

Abstract
Activation of the Akt/protein kinase B (PKB) kinase pathway can be neuroprotective after stroke. Akt is activated by growth factors via a phosphorylation-dependent pathway involving the kinases phosphoinositide 3 (PI3) kinase and phosphoinositide-dependent protein kinase-1 (PDK1) and is negatively regulated by phosphatase and tensin homolog deleted on chromosome 10 (PTEN). Akt kinase blocks apoptosis by phosphorylating the substrates forkhead transcription factor (FKHR) and glycogen synthase kinase 3β (GSK3β). We found that intra-ischemic hypothermia (30°C) reduced infarct size and improved functional outcomes up to 2 months. Changes in phosphorylation levels of Akt, as measured by Western blots and immunostaining, differed from levels of Akt activity measured in an in vitro assay in normothermic animals. Hypothermia blocked most of these changes and maintained Akt activity. Inhibition of PI3/Akt enlarged infarct size in hypothermic animals. Hypothermia improved phosphorylation of PDK1, PTEN, and FKHR. Hypothermia did not improve GSK3β (Ser9) phosphorylation but blocked the nuclear translocation of phosphorylated β-catenin (Ser33/37/Thr41) downstream of GSK3β. Phosphorylation levels of PTEN, Akt, and Akt substrate decreased before apoptotic cytochrome c release and degradation of microtubule-associated protein-2, a marker of neuronal survival. Hypothermia may protect from ischemic damage in part by preserving Akt activity and attenuating the apoptotic effects of PTEN, PDK1, and FKHR.
Keywords: Akt/PKB, hypothermia, cerebral ischemia, GSK3β, PTEN, β-catenin
Introduction
Neuronal fate after ischemia is determined by a balance between apoptotic and cell survival signals. Caspase-dependent and caspase-independent apoptotic pathways contribute to ischemic damage (Sugawara et al., 1999; Cao et al., 2001, 2003; Zhao et al., 2003, 2004b), and activation of the Akt kinase [also known as protein kinase B (PKB)] signal pathways enhances cell survival (Brunet et al., 2001; Franke et al., 2003) (Fig. 1). During Akt activation, membrane-bound receptor tyrosine kinase (RTK) is phosphorylated and activated by growth factors, including insulin-like growth factor (IGF) and brain-derived growth factor (Franke et al., 2003; Fresno Vara et al., 2004). RTK activates phosphoinositide 3-kinase (PI3K), which generates phosphatidylinositol-3,4,5-trisphosphate (PIP3) from phosphatidylinositol 4,5-bisphosphate (PIP2). Akt is recruited to the membrane and activated by phosphorylation of Ser473 and Thr308. Phosphoinositide-dependent protein kinase-1 (PDK1) is needed for the Thr308 phosphorylation. PDK1 may indirectly phosphorylate Akt at Ser473, probably by interacting with an uncharacterized protein PDK2. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) inactivates Akt by dephosphorylating PIP3 to PIP2 (Franke et al., 2003; Fresno Vara et al., 2004). Activated Akt promotes cell survival by phosphorylating and thus inactivating most of its substrates (Franke et al., 2003; Hanada et al., 2004), such as forkhead transcription factor (FKHR), Bcl-2 associated death protein (Bad), and glycogen synthase kinase 3β (GSK3β). Bad and FKHR, which are cytosolic, are normally phosphorylated by Akt. Dephosphorylation of Bad causes its translocation into mitochondria, triggering cytochrome c release. Cytochrome c then activates caspase-3, leading to apoptosis (Jiang and Wang, 2004). Dephosphorylated FKHR translocates into nuclei and acts as a transcription factor, promoting overexpression of Bcl-2 interacting mediator of cell death (Bim) and Fas ligand (Brunet et al., 2001; Hanada et al., 2004). Bim causes cytochrome c release, whereas Fas ligand activates caspase-8 and caspase-3 activity (Graham and Chen, 2001). Akt also downregulates GSK3β activity by phosphorylating it at Ser9 (Bhat et al., 2000). Dephosphorylation of GSK3β leads to its activation, which phosphorylates β-catenin, a transcription factor playing key roles in cell survival (Nusse, 2003).
Figure 1.
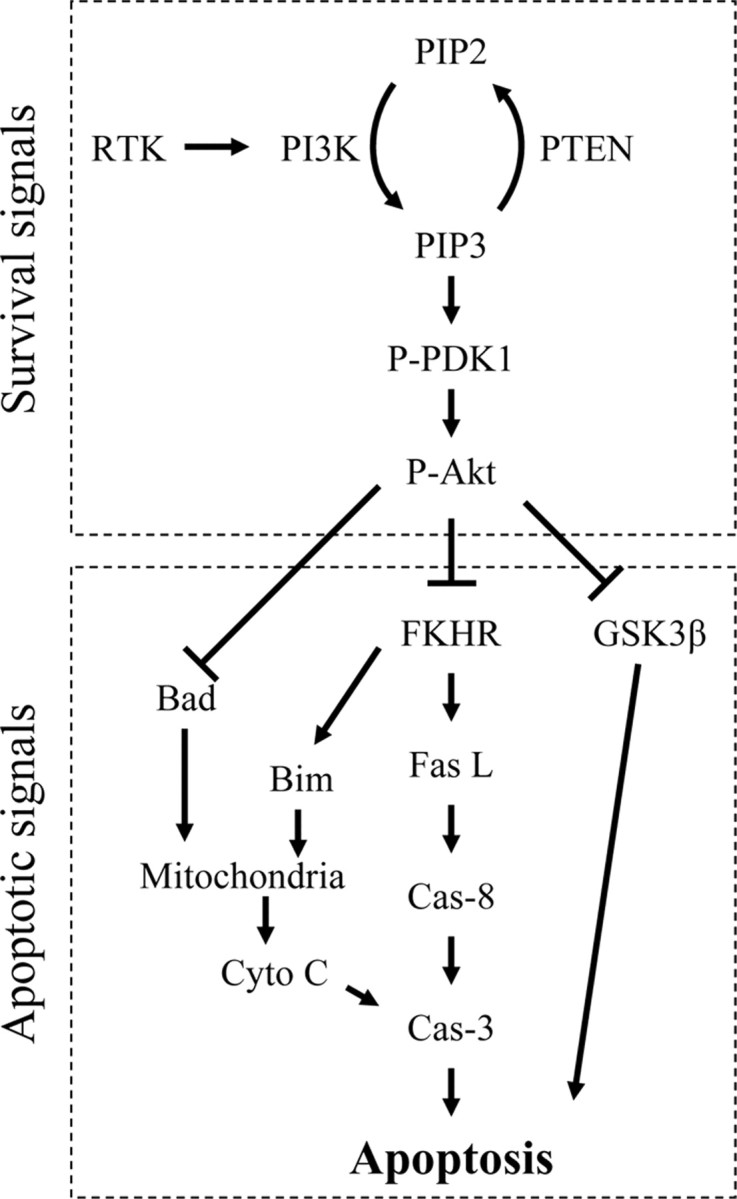
A diagram of Akt/PKB survival signal pathways. Fas L, Fas ligand; Cyto C, cytochrome c; Cas-3, caspase-3; Cas-8, caspase-8.
Akt may promote neuron survival after ischemia (Chan, 2004). Most reports agree that P-Akt (Ser473) transiently increases within hours but decreases after 24 h or more after ischemia (Noshita et al., 2001; Friguls et al., 2002; Jin et al., 2003). Moreover, protection by some agents and in transgenic and knock-out models involves maintained Akt activity after stroke (Limbourg et al., 2002; Jin et al., 2003; Noshita et al., 2003). Protection by ischemic preconditioning also requires Akt activation (Yano et al., 2001; Hashiguchi et al., 2004). However, the contributions of key components of the Akt pathway in stroke, including PTEN, PDK, FKHR, and GSK3β, have not been intensively studied in a single experiment.
Mild or moderate hypothermia protects against experimental stroke (Busto et al., 1989). Hypothermia has been shown to lessen the increase in Akt phosphorylation after hypoxia-ischemia (Tomimatsu et al., 2001), arguing against a role for Akt in hypothermic protection. Here we report the effects of focal ischemia and hypothermia on Akt pathways, infarct volumes, and neurological recovery.
Materials and Methods
Focal cerebral ischemia. Experimental protocols were approved by the Stanford University Administrative Panel on Laboratory Animal Care. Focal cerebral ischemia was generated as described previously (Chen et al., 1986; Zhao et al., 2003, 2004b). Male Sprague Dawley rats (350-450 g) were used. Anesthesia was induced by 5% isoflurane and maintained with 2-3% isoflurane during surgery. Core body temperatures were monitored with a rectal probe. The distal middle cerebral artery (dMCA) was exposed and cauterized above the rhinal fissure. The bilateral common carotid arteries (CCAs) were occluded for 1 h and then released, whereas the dMCA remained occluded. Animals were divided into normothermic or hypothermic groups (or sham surgery controls). Hypothermic rats were maintained at a core temperature of 30°C by spraying 100% alcohol onto the rat's body. Temperature was returned to normal with a lamp and a heating pad. Hypothermia was induced 10 min before ischemia and was maintained for 1 h after ischemia onset. Core temperature of rats in the normothermic group was maintained at 37°C throughout the experiment. We have observed previously a high correlation between rectal temperature and brain temperature in hypothermic rats (Zhao et al., 2004a).
General histology and infarct size measurement. Two or 60 d after ischemia, rats were perfused transcardially with normal saline, followed with 4% paraformaldehyde (PFA). Brains were postfixed with 4% PFA, 20% sucrose for 24 h, and sectioned into four coronal blocks rostral (level 1) to caudal (level 4). Thirty micrometer sections were cut onto glass slides in the coronal plane using a cryostat. Infarct size at 60 d was measured at all four levels; a section from each block was selected and stained with cresyl violet. The percentage of infarct cortex was measured by normalizing to the entire ipsilateral cortex from animals survived for 2 d as described previously (Zhao et al., 2003). Infarct size at 60 d was expressed as a percentage of the area of the intact, non-ischemic cortex according to the following formula: [(area of non-ischemic cortex - area of remaining ischemic cortex)/area of non-ischemic cortex] × 100.
Behavioral testing. Rats with a unilateral cortical stroke exhibit motor asymmetry. Four behavior tests were used to quantify this asymmetry. All behavior tests were performed by a person who was blinded to the experimental conditions. Most tests were performed before dMCA occlusion (dMCAo) and then on days 2, 3, 7, 14, 30, and 60 d after dMCA occlusion, except for the tail hang test, which occurred only at 30 and 60 d. Three days before surgery, rats were gently handled and trained, and baseline testing occurred on the day before surgery. All rats were able to perform the tests and did not exhibit substantial asymmetries before stroke; thus, no rats were excluded from experiments.
Vibrissa-elicited forelimb placing test. Forelimb placing (Schallert et al., 2000) was induced by gently brushing the rats' vibrissas on each side against the edge of a table; rats respond reflexively by placing their forelimb on that side onto the countertop. Reflexive placing is disrupted contralateral to the cortical injury. The reflex was tested 10 times on each side per trial, and two trials occurred per test session. If muscle tension or struggling occurred, rats were held and stroked until they relaxed and the trial was resumed. The percentage of vibrissa stimulations in which a paw placement occurred was calculated.
Postural reflex test. Postural reflexes were assessed using a method modified from Bederson et al. (1986). The animal was placed on a table, and its tail was held by one hand while the other hand gently pushed the animal's shoulder, moving it laterally ∼20 cm. The test was repeated in the opposite direction. This process was repeated several times. The use of the forelimbs to resist the lateral movement was scored. Normal, non-ischemic rats gripped the table and braced frequently during the push, which was scored as 0. The contralateral forelimb of an ischemic rat may show less resistance and become stiff during the push, which was scored as 1. If the forelimb offered no resistance, i.e., did not brace and dragged under the body during the push, this was scored as 2.
Tail hang test. Tail hang test was performed at 30 and 60 d after stroke as described by Borlongan et al. (1998) with slight modification. The rat was lifted 5-10 cm above the table. An ischemia-damaged rat will immediately turn to the contralateral (right) side. This large right turn is accompanied by a twisting of the body and usually a raising of the head toward the holding hand. “Turns” were counted when the angle reached 90° or more. Smaller turns were not counted. The rat was lifted no more than 5 s on each trial and was released for a few seconds before the next trial. The test was repeated 20 times each testing day. The percentage of trials on which a right turn occurred was calculated.
Home cage limb use test. After completing the other behavior tests, the animal was returned to its home cage. Rats usually rear up and explore the wall of the cage (Schallert et al., 2000). We counted the number of times the animal used its forelimbs to brace itself against the wall, counting separately for the ipsilateral, contralateral, or both forelimbs, until 20 such contacts were reached. The percentage of times out of 20 that the ipsilateral forelimb was used was computed using this formula: (ipsilateral + (both/2))/20 × 100%.
Drug delivery. For PI3K inhibitor delivery, LY294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one] was dissolved in DMSO and ethanol to a final concentration of 10 mm. Animals were anesthetized and placed in stereotactic frames. Ten microliters of drug solution or vehicle was infused into the ventricular space ipsilateral to the ischemia (from bregma: anteroposterior, -0.92 mm; mediolateral, 1.5 mm; dorsoventral, 3.5 mm), 1 h before ischemia and 24 h after ischemia. Infarct size was measured 2 d after stroke as described above.
Immunofluorescence staining and confocal microscopy. Rats were perfused transcardially with normal saline, followed by fixation with 4% PFA for 24 h. Brains were cut on a vibratome into slices of 50 μm and stored in antifreeze solution at -20°C. Free-floating immunostaining was performed. Generally, after washing three times in PBS, sections were blocked in PBS containing 5% donkey serum (Sigma, St. Louis, MO) and 0.3% Triton X-100 for 2 h at room temperature and then incubated in the primary antibodies diluted in blocking solution at 4°C overnight. Sections were washed with PBS and incubated for 2 h at room temperature (light shielded) in the secondary antibodies diluted in blocking solution. For double staining, tissues were incubated consecutively with a first pair of primary and secondary antibodies, followed with another pair of primary and secondary antibodies. The manufacturers, catalog numbers, and applications of all primary antibodies used are listed in Table 1. The combination of primary and secondary antibodies is shown in Table 2. All FITC or cyanine 3 (Cy3)-conjugated secondary anti-mouse or anti-rabbit antibodies were purchased from Jackson ImmunoResearch (West Grove, PA) and diluted 1:200 for applications.
Table 1.
Antibodies and their concentrations, manufacturers, and application in the present studies
|
Antibodies |
Source |
Dilutions |
Manufacturer |
Catalog # |
Appl |
|---|---|---|---|---|---|
| P-Akt (Ser473) | Rabbit | 1:50/1:1000 | Cell Signaling Technology | 9271 | IF/WB |
| P-Akt (Thr308) | Rabbit | 1:1000 | Cell Signaling Technology | 9275 | WB |
| P-Akt substrate | Rabbit | 1:200 | Cell Signaling Technology | 9611 | IF |
| P-PTEN (Ser380) | Rabbit | 1:200/1:1000 | Cell Signaling Technology | 9551 | IF/WB |
| P-β-Catenin (Ser33/37/Thr41) | Rabbit | 1:100 | Cell Signaling Technology | 9561 | IF |
| Cytochrome c | Mouse | 1:500 | PharMingen (San Diego, CA) | 556432 | IF |
| CD-31 | Mouse | 1:30 | PharMingen | 550300 | IF |
| MAP-2 | Mouse | 1:500 | Sigma | M4403 | IF |
| AKT | Rabbit | 1:1000 | Cell Signaling Technology | 9272 | WB |
| P-FKHR (Ser256) | Rabbit | 1:1000 | Cell Signaling Technology | 9461 | WB |
| P-GSK3β (Ser9) | Rabbit | 1:1000 | Cell Signaling Technology | 9336 | WB |
| P-GSK3β (Tyr216) | Rabbit | 1:1000 | Biosource International (Camarillo, CA) | 44-604G | WB |
| P-PDK1 (Ser241) | Rabbit | 1:1000 | Cell Signaling Technology | 3061 | WB |
| P-AKT (Tyr308) | Rabbit | 1:1000 | Cell Signaling Technology | 9275 | WB |
| β-Actin |
Mouse |
1:5000 |
Sigma |
A-5441 |
WB |
Appl, Application; IF, immunofluorescence; WB, Western blots.
Table 2.
Paris of primary and secondary antibodies used for double staining
|
First couple |
Second couple |
||||
|---|---|---|---|---|---|
| First Ab |
Second Ab |
First Ab |
Second Ab |
||
| P-AKT (Ser473) | Cy3-Ar | MAP-2 | FITC-AM | ||
| P-AKT substrate | FITC-AR | MAP-2 | Cy3-AM | ||
| P-PTEN | FITC-AR | MAP-2 | Cy3-AM | ||
| CD-31 | Cy3-AM | P-PTEN | FITC-AR | ||
| P-β-Catenin | Cy3-AR | MAP-2 | FITC-AM | ||
| P-AKT (Ser473) | Cy3-AR | Cytochrome c | FITC-AM | ||
| P-AKT substrate | Cy3-AR | Cytochrome c | FITC-AM | ||
| P-PTEN |
Cy3-AR |
Cytochrome c
|
FITC-AM |
||
Ab, Antibody; Cy3-AR, Cy3-conjugated anti-rabbit; FITC-AR, FITC-conjugated anti-rabbit; Cy3-AM, Cy3-conjugated anti-mouse; FITC-AM, FITC-conjugated anti-mouse.
Western blots. Rats survived for 0.5, 1.5, 5, 9, 24, and 48 h after stroke onset were killed by overdose of isoflurane and transcardially perfused with cold PBS. Rat brains were removed, and tissue corresponding to the ischemic penumbra was dissected for Western blot (Fig. 2). The ischemic penumbra was defined as the tissue saved by hypothermia 2 d after stroke, and the corresponding region from the normothermic brain was dissected for comparison. The fresh brain tissue was used for preparation of whole-cell protein extraction as described previously (Noshita et al., 2001). Briefly, brain tissues were cut into small pieces and homogenized in a glass homogenizer using 7 vol of the cold suspension buffer. The homogenate was centrifuged at 10,000 × g for 20 min, and the supernatant was taken for protein detection. Western blot was performed as described previously (Zhao et al., 2003, 2004b, 2005). Twenty micro-grams of protein in each lane was subjected to SDS-PAGE using 4-15% Ready Gel (catalog #L050505A2; Bio-Rad, Hercules, CA) for 1.5 h. Protein bands were transferred from the gel to polyvinylidinene fluoride (Millipore, Bedford, MA) membranes for 1 h. After transfer, Ponceau red staining was performed to confirm even loading of proteins in each membrane. Primary antibodies (Table 1) were incubated overnight at 4°C followed by a horse-radish peroxidase-conjugated secondary anti-rabbit antibody (1:2000; Cell Signaling Technology, Beverly, MA) for 1 h. Protein bands were detected using an enhanced chemiluminescence system (Amersham Biosciences, Arlington Heights, IL) and exposed to Hyperfilm (Amersham Biosciences, Little Chalfont, UK). To further confirm even loading of protein, membranes were stripped by incubating in 0.2 m NaOH solution for 5 min and probed for β-actin (1:5000, A-5441; Sigma). Films were scanned by an optical densitometer and analyzed using Multi-Analyst software (Bio-Rad).
Figure 2.

Tissue corresponding to the ischemic penumbra was dissected for Western blot. Ischemic lesion in a normothermic rat is represented as black plus gray, and black alone represents ischemic infarct in a hypothermic rat. The region spared by hypothermia is defined as the penumbra (gray), and the black area is the ischemic core. The region of the penumbra (within the solid lines, I) was dissected for Western blotting. The corresponding contralateral non-ischemic cortex (II) or similar cortex from sham animal without ischemia was dissected as a control.
In vitro Akt kinase assay. We followed instructions in the Akt kinase assay kit from Cell Signaling Technology (catalog #9840). Rats were killed with an overdose of isoflurane, perfused with iced PBS. Brains were removed, and the ischemic penumbra was dissected on ice. Brain tissue (100-120 mg) was added to seven times the amount of cell lysis buffer and homogenized. The solution was centrifuged at 14,000 × g, and the pellet was discarded while the supernatant (whole-cell extraction) was taken for kinase assay. Twenty microliters of immobilized Akt primary antibody bead slurry was added to 200 μl of whole-cell extraction (150 μg of protein) overnight at 4°C. After centrifugation, the upper solution (solution I) was saved for β-actin detection by Western blot to confirm that the same amount of whole-cell extractions were used for the Akt kinase assay. The Akt antibody pellet was washed with cell lysis buffer and suspended in 50 μl of kinase buffer supplemented with 1 μl of 10 mm ATP and 1 μg of GSK3 fusion protein, which was allowed to incubate for 30 min at 30°C. The reaction was terminated with 25 μl of 3 × SDS sample buffer. The solution was mixed with Vortex and centrifuged. The supernatant was taken (solution II) for Western blot detection of phosphorylated GSK3 fusion protein. Before doing this, to confirm that solution II was not contaminated by unwanted precipitation from the whole-cell extraction, a control experiment was performed in which Akt antibody was omitted (see Fig. 5C). Additionally, a Western blot for both solutions I and II were run in a single set to confirm that solution II was not contaminated by solution I (see Fig. 5C).
Figure 5.

The effect of hypothermia on Akt activity after stroke. A, Representative protein bands from Western blots for P-Akt (Ser473), P-Akt (Thr308), β-actin, and total Akt. Biphasic changes in P-Akt (Ser473) were observed in normothermic rats, whereas hypothermia blocked all such changes except at 24 h. No changes in P-Akt (Thr308) were detected in normothermic animals until 48 h or hypothermic animals at all time points. Total Akt did not decrease after ischemia until 48 h, an effect blocked by hypothermia.β-Actin was used to show equal protein loading of each lane. The same samples were used for the subsequent Western blot for detecting GSK3β, PDK1, and PTEN. For the sake of brevity, bands of β-actin confirming equal loading of each lane are not shown. B, Relative optical densities for P-Akt (Ser473). Optical densities indicate that P-Akt (Ser473) decreased 30 min into dMCAo, increased from 1.5 to 5 h after ischemia onset, decreased at 9 and 24 h, and recovered at 48 h. Hypothermia maintained P-Akt (Ser473) levels at most time points, with a transient decrease observed at 24 h. n = 3-5 per group. *p < 0.05 versus control (con); **p < 0.01 versus control and other time points at 30°C; #p < 0.05 and ##p < 0.01 versus 30°C at 1.5 and 5 h, respectively. Black bars, 37°C; gray bars, 30°C. n = 3-5 per group. C, An experiment was performed to verify that a pure Akt kinase extraction was obtained for the Akt kinase assay. Western blots indicate that β-actin was not seen in solution II containing Akt extraction, suggesting that the Akt kinase assay was not contaminated by the supernatant (solution I) and potential unwanted precipitation from the whole-cell extraction itself (a control solution from sham animal was used). In addition, no GSK3 fusion protein band was observed in the whole-cell extraction, suggesting that it did not contain the endogenous GSK3 protein to confound the Akt kinase assay. D, Representative protein bands from an in vitro Akt kinase assay. The solution used for Akt immunoprecipitation was subjected to β-actin measurement to confirm that equal amounts of protein were used for this assay. E, Akt kinase activity was expressed as percentages of P-GSK3 band compared with that from sham animals. Akt kinase activity decreased at 5 and 24 h after ischemia onset at 37°C; this effect was inhibited by hypothermia. *p < 0.001 versus control and 30°C, 24 h; **p < 0.001 versus control; #p < 0.05 versus 37°C, 24 h; ##p < 0.001 versus 37°C, 5 h; +p < 0.001 versus control. Black bars, 37°C; gray bars, 30°C.
Statistical analyses. Two-way ANOVA was used to compare the protective effect of hypothermia on infarct size, behavioral scores, and optical densities of protein bands from Western blots. Differences in protein bands from Western blots at the same temperature were analyzed using one-way ANOVA, followed by Fisher least significant difference post hoc test. Tests were considered statistically significant at p values <0.05. Data are presented as means ± SEM.
Results
Moderate hypothermia reduces infarct size and attenuates functional disorders up to 2 months after stroke
Hypothermia robustly reduced infarct size compared with normothermia at 2 d after stroke (Fig. 3A,B). Two months after stroke, a large part of the ischemic cortex is lost in normothermic rats, so we measure infarct size by subtracting the area of tissue remaining from the area of the nonischemic side and expressing the difference as a percentage of the area of the nonischemic side. As a result, infarct size at 60 d is similar to that measured at 2 d (Fig. 3C,D), suggesting that hypothermia decreases ischemic damage over the long term rather than merely delaying its emergence.
Figure 3.

Hypothermia reduces infarct size after permanent dMCAo. A, Representative infarcts stained with cresyl violet from level 3 in rats killed 2 d after stroke. The pale area with asterisks represents the infarct region. Normothermic ischemia damaged the cortex ipsilateral to the occluded MCA, whereas hypothermia spared all or most of the injured cortex. Only a small lesion was observed in the presented section from a hypothermic rat. B, Statistical analysis of infarct size 2 d after stroke. Two-way ANOVA (two factors, temperature and brain section level) was used to compare the effect of temperature on the infarct size at each level (data not shown) and on the mean of all four levels. Hypothermia (n = 7) reduced the mean infarct size by >80% compared with normothermia (n = 7; p < 0.001). C, Representative sections stained with cresyl violet from animals surviving 2 months after stroke. Most of the cortex in the infarcted hemisphere was lost in normothermic but not hypothermic rats. D, Infarct size 60 d after stroke. Hypothermia (n = 9) reduced infarct size 60 d after stroke compared with normothermia (n = 8; p < 0.001).
Effects of hypothermia on behavioral deficits after stroke were also detected. Four standard behavioral tests were performed (Fig. 4). Placing of the forelimb contralateral to the injury was disrupted at 2 and 3 d after stroke in normothermic animals and gradually recovered from 7 to 60 d; hypothermia decreased this deficit in the first 7 d after stroke, but no significant differences were observed thereafter (Fig. 4A). The scores for the postural reflex test increased from 2 to 60 d in normothermic animals, but performance of hypothermic rats was significantly less affected at 2 and 3 d, and deficits were eliminated from 7 to 60 d (Fig. 4B). Hypothermia also significantly attenuated the percentage of large right turns in normothermic animals in the tail hang test (Fig. 4C). Finally, a bias in favor of the ipsilateral forelimb was observed in the home cage test from 2 to 60 d; hypothermia robustly decreased this bias throughout the study (Fig. 4D). Infarct sizes and behavioral outcomes were usually closely correlated (Table 3), with poor behavioral performance associated with larger infarct sizes. However, performance on the vibrissa tests at 30 and 60 d were not correlated with infarct size, consistent with Figure 4A, which shows considerable spontaneous recovery of this behavior after stroke in both temperature groups.
Figure 4.

Hypothermia attenuates behavioral deficits up to 2 months after ischemia. A, Vibrissa test. All sham animals (n = 6) showed normal forelimb placing. There was unsuccessful placing of the contralateral forelimb at 2, 3, and 7 d after stroke in normothermic animals (n = 8) but gradual recovery to normal levels at 1 month. Hypothermia (n = 9) attenuated the deficit from 2 to 7 d after stroke. *p < 0.05 and **p < 0.001 versus sham; #p < 0.05 and ##p < 0.001 versus 30°C; +p < 0.05 versus sham.B, Postural reflex test. Scores significantly increased at all time points in normothermic animals. Hypothermia decreased the scores at 2, 3, 7, 14, and 60 d after stroke. *p < 0.01 and **p < 0.001 versus sham; #p < 0.01 and ##p < 0.001 versus 30°C; +p < 0.05 versus sham. C, Tail hang test. The number of large right turns increased in normothermic animals, an effect blocked by hypothermia. #p < 0.01 versus sham and 30°C; *p = 0.005 and **p < 0.001 versus 37°C. D, Home cage test. The percentage of the ipsilateral forelimb placing increased 2, 3, 7, 14, 30, and 60 d after ischemia in normothermic animals. Hypothermia decreased this biased usage of the forelimb at all time points. #p < 0.05 and ##p < 0.001 versus sham; *p < 0.05 and **p < 0.001 versus 30°C.
Table 3.
Correlation between infarct size at 60 d after stroke and functional outcomes
|
Tests |
|
0 d |
2 d |
3 d |
7 d |
14 d |
30 d |
60 d |
|---|---|---|---|---|---|---|---|---|
| p | 0.438 | ** | * | * | 0.0758 | 0.278 | 0.174 | |
| Vibrissa | r | 0.202 | −0.759 | −0.524 | −0.520 | −0.442 | −0.279 | −0.346 |
| p | 0.0589 | ** | *** | *** | 0.0903 | * | ||
| Reflex | r | 0.516 | 0.621 | 0.786 | 0.775 | 0.423 | 0.600 | |
| p | ** | * | ||||||
| Tail | r | 0.657 | 0.582 | |||||
| p | 0.716 | 0.105 | * | * | ** | * | 0.0517 | |
| Cage |
r
|
0.095 |
0.435 |
0.507 |
0.562 |
0.659 |
0.574 |
0.479 |
*p < 0.05; **p < 0.01; ***p < 0.001. The actual p values were shown when they did not reach significant levels. r, Correlation coefficient value.
The effect of hypothermia on Akt activity after cerebral ischemia
Western blots indicated that, in normothermic animals, phosphorylated Akt (P-Akt) (Ser473) decreases at 30 min, increases at 1.5 and 5 h, decreases at 9 and 24 h, and recovers at 48 h (Fig. 5). Such a transient increase in P-Akt (Ser473) is consistent with previous reports (Noshita et al., 2001), except that the initial decline has not been reported previously. Hypothermia prevents all post-ischemic changes in P-Akt (Ser473) seen in normothermic animals except at 24 h (Fig. 5A,B). However, changes in P-Akt (Thr308) were not detected until 48 h, when it decreased. Hypothermia also did not alter the effect of stroke on P-Akt (Thr308).
Because phosphorylation levels of Akt may not entirely represent its kinase activity, we performed an in vitro kinase assay to directly determine Akt kinase activity. First, an experiment was performed to confirm that pure Akt kinase extraction from the whole-cell homogenate was obtained for the Akt kinase assay (Fig. 5C). To our surprise, the result of the in vitro Akt kinase assay was not consistent with the in vivo phosphorylation levels of P-Akt (Ser473), in that the actual Akt activity decreased 5 and 24 h after cerebral ischemia (Fig. 5D), whereas P-Akt (Ser473) levels increased at 5 h. Hypothermia maintained post-ischemic P-Akt (Ser473) levels (Fig. 5B), but, perhaps more importantly, it attenuated the decline in Akt kinase activity at 5 and 24 h (Fig. 5D,E).
Double-immunofluorescence staining of microtubule-associated protein-2 (MAP-2) and P-Akt was performed and examined under a confocal microscope (Fig. 6). MAP-2 is a marker for both neuronal type and neuronal survival (Matesic and Lin, 1994; Blomgren et al., 1995). MAP-2 expression was significantly decreased in the ischemic core but was attenuated in the ischemic penumbra at 5 h and was almost undetectable at 24 h in the corresponding brain regions after normothermic ischemia. Nevertheless, MAP-2 expression in the ischemic penumbra was primarily maintained in hypothermic rats 24 h after stroke. Because the dynamic pattern of MAP-2 expression in normothermic and hypothermic brains after stroke was correlated with infarct size, as detected by staining with cresyl violet, MAP-2 served as a marker for the ischemic core and penumbra in this study. P-Akt (Ser473) normally expresses in neurons of non-ischemic cortex and remains in the ischemic penumbra at 5 h but not 24 h in normothermic brains. Although Western blot suggests that P-Akt (Ser473) transiently overexpressed at 5 h, no obvious increase in immunostaining in the penumbra was observed. This may simply reflect the fact that immunofluorescence staining is not sensitive enough to detect its transient increase after stroke. In hypothermic animals, consistent with the results of Western blots, the intensity of P-Akt (Ser473) immunostaining did not decrease at 5 h but decreased at 24 h. Despite the decrease in P-Akt (Ser473) at 24 h, MAP-2 staining did not significantly decrease, suggesting that the level of P-Akt (Ser473) does not reflect neuronal survival in the hypothermic penumbra.
Figure 6.
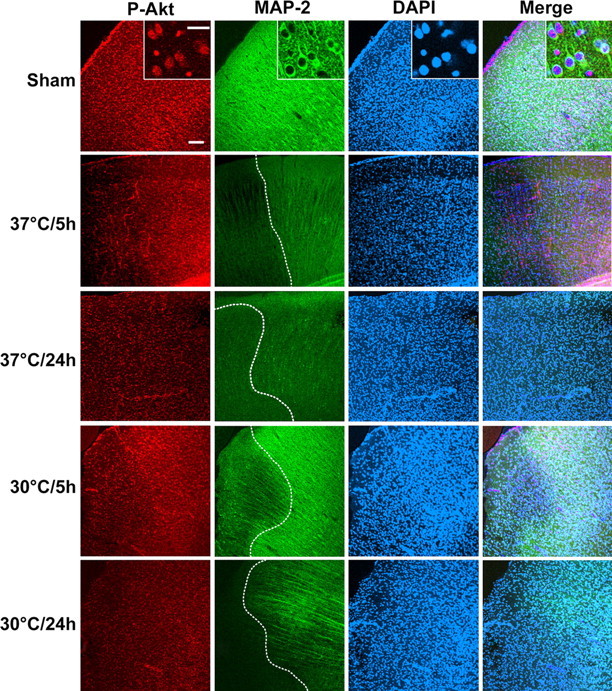
Triple staining of P-Akt (Ser473), MAP-2, and 4′, 6′-diamidino-2-phenylindole (DAPI). Overview pictures were taken at the border between the ischemic core and the penumbra defined by MAP-2 staining (dashed line in MAP staining). Both P-Akt (Ser473) and MAP-2 expressed in non-ischemic cortex (Sham). P-Akt (Ser473) colocalized with MAP-2 as shown in the high-magnification inset. MAP-2 expression decreased in the ischemic core at 5 h in normothermic animals, was slightly decreased in the penumbra, and disappeared at 24 h. P-Akt (Ser473) expression decreased in the core at 5 h but was maintained in the penumbra and decreased 24 h after normothermic stroke. Both MAP-2 and P-Akt (Ser473) expression decreased in the core even in the hypothermic animals, but MAP-2 still expressed in the penumbra at 24 h whereas P-Akt (Ser473) decreased. Scale bars: low magnification, 100 μm; inset, 10 μm.
The fact that hypothermia attenuated the decrease in Akt activity after stroke suggests that it should attenuate the post-insult decline in phosphorylation of Akt substrates as well. Therefore, we did double immunostaining of P-Akt substrate and MAP-2 (Fig. 7). The P-Akt substrate decreased at 5 h in the ischemic penumbra of normothermic brains, suggesting that the transient increase in P-Akt (Ser473) does not lead to phosphorylation of its substrates. Nevertheless, hypothermia improved expression of P-Akt substrate in the penumbra at 5 and 24 h, consistent with the attenuated decline in Akt activity.
Figure 7.
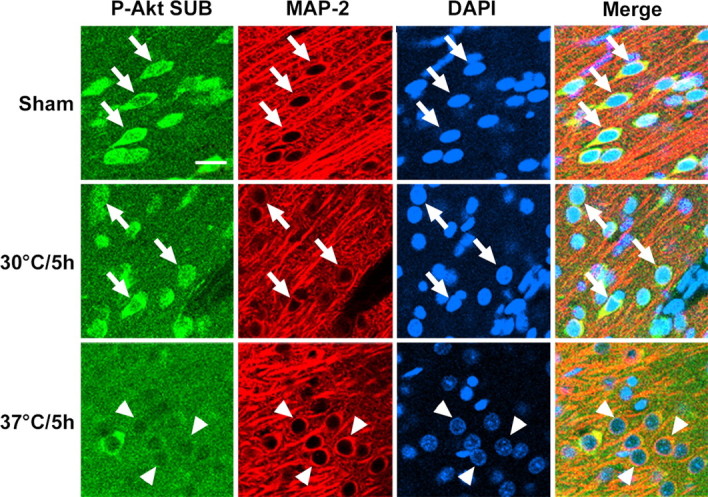
Triple staining of P-Akt substrate (P-Akt SUB), MAP-2, and DAPI. P-Akt substrate strongly expressed in the non-ischemic contralateral cortex of ischemic animals and cortex of sham animals and colocalized with MAP-2 (arrows). In normothermic animals, immunoreactivity of P-Akt substrate decreased at 5 h (arrowheads) and 24 h (data not shown) after stroke. Hypothermia improved its expression both at 5 and 24 h (see also Fig. 12). Scale bar, 20 μm.
PI3K inhibitor worsens ischemic damage in hypothermic animals
To provide direct evidence about the contribution of Akt activity to the protective effect of hypothermia, we examined the effects of inhibiting the activity of the upstream activator of Akt, PI3 kinase, with the specific inhibitor LY294002, on infarct size after hypothermic ischemia. Larger infarcts were observed in LY294002-treated animals (44.6 ± 5.4%) compared with vehicle-treated rats (21.5 ± 4.8%; p < 0.01).
Hypothermia attenuates dephosphorylation of PDK1 and PTEN
The effects of hypothermia on events upstream of Akt were investigated. P-PDK1 (Ser241) decreased after ischemia onset at all time points (Fig. 8A,B). Although levels of P-PDK1 decreased at 5 and 9 h in hypothermic animals, it recovered at 24 h (Fig. 8A,B). P-PTEN (Ser380) decreased from 0.5 to 9 h after ischemia onset and then recovered at 24-48 h (Fig. 8A,C). It was striking that hypothermia blocked the decrease in P-PTEN at all time points (Fig. 8A,C).
Figure 8.
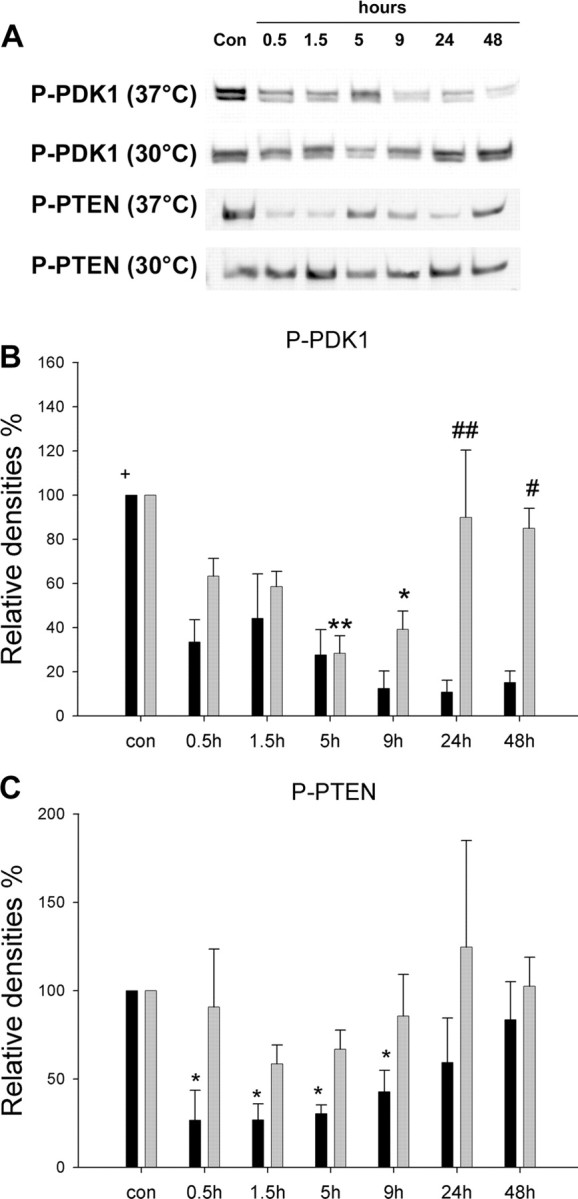
Hypothermia attenuated decreases in P-PDK1 and P-PTEN after stroke. A, Representative protein bands from Western blots of P-PDK1 and P-PTEN. B, Optical densities of P-PDK1. P-PDK1 protein significantly decreased at all time points after normothermic ischemia (black bars). Although P-PDK1 decreased at 5 and 9 h in hypothermic animals (gray bars), it recovered from 24 h, and total levels of P-PDK1 across all time points were substantially higher under hypothermia. +p < 0.001 versus all groups, 37°C; *p < 0.05 and **p < 0.01 versus control (con); #p < 0.05 and ##p<0.001 versus 37°C at the same time point. n = 3-5 per group. C, Mean optical densities demonstrate that P-PTEN decreased from 0.5 to 9 h after ischemia and started to recover at 24 h. No decreases in P-PTEN were observed in hypothermic animals after ischemia, and total level of P-PTEN in hypothermic rats were higher than in normothermic animals (p < 0.05), suggesting that hypothermia maintained protein levels of P-PTEN after stroke. *p < 0.05 versus control. n = 3-5 per group. Black bars, 37°C; gray bars, 30°C.
Double staining of P-PTEN and MAP-2 suggests that P-PTEN expressed in non-ischemic neurons and decreased at 5 h after ischemic onset in normothermic animals (Fig. 9A). However, P-PTEN overexpressed in some blood vessels (Fig. 9A,B) in the ischemic cortex at 24 h, whereas it decreased in neurons, which may explain the finding on Western blot that P-PTEN levels start to recover at 24 h. Hypothermia attenuated the decrease in P-PTEN expression in neurons at 5 and 24 h (Fig. 9A), which is consistent with the results of Western blot.
Figure 9.
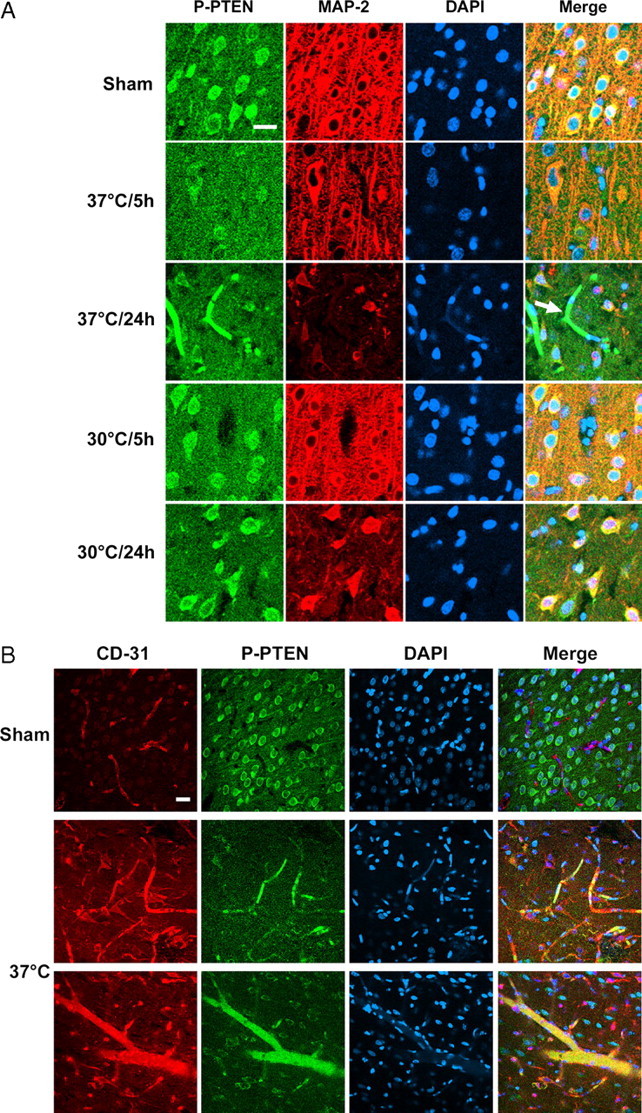
A, Triple staining of P-PTEN, MAP-2, and DAPI confirms that hypothermia maintained P-PTEN expression after ischemia. The photomicrographis of the ischemic penumbra. P-PTEN ubiquitously expressed in cortical neurons and decreased at 5 and 24 h in neurons both in the ischemic core (data not shown) and penumbra of normothermic brains. However, P-PTEN overexpressed in capillaries at 24 h (arrow). Hypothermia attenuated the decrease in immunoreactivity of P-PTEN both at 5 and 24 h after stroke. Scale bar, 20 μm. B, Triple staining of P-PTEN, CD-31 (an endothelial cell marker), and DAPI, indicating that P-PTEN expressed in capillaries and some larger vessels 24 h after stroke but not in blood vessels of the non-ischemic cortex. Scale bar, 20 μm.
Hypothermia improves phosphorylation of FKHR but does not enhance phosphorylation of GSK3β (Ser 9) and may inhibit activity of β-catenin
We then studied the effect of hypothermia on downstream targets of Akt. Western blots demonstrate that P-FKHR (Ser256) decreased after ischemia at all time points (Fig. 10A,B). In contrast, hypothermia transiently preserved P-FKHR (Ser256) at 0.5 h and increased it at 1.5 h, followed by a transient decrease at 5 and 9 h and a recovery beginning at 24 h (Fig. 10A,B).
Figure 10.
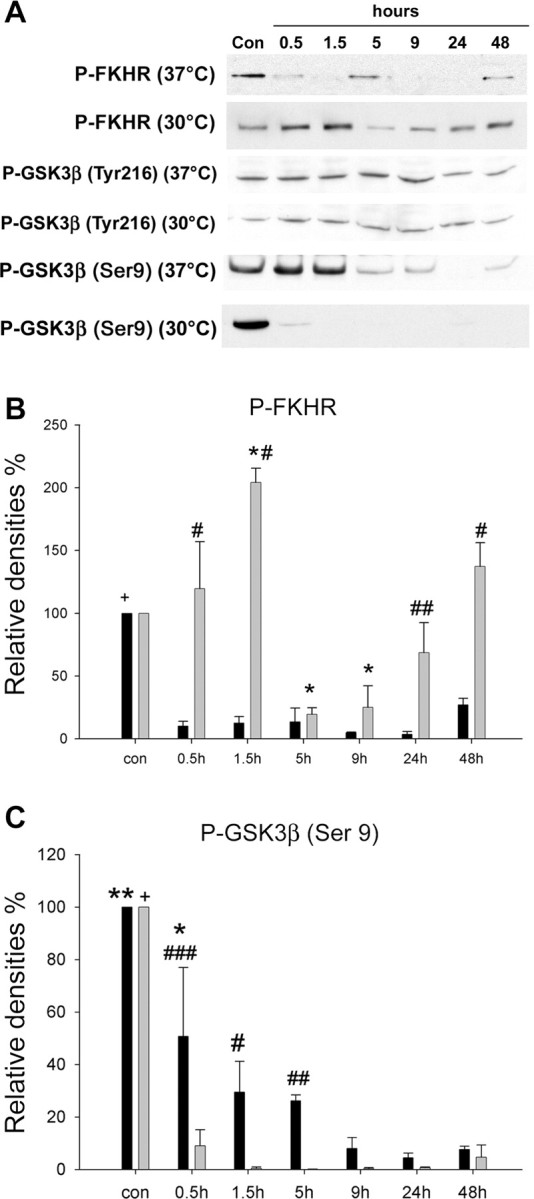
Phosphorylation levels of FKHR and GSK3β after cerebral ischemia. A, Representative protein bands from Western blots for P-FKHR, P-GSK3β (Tyr216), and P-GSK3β (Ser9). P-GSK3β (Tyr216) did not change after cerebral ischemia in either normothermic or hypothermic animals. B, Optical densities indicate that P-FKHR significantly decreased at all time points after stroke, whereas hypothermia transiently preserved P-FKHR at 0.5 h and enhanced P-FKHR at 1.5 h. Although P-FKHR decreased to low levels at 5 and 9 h with hypothermia, it began to recover earlier than under normothermia. +p < 0.001 versus all groups of 37°C; *p < 0.01 versus control (con); #p < 0.001 and ##p < 0.01 versus 37°C at the same time point. n = 3-5 per group. Black bars, 37°C; gray bars, 30°C. C, Optical densities of P-GSK3β (Ser9) bands shows that P-GSK3β (Ser9) gradually decreased after ischemia, and hypothermia did not block this. In fact, hypothermia seemed to magnify the decrease in P-GSK3β, suggesting that dephosphorylation of GSK3β may not play a role in ischemic cell damage. +p < 0.001 versus all groups, 30°C; *p < 0.05 versus control; **p < 0.001 versus all groups except 0.5 h, 37°C; #p < 0.05, ##p < 0.01, and ###p < 0.001 versus 30°C. n = 3-5 per group. Black bars, 37°C; gray bars, 30°C.
P-GSK3β (Tyr216) levels did not change after cerebral ischemia in either group (Fig. 10A). P-GSK3β (Ser9) significantly decreased after ischemia at all time points in normothermic animals (Fig. 10A,B), an effect which was worsened further under hypothermia. It is suggested that dephosphorylation of GSK3β (Ser9) leads to increased activity of GSK3β and apoptosis, but that this does not occur under hypothermia. We hypothesized that hypothermia might block an event downstream of GSK3β, β-catenin. Confocal microscopy revealed that P-β-catenin diffusely expressed in neuronal cytosol in non-ischemic cortex, but it started to express in the nuclei at 5 h and intensively stained in the nuclei in the penumbra 24 h after normothermic ischemia, suggesting that subcellular redistribution of P-β-catenin might be involved in ischemic neuronal death (Fig. 11). Interestingly, P-β-catenin-positive neurons were rarely seen in the ischemic core, probably because few neurons survived. In contrast, after hypothermia, P-β-catenin was detected in the nuclei of almost all neurons in the ischemic core as early as 5 h but not in the ischemic penumbra.
Figure 11.
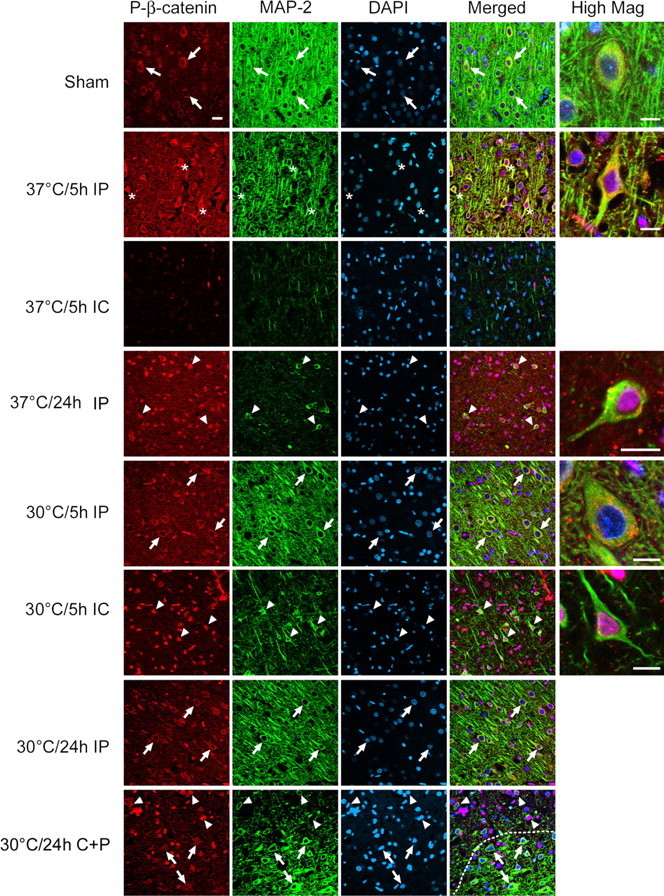
Triple staining of P-β-catenin, MAP-2, and DAPI. Pictures were taken from sham, normothermic, and hypothermic animals at 5 and 24 h after ischemia, and some merged higher-magnification images are presented. Immunostainings in both ischemic penumbra (IP) and ischemic core (IC) were also compared. P-β-Catenin normally expresses in the cytosol (arrows) in non-ischemic brain but translocated into the nuclei (asterisks) of neurons in the penumbra of normothermic brains at 5 h, although few positive cells in the core were detected. Twenty-four hours later, nuclear-positive staining (arrowheads) was observed in most cells. However, nuclear translocation of P-β-catenin was detected only in the ischemic core (arrowheads) in the hypothermic brain both at 5 and 24 h. A picture showing the transitional pattern of subcellular distribution of P-β-catenin from ischemic penumbra to core was taken in a hypothermic rat at 24 h after ischemia, indicating that P-β-catenin mainly expressed in the cytosol in the penumbra but in the nuclei in the ischemic core. A dotted line indicates the border between the ischemic core, with less MAP-2 staining, and the penumbra, with more MAP-2 staining. Scale bars: low magnification, 20 μm; higher magnification, 10 μm. con, Control.
Dephosphorylation of P-Akt, P-Akt substrate, and P-PTEN precede cytochrome c release
We have shown that disorders of the Akt pathway occur before the MAP-2 decrease in the penumbra of normothermic brains, and hypothermia maintains MAP-2 expression by improving disorders of Akt pathway (Figs. 6, 7, 9, 11). This suggests that Akt pathways are closely associated with neuronal survival. We further asked whether some of these events are linked to cytochrome c release, a cascade leading to apoptosis. We found cytochrome c release in a few neurons at 5 h (data not shown) and in many neurons in the penumbra at 24 h after stroke in normothermic brains but not in hypothermic animals. Double staining indicates that decreases in P-Akt, P-Akt substrates, and P-PTEN all preceded cytochrome c release in normothermic animals (Fig. 12). Although hypothermia did not block Akt dephosphorylation at 24 h, it did inhibit both decreases in P-Akt substrate, P-PTEN.
Figure 12.
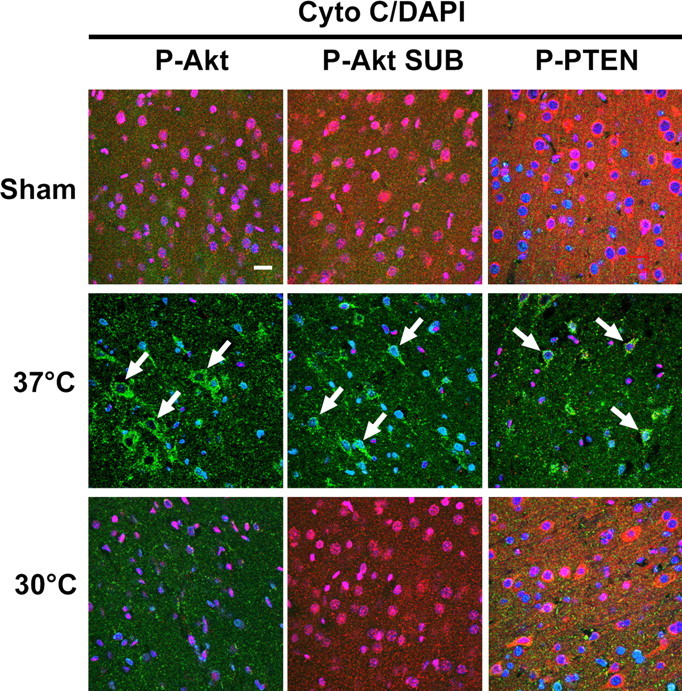
Cytochrome c (Cyto C) releases only after P-Akt, P-Akt substrate (P-Akt SUB), and P-PTEN decrease in normothermic but not in hypothermic animals. Triple staining of P-Akt (Ser473) (red), P-Akt substrate (red), and P-PTEN (red) with cytochrome c (green) and DAPI (blue) was performed. Cytochrome c immunostaining was not observed in the cortex of sham animals and was detected in a few cells at 5 h after stroke (data not shown). Strong immunostaining of cytochrome c (arrows) was detected in the ischemic cortex penumbra 24 h after normothermic ischemia when P-Akt (Ser473), P-Akt substrate, and P-PTEN substantially decreased. Hypothermic blocked the decreases in P-Akt substrate and P-PTEN but not the Akt dephosphorylation at 24 h in the penumbra. Scale bar, 20 μm.
Discussion
Despite extensive research, the protective mechanisms of hypothermia are still not well understood. To our knowledge, this is the first clear demonstration that PI3/Akt pathways play critical roles in neuroprotection by hypothermia. Akt activity and phosphorylation level of PTEN, PDK1, GSK3β, and FKHR decreased at early time points in the penumbra after ischemia and preceded degradation of MAP-2 and cytochrome c release, suggesting that dysfunction of Akt pathway might help mediate ischemic damage. Hypothermia attenuated decreases in Akt activity after ischemia onset and improved phosphorylation level of PTEN, PDK1, and FKHR (but not GSK3β), suggesting that maintenance of Akt pathways might be neuroprotective. Additionally, a PI3K inhibitor enlarged infarct size in hypothermic animals, also suggesting that the PI3/Akt pathway contributes to hypothermic neuroprotection. We also observed that the transcription factor P-β-catenin, which acts downstream of GSK3β, translocated from cytosol into the nucleus as early as 5 h after stroke, suggesting a role for β-catenin in ischemic neurotoxicity. Moreover, we observed that hypothermia blocked subcellular redistribution of P-β-catenin in the penumbra. Last, we demonstrated for the first time that P-PTEN overexpressed in cerebral blood vessels after stroke, indicating that it might be involved in regulating the functions of blood vessels.
The well defined ischemic border generated in the distal MCA occlusion model allowed us to dissect penumbral regions for Western blot analysis. The penumbra in the hypothermic brain was defined as the area saved by hypothermia compared with normothermic brain. The corresponding regions in normothermic animals also show characteristics of the penumbra, in that MAP-2 immunoreactivity, a marker of neuronal survival, was transiently maintained here but not in the core at 5 h after stroke. We observed that P-Akt (Ser473) decreased 30 min after ischemia onset, when both bilateral CCA and distal MCA were occluded but, in agreement with other reports (Kawano et al., 2001; Noshita et al., 2001; Abe et al., 2004), increased after reperfusion. Hypothermia blocked these changes but did not prevent the decline in P-Akt (Ser473) at 24 h. Hypothermia blocked the transient increase in P-Akt (Ser473) in a model of neonatal hypoxiaischemia (Tomimatsu et al., 2001), suggesting that Akt is not essential to hypothermic protection. However, complete activation of Akt requires phosphorylation of both Ser473 and Thr308 (Scheid et al., 2002). P-Akt (Thr308) did not change until 48 h, when it decreased, and, although hypothermia prevented that decline, it had no earlier effect on P-Akt (Thr308). It is unlikely that phosphorylation at Ser473 and Thr308 alone is responsible for hypothermic protection. Nevertheless, in vitro assay showed that Akt activity decreased at 5 h in normothermic animals but was maintained in hypothermia animals. However, in this study, we only used a GSK3 fusion protein as a substrate to assay Akt kinase activity, which may not indicate total Akt activity because Akt has numerous substrates. To further test whether the level of P-Akt (Ser473) is not representative of Akt activity, we examined whole phosphorylation levels of Akt substrate. Indeed, confocal microscopy indicated that P-Akt substrate decreased at 5 h after stroke in normothermic animals, an effect that was blocked by hypothermia. Thus, Akt phosphorylation at Ser473 and Thr308 does not necessarily represent Akt activation. Moreover, Ser473 phosphorylation is not sufficient to stimulate Akt activity (Alessi et al., 1996; Bellacosa et al., 1998; Hill et al., 2001), and tyrosine phosphorylation is necessary (Chen et al., 2001). This may explain why transient increases in P-Akt (Ser473) after stroke are not neuroprotective in normothermic animals.
As a novel finding, we observed that P-PDK1, upstream of Akt, decreased immediately after stroke onset. P-Akt (Thr308) is thought to be regulated by P-PDK1, whereas P-Akt (Ser473) may be phosphorylated indirectly by PDK1 (Franke et al., 2003; Fresno Vara et al., 2004). P-Akt (Ser473) may also be autophosphorylated or phosphorylated by other kinases (Scheid et al., 2002). Therefore, it is not surprising that levels of P-PDK1 and P-Akt (Ser473) dissociate after stroke onset. However, despite P-PDK1 decreasing after ischemia, the level of P-Akt (Thr308) was maintained, suggesting the involvement of other kinases.
PTEN is a negative regulator of Akt, dephosphorylating PIP3 to PIP2, thus preventing recruitment of Akt to the membrane for phosphorylation (Franke et al., 2003; Fresno Vara et al., 2004). Reports implicate PTEN in cerebral ischemia (Omori et al., 2002; Lee et al., 2004; Ning et al., 2004). Ning et al. (2004) showed that downregulation of PTEN protein protects against ischemic damage. However, cilostazol lessens ischemic damage by reducing P-PTEN (Lee et al., 2004), in conflict with our results. Our data indicate that P-PTEN decreased after stroke onset, a decline blocked by hypothermia. In fact, P-PTEN was markedly and consistently preserved by hypothermia at early time points (0.5-9 h after ischemia) compared with P-Akt, P-PDK1, P-GSK3β, and P-FKHR. Therefore, hypothermia may mediate its neuroprotective action primarily through its effects on P-PTEN. We also found that P-PTEN was overexpressed in cerebral blood vessels from 24 h after stroke, which might play a role in regulating the functions of blood vessels after ischemia. This may also explain the recovery of P-PTEN at 24 h despite its being decreased in ischemic neurons. P-PTEN has also been shown to increase in active astrocytes in the hippocampus up to 2 weeks after transient forebrain ischemia in rats (Choi et al., 2005). Whether this occurs in the ischemic margin after focal ischemia deserves additional study.
Hypothermia also had effects on downstream events along the Akt pathway. P-FKHR (Ser256) decreases in hippocampal CA1 after global ischemia, leading to FKHR activation, translocation to the nucleus, and induction of Fas ligand and Bim, thereby causing apoptosis (Kawano et al., 2002). We observed a reduction in P-FKHR after ischemia onset and the attenuation of that effect by hypothermia. However, whether this leads to FKHR translocation and induction of Fas ligand and Bim requires additional investigation.
GSK3β, downstream of Akt, is critical for ischemic neurotoxicity. Although GSK3β is downregulated when phosphorylated at Ser9 by Akt, it is activated by phosphorylation at site Tyr216 (Bhat et al., 2000). IGF-1 protects from ischemic damage by reducing GSK3β levels (Wang et al., 2000). We observed that mild hypothermia downregulated GSK3β expression after transient global ischemia and that a GSK3β inhibitor, Chir025, reduced ischemic neurotoxicity in vitro and in vivo (Kelly et al., 2004). Another report suggests that P-GSK3β (Tyr216) increased after ischemia (Bhat et al., 2000). Although hypothermia maintained many steps in the Akt pathway after stroke, it did not improve GSK3β (Ser9) phosphorylation. β-Catenin is a critical transcription factor acting downstream of GSK3β. Phosphorylation of β-catenin by GSK3β leads to its degradation and apoptosis. P-β-Catenin translocates from the cytosol into the nuclei in hippocampal neurons after ischemia (Cappuccio et al., 2005). Similarly, we found nuclear β-catenin translocation in the penumbra but not in the core after ischemia in normothermic animals. Interestingly, hypothermia blocked nuclear β-catenin phosphorylation in the ischemic penumbra, but nuclear translocation was still observed in the core. It is likely that nuclear P-β-catenin translocation is an active process, only occurring in the penumbra during normothermia, in which ATP levels are maintained. Hypothermia may also improve energy supply in the ischemic core, enabling β-catenin translocation into the nuclei. Our results suggest that hypothermia can block events downstream of GSK3β, thereby decreasing damage in the penumbra.
Despite GSK3β being an Akt substrate in vitro, it is unlikely to be in vivo. We observed in vivo that, although Akt kinase activity was improved by hypothermia, P-GSK3β (Ser9) decreased even more in hypothermic animals than in normothermics. Therefore, GSK3β may not be a substrate of Akt kinase during ischemia, GSK3β and Akt may be separated intracellularly, or other protein kinases (such as protein kinase A and C) may regulate of GSK3β phosphorylation state in this case.
Finally, hypothermia not only reduced infarct size but improved neurological function. The postural reflex test, tail hang test, and home cage test demonstrated that hypothermia improved motor asymmetry up to 60 d after stroke. Normothermic animals showed recovery from 7 to 60 d in the vibrissa and postural reflex tests. Recovery was not observed in normothermic rats in the tail hang and home cage tests, suggesting that these tests might be preferable for assessing chronic behavioral deficits in this stroke model. Despite some cortical damage in hypothermic animals, no behavioral deficits were detected at any time point in the home cage test, suggesting that the test might not be sensitive enough to detect the effects of limited ischemic damage. In contrast, deficits were detected 2 and 3 d after stroke in the vibrissa and postural reflex tests. In addition, performance in most behavioral tests correlated with infarct volume at most time points, indicating that these tests can predict infarct size.
In conclusion, hypothermia reduces infarct size and improves functional outcomes after stroke. Such protection may involve maintaining Akt activity and blunting the decreases in phosphorylation of PTEN, PDK1, and FKHR. Although GSK3β activity may not be blocked by hypothermia, a downstream effect of it, β-catenin activity, was attenuated by hypothermia. Collectively, these effects on the Akt pathway may help explain the neuroprotective actions of hypothermia.
Footnotes
This study was supported by National Institute of Neurological Disorders and Stroke Grants R01 NS27292 (G.K.S.) and P01 NS37520 (G.K.S., R.M.S.). We thank Elizabeth Hoyte for assistance preparing the figures and Dr. Tonya Bliss for consultation on the behavioral tests.
Correspondence should be addressed to Dr. Gary K. Steinberg, Department of Neurosurgery, Stanford University School of Medicine, 300 Pasteur Drive, R200, Stanford, CA 94305-5327. E-mail: cerebral@stanford.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259794-13$15.00/0
References
- Abe T, Takagi N, Nakano M, Furuya M, Takeo S (2004) Altered Bad localization and interaction between Bad and Bcl-xL in the hippocampus after transient global ischemia. Brain Res 1009: 159-168. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541-6551. [PMC free article] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H (1986) Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 17: 472-476. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, Mc-Cormick F, Feng J, Tsichlis P (1998) Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene 17: 313-325. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee CM (2000) Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc Natl Acad Sci USA 97: 11074-11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomgren K, McRae A, Bona E, Saido TC, Karlsson JO, Hagberg H (1995) Degradation of fodrin and MAP 2 after neonatal cerebral hypoxic-ischemia. Brain Res 684: 136-142. [DOI] [PubMed] [Google Scholar]
- Borlongan CV, Tajima Y, Trojanowski JQ, Lee VM, Sanberg PR (1998) Transplantation of cryopreserved human embryonal carcinoma-derived neurons (NT2N cells) promotes functional recovery in ischemic rats. Exp Neurol 149: 310-321. [DOI] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME (2001) Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 11: 297-305. [DOI] [PubMed] [Google Scholar]
- Busto R, Dietrich WD, Globus MY, Ginsberg MD (1989) The importance of brain temperature in cerebral ischemic injury. Stroke 20: 1113-1114. [DOI] [PubMed] [Google Scholar]
- Cao G, Pei W, Lan J, Stetler RA, Luo Y, Nagayama T, Graham SH, Yin XM, Simon RP, Chen J (2001) Caspase-activated DNase/DNA fragmentation factor 40 mediates apoptotic DNA fragmentation in transient cerebral ischemia and in neuronal cultures. J Neurosci 21: 4678-4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Clark RS, Pei W, Yin W, Zhang F, Sun FY, Graham SH, Chen J (2003) Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J Cereb Blood Flow Metab 23: 1137-1150. [DOI] [PubMed] [Google Scholar]
- Cappuccio I, Calderone A, Busceti CL, Biagioni F, Pontarelli F, Bruno V, Storto M, Terstappen GT, Gaviraghi G, Fornai F, Battaglia G, Melchiorri D, Zukin S, Nicoletti F, Caricasole A (2005) Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is required for the development of ischemic neuronal death. J Neurosci 25: 2647-2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PH (2004) Future targets and cascades for neuroprotective strategies. Stroke 35: 2748-2750. [DOI] [PubMed] [Google Scholar]
- Chen R, Kim O, Yang J, Sato K, Eisenmann KM, McCarthy J, Chen H, Qiu Y (2001) Regulation of Akt/PKB activation by tyrosine phosphorylation. J Biol Chem 276: 31858-31862. [DOI] [PubMed] [Google Scholar]
- Chen ST, Hsu CY, Hogan EL, Maricq H, Balentine JD (1986) A model of focal ischemic stroke in the rat: reproducible extensive cortical infarction. Stroke 17: 738-743. [DOI] [PubMed] [Google Scholar]
- Choi JS, Park HJ, Kim HY, Kim SY, Lee JE, Choi YS, Chun MH, Chung JW, Lee MY (2005) Phosphorylation of PTEN and Akt in astrocytes of the rat hippocampus following transient forebrain ischemia. Cell Tissue Res 319: 359-366. [DOI] [PubMed] [Google Scholar]
- Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C (2003) PI3K/Akt and apoptosis: size matters. Oncogene 22: 8983-8998. [DOI] [PubMed] [Google Scholar]
- Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M (2004) PI3K/Akt signalling pathway and cancer. Cancer Treat Rev 30: 193-204. [DOI] [PubMed] [Google Scholar]
- Friguls B, Petegnief V, Justicia C, Pallas M, Planas AM (2002) Activation of ERK and Akt signaling in focal cerebral ischemia: modulation by TGF-alpha and involvement of NMDA receptor. Neurobiol Dis 11: 443-456. [DOI] [PubMed] [Google Scholar]
- Graham SH, Chen J (2001) Programmed cell death in cerebral ischemia. J Cereb Blood Flow Metab 21: 99-109. [DOI] [PubMed] [Google Scholar]
- Hanada M, Feng J, Hemmings BA (2004) Structure, regulation and function of PKB/AKT—a major therapeutic target. Biochim Biophys Acta 1697: 3-16. [DOI] [PubMed] [Google Scholar]
- Hashiguchi A, Yano S, Morioka M, Hamada J, Ushio Y, Takeuchi Y, Fukunaga K (2004) Up-regulation of endothelial nitric oxide synthase via phosphatidylinositol 3-kinase pathway contributes to ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab 24: 271-279. [DOI] [PubMed] [Google Scholar]
- Hill MM, Andjelkovic M, Brazil DP, Ferrari S, Fabbro D, Hemmings BA (2001) Insulin-stimulated protein kinase B phosphorylation on Ser-473 is independent of its activity and occurs through a staurosporine-insensitive kinase. J Biol Chem 276: 25643-25646. [DOI] [PubMed] [Google Scholar]
- Jiang X, Wang X (2004) Cytochrome C-mediated apoptosis. Annu Rev Biochem 73: 87-106. [DOI] [PubMed] [Google Scholar]
- Jin G, Omori N, Li F, Nagano I, Manabe Y, Shoji M, Abe K (2003) Protection against ischemic brain damage by GDNF affecting cell survival and death signals. Neurol Res 25: 249-253. [DOI] [PubMed] [Google Scholar]
- Kawano T, Fukunaga K, Takeuchi Y, Morioka M, Yano S, Hamada J, Ushio Y, Miyamoto E (2001) Neuroprotective effect of sodium orthovanadate on delayed neuronal death after transient forebrain ischemia in gerbil hippocampus. J Cereb Blood Flow Metab 21: 1268-1280. [DOI] [PubMed] [Google Scholar]
- Kawano T, Morioka M, Yano S, Hamada J, Ushio Y, Miyamoto E, Fukunaga K (2002) Decreased akt activity is associated with activation of forkhead transcription factor after transient forebrain ischemia in gerbil hippocampus. J Cereb Blood Flow Metab 22: 926-934. [DOI] [PubMed] [Google Scholar]
- Kelly S, Zhao H, Hua Sun G, Cheng D, Qiao Y, Luo J, Martin K, Steinberg GK, Harrison SD, Yenari MA (2004) Glycogen synthase kinase 3beta inhibitor Chir025 reduces neuronal death resulting from oxygen-glucose deprivation, glutamate excitotoxicity, and cerebral ischemia. Exp Neurol 188: 378-386. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kim KY, Lee YK, Park SY, Kim CD, Lee WS, Rhim BY, Hong KW (2004) Cilostazol prevents focal cerebral ischemic injury by enhancing casein kinase 2 phosphorylation and suppression of phosphatase and tensin homolog deleted from chromosome 10 phosphorylation in rats. J Pharmacol Exp Ther 308: 896-903. [DOI] [PubMed] [Google Scholar]
- Limbourg FP, Huang Z, Plumier JC, Simoncini T, Fujioka M, Tuckermann J, Schutz G, Moskowitz MA, Liao JK (2002) Rapid nontranscriptional activation of endothelial nitric oxide synthase mediates increased cerebral blood flow and stroke protection by corticosteroids. J Clin Invest 110: 1729-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matesic DF, Lin RC (1994) Microtubule-associated protein 2 as an early indicator of ischemia-induced neurodegeneration in the gerbil forebrain. J Neurochem 63: 1012-1020. [DOI] [PubMed] [Google Scholar]
- Ning K, Pei L, Liao M, Liu B, Zhang Y, Jiang W, Mielke JG, Li L, Chen Y, El-Hayek YH, Fehlings MG, Zhang X, Liu F, Eubanks J, Wan Q (2004) Dual neuroprotective signaling mediated by downregulating two distinct phosphatase activities of PTEN. J Neurosci 24: 4052-4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noshita N, Lewen A, Sugawara T, Chan PH (2001) Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab 21: 1442-1450. [DOI] [PubMed] [Google Scholar]
- Noshita N, Sugawara T, Lewen A, Hayashi T, Chan PH (2003) Copper-zinc superoxide dismutase affects Akt activation after transient focal cerebral ischemia in mice. Stroke 34: 1513-1518. [DOI] [PubMed] [Google Scholar]
- Nusse R (2003) Wnts and Hedgehogs: lipid-modified proteins and similarities in signaling mechanisms at the cell surface. Development 130: 5297-5305. [DOI] [PubMed] [Google Scholar]
- Omori N, Jin G, Li F, Zhang WR, Wang SJ, Hamakawa Y, Nagano I, Manabe Y, Shoji M, Abe K (2002) Enhanced phosphorylation of PTEN in rat brain after transient middle cerebral artery occlusion. Brain Res 954: 317-322. [DOI] [PubMed] [Google Scholar]
- Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST (2000) CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology 39: 777-787. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Marignani PA, Woodgett JR (2002) Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol Cell Biol 22: 6247-6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH (1999) Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci 19: RC39(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomimatsu T, Fukuda H, Endo M, Watanabe N, Mu J, Kohzuki M, Fujii E, Kanzaki T, Murata Y (2001) Effects of hypothermia on neonatal hypoxic-ischemic brain injury in the rat: phosphorylation of Akt, activation of caspase-3-like protease. Neurosci Lett 312: 21-24. [DOI] [PubMed] [Google Scholar]
- Wang JM, Hayashi T, Zhang WR, Sakai K, Shiro Y, Abe K (2000) Reduction of ischemic brain injury by topical application of insulin-like growth factor-I after transient middle cerebral artery occlusion in rats. Brain Res 859: 381-385. [DOI] [PubMed] [Google Scholar]
- Yano S, Morioka M, Fukunaga K, Kawano T, Hara T, Kai Y, Hamada J, Miyamoto E, Ushio Y (2001) Activation of Akt/protein kinase B contributes to induction of ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab 21: 351-360. [DOI] [PubMed] [Google Scholar]
- Zhao H, Yenari MA, Cheng D, Sapolsky RM, Steinberg GK (2003) Bcl-2 overexpression protects against neuron loss within the ischemic margin following experimental stroke and inhibits cytochrome c translocation and caspase-3 activity. J Neurochem 85: 1026-1036. [DOI] [PubMed] [Google Scholar]
- Zhao H, Yenari MA, Sapolsky RM, Steinberg GK (2004a) Mild postischemic hypothermia prolongs the time window for gene therapy by inhibiting cytochrome C release. Stroke 35: 572-577. [DOI] [PubMed] [Google Scholar]
- Zhao H, Yenari MA, Cheng D, Barreto-Chang OL, Sapolsky RM, Steinberg GK (2004b) Bcl-2 transfection via herpes simplex virus blocks apoptosis-inducing factor translocation after focal ischemia in the rat. J Cereb Blood Flow Metab 24: 681-692. [DOI] [PubMed] [Google Scholar]
- Zhao H, Yenari MA, Cheng D, Sapolsky RM, Steinberg GK (2005) Biphasic cytochrome c release after transient global ischemia and its inhibition by hypothermia. J Cereb Blood Flow Metab 25: 1119-1129. [DOI] [PubMed] [Google Scholar]