

Abstract
Amyloid β peptides generate oxidative stress in hippocampal astrocytes through a mechanism sensitive to inhibitors of the NADPH oxidase [diphenylene iodonium (DPI) and apocynin]. Seeking evidence for the expression and function of the enzyme in primary hippocampal astrocytes, we confirmed the expression of the subunits of the phagocyte NADPH oxidase by Western blot analysis and by immunofluorescence and coexpression with the astrocyte-specific marker glial fibrillary acidic protein both in cultures and in vivo. Functional assays using lucigenin luminescence, dihydroethidine, or dicarboxyfluorescein fluorescence to measure the production of reactive oxygen species (ROS) demonstrated DPI and apocynin-sensitive ROS generation in response to the phorbol ester PMA and to raised [Ca2+]c after application of ionomycin or P2u receptor activation. Stimulation by PMA but not Ca2+ was inhibited by the protein kinase C (PKC) inhibitors staurosporine and hispidin. Responses were absent in transgenic mice lacking gp91phox. Expression of gp91phox and p67phox was increased in reactive astrocytes, which showed increased rates of both resting and stimulated ROS generation. NADPH oxidase activity was modulated by intracellular pH, suppressed by intracellular alkalinization, and enhanced by acidification. The protonophore carbonyl cyanide p-trifluoromethoxyphenylhydrazone suppressed basal ROS generation but markedly increased PMA-stimulated ROS generation. This was independent of mitochondrial ROS production, because it was unaffected by mitochondrial depolarization with rotenone and oligomycin. Thus, the NADPH oxidase is expressed in astrocytes and is functional, activated by PKC and intracellular calcium, modulated by pHi, and upregulated by astrocyte activation. The astrocytic NADPH oxidase is likely to play important roles in CNS physiology and pathology.
Keywords: NADPH oxidase, intracellular calcium, free radicals, oxidative stress, Alzheimer's disease, amyloid β peptide
Introduction
The NADPH oxidase (nox) family of enzymes was originally discovered expressed in neutrophils and related immune competent cells (macrophages, basophils, and eosinophils). In these cells, the enzyme activates bacterial killing, possibly through the generation of massive quantities of superoxide, although this mechanism has been questioned (Ahluwalia et al., 2004). More recently, it has become clear that the expression of the enzyme is far more widespread than initially thought (Lambeth, 2004). The expression of several isoforms has been clearly shown throughout the cardiovascular system (Griendling, 2004). Within the CNS, attention has been focused primarily on microglia as sources of immunological activity, and it has long been clear that these cells express the oxidase (Colton, 1994). We recently found that amyloid β (Aβ) peptides cause oxidative stress specifically in astrocytes through a mechanism attributable to activation of an NADPH oxidase, because astrocyte-specific reactive oxygen species (ROS) generation was suppressed by the inhibitors of the enzyme, diphenylene iodonium (DPI) and apocynin (Abramov et al., 2004a). More recently, we have seen inhibition with 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF) (Abramov et al., 2004b). This led us to seek additional evidence for the expression of the oxidase in astrocytes and to explore mechanisms involved in its regulation.
In the experiments described below, we demonstrated the presence of a phagocyte-like NADPH oxidase in pure astrocyte cultures. We also explored the properties and modulation of enzyme activity by measurements of free-radical generation. Because there are technical questions concerning all indicators of ROS generation, we used three different assays to validate findings that have proven consistent between the different assays. The phorbol ester PMA is used as an activator of the oxidase in neutrophils and increases ROS production in astrocytes by all measures that we have used. We found previously that amyloid β promotes a calcium influx pathway and so initiates calcium signals selectively in astrocytes (Abramov et al., 2003, 2004a). Amyloid β also increases the rate of ROS generation and causes glutathione depletion in astrocytes, and these effects were calcium dependent. Therefore, we explored the effect of calcium on ROS generation systematically and found that raising calcium using both ionophores and physiological calcium signals activates the oxidase in astrocytes.
The NADPH oxidase is believed to function as a simple electron transport chain linked to cytosolic NADPH and flavoprotein and seems to translocate a proton from NADPH while transferring an electron to molecular oxygen. In neutrophils, this activity generates a change in membrane potential, but an inevitable consequence of this mode of action is that the rate of activity must be limited by the proton gradient generated across the plasma membrane. The oxidase activity should then be modulated by intracellular pH. Because changes in pHi are a major feature of many pathological states, we explored this issue as well. Together, our observations suggest that the contributions of NADPH oxidase activation in astrocytes should be considered in physiological and pathological states in the mammalian CNS.
Materials and Methods
Cell culture. Isolated cortical astrocytes were prepared as described previously (Boitier et al., 1999). Cerebra taken from 2- to 5-d-old Sprague Dawley rats (University College London breeding colony, London, UK) or 4-d-old mice were used. For some experiments, gp91phox knock-out transgenic mice (obtained from A. M. Shah, King's College London, London, UK) were used, and C57BL/6 pups (the strain used as a background for the transgenic animals) were used as controls. The cerebra were chopped and triturated until homogenous, passed through a 297 μm mesh, and trypsinized (50,000 U/ml porcine pancreas; Sigma, Gillingham, UK) with 336 U/ml DNase 1 (bovine pancreas; Sigma), and 1.033 U/ml collagenase (Sigma) at 37°C for 15 min. After the addition of fetal bovine serum (10% of final volume) and filtering through 140 μm mesh, the tissue was centrifuged through 0.4 m sucrose (400 g; 10 min), and the resulting pellet was transferred to Minimal Essential Medium supplemented with 5% fetal calf serum, 2 mm glutamine, and 1 mm malate in tissue culture flasks precoated with 0.01% poly-d-lysine. The cells reached confluency at 12-14 d in vitro were harvested and reseeded onto 24-mm-diameter glass coverslips (BDH, Poole, UK) precoated with 0.01% poly-d-lysine for fluorescence (fl) measurements and used during 2-4 d.
Western blot analysis. Cells were washed into 10 mm PIPES buffer, pH 7.3, containing 3 mm NaCl, 100 mm KCl, 3.5 mm MgCl2, and 6% sucrose. They were broken by sonication for 3 × 5 s (MSE Sonyprep; MSE, Crawley, Surrey, UK) in the presence of protease inhibitors (1 mm di-isopropyl fluorophosphate, 0.2 mm phenylmethanesulfonyl fluoride, 1 μg/ml leupeptin, and 1 μg/ml tosyl-lysine chloroketone). The lysates were fractionated into cytosol and membrane fractions using a discontinuous sucrose gradient consisting of 34 and 15% (w/w) sucrose and centrifuging at 200,000 × g for 30 min at 4°C in a Beckman TLS 55 rotor (Beckman Instruments, Fullerton, CA). Membranes were collected from the 34/15% interface and sedimented in a Beckman TLA 100.4 rotor at 200,000 × g for 15 min. They were finally resuspended in the above-mentioned buffer at a protein concentration of 70 μg/ml. Protein concentrations were determined with the Bio-Rad Protein Assay (Bio-Rad, München, Germany). Cytosol and membrane proteins were subjected to SDS-PAGE and Western blotting according to standard procedures. Six micrograms of cytosol proteins and 1.2 μg of membrane proteins were applied per lane. Blots were developed using affinity-purified antibodies against the human phox proteins described previously (Wientjes et al., 1993, 1997), horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences, Piscataway, NJ), enhanced chemiluminescence (CL), and a Bio-Rad Chemidoc imaging system. As a negative control, an anti-gp91phox antiserum was used, which had been depleted for gp91phox antibodies by incubating it with recombinant gp91phox bound to beads.
Immunofluorescence studies. Immunofluorescence staining was performed on the same cultures fixed with cold methanol and permeabilized with sodium deoxycholate (4 mm in TBS) and Triton X-100 (0.025% in TBS). The blocking solution contained 1% BSA and 10% each of the normal sera from the animals in which the secondary antibodies were raised (goat and donkey) in TBS. The primary antibodies used were mouse anti-glial fibrillary acidic protein (GFAP) (1:600; Sigma) or mouse anti-OX-42 (CD11b; 1:1000; Serotec, Kidlington, UK) and rabbit anti-gp91 or anti-p67 (both 1:100). Secondary anti-mouse FITC-conjugated (1:500; Abcam, Cambridge, UK) and anti-rabbit cyanine 5 (Cy5)-conjugated (1:500; Chemicon, Chandlers Ford, UK) antibodies were used. Nuclei were stained with 0.01% 4,6-diamidino-2-phenylindole HCl (DAPI; Sigma), and the coverslips were mounted using AF1 (Citifluor, London, UK). Negative controls were performed by the omission of primary antibody or secondary antibody or the omission of cells.
Imaging of [Ca2+]c and ROS generation. Cells were loaded for 30 min at room temperature with 5 μm fura-2 AM (Invitrogen, Eugene, OR) and 0.005% Pluronic or 20 μm 2′,7′-dichlorodihydrofluorescein diacetate (DCF) (Invitrogen) in a HEPES-buffered salt solution (HBSS) composed of the following (in mm): 156 NaCl, 3 KCl, 2MgSO4, 1.25 KH2PO4, 2 CaCl2, 10 glucose, and 10 HEPES, pH adjusted to 7.35 with NaOH. For measurement of hydroethidine (HEt) fluorescence, dihydroethidium (2 μm) was present in all solutions during these experiments, and no preincubation was used, to limit the intracellular accumulation of oxidized product.
Fluorescence measurements were obtained on an epifluorescence inverted microscope equipped with a 20× fluorite objective. [Ca2+]c and HEt were monitored in single cells using excitation light provided by a Xenon arc lamp, the beam passing sequentially through 10 nm bandpass filters centered at 340, 380, and 490 nm housed in computer-controlled filter wheel (Cairn Research, Kent, UK). Cells loaded with DCF were illuminated at 490 nm at low-light levels to avoid auto-oxidation. Changes in the rate of rise of the signal were interpreted as changes in rates of ROS generation and could be fitted with a regression coefficient of >0.98 by a linear regression line (Origin; Microcal Software, Northampton, MA). Ethidium fluorescence was exited at 490 nm, and emitted fluorescence was measured at >515 nm.
Emitted fluorescence light was reflected through a 515 nm long-pass filter to a frame transfer cooled CCD camera (Hamamatsu Orca ER; Hamamatsu, Welwyn Garden City, UK). All imaging data were collected and analyzed using Kinetic Imaging (Wirral, UK) software. The fluorescence data were acquired at intervals of 10-15 s. All presented data were obtained from at least five coverslips and two to three different cell preparations.
Measurement of superoxide production using lucigenin luminescence. Cortical astrocytes cultured in 35 mm dishes were used for CL measurement in a photon-counting device composed of a gallium arsenide photomultiplier tube (Hamamatsu R943) thermoelectrically cooled to -20°C. CL emission from samples in dishes sealed with polythene film, maintained at 37°C in a thermostatic light-sealed chamber, was reflected and focused onto the photomultiplier tube. 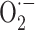 production was measured using lucigenin at subrecycling concentrations (10 μm) (Li et al., 1998). Cells were washed twice with a saline solution, and 1 ml of lucigenin-containing physiological saline (HBSS) was added. CL was measured for 15-20 min after the addition of PMA [500 ng/ml; before the addition of NADPH oxidase inhibitors or superoxide dismutase (SOD)].
production was measured using lucigenin at subrecycling concentrations (10 μm) (Li et al., 1998). Cells were washed twice with a saline solution, and 1 ml of lucigenin-containing physiological saline (HBSS) was added. CL was measured for 15-20 min after the addition of PMA [500 ng/ml; before the addition of NADPH oxidase inhibitors or superoxide dismutase (SOD)].
Results
Expression of NADPH oxidase subunits in astrocytes
Western blots clearly showed the presence of gp91phox and p67phox in rat astrocytes (Fig. 1Ai). The level of expression was much lower than in rat neutrophils (five times less neutrophil material was applied to the gel). The presence of NADPH oxidase in the rodent CNS has been reported previously (Serrano et al., 2003). It must be emphasized that rat gp91phox runs at a different position in the gel (much lower apparent molecular weight) than the human isoform, which is consistent with data reported by Hitt and Kleinberg (1996). It is thought that the rat gp91phox might not be glycosylated and hence runs at a lower apparent molecular weight. It was also consistently observed that gp91phox from astrocytes runs at a slightly different position than that from neutrophils (Fig. 1Ai), which might mean either that there is a difference in posttranslational modification of this protein in these cells or that the protein in astrocytes is not gp91phox but one of its homologues. As a negative control, an anti-gp91phox antiserum was used, which was depleted for anti-gp91phox antibodies by adsorbing them out. The absence of any bands in this lane (data not shown) shows the specificity of reaction.
Figure 1.

An NADPH oxidase is expressed in primary astrocytes. A, Western blots showing the expression of the subunits of the NADPH oxidase. i shows a Western blot of whole-cell extracts showing the expression of gp91phox and p67phox in astrocytes (a) using neutrophils (n) as a reference. PNS material from rat neutrophils and astrocytes was obtained and subjected to Western blot analysis as described in Materials and Methods. Six micrograms of neutrophil and 30 μg of astrocyte protein was applied and probed with antibodies to human gp91phox and p67phox as shown, because there is a high degree of homology between the proteins from human and rat. Arrowheads indicate the positions of the proteins. The figure is representative of three experiments. ii, Cytosol proteins probed for the cytosolic components of the NADPH oxidase, showing the expression of p67phox, p47phox, p40phox, and Rac as indicated. iii shows expression of the flavocytochrome subunits in the membrane fraction, namely gp91phox and p22phox as indicated. For a control and to ensure specificity, a control blot was made using antigp91phox antiserum, which was depleted for anti-gp91phox antibodies by adsorbing them out; this is not shown because the blot was completely blank. B, Immunofluorescence imaging using antibodies to gp91phox and p67phox counterstained with Cy5-labeled rabbit anti-human antibodies reveal the expression of both proteins in astrocyte cultures. Cells were counterstained with antibodies to GFAP (green) as a glial-specific marker and with DAPI to stain nuclei (blue), and images were acquired on a Zeiss (Welwyn Garden City, UK) 510 CLSM. These images are representative of five separate preparations, and staining was invariably seen clearly. The bottom panel shows a slightly higher gain image to show the distribution of the gp91phox. Appropriate controls showed no significant background signal under these conditions. Scale bars, 20 μm.
The neutrophil NADPH oxidase consists of a number of subunits, which redistribute during activation of the enzyme (for review, see Robinson et al., 2004). We therefore sought evidence for the expression of the subunits in subcellular fractions of the astrocytes (Fig. 1Aii,Aiii). As in the neutrophil, we found evidence for the expression of p67phox, p47phox, p40phox, and the small G-protein rac in cytosolic fractions and of gp91phox and p22phox in membrane fractions, strongly suggesting that the enzyme expressed in the astrocytes is similar if not identical to the neutrophil enzyme.
The expression of the enzyme was further established with immunofluorescence studies, using well established antibodies against the human subunits gp91phox and p67phox, which clearly showed coexpression with the astrocyte-specific marker GFAP for both proteins (Fig. 1B). Costaining with ox42 for microglia confirmed that the number of microglia in the culture was extremely low; most fields of view on the microscope (at 20×) showed no cells at all. Microglia that were found in the cultures were very brightly stained with the antibodies, suggesting that expression levels in these cells are significantly higher than in the quiescent astrocytes.
Concerned that the expression of this enzyme system might be restricted to cell culture conditions, we also performed immunofluorescence studies on tissue sections freshly prepared from the hippocampus and cortex of postnatal day 4 (P4) mice. Confocal imaging confirmed the coexpression of both gp91phox and p67phox with GFAP in these sections, strongly suggesting that the enzyme is expressed in astrocytes in situ. Images of these sections are shown in supplemental Figure 1 (available at www.jneurosci.org as supplemental material).
Functional studies: activation of free-radical generation by PMA
A standard tool with which to activate the NADPH oxidase in neutrophils, the exemplar cells expressing the enzyme, is the phorbol ester PMA. We found that application of PMA (1 μg/ml) produced a rapid and significant increase in free-radical generation in astrocytes. Because assays of free-radical production in cells are notoriously capricious, we used three independent assays to measure the rate of ROS generation. Lucigenin is a highly specific detector of superoxide, which emits a chemiluminescent signal in response to superoxide (Faulkner and Fridovich, 1993). The addition of PMA to a culture of astrocytes in the presence of lucigenin caused a prompt increase in luminescence that was instantly reversed after the addition of SOD to scavenge superoxide (Fig. 2A) or by the addition of DPI to inhibit the oxidase (Fig. 2B). The lucigenin does not penetrate well into the cell, and thus this signal measures the extracellular production of superoxide, confirmed by the reversal of chemiluminescence after the addition of SOD. The fluorescent probes, DCF and HEt, are both nonfluorescent derivatives of fluorescent moieties, which are oxidized by ROS (there is some controversy about the precise species involved), but both showed an increased rate of oxidation after exposure to PMA [an increase of 5.3-fold for DCF (from a resting slope of 9.89 ± 3 to 52.4 ± 6 fl U/min; n = 196 cells) and of 4.6-fold for HEt (from a resting slope of 3.7 ± 0.4 to 17.2 ± 2 fl U/min; n = 143)] (Fig. 2C,D). PMA-activated ROS production was completely suppressed by the inhibitors of NADPH oxidase: DPI (0.5 μm; n = 143) (Fig. 2C,D), apocynin (1 mm; n = 99), or AEBSF (20 μm; n = 86). Although DPI at higher concentrations may also affect mitochondrial function, we have shown previously that, at this concentration, it has no effect on mitochondrial function (Abramov et al., 2004a). Thus, PMA increases ROS generation in astrocytes through a mechanism sensitive to inhibitors of the NADPH oxidase.
Figure 2.

PMA stimulates ROS production by the NADPH oxidase in primary astrocyte cultures. Application of PMA induced an increase in ROS generation from astrocyte cultures measured using lucigenin luminescence (A, B), DCF (C), or HEt (D) fluorescence in response to 1 μg/ml PMA. The lucigenin response was reversed by the addition of superoxide dismutase to scavenge superoxide (A), demonstrating the specificity of the response, and was blocked by DPI (B). The gray traces in C and D show data from experiments in which cells were preincubated for 20 min with 0.5 μm DPI.
The astrocyte NADPH oxidase activity is activated by calcium
We reported previously that Aβ increases ROS generation in astrocytes, raises intracellular calcium ([Ca2+]c), and depletes glutathione (GSH), and that both the GSH depletion and increased ROS generation were calcium dependent (Abramov et al., 2003) and prevented by DPI and apocynin (Abramov et al., 2004a). These observations strongly suggested that the effects of Aβ on the NADPH oxidase might be mediated primarily through the effects of Aβ on intracellular calcium concentration. We therefore have now directly examined effects of changing [Ca2+]c on rates of ROS generation by the enzyme. The Ca2+ ionophore ionomycin (5 μm) increased the rate of ROS production in astrocytes, measured by DCF (5.7-fold change in slope, from 9.89 ± 3.1 to 56.8 ± 8.1 arbitrary U/min; n = 99) (Fig. 3A) or HEt (4.89-fold; p < 0.005; n = 91) (Fig. 3C). The effect of ionomycin was blocked by DPI (slope of HEt fluorescence was 10.32 ± 2.1 arbitrary U/min compared with 9.89 ± 3.1; n = 86). Another calcium ionophore, A23187 (20 μm), had a similar effect on ROS production (an increase in the slope of HEt fluorescence by 3.76-fold; from 3.7 ± 0.4 to 13.9 ± 1.1 arbitrary U/min; n = 76; p < 0.01), and this effect was also blocked by 20 min incubation of cortical or hippocampal astrocytes with 0.5 μm DPI (n = 45) or 1 mm apocynin (n = 51). Thus, the NADPH oxidase in cortical or hippocampal astrocytes can be activated by raising [Ca2+]c.
Figure 3.

ROS production in astrocytes is activated by [Ca2+]c. A, The calcium ionophore ionomycin increased ROS generation, measured here with DCF fluorescence. B, Simultaneous measurements of [Ca2+]c (fura-2) and ROS production (HEt) in cortical astrocytes during application of 100 μm ATP. The mean values of the rate of ROS production measured with HEt are shown in C. In this case, signals measured from the rate of increase of HEt fluorescence were normalized to the rate at baseline taken as 100%. DPI (0.5 μm) or apocynin (1 mm) were used as inhibitors of NADPH oxidase (gray columns). D, Cells were bathed in a Ca2+-free saline containing 500 μm EGTA. The addition of thapsigargin (0.5 μm) released Ca2+ from the endoplasmic reticulum and caused small transient increase in ROS production while completely suppressing the response to subsequent application of ATP.
Activation of the oxidase by an ionophore may engage far higher concentrations of calcium than seen physiologically. Therefore, we explored the possible activation of the enzyme by more physiological calcium signals. ATP acts at P2U receptors on astrocytes and releases calcium from the ER through an IP3-dependent pathway (Peuchen et al., 1996). ATP is released in the CNS and is believed to act as a physiological signal to glial cells (Guthrie et al., 1999). ATP (100 μm) routinely increased [Ca2+]c in cortical and hippocampal astrocytes (n = 156) (Fig. 3B) and also routinely produced a significant increase in ROS production: the slope of the HEt signal increased 2.71-fold (±0.23; p < 0.05), which was coincident with the calcium signal (Fig. 3B). ATP could activate the NADPH oxidase either through a direct effect of the calcium signal or through the generation of diacyl glycerol and activation of protein kinase C (PKC) as a consequence of activation of the G-protein-coupled signaling pathway, thus converging with the PMA-activated mechanism. Depletion of the intracellular calcium store by incubation of astrocytes with the smooth endoplasmic reticulum calcium ATPase inhibitor thapsigargin (1 μm) in the absence of extracellular Ca2+ completely prevented the effect of ATP on ROS generation. In the presence of the PKC inhibitors hispidin (20 μm) or staurosporine (100 nm), ATP caused a significant increase in ROS production by 2.4 ± 0.22-fold (n = 49 for hispidin) and 2.1 ± 0.1-fold (n = 44 for staurosporine). This was significantly reduced compared with the control, suggesting that the response is augmented by activation of PKC, but the fact that the response remained robust strongly suggests that NADPH oxidase activation is, in this case, predominantly a direct response to calcium (n = 84) (Fig. 3D). Indeed, note that the modest increase in [Ca2+]c seen on application of thapsigargin also caused a small increase in the rate of DCF oxidation (Fig. 3D).
These data are summarized in Figure 3C, in which measurements using HEt have been normalized to the basal slope for HEt, which is taken as 100%. The responses to PMA and to calcium signals were, in all cases, blocked by DPI (0.5 μm) and/or by apocynin, inhibitors of the NADPH oxidase.
To further characterize the molecular identity of the NADPH oxidase expressed in the astrocytes, we prepared cells from gp91phox knock-out transgenic mice. The mouse astrocytes showed increases in ROS production measured with HEt in response to PMA and A23187, which were indistinguishable from the response of rat astrocytes (n = 54 cells to PMA and 39 cells to A23187 from three preparations) (Fig. 4A,B). However, in cells from the transgenic mice, almost no significant response was seen to either PMA (n = 60 cells; three preparations) or A23187 (n = 58; three preparations). The pooled data are summarized in Figure 4B. These data strongly implicate gp91phox in the responses that we have seen. Activation of the enzyme in neutrophils involves translocation of the subunit p67 to the plasma membrane. Western blots of cells fractionated to separate membrane and cytosolic components failed to reveal any movement of p67phox in response to PMA and showed p67phox already present in membrane fractions in “unstimulated” cells (data not shown), and the same result was seen for p47phox (data not shown). The oxidase is very readily activated by manipulations of the cells and may be activated by shear stresses even in plating the cells (J. Jacobson, personal observations) and thus it may be hard to obtain blots of cells that are genuinely quiescent. Alternatively, it is possible that there is a preassembled complex on the astrocyte membrane, which can then be activated by agonists, as described in endothelial cells (Li and Shah, 2002).
Figure 4.

Astrocytes from gp91phox knock-out transgenic mice do not show increased ROS production in response to PMA or to Ca2+. Astrocyte cultures were prepared from a mouse exactly as described for the rat cells, using control and gp91 transgenic knock-out animals (a generous gift from D. Shah, King's College London, London, UK). Using HEt as described previously, the control cells showed a robust increase in ROS generation in response both to PMA (as illustrated in A; n = 58) and to an increase in [Ca2+] using A23187 (n = 39 cells; pooled data shown in B). In contrast, cells from the transgenic animals showed only minimal response to both stimuli (n = 60 for PMA and 58 for A23187). arb.U, Arbitrary units.
PMA activates the neutrophil oxidase through activation of PKCβ (Dekker et al., 2000), phosphorylation of the cytosolic subunit p47 phox, and its translocation to the plasma membrane (Nauseef, 2004). Calcium may act either directly, as described for the isoform Nox5 (Banfi et al., 2001), which has a calcium binding domain, or through calcium-dependent activation of PKC. To test these alternatives, we used the wide-spectrum PKC inhibitor staurosporine (100 nm) and the more selective PKCβ inhibitor hispidin. Hispidin (20 μm) almost completely suppressed the response to PMA (the slope of the HEt signal was 3.64 ± 0.21 compared with the basal rate 3.7 ± 0.4 arbitrary U/min; n = 64 cells) (Fig. 5A). Staurosporine had a similar effect (HEt slope 3.88 ± 0.43 compared with a resting level of 3.7 ± 0.4 arbitrary U/min; n = 59 cells). However, the response to A23187 was not significantly altered by either PKC inhibitor: the HEt slope in response to the ionophore was increased 3.8-fold (p < 0.001) compared with the resting level; n = 71) (Fig. 5B,C). These data strongly suggest that in these cells, PKCβ is the main PKC isoform responsible for activation of the oxidase, but that the calcium-dependent activation does not operate through the PKC but more likely acts directly on the oxidase.
Figure 5.
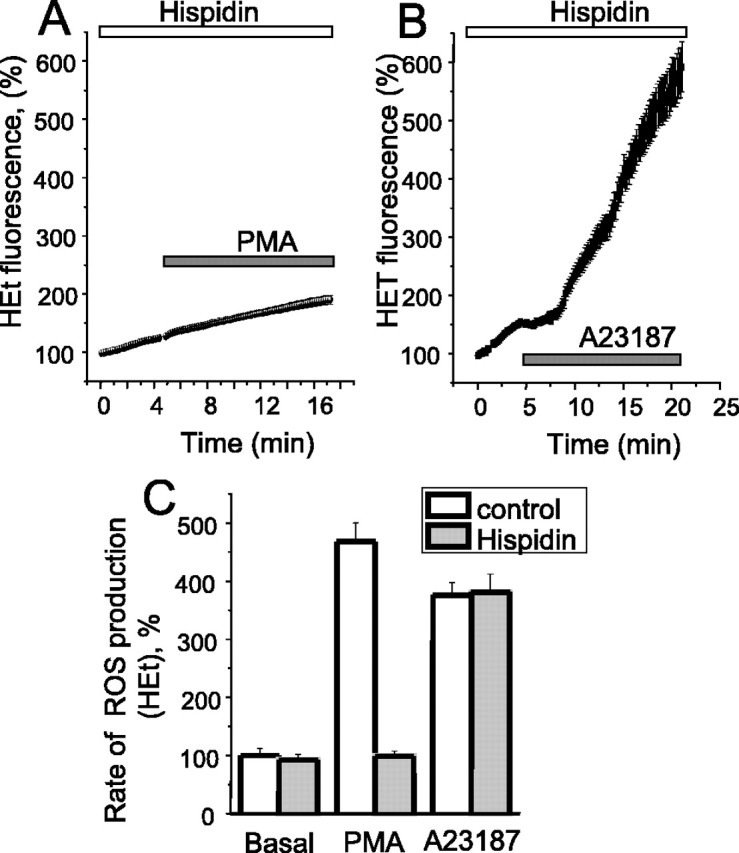
The PKCβ inhibitor hispidin inhibits PMA but not Ca2+-induced ROS production in cortical astrocytes. Cells were preincubated (20 min) with 20 μm hispidin. Hispidin inhibited the effect of 1 μg/ml PMA (A, C) but had no significant effect on the effect of the calcium ionophore 20 μm A23187 (B, C). In C, the mean values of the rates of ROS production measured with HEt are shown.
Astrocyte activation upregulates NADPH oxidase activity
Within any culture of astrocytes, occasional “activated” cells are found. These are characterized by a change in morphology, with extended processes compared with the flatter pavement-like quiescent cells, and by an increased expression of GFAP (Gehrmann et al., 1995). We routinely found that such cells showed an increased expression of both gp91phox and p67phox (Fig. 6A), showing a clear correlation between increased GFAP and increased gp91phox staining (Fig. 6A), and we also noticed that the ROS generation in such cells was greater than in their neighbors. We therefore tested the functional activity of the system after activation of the population with LPS+ γ-interferon (γIFN; n = 121) to achieve more uniform activation of the cells. In this population, the basal rate of HEt fluorescence increase was increased 2.2-fold (p < 0.05). This resting level was suppressed by DPI and by apocynin (n = 67) (Fig. 6B). Therefore, in keeping with the change in immunofluorescence, activation seems to significantly increase the resting activity of the NADPH oxidase. Lipopolysaccharide (LPS) treatment also upregulates the expression of intracellular nitric oxide synthase (iNOS), and we wondered whether NO might scavenge some of the superoxide produced, but the broad spectrum inhibitor of NOS, N-ω-nitro-l-arginine methyl ester (50 μm), did not change the LPS+γ-interferon-induced rate of HEt fluorescence at rest (data not shown; n = 76). The action of ATP on LPS+γIFN-treated astrocytes was insignificant (from a resting slope of 2.22 ± 0.27 to 2.71 ± 0.23%), but application of PMA further increased the rate of change of the signal by an additional 2.3-fold.
Figure 6.

NADPH oxidase activity is upregulated in activated astrocyte (A) immunofluorescence images of gp91phox and p67phox (Cy5; red) counterstained with anti-GFAP antibodies (FITC; green), and DAPI (blue) shows that activated astrocytes have increased intensity of staining for GFAP and for both subunits of the oxidase. B, The increase in ROS production in response to PMA was significantly amplified after overnight treatment of astrocytes with LPS and γ-interferon (measured with HEt fluorescence). The histogram in C summarizes the rates of increase of HEt fluorescence in response to PMA with and without LPS and γ-interferon pretreatment.
Astrocyte NADPH oxidase activity is regulated by membrane proton conductance and by pHi
The NADPH oxidase is thought to translocate protons across the plasma membrane along with the transfer of electrons to oxygen (Henderson et al., 1987; De Coursey, 2004). Therefore, drawing a parallel with the mitochondrial respiratory chain, activity would be limited by the electrochemical potential for protons. It has been suggested that this is overcome through the activity of a proton channel or a K+ channel in neutrophil membranes (Ahluwalia et al., 2004; De Coursey, 2004). We therefore wondered whether a protonophore, such as the mitochondrial uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), would play a permissive role, promote proton shuttling across the membrane, thus allowing the oxidase to cycle freely. In unstimulated quiescent astrocytes, application of FCCP suppressed the resting rate of ROS generation slightly, reducing the rate of change of the HEt signal to 62 ± 34% of the basal rate (n = 67) (Fig. 7A). This suggests that some resting signal might come from mitochondria, the rate of free-radical generation of which is thought to be reduced by mitochondrial depolarization (Starkov and Fiskum, 2003; Fink et al., 2004). However, when FCCP was applied after activation of the oxidase by PMA, the rate of ROS generation was dramatically enhanced by the protonophore (Fig. 7B,D). We were also concerned that the HEt signal in response to FCCP may be contaminated by a signal originating from the mitochondrial accumulation of ethidium (Budd et al., 1997), which shows potential dependent accumulation in mitochondria and is therefore released on mitochondrial depolarization. We therefore promoted mitochondrial depolarization using rotenone combined with oligomycin before the application of PMA (n = 63) (Fig. 7C). This serves both to prevent any mitochondrial ROS production but also to prevent the potential dependent accumulation of ethidium into mitochondria during the course of the experiment. Once again, application of FCCP after PMA activation caused a dramatic increase in the rate of ROS production, under conditions in which FCCP could not have any effect on mitochondrial potential. The origin of this response at the oxidase was again confirmed by its sensitivity to DPI, which reduced the HEt slope to levels lower than the resting value (n = 55; 71 ± 3.6% of the control slope).
Figure 7.

Proton shunting with FCCP increases NADPH oxidase activity in cortical astrocytes. Application of 1 μm FCCP to control (A) cells caused a modest suppression of the basal rate of rise of HEt fluorescence. In contrast, application of FCCP to cortical astrocytes after PMA stimulation (B) caused a considerable increase in ROS generation. In the experiment presented in C, astrocytes were treated with 0.2 μg/ml oligomycin and 20 μm rotenone to cause complete mitochondrial depolarization before application of PMA. In this case, the additional application of FCCP still increased ROS generation, although the mitochondria were already depolarized.
A similar argument predicts that changes in intracellular pH might serve to modulate the rate of activity of the enzyme, which would be expected to be enhanced by acidosis and inhibited by alkalosis. To test this proposal, we used a standard proton-loading protocol using ammonium chloride applied after activation of the enzyme with PMA. Dissociating ammonia crosses the membrane and binds protons in the cytosol, causing a transient alkalosis, after which the cell re-equilibrates, involving an increased accumulation of protons that are buffered by the NH3. This period of intracellular alkalosis suppressed ROS generation (to 52 ± 5.3% of the basal rate; p < 0.05; n = 121) (Fig. 8A). After washout of the NH4Cl, the increased proton load was unmasked as an acidosis. During this period, the rate of ROS generation was dramatically enhanced (to a mean of 6.35-fold increase over the resting rate; p < 0.001; n = 121). These responses were abolished by DPI, and thus again represent modulation of the activity of the enzyme rather than some artifact resulting from pH sensitivity of the dyes or some other form of pH-dependent ROS generation (Fig. 8B). Although it remains possible that NH4Cl has other actions that modulate oxidase activity [it is an endosomal/lysosomal inhibitor (Hart and Young, 1991)], these data suggest that oxidase activity is modulated by transmembrane pH gradients.
Figure 8.
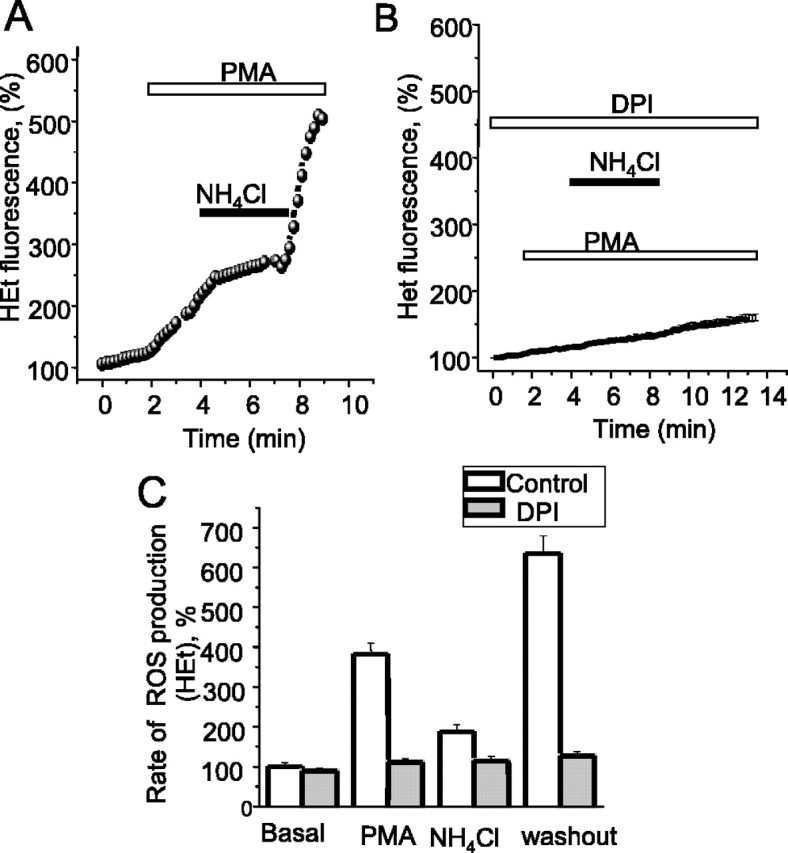
Modulation of NADPH oxidase activity by intracellular pH. [pH]i was manipulated by proton loading using 5 mm NH4Cl. During NH4Cl exposure, the intracellular alkalosis was associated with a suppression of ROS generation, whereas the intracellular acidosis seen on NH4Cl washout enhanced ROS generation (A). All of these effects were abolished by DPI, confirming that these are effects of the changing pHi on oxidase activity and not because of pH sensitivity of the dyes (B). Pooled data are summarized in C.
Discussion
Observations following the effects of amyloid β peptide on astrocytes in culture have drawn our attention to the expression of an NADPH oxidase in astrocytes. This has been surprising and raises all manner of questions about the functional significance of the enzyme. Although NADPH oxidase (nox) expression in microglia has been recognized for some time and presumably reflects the immunological lineage and inflammatory role of the microglia, and nox enzymes have been identified in mouse brain (Serrano et al., 2003), the specific expression of such an enzyme in astrocytes has not been described. Note that the levels of expression in quiescent astrocytes seem to be relatively low; we needed to dilute neutrophil protein substantially to have comparable intensity of signal on Western blots. Nevertheless, stimulation of cells with PMA or with high calcium causes significant increases in free-radical generation, suggesting that even this level of expression is functionally relevant. Expression of nox isoforms has been identified in increasing numbers of nonimmune cell types, and yet, in most cases, the function of the enzyme remains obscure (Griendling, 2004; Lambeth, 2004). It is not at all clear whether the enzyme functions primarily as a generator of superoxide or whether this is a byproduct of some other ionic activity, for example acting as a proton translocator. If its main function is to generate superoxide, does this serve a role in cell signaling or is it involved only in pathology? Our observations here show that even modest stimulation with ATP acting at a P2u receptor seems sufficient to increase the rate of superoxide generation significantly, suggesting that activation of the enzyme to some degree may be commonplace in astrocytes in situ, in which calcium signals are a routine feature of cell signaling. To identify the functional consequences of that pathway seems now to be an important goal. Certainly the activation of the enzyme in response to Aβ seems to have extensive pathological consequences leading to frank oxidative stress, glutathione depletion, and neuronal death.
Three additional observations in this study seem particularly worthy of note: the modulation of the rate of free-radical generation by calcium, transmembrane pH gradients, or pHi; and the upregulation of expression in activated astrocytes. The first is intriguing. One isoform of the NADPH oxidase, Nox5, contains a calcium-binding motif and appears to be directly activated by calcium. However, Nox5 seems to exist in two forms, one similar in size to gp91phox, which has no EF hands, and another that has a greater molecular weight, which expresses two EF hands that underlie its Ca2+ dependence. The longer isoform should be a lot bigger than gp91phox (which the astrocyte isoform is not), and even the shorter form has only 27% identity with gp91phox, which makes it less likely that antibodies against gp91phox would cross-react with it. Although it remains possible that this enzyme represents a novel isoform, the expression of the full complement of subunits that react with antibodies to the neutrophil subunits (p22, p40, p47, p67, and gp91phox) strongly suggests that the enzyme is the same as or similar to the neutrophil enzyme. Additionally, the very blunted or absent responses seen in astrocytes from gp91phox (-/-) mice strongly suggest that this is the enzyme expressed by the astrocytes. Our inability to detect any significant translocation of subunits to the membrane suggests either that the enzyme in these cells may be present as preassembled complexes on cell membranes, as described in endothelial cells (Li and Shah, 2002) or that the enzyme is already activated through the process of handling the cells. This also seems quite possible, because we found that simply touching the cells with a micropipette or plating them out is sufficient to increase DPI-sensitive ROS generation (J. Jacobson, unpublished observations).
Functionally, we have shown previously that Aβ generates oxidative stress in astrocytes and in nearby neurons through a mechanism that is sensitive to inhibitors of the NADPH oxidase and that is also calcium dependent. The present observations would be consistent with direct activation of the oxidase by the calcium signals generated in astrocytes after exposure to Aβ (Abramov et al., 2003) and thus suggests that these properties of the system may be of profound pathological significance. The sensitivity to pHi also seems of potential functional importance, as clearly the enzyme activity will be enhanced by pathological conditions that promote an intracellular acidosis, especially if accompanied by increases in intracellular calcium. Indeed, it remains possible that an intracellular acidification could contribute to the activation of the NADPH oxidase induced by Aβ, given that the peptide changes intracellular pH as well as raising [Ca2+]c (Abramov et al., 2004b).
These observations also have an important practical corollary. We have shown that ROS generation by the enzyme is greatly enhanced by the protonophore FCCP, whereas mitochondrial free-radical generation is reduced by FCCP. There are previous reports of increased ROS generation in response to FCCP, which is usually described as a mitochondrial uncoupler, emphasizing its effect on mitochondrial physiology, and thus effects are often attributed to an effect on mitochondria. We wonder whether, under such conditions, an increase in ROS production in response to a proton ionophore is likely to be diagnostic of the involvement of an NADPH oxidase.
Astrocytes seem to exist in a variety of states and may be activated by a number of pathological conditions. Activated astrocytes express increased levels of a variety of enzymes and secrete cytokines, express iNOS, and are operationally characterized by a change in morphology and an increase in the expression of the marker protein GFAP. The upregulation of the oxidase activity after activation of astrocytes that we noted here is also of great potential importance. Episodes of inflammatory disease are often associated with exacerbations in patients with Alzheimer's disease and other neurodegenerative disorders, and thus it is tempting to speculate that activation of the oxidase in these conditions might provide a mechanism to underlie such episodes. We have shown previously that the vulnerability of astrocytes themselves to Aβ toxicity is significantly increased when they are activated (Casley et al., 2002), and the potential for cytotoxicity in the presence of activated astrocytes seems much enhanced. Thus, the upregulation of the NADPH oxidase in activated astrocytes may have profound consequences if combined with the increased expression of iNOS and NO generation and hence increased generation of the toxic peroxynitrite, suggesting a mechanism whereby astrocyte activation may contribute to the expression of a pathological state in the CNS.
Footnotes
This work was supported by the Wellcome Trust. We thank Prof. Tony Segal for helpful advice, Prof. Ajay M. Shah and Dr. Alison Cave for their helpful provision of gp91phox-/- transgenic mice, and Lynsey Bilsland for her help with histological processing.
Correspondence should be addressed to Michael R. Duchen, Department of Physiology, University College London, Gower Street, London WC1E 6BT, UK. E-mail: m.duchen@ucl.ac.uk.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259176-09$15.00/0
References
- Abramov AY, Canevari L, Duchen MR (2003) Changes in [Ca2+]c and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci 23: 5088-5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov AY, Canevari L, Duchen MR (2004a) Amyloid β-peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 24: 565-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov AY, Canevari L, Duchen MR (2004b) Calcium signals induced by amyloid β peptide and their consequences in neurons and astrocytes in culture. Biochim Biophys Acta 1742: 81-87. [DOI] [PubMed] [Google Scholar]
- Ahluwalia J, Tinker A, Clapp LH, Duchen MR, Abramov AY, Pope S, Nobles M, Segal AW (2004) The large-conductance Ca2+-activated K+ channel is essential for innate immunity. Nature 427: 853-858. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Banfi B, Molnar G, Maturana A, Steger K, Hegedus B, Demaurex N, Krause KH (2001) A Ca2+-activated NADPH oxidase in testis, spleen, and lymph nodes. J Biol Chem 276: 37594-38601. [DOI] [PubMed] [Google Scholar]
- Boitier E, Rea R, Duchen MR (1999) Mitochondria exert a negative feedback on the propagation of intracellular Ca2+ waves in rat cortical astrocytes. J Cell Biol 145: 795-808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Castilho RF, Nicholls DG (1997) Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett 415: 21-24. [DOI] [PubMed] [Google Scholar]
- Casley C, Land J, Sharpe M, Clark J, Duchen M, Canevari L (2002) β-Amyloid fragment 25-35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol Dis 10: 258-267. [DOI] [PubMed] [Google Scholar]
- Colton CA (1994) Microglial oxyradical production: causes and consequences. Neuropathol Appl Neurobiol 20: 208-209. [PubMed] [Google Scholar]
- De Coursey TE (2004) During the respiratory burst, do phagocytes need proton channels or potassium channels, or both? Sci STKE 233: pe21. [DOI] [PubMed] [Google Scholar]
- Dekker LV, Leitges M, Altschuler G, Mistry N, McDermott A, Roes J, Segal AW (2000) Protein kinase C-beta contributes to NADPH oxidase activation in neutrophils. Biochem J 347: 285-289. [PMC free article] [PubMed] [Google Scholar]
- Faulkner K, Fridovich I (1993) Luminol and lucigenin as detectors for O2-. Free Radic Biol Med 15: 447-451. [DOI] [PubMed] [Google Scholar]
- Fink BD, Reszka KJ, Herlein JA, Mathahs MM, Sivitz WI (2004) Respiratory uncoupling by UCP1 and UCP2 and superoxide generation in endothelial cell mitochondria. Am J Physiol Endocrinol Metab 288: E71-E79. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Yao DL, Bonetti B, Brenner M, Bondy C, Wekerle H, Kreutzberg GW, Webster HF (1995) Astrocytes upregulate glial fibrillary acidic protein (GFAP), but not insulin-like growth factor-I (IGF-I) during experimental autoimmune neuritis (EAN). Brain Pathol 5: 1-10. [DOI] [PubMed] [Google Scholar]
- Griendling KK (2004) Novel NAD(P)H oxidases in the cardiovascular system. Heart 90: 491-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie PB, Knappenberger J, Segal M, Bennett MV, Charles AC, Kater SB (1999) ATP released from astrocytes mediates glial calcium waves. J Neurosci 19: 520-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart PD, Young MR (1991) Ammonium chloride, an inhibitor of phagosome-lysosome fusion in macrophages, concurrently induces phagosome-endosome fusion, and opens a novel pathway: studies of a pathogenic mycobacterium and a nonpathogenic yeast. J Exp Med 174: 881-889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LM, Chappell JB, Jones OTG (1987) The superoxide-generating NADPH oxidase of human neutrophils is electrogenic and associated with an H+ channel. Biochem J 246: 325-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitt ND, Kleinberg ME (1996) Identification of neutrophil NADPH oxidase proteins gp91phox, p22phox p67phox and p47phox in mammalian species. Am J Vet Res 57: 672-676. [PubMed] [Google Scholar]
- Lambeth JD (2004) NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4: 181-189. [DOI] [PubMed] [Google Scholar]
- Li J-M, Shah AM (2002) Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem 277: 19952-19960. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhu H, Kuppusamy P, Roubaud V, Zweier JL, Trush MA (1998) Validation of lucigenin (bis-N-methylacridinium) as a chemilumigenic probe for detecting superoxide anion radical production by enzymatic and cellular systems. J Biol Chem 273: 2015-2023. [DOI] [PubMed] [Google Scholar]
- Nauseef WM (2004) Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol 122: 277-291. [DOI] [PubMed] [Google Scholar]
- Peuchen S, Clark JB, Duchen MR (1996) Mechanisms of intracellular calcium regulation in adult astrocytes. Neuroscience 71: 871-883. [DOI] [PubMed] [Google Scholar]
- Robinson JM, Ohira T, Badwey JA (2004) Regulation of the NADPH-oxidase complex of phagocytic leukocytes. Recent insights from structural biology, molecular genetics, and microscopy. Histochem Cell Biol 22: 293-304. [DOI] [PubMed] [Google Scholar]
- Serrano F, Kolluri NS, Wientjes FB, Card JP, Klann E (2003) NADPH oxidase immunoreactivity in the mouse brain. Brain Res 988: 193-198. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G (2003) Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem 86: 1101-1107. [DOI] [PubMed] [Google Scholar]
- Wientjes FB, Hsuan JJ, Totty NF, Segal AW (1993) p40phox, a third cytosolic component of the activation complex of the NADPH oxidase to contain src homology 3 domains. Biochem J 296: 557-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wientjes FB, Segal AW, Hartwig JH (1997) Immunoelectron microscopy shows a clustered distribution of NADPH oxidase components in the human neutrophil plasma membrane. J Leukoc Biol 61: 303-312. [DOI] [PubMed] [Google Scholar]