against mutant htt in a similar mouse model (Harper et al., 2005) support silencing mutant-htt expression as a valid therapeutic approach that has the advantage of aiming toxicity at its root. The present work addresses the issue of whether silencing mutant-htt strategies such as RNAi delivery should be undertaken only in the early stages of disease or if, on the contrary, it might also have good chances to work in the advanced stages of disease. Importantly, our results support a beneficial effect in late disease even after significant loss of striatal neurons might have taken place.
Interestingly, the therapeutic potential of gene silencing has recently been extended to another polyQ disease, namely spinocerebellar ataxia type 1 (SCA-1). This has been done in mouse models either by viral delivery of RNA interference (Xia et al., 2004) and by a conditional transgenic approach (Zu et al., 2004). However, conditional SCA-1 transgenic mice recover motor function in early stages of disease but lose this ability at the age of 5.5 months (although no noticeable Purkinje cell loss has taken place yet at this age). These results, together with ours, suggest that striatum, or more generally the basal ganglia, have a higher potential for functional recovery than cerebellar circuits.
In this study, we also report for the first time the presence of amyloid-like inclusions in a mouse model of HD. This type of inclusions had been described in the postmortem brain of HD patients (McGowan et al., 2000). However, in the R6/2 transgenic mouse model, they appear to be missing (McGowan et al., 2000). A possible explanation for the lack of amyloid-like inclusions in other transgenic mouse models could simply be that they are late events in HD neuropathology. Accordingly, the rapidly progressing R6/2 mice would not have the chance to develop them in their short life span.
Regarding the genesis of amyloid-like inclusions in old Tet/HD94 mice, it is possible that preexisting nonamyloid inclusions mature to a more ordered status. On the contrary, it could also happen that, at the age of 17 months, Tet/HD94 neurons fulfill the requirement for developing de novo a new type of aggregate with an amyloid-like structure. The first scenario seems more likely in view of the ordered core inside inclusions that are very similar to the predominant ones. This suggests that a higher degree of compaction occurs inside inclusions probably attributable to the high local concentration of N-term-htt.
Regarding the relevance of these late-appearing amyloid-like inclusions to HD pathogenesis, the fact that they remain in gene-off mice that fully recover their motor ability and in which significant additional neuronal loss is prevented, strongly suggest that they are not responsible for motor dysfunction or neuronal loss. These results therefore rule out amyloid-like mature inclusions as the main toxic species of mutant htt.
The late appearance of neuronal loss and irreversible amyloid-like inclusions in Tet/HD94 mice confirm these mice as an HD model with slow phenotype progression. In view of this property, Tet/HD94 mice are a good tool to study both early aspects of disease and also the age-related worsening. In this regard, the late onset of neuronal death after many months of striatal atrophy and motor symptoms suggests that neuronal loss could be an aging-related phenomenon. Some age-related factors that might contribute are the decline proteasome activity (Keller et al., 2000a,b) and the onset of neuroinflammatory processes. Accordingly, the age-dependent decrease in proteasome activity has been suggested to contribute to the late onset of htt toxicity (Zhou et al., 2003). Interestingly, we previously showed changes in the subunit composition of the proteasome of 14-month-old Tet/HD94 mice (Diaz-Hernandez et al., 2003) that seem to be secondary to inflammatory processes (Diaz-Hernandez et al., 2004a).
Finally, during the review process of this article, the generation of a conditional mouse model of tauopathy was reported (SantaCruz et al., 2005). For this, the same experimental design that we used to generate our conditional mouse model of HD was used but to express mutant tau instead. This Alzheimer's disease-related experiment is therefore equivalent to that reported here in the Huntington's disease conditional model (at ages in which neuronal loss and amyloid-like inclusion are detectable). Briefly, SantaCruz et al. find that suppression of mutant-tau expression in the mutant-tau conditional mouse leads to behavioral recovery and stabilization of neuron numbers, whereas amyloid-like tau deposits remain. Therefore, both studies reach almost identical conclusions in two different neurodegenerative diseases.
In summary, the data presented here rule out the newly described and late-appearing amyloid-like inclusions as the main pathogenic species in HD and strongly suggest that halting mutant-htt expression can be of therapeutic value not only in early stages of pathology (when only neuronal dysfunction is responsible for the disease) but also in more advanced stages when striatal neuron loss has taken place.
Footnotes
This work was supported by grants from Comunidad Autónoma de Madrid, Fundación “La Caixa,” Spanish Ministerio de Educación y Ciencia, and by institutional grants from Fundación Ramón Areces and from Fondo de Investigaciones Sanitarias. M.D.-H. was the recipient of a postdoctoral fellowship from Comunidad Autónoma de Madrid and is currently there cipientofa “JuandelaCierva” grant from Spanish Ministerio de Cienciay Tecnología. We thank Drs. Félix Hernández, Jesús Avila, and Josep M. Canals for helpful discussion and comments, and Dr. Erich E. Wanker for kindly providing CAG53b antibody. We are also grateful to Javier Palacín, Carlos Sánchez, Virginia García de Yébenes, Raquel Cuadros, and Elena Langa for technical assistance.
Correspondence should be addressed to José J. Lucas, Centro de Biología Molecular “Severo Ochoa,” Consejo Superior de Investigaciones Científicas/Universidad Autónoma de Madrid, Facultad Ciencias, Universidad Autónoma de Madrid, Cantoblanco, 28049 Madrid, Spain. E-mail: jjlucas@cbm.uam.es.
M. Díaz-Hernández's present address: Departamento de Bioquímica y Biología Molecular IV, Facultad de Veterinaria, Universidad Complutense de Madrid, 28040 Madrid, Spain.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259773-09$15.00/0
References
- Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Thomas M, Friedlich A, Browne SE, Schilling G, Borchelt DR, Hersch SM, Ross CA, Beal MF (2001) Creatine increase survival and delays motor symptoms in a transgenic animal model of Huntington's disease. Neurobiol Dis 8: 479-491. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431: 805-810. [DOI] [PubMed] [Google Scholar]
- Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martin-Ibanez R, Munoz MT, Mengod G, Ernfors P, Alberch J (2004) Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington's disease. J Neurosci 24: 7727-7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Kelz MB, Zeng G, Sakai N, Steffen C, Shockett PE, Picciotto MR, Duman RS, Nestler EJ (1998) Transgenic animals with inducible, targeted gene expression in brain. Mol Pharmacol 54: 495-503. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Penney EB, Pearson AG, van Roon-Mom WM, Butterworth NJ, Dragunow M, Connor B, Faull RL (2003) Increased cell proliferation and neurogenesis in the adult human Huntington's disease brain. Proc Natl Acad Sci USA 100: 9023-9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Hernandez M, Hernandez F, Martin-Aparicio E, Gomez-Ramos P, Moran MA, Castano JG, Ferrer I, Avila J, Lucas JJ (2003) Neuronal induction of the immunoproteasome in Huntington's disease. J Neurosci 23: 11653-11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Hernandez M, Martin-Aparicio E, Avila J, Hernandez F, Lucas JJ (2004a) Enhaced induction of the immunoproteasome by interferon-gamma in neurons expressing mutant huntingtin. Neurotox Res 6: 463-468. [DOI] [PubMed] [Google Scholar]
- Diaz-Hernandez M, Moreno-Herrero F, Gomez-Ramos P, Moran MA, Ferrer I, Baro AM, Avila J, Hernandez F, Lucas JJ (2004b) Biochemical, ultrastructural, and reversibility studies on huntingtin filaments isolated from mouse and human brain. J Neurosci 24: 9361-9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF (2002) Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci 22: 1592-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM (2003) Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci 23: 9418-9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidetti P, Charles V, Chen EY, Reddy PH, Kordower JH, Whetsell Jr WO, Schwarcz R, Tagle DA (2001) Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol 169: 340-350. [DOI] [PubMed] [Google Scholar]
- Hansson O, Peters A, Leist M, Nicotera P, Castilho RF, Brundin P (1999) Transgenic mice expressing a Huntington's disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc Natl Acad Sci USA 96: 8727-8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper SQ, Staber PD, He X, Eliason SL, Martins IH, Mao Q, Yang L, Kotin RM, Paulson HL, Davidson BL (2005) From the cover: RNA interference improves motor and neuropathological abnormalities in a Huntington's disease mouse model. Proc Natl Acad Sci USA 102: 5820-5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Cordery PM, Woodman B, Mahal A, van Dellen A, Blakemore C, Lewis CM, Hannan AJ, Bates GP (2002) Environmental enrichment slows disease progression in R6/2 Huntington's disease mice. Ann Neurol 51: 235-242. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR (1999) A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 23: 181-192. [DOI] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971-983. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR (2000a) Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech Ageing Dev 113: 61-70. [DOI] [PubMed] [Google Scholar]
- Keller JN, Huang FF, Markesbery WR (2000b) Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience 98: 149-156. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH (1997) More hippocampal neurons in adult mice living in an enriched environment. Nature 386: 493-495. [DOI] [PubMed] [Google Scholar]
- Landles C, Bates GP (2004) Huntingtin and the molecular pathogenesis of Huntington's disease. Fourth in molecular medicine review series. EMBO Rep 5: 958-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazic SE, Grote H, Armstrong RJ, Blakemore C, Hannan AJ, van Dellen A, Barker RA (2004) Decreased hippocampal cell proliferation in R6/1 Huntington's mice. NeuroReport 15: 811-813. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J (2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J 20: 27-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Aparicio E, Yamamoto A, Hernandez F, Hen R, Avila J, Lucas JJ (2001) Proteasomal-dependent aggregate reversal and absence of cell death in a conditional mouse model of Huntington's disease. J Neurosci 21: 8772-8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan DP, van Roon-Mom W, Holloway H, Bates GP, Mangiarini L, Cooper GJ, Faull RL, Snell RG (2000) Amyloid-like inclusions in Huntington's disease. Neuroscience 100: 677-680. [DOI] [PubMed] [Google Scholar]
- Mizuno H, Shibayama H, Tanaka F, Doyu M, Sobue G, Iwata H, Kobayashi H, Yamada K, Iwai K, Takeuchi T, Hashimoto N, Ishihara R, Ibuki Y, Ogasawara S, Ozeki M (2000) An autopsy case with clinically and molecular genetically diagnosed Huntington's disease with only minimal nonspecific neuropathological findings. Clin Neuropathol 19: 94-103. [PubMed] [Google Scholar]
- Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10: 1441-1448. [DOI] [PubMed] [Google Scholar]
- Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Fried-lander RM (1999) Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature 399: 263-267. [DOI] [PubMed] [Google Scholar]
- Oorschot DE (1996) Total number of neurons in the neostriatal, pallidal, subthalamic, and substantia nigral nuclei of the rat basal ganglia: a stereological study using the cavalieri and optical disector methods. J Comp Neurol 366: 580-599. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2001) Mouse brain in stereotaxic coordinates, Ed 2. San Diego: Academic.
- Poirier MA, Li H, Macosko J, Cai S, Amzel M, Ross CA (2002) Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J Biol Chem 277: 41032-41037. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE (1995) Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci 15: 3775-3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Williams M, Charles V, Garrett L, Pike-Buchanan L, Whetsell Jr WO, Miller G, Tagle DA (1998) Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet 20: 198-202. [DOI] [PubMed] [Google Scholar]
- Ross CA (1997) Intranuclear neuronal inclusions: a common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron 19: 1147-1150. [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA (2004) Protein aggregation and neurodegenerative disease. Nat Med 10 [Suppl]: S10-S17. [DOI] [PubMed] [Google Scholar]
- Sanchez I, Mahlke C, Yuan J (2003) Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 421: 373-379. [DOI] [PubMed] [Google Scholar]
- SantaCruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309: 476-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE (1997) Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90: 549-558. [DOI] [PubMed] [Google Scholar]
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR (1999) Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet 8: 397-407. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M, Nukina N (2004) Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 10: 148-154. [DOI] [PubMed] [Google Scholar]
- Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW (2000) Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci USA 97: 8093-8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dellen A, Welch J, Dixon RM, Cordery P, York D, Styles P, Blakemore C, Hannan AJ (2000) N-Acetylaspartate and DARPP-32 levels decrease in the corpus striatum of Huntington's disease mice. NeuroReport 11: 3751-3757. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57: 369-384. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson Jr EP (1985) Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44: 559-577. [DOI] [PubMed] [Google Scholar]
- Xia H, Mao Q, Eliason SL, Harper SQ, Martins IH, Orr HT, Paulson HL, Yang L, Kotin RM, Davidson BL (2004) RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med 10: 816-820. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R (2000) Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101: 57-66. [DOI] [PubMed] [Google Scholar]
- Zhou H, Cao F, Wang Z, Yu ZX, Nguyen HP, Evans J, Li SH, Li XJ (2003) Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J Cell Biol 163: 109-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT (2000) Glutamine repeats and neurodegeneration. Annu Rev Neurosci 23: 217-247. [DOI] [PubMed] [Google Scholar]
- Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB, Orr HT (2004) Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci 24: 8853-8861. [DOI] [PMC free article] [PubMed] [Google Scholar]