

Abstract
The contribution of inflammation to the progression of neurodegenerative diseases such as Alzheimer's, Parkinson's, and prion diseases is poorly understood. Brain inflammation in animal models of these diseases is dominated by chronic microglial activation with minimal proinflammatory cytokine expression. However, these inflammatory cells are “primed” to produce exaggerated inflammatory responses to subsequent lipopolysaccharide (LPS) challenges. We show that, using the ME7 model of prion disease, intracerebral challenge with LPS results in dramatic interleukin-1β (IL-1β) expression, neutrophil infiltration, and inducible nitric oxide synthase expression in the brain parenchyma of prion-diseased mice compared with the same challenge in normal mice. Systemic inflammation evoked by LPS also produced greater increases in proinflammatory cytokines, pentraxin 3, and inducible nitric oxide synthase transcription in prion-diseased mice than in control mice and induced microglial expression of IL-1β. These systemic challenges also increased neuronal apoptosis in the brains of ME7 animals. Thus, both central and peripheral inflammation can exacerbate local brain inflammation and neuronal death. The finding that a single acute systemic inflammatory event can induce neuronal death in the CNS has implications for therapy in neurodegenerative diseases.
Keywords: microglia, cytokines, prion disease, Alzheimer's disease, peripheral infection, neurodegeneration, acute phase proteins
Introduction
There is much speculation on the role of CNS inflammation in the progression of neurodegenerative diseases such as Alzheimer's disease (AD) (Akiyama et al., 2000; Mrak and Griffin, 2001; McGeer and McGeer, 2003). The animal models available for AD do not show significant neurodegeneration (Irizarry et al., 1997) and, hence, although they show some evidence of inflammation, they do not exhibit an inflammatory response that can be a response to neurodegeneration. The prion diseases (transmissible spongiform encephalopathies) share many pathological features with AD and other chronic neurodegenerative conditions in that they are typified by the extracellular deposition of insoluble plaques, show neurodegeneration along neuroanatomical pathways, and display marked astrocytosis and microglial activation (DeArmond, 1993; Price et al., 1993; Soto et al., 2003). We have shown that during ME7-induced prion disease, there is an atypical CNS inflammatory response (Perry et al., 2002) dominated by increases in TGFβ1 with very little contribution from the proinflammatory cytokines interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6) (Betmouni et al., 1996; Walsh et al., 2001; Cunningham et al., 2002). Similarly, low expression of these cytokines has been reported in animal models of AD and Parkinson's disease (Sly et al., 2001; Depino et al., 2003).
However, it is also known that peripheral infection can initiate the synthesis of cytokines within the CNS (Laye et al., 1994; Pitossi et al., 1997). We have shown recently that systemic challenge with bacterial endotoxin [lipopolysaccharide (LPS)] provokes exaggerated CNS IL-1β synthesis and sickness behavior responses in animals with ongoing chronic inflammation (Combrinck et al., 2002), and this also appears to be true in transgenic models of AD (Sly et al., 2001; Lee et al., 2002). In a previous study, we proposed that microglia in the prion-diseased brain are in a “primed” state, and subsequent challenges, either centrally or systemically, result in an amplified cytokine and inflammatory response. This acutely exacerbated local inflammation may lead to increased neuronal loss and functional deficit (Perry et al., 2003).
In the current study, we used LPS to mimic aspects of central or systemic infection and the ME7 model of murine prion disease to model chronic neurodegeneration to address the hypothesis that microglial cells, activated by chronic neurodegeneration, are primed to produce greater proinflammatory responses to subsequent central and systemic inflammatory challenges. To achieve this, we assessed proinflammatory cytokine expression, inflammatory cell infiltration, and expression of effector enzymes such as cyclooxygenase 2 (COX-2) and inducible nitric oxide (NO) synthase (iNOS) after intracerebral and intraperitoneal LPS challenges. In addition, using immunohistochemical staining and quantification of terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)-positive and activated caspase-3-positive cell death after intraperitoneal LPS challenges, we addressed the hypothesis that this exaggerated inflammation exacerbates the neurodegenerative process.
Together, these proposals comprise the “microglial priming hypothesis,” which predicts that microglia, primed by an existing pathology such as synapse, axon, or neuronal cell body degeneration or by amyloid deposition can be stimulated to an aggressive state of inflammation by secondary influences, such as infection and injury, and during this window of activation can exacerbate neuronal death.
Materials and Methods
Animals and stereotaxic surgery. Female C57BL/6 mice were obtained from Harlan (Bicester, UK), housed in groups of five or six under standard light and temperature regimes, and fed pelleted food and water ad libitum. The mice were anesthetized intraperitoneally with Avertin (2,2,2-tribromoethanol) and positioned in a stereotaxic frame. Two small holes were drilled in the skull, on either side of the midline, to allow the bilateral injection of 1 μl of a 10% w/v ME7-infected C57BL/6 brain homogenate made in PBS. These injections were made directly into the dorsal hippocampus (coordinates from bregma: anteroposterior, -2.0 mm; lateral, -1.6 mm; depth, -1.5 mm) using a 10 μl Hamilton syringe with a 26 gauge needle. Control animals were injected with a 10% w/v normal brain homogenate (NBH) in PBS, derived from a naive C57BL/6 mouse. All procedures were performed in accordance with a United Kingdom Home Office license.
Peripheral and central LPS challenges. ME7 prion-diseased animals (which will be referred to as ME7 animals throughout) and NBH-injected animals (referred to as NBH throughout) were subjected to acute intracerebral or intraperitoneal inflammatory challenges (referred to as ME7+LPS, ME7+saline, NBH+LPS, intracerebrally or intraperitoneally, where appropriate). Nineteen weeks after ME7 or NBH injection, just before overt clinical signs (Betmouni et al., 1999), a group of animals was injected intrahippocampally with 2 μg of LPS (NBH and ME7) or with sterile saline (ME7) under avertin anesthesia as described previously and returned to their home cages once they had recovered their righting reflex. An additional group of mice was injected intraperitoneally with 500 μg/kg LPS (Salmonella equine abortus; Sigma, Poole, UK) in sterile saline or with saline alone and returned to their cages. The choice of 19 weeks post-ME7 inoculation for all LPS or saline challenges was made based on our previous knowledge of the degree and regional distribution of microglial activation in the animals at this time in disease and on our previous studies showing exaggerated sickness behavior responses and hippocampal IL-1β expression in animals at this stage in prion disease (Combrinck et al., 2002). The intracerebral LPS dose used in these studies was based on its ability to induce robust microglial IL-1β expression in normal animals and, for the systemic challenges, was based on the dose required to produce a robust febrile response (Combrinck et al., 2002).
Tissue preparation. Animals challenged intracerebrally with LPS were terminally anesthetized at 18 h after LPS and examined for evidence of cell infiltration/inflammatory activation. LPS- and saline-injected animals used for RNA extraction were terminally anesthetized 6 h after intraperitoneal injection, transcardially perfused with heparinized saline, and the brains were removed (n = 5 for each animal group). Thick coronal sections (∼2 mm) were taken at approximately -1.0 to -3.0 mm from bregma, and the hippocampus and thalamus were quickly removed, immediately frozen in liquid nitrogen, and stored at -80°C. Additional groups of intraperitoneally challenged animals were terminally anesthetized at 18 h after injection with LPS or saline and transcardially perfused and fixed in 10% formalin solution to investigate neutrophil infiltration, apoptotic cell death and other aspects of the pathology (n = 9 for ME7+LPS intraperitoneally; n = 5 for ME7+saline intraperitoneally; n = 3 for ME7; n = 3 for NBH; n = 5 for NBH+LPS intraperitoneally). Additional ME7 and NBH animals were challenged intraperito-neally with LPS or saline and perfused with heparinized saline and 10% formalin at 6, 9, and 13 h after systemic challenge to examine the time course of caspase-positive cell death. At least three animals were used at each time point for these studies.
Immunocytochemistry. Ten percent formalin-fixed tissue was postfixed over 2 d in the same fixative and then paraffin embedded. Sections (10 μm) were cut on a microtome and floated onto electrostatically charged slides. Immunocytochemistry was performed for neutrophils (rabbit polyclonal anti-neutrophil serum, MBS1, generated in-house), apoptotic cells [TUNEL (“Dead End” staining kit; Promega, Southampton, UK) and cleaved caspase-3 (Chemicon, Southampton, UK)], IL-1β (Peprotech, London, UK), iNOS (BD Biosciences, Oxford, UK), glial fibrillary acidic protein (GFAP; Dako, High Wycombe, UK), COX-2 (Santa Cruz Biotechnology, Santa Cruz, CA), neurons [neuron-specific nuclear protein (NeuN) (Chemicon) and neurofilament heavy chain (NFH; Abcam, Cambridge, UK)], and microglia (tomato lectin; Sigma). All biotinylated secondary antibodies were supplied by Vector Laboratories (Peterborough, UK), and all staining was visualized using the ABC method using peroxidase as enzyme, 0.015% v/v hydrogen peroxide as substrate, and diaminobenzidine as chromagen.
Tissues were dewaxed and rehydrated through xylene and alcohols. Endogenous peroxidase was eliminated by incubating sections in 1% H2O2/methanol for 10 min.
Neutrophils. Sections for MBS-1 staining were treated with pepsin 0.04% in 0.1 m HCl for 20 min at room temperature before PBS washing, blocked in 10% normal goat serum, and incubated with the primary antibody (Ab) at 1:200 for 2 h at room temperature. After additional washing, the sections were incubated with biotinylated goat anti-rabbit IgG and continued as described above.
TUNEL. Sections for TUNEL staining were washed in 0.85% NaCl and then PBS, pretreated with proteinase K (20 μg/ml), and permeabilized using 0.1% Triton X-100 for 10 min at room temperature. Sections were preincubated with equilibration buffer for 10 min and then with incubation medium for 2 h at 37°C, labeling apoptotic cells with fluorescein. The reaction was stopped using a sodium citrate solution. Sections were blocked with 10% normal goat serum and incubated with biotinylated goat anti-fluorescein antibody before continuing as described above. TUNEL-positive cells were counted in full coronal sections and included only if displaying shrunken morphology and condensed chromatin as well as being TUNEL labeled.
IL-1β. Sections for IL-1β were incubated in citrate buffer, pH 6, and microwaved two times for 3 min with 5 min cooling between incubations. They were washed in TBS and blocked with 50% normal goat serum before overnight incubation at 4°C with the primary antibody at 1:50, prepared in 50% serum to block nonspecific interactions with activated astrocytes. The sections were washed and then incubated with biotinylated goat anti-rabbit antibody and continued as described above.
Microglia. Sections for tomato lectin staining were washed three times in TBS 1% Triton X-100 containing 1 mm magnesium, manganese, and calcium chlorides. Biotinylated tomato lectin was prepared at 50 μg/ml in the same buffer, and sections were incubated overnight at 4°C. After thorough washing, the protocol continued as described above.
COX-2. Sections for cyclooxygenase-2 staining were quenched with 1% H2O2 in absolute methanol for 20 min and washed briefly in PBS before antigen retrieval by microwaving in citrate buffer two times for 3 min with 5 min cooling between incubations. After washing and blocking with 10% horse serum in PBS for 2 h, the primary Ab (goat polyclonal anti-Cox-2 antibody; Santa Cruz Biotechnology) was diluted 1:1000 in PBS and incubated overnight at room temperature. The sections were then washed in PBS, incubated with biotinylated affinity-purified horse anti-goat IgG and processed for ABC peroxidase reaction as above.
iNOS. Sections were quenched, pretreated with citrate buffer and microwaving, and digested with 0.04% pepsin as outlined above and then washed in PBS before blocking in 10% normal goat serum and incubation overnight at 4°C with a 1:100 dilution of a rabbit polyclonal antibody against iNOS. After additional washing with PBS, the sections were incubated with a goat anti-rabbit secondary antibody, and the ABC and DAB reactions were completed as above.
NeuN. NeuN staining was achieved using the same citrate buffer pretreatment as above, followed by 30 min in normal horse serum (10%), overnight incubation with the primary antibody at 1:5000, incubation with a horse anti-mouse secondary at 1:100 for 40 min, and completion of the ABC and DAB reactions as above.
Cleaved caspase-3. Activated caspase-3 was examined to determine the mechanism of cell death and also to facilitate double-labeling of dying cells by examining cells earlier in the cell death cascade. After citrate buffer and microwave pretreatment, sections were incubated with pepsin 0.04% in 0.1 m HCl for 20 min at room temperature and then washed before application of 10% normal goat serum for 30 min followed by overnight incubation at room temperature with the primary antibody at a dilution of 1:50. Goat anti-rabbit secondary antibody was applied for 40 min at room temperature, and the reaction was completed as above.
Double staining of NFH with caspase-3. To facilitate double staining of NFH and cleaved caspase-3, it was necessary to prelabel the rabbit anti-NFH antibody to avoid cross-reactivity between successive goat anti-rabbit secondary antibodies. Thus, anti-cleaved caspase-3 staining was performed first, as above with the substitution of an Alexa Fluor 488-linked goat anti-rabbit secondary antibody for the biotinylated one used above. Sections were then incubated with an NFH antibody prelabeled with a Zenon Alexa Fluor 546 kit (Invitrogen, Paisley, UK). The labeling simply attaches the fluorchrome directly to the primary antibody, obviating the need for a secondary antibody. This labeling was performed according to the instructions of the manufacturer, using a label to antibody ratio of 4:1 and a final antibody dilution of 1:250. This ratio and antibody concentration had been observed previously to give good-quality NFH staining in a single labeling experiment. For the purposes of nuclear counterstaining, we used TOPRO-3 to give fluorescence at 633 nm.
Confocal imaging. Triple-stained slides were imaged on a Leica (Nussloch, Germany) SP2 confocal laser-scanning microscope, using simultaneous excitation at 488, 546, and 633 nm. A Z-series was obtained through the thickness of the tissue (10 μm) from which representative 1 μm optical sections were extracted. Fluorochromes were also excited sequentially rather than simultaneously to exclude the possibility of “bleed through” from fluorochromes with overlapping spectra.
RNA extraction. Total RNA was extracted from brain samples using Qiagen (Crawley, UK) RNeasy mini columns according to the instructions of the manufacturer. Contaminating genomic DNA was degraded during extraction with Qiagen DNase1 enzyme. The typical yield for brain tissue was ∼5 μg per extraction. RNA was stored at -80°C until cDNA synthesis and PCR assay.
Taqman reverse transcription-PCR assay. All equipment and reagents were supplied by Applied Biosystems (Warrington, UK) unless otherwise stated. Assays for serum amyloid A (SAA), α2 antiplasmin (α2-AP), and complement 3 (C3) were performed as described previously (Wilcockson et al., 2002). Assays for pentraxin 3 (PTX3), IL-1β, IL-6, and TNFα were designed using the published sequences for these genes, applied to Primer Express software. Where possible, probes were designed to cross an intron such that they were cDNA specific. Table 1 lists the sequences for primers and probes for each assay. All primer pairs were checked for specificity by standard reverse transcription (RT)-PCR using Promega PCR reagents followed by gel electrophoresis. Each primer pair produced a discrete band of the expected amplicon size (data not shown).
Table 1.
Mouse cytokine and acute phase protein Taqman primer and probe sequences
|
Target |
Accession number |
Oligonucleotide |
Sequence |
Amplicon size (bp) |
|---|---|---|---|---|
| SAA | M13522 | Forward primer | 5′-GCCATGGAGGGTTTTTTTCATT-3′ | 80 |
| Reverse primer | 5′-CCTTTGGGCAGCATCATAGTTC-3′ | |||
| Probe | 5′-CACATGTCTCCAGCCCCTTGGAAAGC-3′ | |||
| α2-AP | Y12312 | Forward primer | 5′GCGGTTCACAGTGTCGGT-3′ | 66 |
| Reverse primer | 5′-TCCAGCAGGAACCATCGAA-3′ | |||
| Probe | 5′-ACATGATGCACGCGGTGTCATATCCT-3′ | |||
| C3 | J00367 | Forward primer | 5′-CCATGTATTCCATCATTACTCCCAA-3′ | 72 |
| Reverse primer | 5′-CGTGGGCCTCCAGTCAGA-3′ | |||
| Probe | 5′-CCTACGGCTGGAGAGCGAAGAGACC-3′ | |||
| PTX3 | X83601 | Forward primer | 5′-ACAACGAAATAGACAATGGACTTCAT-3′ | 62 |
| Reverse primer | 5′-CTGGCGGCAGTCGCA-3′ | |||
| Probe | 5′-CCACCGAGGACCCCACGCC-3′ | |||
| IL-1β | M15131 | Forward primer | 5′-GCCACCCACCCTGCA-3′ | 69 |
| Reverse primer | 5′-ACCGCTTTTCCATCTTCTTCTT-3′ | |||
| Probe | 5′-TGGAGAGTCTGGATCCCAAGCAATACCC-3′ | |||
| TNFα | M11731 | Forward primer | 5′-CTCCAGGCGGTGCCTATG-3′ | 149 |
| Reverse primer | 5′-GGGCCATAGAACTGATGAGAGG-3′ | |||
| Probe | 5′-TCAGCCTCTTCTCATTCCTGCTTGTGG-3′ | |||
| IL-6 | NM_031168 | Forward primer | 5′-TCCAGAAACCGCTATGAAGTTC-3′ | 72 |
| Reverse primer | 5′-CACCAGCATCAGTCCCAAGA-3′ | |||
| Probe | 5′-CTCTGCAAGAGACTTCCATCCAGTTGCC-3′ | |||
| TGFβ1 | AJ009862 | Forward primer | 5′-CGTGGAAATCAACGGGATCA-3′ | 84 |
| Reverse primer | 5′-GGCCATGAGGAGCAGGAA-3′ | |||
| Probe | 5′-ACCTGGGCACCATCCATGACATGA-3′ | |||
| iN0S | NM_010927 | Forward primer | 5′-CAGCTGGGCTGTACAAACCTT-3′ | 95 |
| Reverse primer | 5′-CATTGGAAGTGAAGCGTTTCG-3′ | |||
|
|
|
Probe |
5′-CGGGCAGCCTGTGAGACCTTTGA-3′ |
|
For Taqman PCR, cDNA was generated from total RNA using Taqman Gold RT reagents. Two hundred nanograms of total RNA were reverse transcribed in a 10 μl reaction volume. One microliter of the RT reaction (equivalent to 20 ng of RNA) was subsequently used for the PCR, performed as described previously (Wilcockson et al., 2002). The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was measured in each sample using Applied Biosystems Rodent GAPDH Taqman kit.
For the acute-phase reactants SAA, α2-AP, C3, and PTX3 assays, standard curves were made from in vitro transcripts of each gene as described previously (Wilcockson et al., 2002). Acute-phase protein mRNA levels were thus expressed as copies per nanogram of total RNA (as determined by normalization with GAPDH). Cytokine mRNA expression in prion-diseased and normal animals was assessed by relative quantitation as described previously (Cunningham et al., 2005). A standard curve was constructed from total RNA isolated from mouse brain tissue after intracerebral challenge with 2.5 μg of LPS, which is known to upregulate all target transcripts of interest in this study. This standard curve was constructed using a higher concentration of RNA in the reverse-transcriptase reaction than for samples for analysis to ensure that the cytokine transcription of all animal groups will fall within the range of the standard curve constructed. Serial one in five dilutions of the cDNA synthesized from brains of LPS-injected mice were made and a curve plotted of the Ct value (the cycle number at which the transcribed gene crosses the threshold of detection) versus the log of the concentration (assigned an arbitrary value, because the absolute concentration of cytokine transcripts is not known). Thus, the standard curve generates arbitrary values for concentration. These data were then normalized for GAPDH concentration in each sample.
Statistics. Animal groups in the peripheral LPS challenge groups were compared by ANOVA, and data were treated as significant at the 95% confidence interval. Cell numbers after TUNEL and caspase-3 stains were compared by ANOVA with Bonferroni's post hoc test.
Results
Microglial activation after intracerebral LPS
We previously used CD68 and major histocompatibility complex class I and II to demonstrate that microglial cells show activation in prion disease, as defined by altered morphology and upregulation of cell surface markers (Betmouni et al., 1996). Here, we used tomato lectin immunohistochemistry and found that this marker also reveals microglial cells with altered morphology in the disease (Fig. 1b). In addition, all features appropriate to prion disease pathology at 19 weeks after inoculation were evident in the tissue [vacuolation and CA1 neuronal loss (Fig. 1b,c) and PrPSc deposition (data not shown)]. Tomato lectin also labels the vasculature. NBH+LPS intracerebral animals show clear staining of the vasculature and a small number of microglial cells with thin, relatively ramified processes (Fig. 1a). The staining in the case of ME7 animals injected intracerebrally with saline shows many microglial cells that are more densely stained with a less ramified and characteristic activated morphology (Fig. 1b). ME7 animals injected intracerebrally with LPS also show clear activation by these criteria, but the morphological appearance is not significantly different to those not injected with LPS (Fig. 1b,c). Thus, at a morphological level, we observed no clear difference between microglia in ME7 animals with or without a subsequent LPS challenge.
Figure 1.
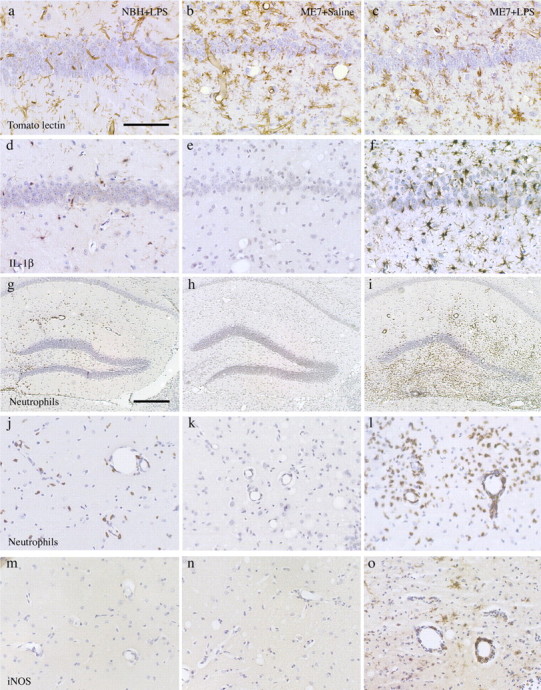
Neuropathological and inflammatory changes after intracerebral LPS. a-c, Tomato lectin staining for microglial cells in brains from NBH+LPS (a), ME7+saline (b), and ME7+LPS (c) animals. d-f, IL-1β immunostaining in NBH+LPS (d), ME7+saline (e), and ME7+LPS (f) brains. MBS-1 immunostaining for infiltrating neutrophils in NBH+LPS (g, j), ME7+saline (h, k), and ME7+LPS (i, l) brains is shown at 10× (g, h, i) and 40× (j, k, l), respectively. m-o, Immunostaining for iNOS in NBH+LPS (m), ME7+saline (n), and ME7+LPS (o) brains. Scale bar: (in a) a-f, j-o, 100 μm; (in g) g, h, i, 500 μm.
IL-1β expression after LPS challenge
Immunohistochemical staining after intracerebral LPS challenge in NBH animals showed some staining for IL-1β. This staining was clearly localized to activated microglia both in the vicinity of the intracerebral LPS injection and also the overlying ipsilateral cortex and underlying dorsal thalamus (Fig. 1d). Infiltrating neutrophils (see below) also showed some IL-1β expression. ME7 animals injected with saline did not show any IL-1β staining (Fig. 1e), as we described previously for ME7-infected mice (Walsh et al., 2001). LPS-injected ME7 animals showed a greatly increased density of IL-1β staining, both in the number of positively stained cells and in the intensity of staining in each activated cell (Fig. 1f). IL-1β staining was observed in the injected hippocampus but showed a wider distribution in adjacent cortex and thalamus and in the contralateral hippocampus. Thus, at a functional level, the microglial cells from ME7 animals injected with saline or LPS are markedly different despite showing the same morphology.
Intracerebral challenge with LPS, neutrophil infiltration
Intracerebral challenge with 2 μg of LPS provoked some neutrophil infiltration into the parenchyma of NBH animals (Fig. 1g,j). This infiltration was relatively dispersed, and the extent of infiltration was limited, although there was evidence of neutrophil adherence to the vasculature. There was no infiltration in ME7 animals challenged with saline (Fig. 1h,k). LPS challenge of ME7-infected animals produced massive neutrophil infiltration, and this influx was centered on the site of intracerebral challenge (Fig. 1i,l). Blood vessels and pial membrane-bound spaces were filled with neutrophils in addition to the very considerable numbers in the parenchyma. There was also some evidence for hemorrhage, which was not evident in NBH animals challenged intracerebrally with this dose of LPS.
iNOS staining after intracerebral challenge
We have shown previously that iNOS is not expressed in the brains of ME7-infected animals (Walsh et al., 2001). There was no detectable expression of iNOS in the brains of ME7 animals injected intracerebrally with saline (Fig. 1m) or in NBH+LPS intracerebral animals (Fig. 1n). In contrast, intracerebral LPS challenge to ME7 animals induced clear expression of iNOS in microglial cells and in some infiltrating neutrophils (Fig. 1o).
CNS cytokine and acute-phase responses to peripheral inflammation
To investigate whether ME7 animals showed exaggerated brain cytokine and acute-phase reactant mRNA levels after peripheral inflammation, we injected ME7 and NBH-injected mice intraperitoneally with LPS or saline and measured cytokine transcripts after 6 h.
Cytokine expression in LPS-injected NBH and ME7 mice
ME7 animals challenged with LPS intraperitoneally showed greater increases in IL-1β than NBH animals given the same LPS challenge (Fig. 2a) (p < 0.001). ME7 mice show slightly higher IL-1β transcript levels than NBH mice, but this cannot account for the difference between the LPS challenges; the level of expression of IL-1β mRNA in the ME7+LPS group was more than the sum of NBH+LPS and ME7 alone. The IL-1β increase, induced by LPS in ME7 animals, was approximately threefold greater than that induced by LPS in NBH animals.
Figure 2.
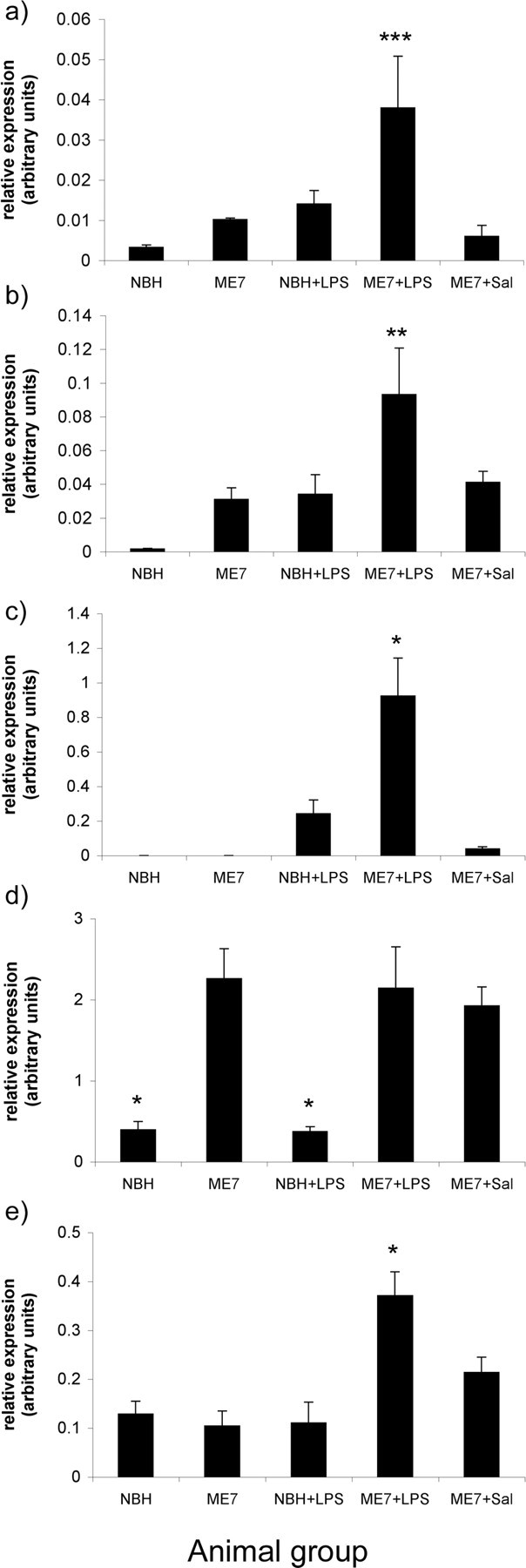
The impact of systemic challenge with LPS on cytokine and inflammatory gene transcription in ME7 and NBH mice. LPS produced greater IL-1β (a), TNFα (b), IL-6 (c), and iNOS (e) mRNA increases in ME7 animals than in NBH animals. The difference between the IL-1β, TNFα, IL-6, and iNOS expression in NBH+LPS and ME7+LPS was statistically significant (p < 0.001, p < 0.01, and p < 0.05, respectively). All ME7 groups were statistically significantly different from both NBH groups in TGFβ1 transcription (d), but LPS (intraperitoneal) did not produce increases in TGFβ1 in either NBH or ME7 animals. Black bars represent mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ANOVA. NBH+LPS, n = 4; ME7+saline, ME7+LPS, n = 5. Sal, Saline.
The pattern of TNFα expression was similar to that of IL-1β expression in the same experiment (Fig. 2b). ME7 mice showed increased levels of TNFα transcripts compared with NBH mice. TNFα expression in the ME7+LPS group was greater than that in the NBH+LPS group (p < 0.01). Once again, this difference was not accounted for by the baseline expression observed in ME7-infected mice (i.e., the effect was more than additive). The increase in TNFα induced by LPS in ME7 animals is 1.7-fold greater than that induced by LPS in NBH animals.
IL-6 transcripts were below the assay limit in both the NBH and ME7 groups. Peripheral challenge with LPS induced quantifiable levels of IL-6 in the brains of both NBH and ME7-infected mice (Fig. 2c). The IL-6 increase induced by LPS in ME7 animals is approximately threefold greater than that induced by LPS in NBH animals (p < 0.01). This pattern was not observed for TGFβ1, which is clearly elevated in ME7, ME7+saline and ME7+LPS but not significantly different between these groups. Intraperitoneal LPS does not induce TGFβ1 in the hippocampus of either ME7 or NBH animals (Fig. 2d). Transcription of the gene for iNOS is consistent and low among NBH, ME7, NBH+LPS, and ME7+saline groups and shows elevation only in the ME7+LPS intraperitoneally challenged group (p < 0.01) (Fig. 2e).
Acute-phase reactant expression in NBH and ME7 mice challenged intraperitoneally with LPS
A peripheral challenge with LPS produced increases in acutephase reactant mRNA expression in the brains of NBH and ME7-infected mice. In the ME7+saline and NBH+LPS intraperitoneal groups, there was no difference in the level of PTX3 mRNA (Fig. 3a). However, when the ME7 group was challenged with LPS, there was a dramatic increase in PTX3 gene expression, more than could be accounted for by the ME7 and LPS treatments alone. The difference between ME7-infected and ME7+LPS intraperitoneally was statistically significant (p < 0.001), as was the difference between ME7+LPS intraperitoneally and NBH+LPS intraperitoneally (p < 0.01). There was no statistical difference in α2-AP mRNA between the ME7+saline intraperitoneal, ME7+LPS intraperitoneal, and NBH+LPS intraperitoneal groups (Fig. 3b), although there was a trend toward increased α2-AP expression in the two LPS-injected groups. There were also no statistical differences in C3 mRNA expression in any of the treatments (Fig. 3c), although ME7+LPS intraperitoneal animals showed a trend toward increased expression compared with the ME7+saline intraperitoneal and NBH+LPS intraperitoneal groups. SAA mRNA expression in ME7+LPS intraperitoneal and NBH+LPS intraperitoneal brains was markedly elevated compared with ME7+saline intraperitoneal animals (Fig. 3d) (p < 0.01), but there was no difference between SAA expression in the two LPS-injected groups.
Figure 3.

Impact of systemic challenge with LPS on CNS acute phase reactant transcription expression in ME7 and NBH animals. PTX3 (a), α2-AP (b), C3 (c), and SAA (d) mRNA were all increased by peripheral LPS injection, but only PTX3 showed a statistically significant increase in the ME7+LPS group relative to NBH+LPS. All data are presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ANOVA. NBH+LPS, n = 4; ME7+saline, ME7+LPS, n = 5. Sal, Saline.
Microglial phenotype, transduction of peripheral inflammatory signal
The morphological appearance of microglial cells in both ME7 and ME7+LPS intraperitoneal groups is characteristic of activated cells compared with that in NBH animals challenged intraperitoneally with LPS (Fig. 4a,b). However, as was evident after intracerebral LPS challenge, intraperitoneal LPS did not induce any additional morphological change in these microglia as judged by tomato lectin staining (Fig. 4c). After LPS challenge intraperitoneally, perivascular macrophages and endothelial cells of the hippocampal vasculature are shown to express IL-1β (Fig. 4d). No positively stained microglia could be observed in this NBH group nor in the ME7 group injected with saline (Fig. 4d,e). However, endothelial cells, perivascular macrophages, and microglial cells in the vicinity of the hippocampal vasculature showed clear IL-1β staining after intraperitoneal LPS in ME7 animals (Fig. 4f, inset). Thus, the activated microglia present in prion-diseased brains are primed to produce a greater IL-1β response to the peripheral LPS-induced inflammatory signal.
Figure 4.
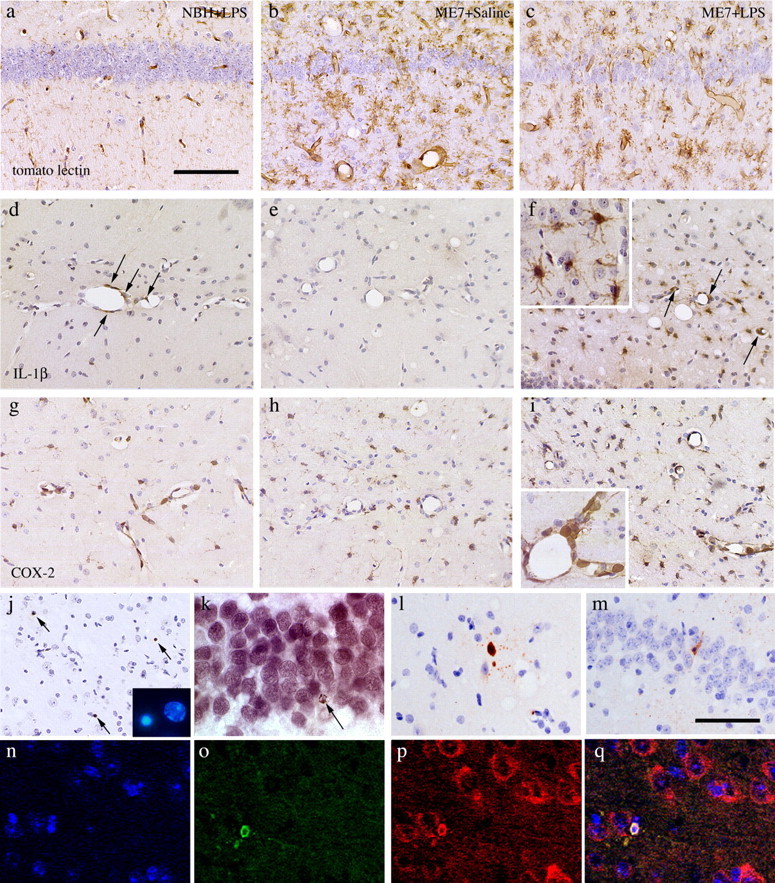
Neuropathological and inflammatory changes after intraperitoneal LPS. a-c, Tomato lectin staining for microglial cells in brains from NBH+LPS (a), ME7+saline (b), and ME7+LPS (c) animals. d-f, IL-1β immunostaining in NBH+LPS (d), ME7+saline (e), and ME7+LPS (f) brains (inset illustrates clear IL-1β-positive staining in cells with microglial morphology at 63× magnification). Immunostaining for cyclooxygenase-2 in NBH+LPS (g), ME7+saline (h), and ME7+LPS (i) brains is shown at 40× (g, h, i) and 63× (inset), respectively. j, TUNEL-positive staining in three cells (arrows) in a single field in the thalamus of an ME7+LPS brain. Inset, Hoescht 33352-stained nucleus showing shrunken and condensed chromatin (left) compared with normal (right). TUNEL-positive DAB staining in a CA3 neuron is shown (k), as identified by morphology and location, with failure to clearly double stain for NeuN (VIP chromagen). l, m, Activated caspase-3-positive cells showing neuronal morphology. An activated caspase-3-positive neuron (o), double stained using immunolabeling for neurofilament heavy chain (p) with overlay (q) in a 1 μm section by confocal microscopy is shown. n shows TOPRO-3 nuclear stain. Scale bars: (in a) a-i, 100 μm; (in m) l, m, 80 μm; n-q, 50 μm.
The endothelial expression of cyclooxygenase and consequent secretion of prostaglandin E2 is believed to be one common mechanism of the transduction of peripheral inflammatory signals across the blood-brain barrier (Ek et al., 2001). We observed very marked upregulation of COX-2 on the endothelial cells and perivascular macrophages (Fig. 4g,i). Prion disease per se does induce microglial COX-2 expression (Fig. 4h), as described previously (Walsh et al., 2000).
Consequences of increased CNS inflammation after peripheral LPS
To examine the consequences of increased brain inflammation resulting from peripheral LPS challenge, we examined cell death. We used TUNEL staining to label cells undergoing DNA fragmentation. This method may reveal cells that are positively stained but that do not subsequently undergo cell death. For this reason, we included in our counts only cells that stain positive and show a shrunken morphology and clearly condensed chromatin in the nucleus, consistent with irreversible cell damage (Fig. 4j, inset). There is considerable apoptotic cell death occurring in ME7 animals at 19 weeks after injection and indeed a very low level in normal animals. As shown in Table 2, there were increased numbers of TUNEL-positive cells showing apoptotic morphology in ME7 animals with respect to NBH animals. Systemic injection of saline had no significant effect on the number of apoptotic cells in ME7 animals at 19 weeks after inoculation. Similarly, the intraperitoneal injection of LPS had no significant effect on the numbers of apoptotic cells in NBH animals. However, LPS challenge produced a statistically significant increase in the number of apoptotic cells in ME7 animals (Fig. 4j; Table 2). When corrected for baseline TUNEL-positive numbers (i.e., that in NBH animals), cell death in prion-diseased animals is more than twice as high after LPS challenge than in unchallenged or saline-challenged ME7 animals (Table 2). We attempted to use double immunocytochemistry to identify these dying cells as neuronal, but because of the advanced stage of degeneration at 18 h, these cells could not be identified morphologically and express no detectable cellular antigens such as GFAP or NeuN (Fig. 4k). For this reason, we also stained sections from these animals and others taken from animals 3, 6, 9, and 13 h after LPS with anti-activated caspase-3 antibodies. This staining revealed less dying cells than the TUNEL technique, consistent with the shorter temporal window of expression of activated caspase-3 (Brecht et al., 2001). However, in these studies, ME7 animals challenged with LPS intraperitoneally contained approximately twice as many activated caspase-3-positive cells as ME7 animals challenged with saline (Table 2). Many of the activated caspase-3-positive cells had morphology or localization, suggesting that they were neurons (Fig. 4l,m), but double staining of these cells also presented some technical difficulties because of the altered antigen expression of degenerating cells (i.e., these dying cells were also negative for GFAP, NeuN, and tomato lectin). However, the vast majority of these cells stained positive for activated caspase-3 and the neuronal marker neurofilament heavy chain (Fig. 4o-q). This protein was selected on the basis of its biochemical stability, which we judged likely to ensure its preservation late into the process of cell degeneration.
Table 2.
Numbers of apoptotic cellsa counted per coronal section at hippocampal level
|
Animal group |
TUNEL-positive cells |
Activated caspase-3-positive cells |
|---|---|---|
| NBH | 5.3 ± 0.2 (n= 3) | ND |
| ME7 | 26.8 ± 1.6 (n= 3) | ND |
| NBH+LPS | 6.8 ± 0.5 (n= 5) | 1.4 ± 0.7 (n= 5) |
| ME7+LPS | 50.2 ± 2.3 (n=9)b | 12.2 ± 2.0 (n=5)b |
| ME7+saline |
27.8 ± 3.0 (n= 5) |
6.6 ± 1.3 (n= 6) |
Sections were taken between bregma −2.0 and bregma −2.5 mm, and all apoptotic cells in coronal sections were counted by two blinded observers. Counts in animals challenged intraperitoneally with saline or LPS were performed in brains perfused at 18 h after challenge. Apoptotic cells clearly identifiable as ependymal/endothelial/vascular were omitted. Counts were averaged across four coronal sections per animal and averaged across at least five animals in all key comparisons. Groups were compared statistically by ANOVA with Bonferroni's post hoc test.
As identified by both positive staining and by cell and nuclear morphology.
Denotes significant difference from ME7 alone or ME7 + saline, p<0.0167.
Discussion
In the current study, we demonstrated that in prion disease-induced chronic neurodegeneration, microglia show characteristic alterations of morphology and antigen expression typical of activated microglia but do not produce the inflammatory mediators IL-1β or iNOS. However, these cells are primed to produce a greater response to a subsequent inflammatory challenge, whether administered intracerebrally or intraperitoneally. The inflammatory response to intracerebral LPS in ME7 animals with previous microglial activation is severe, with marked microglial IL-1β and iNOS expression and massive neutrophil infiltration. Similarly, the intraperitoneal LPS challenge produced marked microglial IL-1β expression and transcription of other inflammatory genes such as IL-6, TNF-α, PTX3, and iNOS in prion-diseased animals. Significantly, in animals with ongoing neurodegeneration and microglial activation, this single systemic LPS challenge induced neuronal death.
Consequences of intracerebral LPS challenge
Labeling with tomato lectin clearly shows that prion disease induces microglial activation on the basis of increased cell numbers, increased expression of lectin-targeted antigen, and morphological features of activation. However, these cells do not synthesize detectable amounts of IL-1β protein. After intracerebral stimulation with LPS, these microglial cells produce readily detectable IL-1β in the absence of any additional change in morphology or numbers. Thus, microglial morphology is insufficient for analysis of the phenotype of these cells: activated microglial cells can show clear divergence in function in vivo.
Intraparenchymal injection of LPS in the normal brain provokes limited neutrophil recruitment into the parenchyma (Andersson et al., 1992). LPS challenge to the brain parenchyma of ME7 animals provoked very marked neutrophil infiltration not apparent in LPS-challenged NBH animals. Thus, the microglial IL-1β synthesis has clear biological consequences. This neutrophil infiltration is probably induced via the expression of the CXC chemokine macrophage inflammatory protein-2. This chemokine is induced by IL-1β (Xu et al., 1995), and neutrophil infiltration to the CNS can be blocked by neutralization of either IL-1β or the rat equivalent CXC chemokine, cytokine-induced neutrophil chemoattractant 1 (Anthony et al., 1998). IL-1β expression is also known to induce iNOS (Serou et al., 1999) and thus may underlie the microglial expression of iNOS seen here. The production of the oxidant NO has been demonstrated previously to be involved in neuronal death and damage (Takeuchi et al., 1998; Smith et al., 2001).
Intracerebral challenge with LPS in the current study demonstrates the degree to which the brain microenvironment and its cellular constituents are primed during chronic neurodegeneration to produce exaggerated responses to subsequent inflammatory challenges. Our data suggest that the inflammatory response to insults such as CNS infection, stroke, and head injury would be exacerbated in those already experiencing neurodegeneration-associated microglial activation. Given the evidence that IL-1β exacerbates ischemic neuronal injury (Stroemer and Rothwell, 1998), we predict that strokes and CNS infections should have more severe consequences in persons with ongoing neurodegeneration. There are few studies on the impact of CNS infection on dementia, but there are a number of studies showing that cerebral ischemia has more severe consequences in patients with ongoing dementia (Appelros et al., 2002; Koistinaho and Koistinaho, 2005). The results presented here provide a possible explanation of these clinical findings.
CNS consequences of systemic inflammation
It is known that peripheral inflammation can stimulate central inflammatory cytokine mRNA and protein synthesis (Laye et al., 1994; Pitossi et al., 1997) and that these cytokines coordinate a group of adaptive behavioral changes collectively known as sickness behavior (Konsman et al., 2002). In prion-diseased mice, in which there is prior microglial activation, LPS induces exaggerated sickness behavior and increased central synthesis of IL-1β (Combrinck et al., 2002). Using the same ME7 model, we now show that LPS also induces exaggerated changes in a number of cytokines and acute-phase reactants in degenerating brains. Proinflammatory cytokine mRNAs for IL-1β, IL-6, and TNFα were induced to higher levels by LPS in ME7 animals than in NBH animals, and this could not be accounted for by the additive effect of the individual treatments. This LPS challenge did not result in acute elevation of TGFβ1, a gene already highly expressed in this model (Cunningham et al., 2002). This results in a “switch” in the balance between proinflammatory and anti-inflammatory cytokines and may allow the activation of different downstream cascades during the acute phase after systemic inflammation. These results are consistent with increased CNS cytokine mRNA expression after i.p. LPS in both amyloid precursor protein overexpressing mice (Sly et al., 2001) and presenilin-1 mutant mice (Lee et al., 2002), indicating that this “microglial priming” is not specific to murine prion disease.
Acute-phase reactant gene expression following LPS challenge was generally similar in ME7 and NBH animals, but PTX3 was highly induced by LPS in prion-diseased mice compared with control animals. PTX3 is a member of the pentraxins, which are thought to opsonize apoptotic cells, pathogens, and debris for phagocytosis by Fcγ receptor-expressing cells (Bharadwaj et al., 2001; Mold et al., 2002a,b). Like C-reactive protein, PTX3 has been shown to exert control over the degree of activation of macrophages: PTX3 overexpressing macrophages produce more robust responses to LPS challenges than controls (Dias et al., 2001). The observed enhanced levels of PTX3 in the current study may play a role in the exaggerated response to LPS.
If LPS does have effects on opsonization or phagocytosis, it is possible that this systemic stimulation could also have some beneficial roles in clearance of amyloid proteins as has been shown in animal models of AD (DiCarlo et al., 2001; Herber et al., 2004). However, a thorough discussion of this area is beyond the scope of this study.
Thus, systemic inflammation in an animal with chronic neurodegeneration induces an inflammatory phenotype that is distinct from that produced by either of these conditions alone but which is also distinct from the additive effects of the individual profiles. The peripheral cytokine responses are not different in prion-diseased and normal animals (Cunningham et al., 2005), and thus the exacerbation of the brain inflammatory response is occurring at the CNS level. An increase in numbers and sensitivity of the microglial population is the most likely explanation for this, but there are also more perivascular macrophages present at the vasculature of prion-diseased animals than in normal animals (Galea et al., 2005) and also activation of astrocytes in areas of pathology, and these may also contribute to the enhanced CNS response to the peripheral challenge.
Mechanisms and implications
We show that a single systemic LPS challenge of 500 μg/kg is sufficient to induce acute cell death in the brains of animals with ongoing brain inflammation and that these dying cells are neurons. It follows that serial acute inflammatory insults superimposed on ongoing chronic neurodegeneration may produce a series of acute episodes of damage over the course of a chronic neurodegenerative disease, and this may have a major impact on the rate of degeneration. Repeated stimulations with LPS have not been performed in these studies, because these animals are known to become tolerant to LPS after repeated challenge (Greer and Rietschel, 1978; Ziegler-Heitbrock, 1995). However, it has been shown in the superoxide dismutase-1G37R model of amyotrophic lateral sclerosis that a relatively severe chronic stimulation with LPS exacerbates neuronal death and motor neuron axon degeneration (Nguyen et al., 2004). That a less severe inflammatory challenge is also sufficient to induce acute neuronal death may have important consequences for patients with chronic neurodegenerative disease subsequently exposed to common low-grade peripheral infections. Proinflammatory cytokines, including IL-1β and TNFα, are known to exacerbate neuronal damage in acute injury models (Stroemer and Rothwell, 1998), and this may occur through IL-1β-activated pathways such as induction of inducible COX-2 (Serou et al., 1999), tissue plasminogen activator (Eberhardt et al., 2002), iNOS (Tamatani et al., 1998), and increased synthesis of the damaging reactive oxygen species superoxide and peroxynitrite (Fink et al., 1999).
There is already clinical evidence that infections are deleterious in dementia. Delirium is a transient disorder of cognition and attention and is a common consequence of systemic infection in the elderly and demented (Elie et al., 1998). Episodes of delirium significantly increase the risk of dementia (Rockwood et al., 1999), shorten the time to permanent institutionalization (Pitkala et al., 2005), and are associated with long-term functional decline (Murray et al., 1993) and increased mortality (Rockwood et al., 1999). There is also epidemiological and clinical evidence that infections accelerate the progression of AD (Holmes et al., 2003; Dunn et al., 2005). Together, these studies indicate that insults that trigger delirium, such as infection, accelerate the neurodegenerative process.
Conclusion
In closing, it seems important to reiterate that we currently have little evidence that inflammation has a direct role in prion disease progression per se: the microglial response in prion disease and in other animal models of neurodegeneration appears to be tightly controlled to prevent exacerbation of the degenerative process. But this inflammatory response alters the number, morphology, and sensitivity of microglia such that the brain becomes primed and more vulnerable to subsequent inflammatory insults (Sly et al., 2001; Lee et al., 2002; Nguyen et al., 2004). These insults may then impact on the rate of neurodegeneration. Suppression of peripheral inflammation may be a relevant therapeutic target in chronic neurodegeneration and may be a component of the protection of nonsteroidal anti-inflammatory drugs against the development of AD.
Footnotes
This work was supported by the Wellcome Trust, the Medical Research Council, and the Nuffield Foundation. We thank Anton Page (Biomedical Imaging Unit, Southampton General Hospital) for assistance with confocal microscopy.
Correspondence should be addressed to Dr. Colm Cunningham, CNS Inflammation Group, Southampton Neuroscience Group, School of Biological Sciences, Bassett Crescent East, Southampton, Hampshire SO16 7PX, UK. E-mail: C.Cunningham@soton.ac.uk.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259275-10$15.00/0
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson PB, Perry VH, Gordon S (1992) The acute inflammatory response to lipopolysaccharide in CNS parenchyma differs from that in other body tissues. Neuroscience 48: 169-186. [DOI] [PubMed] [Google Scholar]
- Anthony D, Dempster R, Fearn S, Clements J, Wells G, Perry VH, Walker K (1998) CXC chemokines generate age-related increases in neutrophil-mediated brain inflammation and blood-brain barrier breakdown. Curr Biol 8: 923-926. [DOI] [PubMed] [Google Scholar]
- Appelros P, Nydevik I, Seiger A, Terent A (2002) Predictors of severe stroke: influence of preexisting dementia and cardiac disorders. Stroke 33: 2357-2362. [DOI] [PubMed] [Google Scholar]
- Betmouni S, Perry VH, Gordon JL (1996) Evidence for an early inflammatory response in the central nervous system of mice with scrapie. Neuroscience 74: 1-5. [DOI] [PubMed] [Google Scholar]
- Betmouni S, Deacon RMJ, Rawlins JNP, Perry VH (1999) Behavioural consequences of prion disease targeted to the hippocampus in a mouse model of scrapie. Psychobiology 27: 63-71. [Google Scholar]
- Bharadwaj D, Mold C, Markham E, Du Clos TW (2001) Serum amyloid P component binds to Fc gamma receptors and opsonizes particles for phagocytosis. J Immunol 166: 6735-6741. [DOI] [PubMed] [Google Scholar]
- Brecht S, Gelderblom M, Srinivasan A, Mielke K, Dityateva G, Herdegen T (2001) Caspase-3 activation and DNA fragmentation in primary hippocampal neurons following glutamate excitotoxicity. Brain Res Mol Brain Res 94: 25-34. [DOI] [PubMed] [Google Scholar]
- Combrinck MI, Perry VH, Cunningham C (2002) Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience 112: 7-11. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Boche D, Perry VH (2002) Transforming growth factor beta1, the dominant cytokine in murine prion disease: influence on inflammatory cytokine synthesis and alteration of vascular extracellular matrix. Neuropathol Appl Neurobiol 28: 107-119. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Wilcockson DC, Boche D, Perry VH (2005) Comparison of inflammatory and acute-phase responses in the brain and peripheral organs of the ME7 model of prion disease. J Virol 79: 5174-5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeArmond SJ (1993) Alzheimer's disease and Creutzfeldt-Jakob disease: overlap of pathogenic mechanisms. Curr Opin Neurol 6: 872-881. [DOI] [PubMed] [Google Scholar]
- Depino AM, Earl C, Kaczmarczyk E, Ferrari C, Besedovsky H, del Rey A, Pitossi FJ, Oertel WH (2003) Microglial activation with atypical proinflammatory cytokine expression in a rat model of Parkinson's disease. Eur J Neurosci 18: 2731-2742. [DOI] [PubMed] [Google Scholar]
- Dias AA, Goodman AR, Dos Santos JL, Gomes RN, Altmeyer A, Bozza PT, Horta MF, Vilcek J, Reis LF (2001) TSG-14 transgenic mice have improved survival to endotoxemia and to CLP-induced sepsis. J Leukoc Biol 69: 928-936. [PubMed] [Google Scholar]
- DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D (2001) Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging 22: 1007-1012. [DOI] [PubMed] [Google Scholar]
- Dunn N, Mullee M, Perry VH, Holmes C (2005) Association between dementia and infectious disease: evidence from a case-control study. Alzheimer Dis Assoc Disord 19: 91-94. [DOI] [PubMed] [Google Scholar]
- Eberhardt W, Beck KF, Pfeilschifter J (2002) Cytokine-induced expression of tPA is differentially modulated by NO and ROS in rat mesangial cells. Kidney Int 61: 20-30. [DOI] [PubMed] [Google Scholar]
- Ek M, Engblom D, Saha S, Blomqvist A, Jakobsson PJ, Ericsson-Dahlstrand A (2001) Inflammatory response: pathway across the blood-brain barrier. Nature 410: 430-431. [DOI] [PubMed] [Google Scholar]
- Elie M, Cole MG, Primeau FJ, Bellavance F (1998) Delirium risk factors in elderly hospitalized patients. J Gen Intern Med 13: 204-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink KB, Andrews LJ, Butler WE, Ona VO, Li M, Bogdanov M, Endres M, Khan SQ, Namura S, Stieg PE, Beal MF, Moskowitz MA, Yuan J, Friedlander RM (1999) Reduction of post-traumatic brain injury and free radical production by inhibition of the caspase-1 cascade. Neuroscience 94: 1213-1218. [DOI] [PubMed] [Google Scholar]
- Galea I, Palin K, Newman TA, Van Rooijen N, Perry VH, Boche D (2005) Mannose receptor expression specifically reveals perivascular macrophages in normal, injured, and diseased mouse brain. Glia 49: 375-384. [DOI] [PubMed] [Google Scholar]
- Greer GG, Rietschel ET (1978) Lipid A-induced tolerance and hyperreactivity to hypothermia in mice. Infect Immun 19: 357-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herber DL, Roth LM, Wilson D, Wilson N, Mason JE, Morgan D, Gordon MN (2004) Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp Neurol 190: 245-253. [DOI] [PubMed] [Google Scholar]
- Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH (2003) Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer's disease. J Neurol Neurosurg Psychiatry 74: 788-789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT (1997) APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol 56: 965-973. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J (2005) Interactions between Alzheimer's disease and cerebral ischemia-focus on inflammation. Brain Res Brain Res Rev 48: 240-250. [DOI] [PubMed] [Google Scholar]
- Konsman JP, Parnet P, Dantzer R (2002) Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci 25: 154-159. [DOI] [PubMed] [Google Scholar]
- Laye S, Parnet P, Goujon E, Dantzer R (1994) Peripheral administration of lipopolysaccharide induces the expression of cytokine transcripts in the brain and pituitary of mice. Brain Res Mol Brain Res 27: 157-162. [DOI] [PubMed] [Google Scholar]
- Lee J, Chan SL, Mattson MP (2002) Adverse effect of a presenilin-1 mutation in microglia results in enhanced nitric oxide and inflammatory cytokine responses to immune challenge in the brain. Neuromolecular Med 2: 29-45. [DOI] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL (2003) Inflammatory processes in Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry 27: 741-749. [DOI] [PubMed] [Google Scholar]
- Mold C, Baca R, Du Clos TW (2002a) Serum amyloid P component and C-reactive protein opsonize apoptotic cells for phagocytosis through Fc-gamma receptors. J Autoimmun 19: 147-154. [DOI] [PubMed] [Google Scholar]
- Mold C, Rodriguez W, Rodic-Polic B, Du Clos TW (2002b) C-reactive protein mediates protection from lipopolysaccharide through interactions with Fc gamma R. J Immunol 169: 7019-7025. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS (2001) Interleukin-1, neuroinflammation, and Alzheimer's disease. Neurobiol Aging 22: 903-908. [DOI] [PubMed] [Google Scholar]
- Murray AM, Levkoff SE, Wetle TT, Beckett L, Cleary PD, Schor JD, Lipsitz LA, Rowe JW, Evans DA (1993) Acute delirium and functional decline in the hospitalized elderly patient. J Gerontol 48: M181-M186. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, D'Aigle T, Gowing G, Julien JP, Rivest S (2004) Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J Neurosci 24: 1340-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Cunningham C, Boche D (2002) Atypical inflammation in the central nervous system in prion disease. Curr Opin Neurol 15: 349-354. [DOI] [PubMed] [Google Scholar]
- Perry VH, Newman TA, Cunningham C (2003) The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci 4: 103-112. [DOI] [PubMed] [Google Scholar]
- Pitkala KH, Laurila JV, Strandberg TE, Tilvis RS (2005) Prognostic significance of delirium in frail older people. Dement Geriatr Cogn Disord 19: 158-163. [DOI] [PubMed] [Google Scholar]
- Pitossi F, del Rey A, Kabiersch A, Besedovsky H (1997) Induction of cytokine transcripts in the central nervous system and pituitary following peripheral administration of endotoxin to mice. J Neurosci Res 48: 287-298. [DOI] [PubMed] [Google Scholar]
- Price DL, Borchelt DR, Sisodia SS (1993) Alzheimer disease and the prion disorders amyloid beta-protein and prion protein amyloidoses. Proc Natl Acad Sci USA 90: 6381-6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwood K, Cosway S, Carver D, Jarrett P, Stadnyk K, Fisk J (1999) The risk of dementia and death after delirium. Age Ageing 28: 551-556. [DOI] [PubMed] [Google Scholar]
- Serou MJ, DeCoster MA, Bazan NG (1999) Interleukin-1 beta activates expression of cyclooxygenase-2 and inducible nitric oxide synthase in primary hippocampal neuronal culture: platelet-activating factor as a preferential mediator of cyclooxygenase-2 expression. J Neurosci Res 58: 593-598. [PubMed] [Google Scholar]
- Sly LM, Krzesicki RF, Brashler JR, Buhl AE, McKinley DD, Carter DB, Chin JE (2001) Endogenous brain cytokine mRNA and inflammatory responses to lipopolysaccharide are elevated in the Tg2576 transgenic mouse model of Alzheimer's disease. Brain Res Bull 56: 581-588. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Kapoor R, Hall SM, Davies M (2001) Electrically active axons degenerate when exposed to nitric oxide. Ann Neurol 49: 470-476. [PubMed] [Google Scholar]
- Soto C, Volkel D, Zimmermann K, Zerr I, Lindner T, Bodemer M, Poser S, Schwarz HP (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 4: 49-60. [DOI] [PubMed] [Google Scholar]
- Stroemer RP, Rothwell NJ (1998) Exacerbation of ischemic brain damage by localized striatal injection of interleukin-1beta in the rat. J Cereb Blood Flow Metab 18: 833-839. [DOI] [PubMed] [Google Scholar]
- Takeuchi A, Isobe KI, Miyaishi O, Sawada M, Fan ZH, Nakashima I, Kiuchi K (1998) Microglial NO induces delayed neuronal death following acute injury in the striatum. Eur J Neurosci 10: 1613-1620. [DOI] [PubMed] [Google Scholar]
- Tamatani M, Ogawa S, Niitsu Y, Tohyama M (1998) Involvement of Bcl-2 family and caspase-3-like protease in NO-mediated neuronal apoptosis. J Neurochem 71: 1588-1596. [DOI] [PubMed] [Google Scholar]
- Walsh DT, Perry VH, Minghetti L (2000) Cyclooxygenase-2 is highly expressed in microglial-like cells in a murine model of prion disease. Glia 29: 392-396. [PubMed] [Google Scholar]
- Walsh DT, Betmouni S, Perry VH (2001) Absence of detectable IL-1beta production in murine prion disease: a model of chronic neurodegeneration. J Neuropathol Exp Neurol 60: 173-182. [DOI] [PubMed] [Google Scholar]
- Wilcockson DC, Campbell SJ, Anthony DC, Perry VH (2002) The systemic and local acute phase response following acute brain injury. J Cereb Blood Flow Metab 22: 318-326. [DOI] [PubMed] [Google Scholar]
- Xu WB, Haddad EB, Tsukagoshi H, Adcock I, Barnes PJ, Chung KF (1995) Induction of macrophage inflammatory protein 2 gene expression by interleukin 1 beta in rat lung. Thorax 50: 1136-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler-Heitbrock HW (1995) Molecular mechanism in tolerance to lipopolysaccharide. J Inflamm 45: 13-26. [PubMed] [Google Scholar]