

Abstract
Although the role of the amygdala in acquisition of conditioned fear is well established, there is debate concerning the intra-amygdala circuits involved. The lateral nucleus of the amygdala (LA) is thought to be an essential site of plasticity in fear conditioning. The LA has both direct and indirect [via the basal nuclei; basal amygdala (BA)] projections to the central nucleus (Ce) of the amygdala, an essential output for fear behaviors. Lesions of the LA or Ce prevent acquisition of conditioned freezing to a conditioned stimulus, but BA lesions do not, suggesting that the BA is not normally involved in fear conditioning. If true, posttraining BA lesions should also have no effect. Replicating previous studies, we found that rats given electrolytic BA lesions before training acquired conditioned fear normally. They also showed normal long-term retention and extinction of conditioned fear. Unexpectedly, BA lesions made after training completely blocked expression of conditioned fear. Despite this deficit, lesioned rats were able to learn a new tone-shock association. Thus, although the LA-Ce system is sufficient for fear acquisition in the absence of the BA, it is not sufficient when the BA is present, suggesting that the BA is an important site of plasticity in fear conditioning. The pattern of lesion deficits we observed (after but not before training) might be explained by homeostatic mechanisms that balance plasticity over multiple inputs, regulating the influence of the BA and LA onto Ce output neurons.
Keywords: prefrontal cortex, central nucleus, consolidation, intercalated cells, infralimbic, lateral amygdala
Introduction
It is well established that the amygdala is critical for the acquisition of conditioned fear associations, but the intra-amygdala circuits involved in this process are unclear (Nader et al., 2001; Koo et al., 2004). The lateral nucleus of the amygdala (LA) receives information about conditioned and unconditioned stimuli (CSs and USs) from thalamic and cortical sources and is an important site of plasticity in fear conditioning (Ressler et al., 2002; Lamprecht and LeDoux, 2004; Maren and Quirk, 2004). Potentiated LA outputs are thought to drive neurons in the medial subdivision of the central nucleus (CeM), which project to the hypothalamus and brainstem sites that generate fear responses (LeDoux et al., 1988; De Oca et al., 1998; Davis, 2000).
There has been debate, however, concerning the route by which the LA influences the CeM (Paré et al., 2004). The LA lacks direct projections to the CeM but projects to the basal amygdala [BA; defined as the basolateral (BL), basomedial (BM), and accessory basal (AB) nuclei], which in turn projects to the CeM (Pitkanen et al., 1997; Paré and Smith, 1998). This suggests that the LA influences the CeM dysynaptically via the BA (LeDoux, 1995). However, pretraining BA lesions have little effect on fear conditioning (Amorapanth et al., 2000; Goosens and Maren, 2001; Nader et al., 2001), suggesting that the LA can also influence the CeM through other routes (LeDoux, 2000). Based on these pretraining lesion data, it was concluded that the BA is not involved in simple fear conditioning (LeDoux, 2000; Amorapanth et al., 2000). Although one might expect posttraining lesions to have similar effects to pretraining lesions, this is not always the case (Rosen et al., 1992; Campeau and Davis, 1995; Corodimas and LeDoux, 1995; Rudy et al., 2004). To fully evaluate the role of the BA in fear conditioning, we compared the effect of pretraining and posttraining lesions of BA.
Although lesion data suggest that the BA is not involved in the acquisition of conditioned fear, it may be involved in extinction of fear. Inhibition of NMDA receptors (Falls et al., 1992; Lee and Kim, 1998), mitogen-activated protein kinase (Lu et al., 2001), phosphatidylinositol 3′ kinase (Lin et al., 2003), or protein synthesis (Lin et al., 2003) in the BA and LA prevents extinction. In addition, the BA is the main source of amygdala projections to the infralimbic prefrontal cortex (IL) (McDonald, 1991; Condé et al., 1995), a structure implicated in consolidation and storage of extinction memory (Morgan et al., 1993; Herry and Garcia, 2002; Milad and Quirk, 2002; Hugues et al., 2004; Santini et al., 2004). We therefore examined the effect of BA lesions on extinction of conditioned fear with an experimental design used previously to investigate the role of the IL in fear extinction (Quirk et al., 2000; Lebron et al., 2004). Unexpectedly, we found that BA lesions had no effect on extinction but severely attenuated expression of previously acquired fear memory.
Materials and Methods
All experimental protocols for this study were approved by the Institutional Animal Care and Use Committee at the Ponce School of Medicine in compliance with the National Institutes of Health guidelines for the care and use of laboratory animals.
Subjects. Rats used in this study were male Sprague Dawley rats weighing ∼300 g. They were housed individually in transparent polyethylene cages in a negative-pressure Biobubble (Colorado Clean Room, Fort Collins, CO) and kept on a 12 h light/dark cycle with ad libitum access to water. Food was restricted to 18 g per day of rat chow until they reached 85% of their body weight, at which time they were trained to bar press for food on a variable interval schedule (VI-60). Bar pressing on a variable reinforcement schedule maintains a constant behavioral background against which freezing responses can be measured more reliably (Quirk et al., 2000; Lebron et al., 2004).
Behavioral apparatus. All phases of the experiment, including bar press training, were performed in the same chamber. The chamber was 25 × 29 × 28 cm, with aluminum and Plexiglas walls (Coulbourn Instruments, Allentown, PA). The floor consisted of 0.5-cm-diameter stainless steel bars spaced at 1.8 cm through which a mild footshock US was delivered. A response lever was positioned 6.5 cm above the floor, and a speaker was mounted on the outside wall opposite the lever for the delivery of tone CSs. The chamber was situated inside a sound-attenuating box, which contained a ventilation fan, and was illuminated by a single house light.
Surgery. Rats were anesthetized with ketamine (90 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) and placed into a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA). Supplemental doses of ketamine were given as needed to maintain a deep level of anesthesia, as indicated by a slow respiratory rate and lack of response to tail pinch. Electrolytic lesions were made using an insulated wire electrode (0.25 mm diameter) with a 0.2 mm exposed tip, connected to a Grass stimulator (S-48) in DC mode with constant current output. Four lesions were made on each side of the amygdala, modified from Amorapanth et al. (2000). The coordinates for each lesion were as follows (in mm): anteroposterior: 2.1, 2.8, 3.3, 4.1; mediolateral: 4.9, 4.9, 5.3, 5.3; dorsoventral: 9.1, 9.3, 9.2, 9.3. A current intensity of 0.5 mA and a duration of 10 s was used for each lesion. For sham-operated rats, the electrode was lowered 3 mm ventral to bregma, and no current was passed. To manage postoperative pain, Buprenex (0.025 mg/kg) was injected intramuscularly.
Fear conditioning. The CS was a 30 s tone with a loudness of 75 dB and a frequency of 4 or 1 kHz. During conditioning, each tone coterminated with a 0.5 mA, 0.5 s footshock US. In a single day, rats received 5 habituation tones, followed by 7 conditioning tones, followed by 20 extinction tones. Depending on the experiment, extinction was given either 1 h or 8 d after conditioning. The mean intertrial interval was 4-6 min throughout the experiment.
Histology. After each experiment, rats received an overdose of sodium pentobarbital (100 mg/kg) and were perfused intracardially with 0.9% saline, followed by buffered formalin. The brains were removed and stored for 24 h in a 30% sucrose formalin solution. Frozen sections (40 μm thick) were cut with a microtome and stained with cresyl violet. Digitized images of the tissue were acquired with an Olympus (Melville, NY) BX51 microscope, and lesion contours were traced onto drawings from a stereotaxic atlas (Paxinos and Watson, 1998). The percentage of damage to the BA for each rat was calculated with MetaMorph image analysis software (Universal Imaging, Downington, PA). Decisions to include animals based on anatomical criteria were made blind with respect to the experimental results.
Data analysis. Freezing to the CS was measured with a stopwatch from different video files by an observer blind with respect to lesion group during the 30 s CS. The data were analyzed in blocks of two trials. Suppression of bar pressing was calculated as follows: suppression ratio = (pretone rate - tone rate)/(pretone rate + tone rate). A ratio of 0 indicates no suppression, and 1 indicates maximal suppression. Freezing and suppression values were compared with a Student's t test.
Results
Histology
The BA was defined to include the BL, BM, and AB nuclei (Paxinos and Watson, 1998). The criteria for inclusion of rats in the BA lesion group were (1) greater than ∼40% destruction of the BA bilaterally and (2) no damage to either the LA or the Ce. Twentytwo of 96 lesioned rats satisfied these criteria. The average percentage of damage to the BA (bilaterally) in these 22 rats was 60.7% (36.3-91.1%). In most cases, the BM and AB nuclei were completely destroyed, with some sparing of the BL nucleus (Fig. 1).
Figure 1.
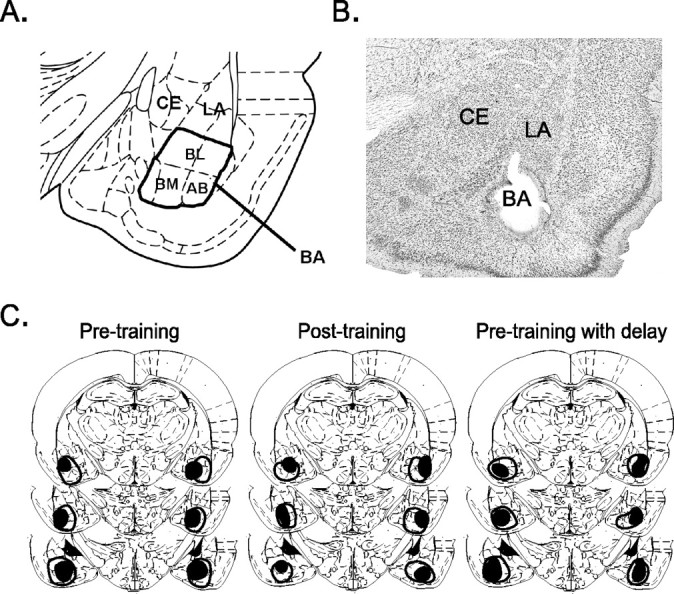
Examples of electrolytic BA lesions. A, The BA was defined to include the BL, BM, and AB nuclei, modified from Paxinos and Watson (1998). B, Micrograph showing a representative BA lesion. C, Coronal sections through the rostrocaudal extent of the amygdala in the three experimental groups: (1) pretraining lesions; (2) posttraining lesions; (3) pretraining lesions with delay. The smallest lesion is represented by the dark shading, whereas the largest lesion is outlined but not shaded. CE, Central nucleus; LA, lateral nucleus.
Pretraining BA lesions had no effect on acquisition or extinction
BA lesions made before training had no effect on acquisition of conditioned fear. As shown in Figure 2A, sham-operated and BA-lesioned rats exhibited similar levels of conditioned freezing (58 and 59%) and suppression of bar pressing (0.69 and 0.68) at the start of extinction, confirming previous reports that BA lesions have no effect on acquisition of fear conditioning (Goosens and Maren, 2001; Nader et al., 2001). In addition, both groups showed equivalent extinction, reaching negligible levels of freezing and suppression in the final block of extinction (lesioned, 19% freezing; sham, 13% freezing; t(17) = 0.75; p = 0.46). On day 2, spontaneous recovery of freezing (measured as a percentage of maximal freezing acquired on day 1) was similar in sham and lesioned rats (41 and 50%, respectively; t(17) = 0.56; p = 0.58). Thus, BA lesions did not prevent acquisition or extinction of conditioned fear.
Figure 2.

Pretraining BA lesions had no effect on conditioning or extinction, but posttraining lesions blocked recall of conditioned fear. A, Percentage of freezing to the tone (top) and suppression of bar pressing (bottom) for BA-lesioned rats (n = 8) and sham-operated rats (n = 11). Pretraining BA lesions had no effect on acquisition or extinction of conditioned fear. The groups were not statistically different in any phase. B, Percentage of freezing to the tone (top) and suppression ratio (bottom) for BA-lesioned rats (n = 9) and sham-operated rats (n = 13). Postconditioning BA lesions blocked expression of conditioned fear. Blocks of two trials are plotted. Error bars indicate SEM. Hab, Habituation; Cond, conditioning; d1, d2, and d10, days 1, 2, and 10, respectively; L, lesion.
Posttraining BA lesions blocked recall of conditioned fear
We next performed posttraining BA lesions, reasoning that previous conditioning might alter the function of BA with respect to expression of conditioning or extinction (Campeau and Davis, 1995). Surgery was performed 2 d after conditioning, and rats were allowed 7 d recovery before testing on day 10. Posttraining BA lesions dramatically reduced the expression of conditioned fear to the tone (Fig. 2B). Lesioned rats dropped from 62% freezing before surgery to 13% freezing after surgery (t(8) = 6.45; p < 0.001), whereas sham-operated rats remained the same (before surgery, 75%; after surgery, 76%). Similarly, bar-press suppression was reduced in lesioned rats after surgery (before surgery, 0.89; after surgery, 0.39; t(8) = 3.66; p = 0.006), whereas sham-operated rats were unchanged (before surgery, 0.97; after surgery, 0.94). BA lesions did not affect the rate of spontaneous bar pressing (sham, 18 per minute; lesioned, 14 per minute; t(20) = 0.67; p = 0.51), indicating that reduced freezing was not attributable to increased motivation to press for food. Thus, posttraining lesions revealed a role for the BA in the recall of previously acquired fear associations.
Despite their inability to express previously learned fear, BA-lesioned rats were able to recondition to a new CS (Fig. 3A). Sham-operated and BA-lesioned animals showed similar levels of acquired freezing (50 and 60%, respectively; t(11) = 0.98; p = 0.34) and suppression (0.57 and 0.62; t(11) = 0.21; p = 0.84) at the end of reconditioning, which did not differ significantly between groups. Furthermore, both lesioned and sham-operated rats showed savings in the rate of reconditioning (Fig. 3B), suggesting generalization between tones and some degree of preserved memory for initial training in lesioned rats. There was a trend toward faster re-extinction in lesioned rats; however, neither freezing nor suppression at the end of extinction differed significantly between groups (shams, 22%; lesions, 3%; t(11) = 1.67; p = 0.12; shams, 0.33; lesions, 0.12; t(11) = 1.25; p = 0.24). The trend toward faster re-extinction in lesioned rats is consistent with loss of memory for the original conditioning.
Figure 3.

Rats with posttraining BA lesions were able to recondition. A, A subset of lesioned (n = 6) and sham-operated (n = 7) rats shown in Figure 2 B were reconditioned with a new tone CS (1 kHz). Both groups conditioned to the new tone, ruling out damage to the LA as the cause of blocked expression (blocks of two trials are plotted). B, Rates of conditioning (day 1) and reconditioning (day 18) are compared for sham-operated (top) and BA-lesioned (bottom) rats. In both groups, savings was observed in the rate of reconditioning, suggesting generalization between tones and some degree of preserved memory in lesioned rats. Error bars indicate SEM. Hab, Habituation; Cond, conditioning; Recond, reconditioning; d1, d2, d10, and d18, days 1, 2, 10, and 18, respectively; L, lesion.
Blocked expression of conditioned fear was not attributable to LA damage or the delay between training and testing
One possible explanation for the reduced freezing in BA-lesioned rats is track-related damage in the LA, because the electrodes were not lowered into the amygdala in sham-operated rats (see Materials and Methods). To investigate this possibility, we prepared a separate group of rats with posttraining sham operations, lowering the electrode through the LA into the BA (n = 5). Sham-operated rats prepared in this way showed no significant decrease in expression of freezing after surgery (before surgery, 80%; after surgery, 76%; t(4) = 1.47; p = 0.22), indicating that track-related damage in the LA was not responsible for the reduced freezing seen with posttraining BA lesions.
Another possible explanation for the BA lesion deficit concerns the delay between training and testing. This delay was 1 h in the first experiment but 8 d in the second experiment, because of postsurgery recovery. It is possible, therefore, that rats with pretraining BA lesions could acquire conditioned fear but could not retain fear memory for 8 d. If so, this would suggest that posttraining lesions of the BA might have removed an essential long-term storage site. Because previous studies of BA lesions measured retention at 24 h only (Amorapanth et al., 2000; Goosens and Maren, 2001; Nader et al., 2001), it is not known whether the rats with BA lesions are capable of retaining fear memory over a longer period.
We therefore repeated the pretraining BA lesions but inserted an 8 d delay between training and testing. BA lesions under these conditions had no effect on expression of conditioned fear on day 10 (Fig. 4). Sham-operated (n = 8) and lesioned (n = 5) rats acquired similar levels of conditioned freezing to the tone (91 and 89%, respectively; t(11) = 0.40; p = 0.69). Eight days later, freezing levels in both groups remained unchanged (sham, 84%; lesion, 85%; t(11) = 0.10; p = 0.92), indicating that the passage of time did not cause a loss of fear memory in BA-lesioned rats. The fact that rats with pretraining BA lesions can form long-lasting fear associations suggests that deficits caused by posttraining BA lesions are not attributable to an inability of the LA-Ce system to retain fear memory over the recovery period.
Figure 4.

Rats with pretraining BA lesions could retain fear memory for 8 d. The percentages offreezing to the tone on test days 2 and 10 for the pretraining with delay (left) and post training (right) experiments are shown. Rats with pretraining BA lesions (n = 5) could retain conditioning memory for 8 d, ruling out the passage of time after recovery from surgery as the cause of blocked expression with posttraining lesions. Error bars indicate SEM. L, lesion.
Discussion
We have revisited the issue of the intra-amygdala circuits in fear conditioning, by comparing the effects of pretraining and posttraining lesions of the BA on the acquisition and expression of conditioned fear. Replicating previous reports, we found that pretraining BA lesions had no effect on acquisition of conditioned fear. We extended those findings by showing that rats with BA lesions can retain fear memory for at least 8 d. Unexpectedly, posttraining BA lesions completely abolished conditioned fear, suggesting that in the intact brain, the BA serves a critical role in fear conditioning.
Pretraining lesions of the BA also had no effect on the extinction of conditioned fear. BA-lesioned rats showed normal within-session extinction and recall of extinction the following day. Similar results were reported recently by LeDoux and colleagues (Sotres-Bayon et al., 2004). Thus, it appears that BA is not an essential site of extinction-related plasticity because rats with damage to the BA can still learn extinction. However, the role of the BA in extinction in the intact brain cannot be assessed because posttraining lesions of the BA blocked expression of conditioned fear. For this reason, negative findings concerning the effect of BA lesions on extinction must be viewed with caution.
Another possible site of extinction-related plasticity in the amygdala is the LA (Sotres-Bayon et al., 2004). Pharmacological manipulations of the BLA (BA plus LA) prevent extinction (Falls et al., 1992; Lu et al., 2001; Lin et al., 2003), and the LA shows extinction-induced upregulation of immediate-early genes (Herry and Mons, 2004) and a GABA receptor clustering protein (Chhatwal et al., 2005). The LA and IL are reciprocally connected (Condé et al., 1995; Vertes, 2004), and recent data suggest that the IL is a site of consolidation of extinction memory (Milad and Quirk, 2002; Hugues et al., 2004; Santini et al., 2004) (but see Gewirtz et al., 1997). These two structures may work together to acquire, store, and express extinction (Maren and Quirk, 2004). Another possible source of extinction plasticity in the amygdala are GABAergic intercalated (ITC) cells, which receive glutamatergic inputs from the LA and BA (Royer et al., 1999) and inhibit CeM output neurons (Paré et al., 2004). ITC cells receive a prominent projection from the IL (McDonald et al., 1996; Pinto and Sesack, 2003) and express NMDA-mediated long-term potentiation (Royer and Paré, 2003), consistent with a role in extinction expression and storage.
The most surprising finding from this study was that posttraining BA lesions completely blocked expression of conditioned fear, whereas pretraining lesions had no effect. One possible explanation is that the BA is the preferred output pathway for fear memory stored in the LA. However, the fact that pretraining lesions of the BA have no effect demonstrates that the LA can access the Ce independently of the BA. If the LA is the main site of conditioning-related plasticity and can access the CeM independently of the BA, there should be no effect of pretraining or posttraining BA lesions. It is also unlikely that our effects are attributable to interruption of fibers of passage originating in adjacent cortical areas such as the perirhinal cortex or auditory cortex, because tracing studies show that these areas project either exclusively to the LA (Romanski and LeDoux, 1993) or to both the LA and BA (McDonald and Jackson, 1987; Shi and Cassell, 1999).
Instead, our findings suggest that the LA-Ce system is not sufficient to support fear conditioning in the intact brain, although it is sufficient in the damaged brain. Under normal conditions, plasticity is necessary in the BA and/or its terminals in the CeM. In support of this, neurons in the BA show associative plasticity during fear conditioning (Maren et al., 1991; Toyomitsu et al., 2002). This suggests that infusions of the NMDA receptor antagonist APV into the BLA, which prevents acquisition of fear conditioning (Miserendino et al., 1990; Fanselow et al., 1994), could be exerting its effects via the BA in addition to the LA. Our findings are also consistent with plasticity in BA outputs to the CeM. The CeM receives projections from the BA, the medial geniculate nucleus (MGm) of the thalamus (Paré et al., 2004), and the LA via ITC cell-mediated disinhibition (Paré et al., 2004). Recent studies suggest that the CeM is an essential site of plasticity in fear conditioning (Wilensky et al., 2001; Goosens and Maren, 2003; Paré et al., 2004). Long-term potentiation has been observed in the projections from the BA to the CeM (Fu and Shinnick-Gallagher, 2004) and from the MGm to the CeM (Samson and Paré, 2005). Thus, the CeM appears to be a site of convergence of plastic inputs from several structures involved in fear learning.
The pattern of lesion effects we observed (after training but not before training) has been observed in other systems, such as the perirhinal cortex in auditory fear conditioning (Rosen et al., 1992; Corodimas and LeDoux, 1995; Campeau and Davis, 1995), the hippocampus in contextual fear conditioning (Maren et al., 1997), and the prefrontal cortex in eyeblink conditioning (Powell et al., 2001). How could a structure be an essential site of plasticity when present but dispensable if removed before learning? This apparent paradox could be explained by competition between parallel inputs, such that learning in one input inhibits learning in other inputs to the same cell.
In support of this, recent physiological evidence suggests that homeostatic control mechanisms dynamically adjust synaptic strength to promote stability across multiple inputs (Buzsaki et al., 2002; Royer and Paré, 2003; Turrigiano and Nelson, 2004). For example, BA inputs to the CeM might gain a disproportionate amount of the total plasticity accrued during acquisition compared with other inputs (Fig. 5). Posttraining lesions of the BA would therefore render the system subthreshold for expression of the conditioned response. In contrast, pretraining lesions would be compensated for by increased plasticity among the remaining inputs to the CeM, such as the MGm, which could then support fear behavior. The LA also projects to the CeM but indirectly via a chain of ITC cells thought to signal the CS by disinhibiting the CeM (Paré et al., 2004). An interesting prediction of this model is that posttraining lesions of the medial subdivision of the MG, which normally have no effect (Campeau and Davis, 1995), would block the expression of conditioned fear in rats that received pretraining lesions of the BA.
Figure 5.

Model to account for deficits with posttraining, but not pretraining, lesions. We suggest that homeostatic mechanisms that balance plasticity over multiple inputs could regulate the influence of the BA, LA, and MGm onto CeM neurons. 1, The CeM receives inputs from the MGm, BA, and LA via ITC cells (dashed line). 2, After fear conditioning, BA inputs gain a large proportion of the total plasticity, inhibiting the development of plasticity in neighboring inputs. 3, Posttraining lesions of the BA remove this plasticity, leaving the system subthreshold for producing a fear response. 4, With pretraining removal of the BA, however, increased plasticity in the LA and MGm supports fear learning. For simplicity, projections from the MGm to the LA and from the LA to the BA are not shown.
Among the mechanisms that could account for this type of heterosynaptic interaction is synaptic scaling in which decreases in synaptic input to a cell are compensated for by increases in the number of AMPA and NMDA receptors at the remaining synapses (Watt et al., 2000; Turrigiano and Nelson, 2004). This regulatory process is thought to occur over a time period of several days, consistent with postlesion recovery time. If true for the CeM, lesions of the BA would trigger a gradual increase in the number of NMDA receptors at the remaining synapses onto the CeM, making them more likely to exhibit plasticity during subsequent training. Additional experiments are needed to determine whether CeM neurons show such heterosynaptic interactions. Homeostatic mechanisms could be operating in other neural systems in which lesions cause deficits when performed after, but not before, training.
Footnotes
This work was supported by National Institutes of Health Grants R01-MH58883 and S06-GM08239 to G.J.Q. We thank Dr. Mohammed Milad for help with data collection and Drs. Denis Paré and Mohammed Milad for comments on this manuscript. We also thank Dr. Joseph LeDoux and Francisco Sotres-Bayon for helpful discussions.
Correspondence should be addressed to Dr. Gregory J. Quirk, Department of Physiology, Ponce School of Medicine, P.O. Box 7004, Ponce, Puerto Rico 00732. E-mail: gjquirk@yahoo.com.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259680-06$15.00/0
References
- Amorapanth P, LeDoux JE, Nader K (2000) Different lateral amygdala outputs mediate reactions and actions elicited by a fear-arousing stimulus. Nat Neurosci 3: 74-79. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Csicsvari J, Dragoi G, Harris K, Henze D, Hirase H (2002) Homeostatic maintenance of neuronal excitability by burst discharges in vivo. Cereb Cortex 12: 893-899. [DOI] [PubMed] [Google Scholar]
- Campeau S, Davis M (1995) Involvement of subcortical and cortical afferents to the lateral nucleus of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J Neurosci 15: 2312-2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal JP, Myers KM, Ressler KJ, Davis M (2005) Regulation of gephyrin and GABAA receptor binding within the amygdala after fear acquisition and extinction. J Neurosci 25: 502-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condé F, Maire-Lepoivre E, Audinat E, Crepel F (1995) Afferent connections of the medial frontal cortex of the rat. II. Cortical and subcortical afferents. J Comp Neurol 352: 567-593. [DOI] [PubMed] [Google Scholar]
- Corodimas KP, LeDoux JE (1995) Disruptive effects of posttraining perirhinal cortex lesions on conditioned fear: contributions of contextual cues. Behav Neurosci 109: 613-619. [DOI] [PubMed] [Google Scholar]
- Davis M (2000) The role of the amygdala in conditioned and unconditioned fear and anxiety. In: The amygdala (Aggleton JP, ed), pp 213-288. Oxford: Oxford UP.
- De Oca BM, DeCola JP, Maren S, Fanselow MS (1998) Distinct regions of the periaqueductal gray are involved in the acquisition and expression of defensive responses. J Neurosci 18: 3426-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falls WA, Miserendino MJ, Davis M (1992) Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci 12: 854-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, Kim JJ, Yipp J, De Oca B (1994) Differential effects of the N-methyl-d-aspartate antagonist dl-2-amino-5-phosphonovalerate on acquisition of fear of auditory and contextual cues. Behav Neurosci 108: 235-240. [DOI] [PubMed] [Google Scholar]
- Fu Y, Shinnick-Gallagher P (2005) Two intra-amygdaloid pathways to the central amygdala exhibit different mechanisms of long-term potentiation. J Neurophysiol 93: 3012-3015. [DOI] [PubMed] [Google Scholar]
- Gewirtz JC, Falls WA, Davis M (1997) Normal conditioned inhibition and extinction of freezing and fear-potentiated startle following electrolytic lesions of medical prefrontal cortex in rats. Behav Neurosci 111: 712-726. [DOI] [PubMed] [Google Scholar]
- Goosens KA, Maren S (2001) Contextual and auditory fear conditioning are mediated by the lateral, basal, and central amygdaloid nuclei in rats. Learn Mem 8: 148-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goosens KA, Maren S (2003) Pretraining NMDA receptor blockade in the basolateral complex, but not the central nucleus, of the amygdala prevents savings of conditional fear. Behav Neurosci 117: 738-750. [DOI] [PubMed] [Google Scholar]
- Herry C, Garcia R (2002) Prefrontal cortex long-term potentiation, but not long-term depression, is associated with the maintenance of extinction of learned fear in mice. J Neurosci 22: 577-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Mons N (2004) Resistance to extinction is associated with impaired immediate early gene induction in medial prefrontal cortex and amygdala. Eur J Neurosci 20: 781-790. [DOI] [PubMed] [Google Scholar]
- Hugues S, Deschaux O, Garcia R (2004) Postextinction infusion of a mitogen-activated protein kinase inhibitor into the medial prefrontal cortex impairs memory of the extinction of conditioned fear. Learn Mem 11: 540-543. [DOI] [PubMed] [Google Scholar]
- Koo JW, Han JS, Kim JJ (2004) Selective neurotoxic lesions of basolateral and central nuclei of the amygdala produce differential effects on fear conditioning. J Neurosci 24: 7654-7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht R, LeDoux J (2004) Structural plasticity and memory. Nat Rev Neurosci 5: 45-54. [DOI] [PubMed] [Google Scholar]
- Lebron K, Milad MR, Quirk GJ (2004) Delayed recall of fear extinction in rats with lesions of ventral medial prefrontal cortex. Learn Mem 11: 544-548. [DOI] [PubMed] [Google Scholar]
- LeDoux JE (1995) Emotion: clues from the brain. Annu Rev Psychol 46: 209-235. [DOI] [PubMed] [Google Scholar]
- LeDoux JE (2000) Emotion circuits in the brain. Annu Rev Neurosci 23: 155-184. [DOI] [PubMed] [Google Scholar]
- LeDoux JE, Iwata J, Cicchetti P, Reis DJ (1988) Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci 8: 2517-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Kim JJ (1998) Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. J Neurosci 18: 8444-8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Lu HY, Gean PW (2003) The similarities and diversities of signal pathways leading to consolidation of conditioning and consolidation of extinction of fear memory. J Neurosci 23: 8310-8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KT, Walker DL, Davis M (2001) Mitogen-activated protein kinase cascade in the basolateral nucleus of amygdala is involved in extinction of fear-potentiated startle. J Neurosci 21: RC162(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Quirk GJ (2004) Neuronal signalling of fear memory. Nat Rev Neurosci 5: 844-852. [DOI] [PubMed] [Google Scholar]
- Maren S, Poremba A, Gabriel M (1991) Basolateral amygdaloid multi-unit neuronal correlates of discriminative avoidance learning in rabbits. Brain Res 549: 311-316. [DOI] [PubMed] [Google Scholar]
- Maren S, Aharonov G, Fanselow MS (1997) Neurotoxic lesions of the dorsal hippocampus and Pavlovian fear conditioning in rats. Behav Brain Res 88: 261-274. [DOI] [PubMed] [Google Scholar]
- McDonald AJ (1991) Organization of amygdaloid projections to the prefrontal cortex and associated striatum in the rat. Neuroscience 44: 1-14. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Jackson TR (1987) Amygdaloid connections with posterior insular and temporal cortical areas in the rat. J Comp Neurol 262: 59-77. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F, Guo L (1996) Projections of the medial and lateral prefrontal cortices to the amygdala: a Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience 71: 55-75. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ (2002) Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420: 70-74. [DOI] [PubMed] [Google Scholar]
- Miserendino MJ, Sananes CB, Melia KR, Davis M (1990) Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature 345: 716-718. [DOI] [PubMed] [Google Scholar]
- Morgan MA, Romanski LM, LeDoux JE (1993) Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett 163: 109-113. [DOI] [PubMed] [Google Scholar]
- Nader K, Majidishad P, Amorapanth P, LeDoux JE (2001) Damage to the lateral and central, but not other, amygdaloid nuclei prevents the acquisition of auditory fear conditioning. Learn Mem 8: 156-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paré D, Smith Y (1998) Intrinsic circuitry of the amygdaloid complex: common principles of organization in rats and cats. Trends Neurosci 21: 240-241. [DOI] [PubMed] [Google Scholar]
- Paré D, Quirk GJ, LeDoux JE (2004) New vistas on amygdala networks in conditioned fear. J Neurophysiol 92: 1-9. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998) The rat brain in stereotaxic coordinates. San Diego: Academic.
- Pinto AO, Sesack SR (2003) Prefrontal cortex projections to the rat amygdala: spatial relationships to dopamine and serotonin afferents. Ann NY Acad Sci 985: 542-544. [Google Scholar]
- Pitkanen A, Savander V, LeDoux JE (1997) Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci 20: 517-523. [DOI] [PubMed] [Google Scholar]
- Powell DA, Skaggs H, Churchwell J, McLaughlin J (2001) Posttraining lesions of the medial prefrontal cortex impair performance of Pavlovian eyeblink conditioning but have no effect on concomitant heart rate changes in rabbits (Oryctolagus cuniculus). Behav Neurosci 115: 1029-1038. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K (2000) The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci 20: 6225-6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler KJ, Paschall G, Zhou XL, Davis M (2002) Regulation of synaptic plasticity genes during consolidation of fear conditioning. J Neurosci 22: 7892-7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanski LM, LeDoux JE (1993) Information cascade from primary auditory cortex to the amygdala: corticocortical and corticoamygdaloid projections of temporal cortex in the rat. Cereb Cortex 3: 515-532. [DOI] [PubMed] [Google Scholar]
- Rosen JB, Hitchcock JM, Miserendino MJ, Falls WA, Campeau S, Davis M (1992) Lesions of the perirhinal cortex but not of the frontal, medial prefrontal, visual, or insular cortex block fear-potentiated startle using a visual conditioned stimulus. J Neurosci 12: 4624-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Paré D (2003) Conservation of total synaptic weight through balanced synaptic depression and potentiation. Nature 422: 518-522. [DOI] [PubMed] [Google Scholar]
- Royer S, Martina M, Paré D (1999) An inhibitory interface gates impulse traffic between the input and output stations of the amygdala. J Neurosci 19: 10575-10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy JW, Huff NC, Matus-Amat P (2004) Understanding contextual fear conditioning: insights from a two-process model. Neurosci Biobehav Rev 28: 675-685. [DOI] [PubMed] [Google Scholar]
- Samson RD, Paré D (2005) Activity-dependent synaptic plasticity in the central nucleus of the amygdala. J Neurosci 25: 1847-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Ge H, Ren K, Pena DO, Quirk GJ (2004) Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. J Neurosci 24: 5704-5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CJ, Cassell MD (1999) Perirhinal cortex projections to the amygdaloid complex and hippocampal formation in the rat. J Comp Neurol 406: 299-328. [DOI] [PubMed] [Google Scholar]
- Sotres-Bayon F, Bush DE, LeDoux JE (2004) Emotional perseveration: an update on prefrontal-amygdala interactions in fear extinction. Learn Mem 11: 525-535. [DOI] [PubMed] [Google Scholar]
- Toyomitsu Y, Nishijo H, Uwano T, Kuratsu J, Ono T (2002) Neuronal responses of the rat amygdala during extinction and reassociation learning in elementary and configural associative tasks. Eur J Neurosci 15: 753-768. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB (2004) Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci 5: 97-107. [DOI] [PubMed] [Google Scholar]
- Vertes RP (2004) Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse 51: 32-58. [DOI] [PubMed] [Google Scholar]
- Watt AJ, van Rossum MC, MacLeod KM, Nelson SB, Turrigiano GG (2000) Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron 26: 659-670. [DOI] [PubMed] [Google Scholar]
- Wilensky AE, Schafe GE, Le Doux JE (2001) Does the central nucleus of the amygdala contribute to the consolidation of auditory fear conditioning? Soc Neurosci Abstr 27: 187.9. [Google Scholar]