

Abstract
Calcitonin gene-related peptide (CGRP) is a sensory neuropeptide important in inflammatory pain that conveys pain information centrally and dilates blood vessels peripherally. Previous studies indicate that activin A increases CGRP-immunoreactive (IR) sensory neurons in vitro, and following wound, activin A protein increases in the skin and more neurons have detectable CGRP expression in the innervating dorsal root ganglion (DRG). These data suggest some adult sensory neurons respond to activin A or other target-derived factors with increased neuropeptide expression. This study was undertaken to test whether activin contributes to inflammatory pain and increased CGRP and to learn which neurons retained plasticity. After adjuvant-induced inflammation, activin mRNA, but not NGF or glial cell line-derived neurotrophic factor, increased in the skin. To examine which DRG neurons increased CGRP immunoreactivity, retrograde tracer-labeled cutaneous neurons were characterized after inflammation. The proportion and size of tracer-labeled DRG neurons with detectable CGRP increased after inflammation. One-third of CGRP-IR neurons that appear after inflammation also had isolectin B4 binding, suggesting that some mechanoreceptors became CGRP-IR. In contrast, the increased proportion of CGRP-IR neurons did not appear to come from RT97-IR neurons. To learn whether central projections were altered after inflammation, CGRP immunoreactivity in the protein kinase Cγ-IR lamina IIi was quantified and found to increase. Injection of activin A protein alone caused robust tactile allodynia and increased CGRP in the DRG. Together, these data support the hypothesis that inflammation and skin changes involving activin A cause some sensory neurons to increase CGRP expression and pain responses.
Keywords: activin, NGF, CGRP, inflammation, tactile allodynia, sensory neurons
Introduction
The sensory neuropeptide calcitonin gene-related peptide (CGRP) is essential for pain after inflammation, but the factors that regulate its expression are not well understood. After noxious stimulation, neuropeptides are released from sensory nerves to transmit pain information to the spinal cord and to promote vasodilation of the skin (Wallengren and Hakanson, 1987; Holzer, 1998). Mice lacking α-CGRP fail to develop secondary hyperalgesia after inflammation (Zhang et al., 2001). Inflammation increases CGRP in the innervating dorsal root ganglion (DRG), sciatic nerve, and inflamed skin, suggesting that sensory neurons increase CGRP, and the peptide is transported to the inflamed area (Smith et al., 1992; Nahin and Byers, 1994; Neumann et al., 1996; Bulling et al., 2001). However, the factors from inflamed skin that increase CGRP are not clear.
The TGF-β family member activin A is a strong candidate to mediate changes in CGRP expression. Activin A induces CGRP expression in cultured embryonic (Ai et al., 1999; Hall et al., 2001) and adult (Cruise et al., 2004) DRG neurons in a dose-dependent manner. After skin wounds, activin A is upregulated and CGRP increases in the innervating DRG (Hübner et al., 1996; Cruise et al., 2004). Activin A may be a common component in inflammatory injury (Hübner et al., 1997; Munz et al., 1999; Jones et al., 2000, 2004; Phillips et al., 2001)
The sensory neurons that acquire CGRP and presumably nociceptive functions in the DRG after injury have been difficult to identify. Distinctive neurons process specialized sensory information. Some 30-40% of DRG neurons are CGRP-immunoreactive (IR), but neuropeptides are present in 50% of the cutaneous neurons (Bennett et al., 1996). Forty percent of neurons express the NGF receptor tyrosine receptor kinase A (trkA), and most of these neurons also express CGRP (Verge et al., 1992; Mu et al., 1993; Averill et al., 1995). In contrast, other nociceptors derived from late-developing sensory neurons are glial cell line-derived neurotrophic factor (GDNF) sensitive, express the c-Ret receptor, and bind the Griffonia simplicifolia isolectin B4 (IB4) (Molliver et al., 1997; Bradbury et al., 1998). Approximately 30% of sensory neurons express IB4 (Silverman and Kruger, 1990). Proprioceptive afferent neurons are recognized by antibody RT97 (Lawson et al., 1984), approximately half the lumbar DRG neurons are RT97-IR (Scott, 1992), and 18% of trkA-IR neurons express RT97 (Averill et al., 1995). Physiological characteristics also distinguish the fibers delivering pain information. All Aδ fibers and half of the C-fiber nociceptors are NGF dependent, and the remaining C fiber nociceptors are GDNF dependent during development (Priestley et al., 2002). In rat lumbar DRG, 40% of the C-fiber cells, 33% of the Aδ-fiber cells, and 17% of the fast A-fiber cells are CGRP-IR (Scott, 1992).
This study was undertaken to test whether activin A contributes to inflammatory pain and to learn which DRG neurons retain phenotypic plasticity. A retrograde tracer labeled cutaneous neurons, and inflammation was induced. We show that inflammation increases activin A mRNA in the skin and CGRP-IR neurons in the DRG. The increased population of CGRP-IR neurons after inflammation is derived from neurons that are also IB4-IR and others that are trkA-IR. We show for the first time that activin A induces tactile allodynia and CGRP expression in the DRG. These data suggest that inflammatory wounds involve activin A upregulation in the skin and increased neuronal expression of neuropeptides essential for pain.
Materials and Methods
Animals. Sixty-five adult female Sprague Dawley rats (8-10 weeks of age; 200-250 g; Zivic Miller, Pittsburgh, PA) were used in the experiments. All of the rats were housed in cages and maintained on a 12 h light/dark cycle, and the experimental protocols with animals were reviewed and performed in accordance with the Institutional Animal Care and Use Committee at Case Western Reserve University.
Inflammation induction and retrograde label of cutaneous neurons innervating ankle area. Fourteen control and 14 experimental adult rats were anesthetized by an intraperitoneal injection (150 μl) of a mixture of 100 mg/ml ketamine (Fort Dodge Animal Health, Fort Dodge, IA), 20 mg/ml xylazine (Phoenix Scientific, St. Joseph, MO), and 10 mg/ml acepromazine maleate (Boehringer Ingelheim Vetmedica, St. Joseph, MO). Hair was shaved from both of the rear ankles, and 10 μl total volume of a saline solution containing 2.5% hydroxystilbamidine methanesulfonate (HM; similar to FluoroGold; Invitrogen, Eugene, OR) was injected subcutaneously with a 20 μl Hamilton syringe with 27G1/2 needle at four sites around the left ankle to label cutaneous sensory neurons innervating this area. After 4 d, four injections of 80 μl total volume of complete Freund's adjuvant (CFA; Sigma, St. Louis, MO) were injected into the same area to induce inflammation. The circumference of the ankle was measured before tracer injection, before CFA injection, and 2 d after CFA injection. The experiments were done three times independently, with 10 rats in each of two experiments (five control, five CFA) and eight rats in the third (four control, four CFA).
Activin A and NGF ankle injection. Rats were anesthetized by ether, and both ankles were shaved. A dose of 1 μg of carrier-free human recombinant activin A/20 μl (R&D Systems, Minneapolis, MN) or 300 ng of NGF/20 μl (Sigma) in sterile saline (Sigma) was injected in the outer lateral side of the left ankle skin. Saline injection and 20 μg of BSA/20 μl of saline injection were used as negative control. In particular, 1 μg of BSA/20 μl of saline injection was used as the negative control for 1 μg of activin injection at 48 h.
Tactile allodynia. Responses to mechanical stimulation were assessed using calibrated von Frey filaments. A series of filaments with logarithmically incremental stiffness from 1.4-15 g was used to determine the force required to elicit 50% withdrawal response using the up-down method of Dixon (Chaplan et al., 1994). Briefly, rats were placed singly in a wire cage and allowed to acclimate for 15 min. Filaments were applied perpendicularly onto the lateral skin surface of either the left or right ankles with a pressure that caused the filament to buckle. Testing was initiated with the filament that possessed a buckling weight of 2 g followed by consecutive stimuli (ascending or descending). Licking or withdrawal of the paw was recorded as positive responses, and the next lightest filament was chosen for the next measurement. Absence of response after 5 s prompted the use of the next heavier filament. The test was complete when four measurements were made after the initial change in behavior or after five consecutive negative or four positive responses had occurred. The resulting sequence of positive and negative responses was used to interpolate the 50% response thresholds as described by Chaplan et al. (1994). The effects of activin A, NGF, or saline injections on tactile allodynia were assessed by an investigator blind to the treatment at 1, 6, 12, 24, and 48 h after injection. The effect of 20 μg of BSA/20 μl of saline injection was assessed at 1 and 6 h. The effect of 1 μg of BSA/20 μl of saline injection was assessed at 48 h. Control animals routinely displayed paw withdrawal thresholds between 13 and 15 g. Any animals with allodynic responses (≤8 g) in the uninjected right ankle were not used for additional analysis.
Tissue collection. Rats were anesthetized by ether, and the left ankle skin was rapidly collected and fresh frozen at -80°C. Rats were then perfused intracardially with 4% paraformaldehyde in 0.1 m phosphate buffer and the fourth and fifth lumbar DRGs (L4, L5 DRG) and L4 spinal cord segments (identified by the level of dorsal root entry) were collected, postfixed, and frozen, and sections were collected on a cryostat at 10 μm (DRG) or 20 μm (spinal cord) thickness. The sections were collected on gelatin-subbed slides and kept at -20°C until use.
Skin RNA isolation and cDNA synthesis. Fresh frozen skin biopsy material was placed in 1 ml of ice-cold Trizol reagent (Invitrogen, Carlsbad, CA), homogenized, and extracted in chloroform. RNA was precipitated in isopropanol at room temperature for 45 min. RNA was washed with 75% ice-cold ethanol twice and resuspended in 50 μl of DEPC water. RNA quantity was determined using 260 nm absorbance. Extracted RNA was treated with DNase to remove genomic DNA contamination using the DNA-free kit (Ambion, Austin, TX) according to the instructions of the manufacturer and confirmed by testing in real-time PCRs with all sets of the primers. No valuable Ct was detected. For the first-strand cDNA synthesis, 2 μg of RNA was then reverse-transcribed (RT) using the Superscript III RNaseH- reverse transcriptase and random primers (Invitrogen, Carlsbad, CA) in a 20 μl reaction volume according to the instructions of the manufacturer. Any contamination of the reagents was tested by omitting RNA in the RT reactions followed by real-time PCR with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers. No valuable Ct was detected.
Quantitative real-time PCR. Two-step SYBR green PCR was performed using an iCycler (Bio-Rad, Hercules, CA). One microliter of RT reaction from total 20 μl reaction was added to 20 μl of PCR mixture based on SYBR Green PCR buffer. The final concentration of each reagent was as follows: 3 mm MgCl2; 0.2 mm each of dATP, dCTP, and dGTP; 0.4 mm dUTP; 25 U/ml iTaq DNA polymerase; SYBR Green I; ROX reference dye; and 200 nm primers. The amplification conditions were as follows: step 1, 95°C and 3 min to activate the iTaq DNA polymerase; step 2, 95°C and 30 s for denaturation, 61°C and 30 s for annealing and extension; 40 cycles. After amplification, a melting curve protocol was run to detect primer dimers and to ensure that only one product was amplified. Product specificity of the PCR products was confirmed by 2% agarose gel electrophoresis. All samples were run in triplicates for each experiment, and each sample was amplified in two or three independent experiments. PCR without cDNA template was used as a negative control. Standard curves were generated for each gene, and all of the primers were found to have excellent PCR amplification efficiency (90-100%) as determined by the slope of the standard curves. Generally, the Ct of each gene from each sample was normalized against that of GAPDH, and fold changes of RNA levels were calculated by 2-ΔΔC t method (Livak and Schmittgen, 2001), in which the relative changes of genes of interest in the experimental group was calculated as the ratio of normalized data over control group.
The following PCR primers were used: rat activin βA subunit, forward, 5′-tgtgaacagtgccaggaga-3′; reverse, 5′-agaaagacggaagtgacgga-3′ (Becker et al., 2003) with an expected 102 bp fragment; rat NGF, forward, 5′-catggtacaatctccttcaac-3′; reverse, 5′-ccaacccacacactgacactg-3′ [modified from the study by Koh et al. (2004)] with an expected 110 bp fragment; rat GDNF, forward, 5′-acgaaaccaaggaggaactga-3′; reverse, 5′-tttgtcgtacattgtctcggc-3′ (Yamagata et al., 2002) with an expected 74 bp fragment; rat GAPDH, forward, 5′-tcaaggctgagaatgggaag-3′; reverse, 5′-tactcagcaccagcatcacc-3′ (Becker et al., 2003) with an expected 103 bp fragment. GAPDH was used as the internal control.
Immunohistochemistry. Sections were washed in PBS and blocked for 1 h at 25°C in 0.4% Triton X-100 and 3% BSA in PBS (8% BSA for trkA staining). Sections were then incubated in primary antibody diluted in 0.4% Triton X-100, 3% BSA, and 10% donkey serum (Jackson ImmunoResearch, West Grove, PA) in PBS overnight at 4°C. For CGRP/IB4 and CGRP/RT97 double staining, the following antibodies were used: rabbit anti-CGRP (1:4000; Sigma), IB4-Alexa 486 (1:100; Invitrogen), and mono-RT97 (1:20; Chemicon). After rinses with PBS, sections were incubated in secondary antibody in 0.4% Triton X-100, 3% BSA, and 10% donkey serum in PBS for 1.5 h. Donkey anti-rabbit cyanine 3 (Cy3) was used for rabbit anti-CGRP, and donkey anti-mouse Cy2 was used for mono-RT97 (1:100; Jackson ImmunoResearch). For trkA and CGRP double staining, additional antibodies were used: guinea pig anti-CGRP (1:200; Peninsula, San Carlos, CA), followed by donkey anti-guinea pig Cy3 (1:100; Jackson ImmunoResearch). Rabbit anti-trkA (1:200; Upstate Biotechnology, Lake Placid, NY) was amplified with goat anti-rabbit biotin (1:300; Chemicon) and streptavidin Cy2 (1:750; Jackson ImmunoResearch). For PKCγ and CGRP double labeling, rabbit anti-CGRP (1:4000; Sigma) followed by donkey anti-rabbit Cy3 and mono-PKCγ (1:50; BD Transduction Laboratories, San Jose, CA) was amplified with donkey anti-mouse biotin (1:300) and streptavidin Cy2 (1:750; Jackson ImmunoResearch). Sections were mounted in ProLong Antifade kit according to the protocol of the manufacturer (Invitrogen).
Neuronal profile counting and area measurement. All of the slides were blinded as to animal treatment by another investigator in the laboratory. Neuronal images were collected using a Leica (Nussloch, Germany) DMR microscope and a Hamamatsu (C4742-95-12G04; Hamamatsu, Shizuoka, Japan) digital CCD camera. Leica filter cube A was used to collect tracer-labeled neuronal profile, filter cube +L5 was used to collect Cy2 fluorescence labeled neuronal profile, and filter cube N2.1 was used to collect Cy3 fluorescence labeled neuronal profile. Only neurons that contained a visible nucleus were counted, and at least 400 tracer-labeled L4 neurons from each rat were counted in the double-labeling experiments, and at least 400 tracer-labeled L4 and L5 neurons from each rat were counted in 48 h activin or BSA-injected rats. For neuron-size measurement, each neuron with a visible nucleus was drawn with a computer mouse, and the soma area was calculated by OpenLab 3.1.5 (Improvision, Lexington, MA). Small neurons were <400 μm2, medium neurons were 400-800 μm2, and large neurons were >800 μm2 (Fang et al., 2002). For spinal cord measurement, the spinal cord sections (38 sections from three control rats and 60 sections from five CFA rats) were double stained with PKCγ (green) and CGRP (red). Both PKCγ and CGRP images of the same section were taken unsaturated in either green or red channel. The PKCγ-IR area in the spinal cord was detected depending on PKCγ staining (green) on each image. Both PKCγ-IR area and total fluorescence were collected under green channel. The CGRP fluorescence (red) in the PKCγ area was collected under red channel with OpenLab3.1.5.
Statistical analyses used the unpaired t test for two-groups comparison and one-way ANOVA followed by Bonferroni/Dunn's test for multiple group comparison. The Kolmogorov-Smirnov test was used to determine whether neuronal size-frequency distributions differed between populations. ***p < 0.0001; **p < 0.001; *p < 0.05 (Statview 4.1 software; Abacus Concepts, Berkeley, CA). Data are present as mean ± SEM.
Results
CFA injection causes ankle inflammation, tactile allodynia, and activin A increase
Retrograde tracer label was injected intradermally around the ankle to identify cutaneous neurons innervating this area (Bennett et al., 1996). The neurons were labeled 4 d before subsequent experimentation to allow full retrograde accumulation of the dye in DRG neurons. Injections of CFA in the ankle (Grubb et al., 1993; Hanesch and Schaible, 1995) resulted in dramatic increases in ankle diameter as well as increased pain perception. The circumference of the CFA-injected ankles increased significantly as expected (Fig. 1A) (p < 0.0001). Dye injection alone did not increase ankle inflammation, and the contralateral right ankles did not show swelling. To test whether inflammation caused physiological changes, measurements with von Frey filaments were performed on the ankles (Grubb et al., 1993) and, as expected, only CFA-injected ankles were accompanied by pronounced tactile allodynia (Fig. 1B) (p < 0.0001). These data indicate that localized CFA injection caused ipsilateral swelling and tactile allodynia but did not induce a systemic inflammation response that affected the uninjected ankle.
Figure 1.

CFA-induced inflammation. A, Circumference of ipsilateral (light bar) and contralateral (dark bar) ankles 2 d after subcutaneous injection of tracer or CFA, compared with uninjected (No Inj) ankles. Only ankles injected with CFA swelled (***p < 0.0001; n = 14 for each group). B, Withdrawal threshold 2 d after CFA injection. CFA injection reduced leg withdrawal threshold (mean ± SEM; n = 10 for each group; ***p < 0.0001). C, mRNA changes in skin with inflammation. Quantitative real-time PCR reveals mean ± SEM of fold changes in activin, NGF, and GDNF in inflamed skin relative to control (Con) ankle skin. GAPDH was used as the housekeeping gene (n = 14 for control and CFA groups).
Molecular changes in the skin were observed after inflammation. To begin to understand which changes in the skin may affect CGRP expression by sensory afferents after inflammation, the mRNA levels of several growth factors implicated in neuropeptide regulation, including activin A, NGF, and GDNF, were assayed by quantitative real-time PCR in both the inflamed ankle skin and control animals at 2 d. Both NGF and activin A mRNA levels have been shown previously to increase in the skin after excisional wound or inflammation (Hübner et al., 1996; Manni et al., 2002), and these and other skin ligands including GDNF can alter neuropeptide levels (Lindsay and Harmar, 1989; Ramer et al., 2003). Activin A mRNA levels increased more than twofold in inflamed ankle skin compared with control ankle skin, GDNF mRNA levels decreased, and no obvious change in the level of NGF mRNA was found (Fig. 1C).
Ankle inflammation increases the proportion of CGRP-IR cutaneous neurons in innervating DRG
Cutaneous sensory neurons in the L4 dermatome in the ankle area were labeled by injection of retrograde tracer that showed bright yellow fluorescence in the innervating DRG neurons (Fig. 2A). Half of the animals received a CFA injection in the left ankle, and all ipsilateral L4 DRGs were harvested at 2 d, sectioned, and blinded for immunohistochemical analysis. In control animals, 47.8 ± 1.3% of the tracer-labeled neurons were CGRP-IR, whereas in CFA-injected animals, 59.5 ± 1.4% of the tracer-labeled neurons were CGRP-IR (Fig. 2B)(p < 0.0001; 5110 and 4502 tracer-labeled neurons were counted in 10 control and 10 CFA animals). Because the tracer was injected 4 d before the experiment, the same cutaneous neuronal populations were pre-labeled in each animal, and the increase was unlikely to be a result of altered retrograde transport of dye. Thus, among tracer-labeled neurons, there was a 12% increase in the neurons with detectable CGRP after inflammation.
Figure 2.

Tracer-labeled CGRP-IR neurons in the L4 DRG. A, Labeled sensory neurons in DRG after injection of tracer in ankle skin. B, CGRP-IR neurons in the same DRG section. Some tracer-labeled neurons were CGRP-IR (arrow), whereas others were not (arrowhead).
To test which subpopulation of adult DRG neurons contributed to the increased CGRP expression after inflammation, neuronal size and antigenic expression were assayed. Generally, neurons delivering nociceptive information have small to medium soma size and express either CGRP immunoreactivity or IB4 binding (Fig. 3A-D), whereas neurons delivering proprioceptive information have larger soma size and can be identified by bright RT97 antibody reactivity (Fig. 3E,F). Overall, the mean size of tracer-labeled sensory neurons increased after inflammation, from 741.9 ± 8.3 to 777.6 ± 8.6 μm2 (Fig. 4A) (p < 0.01; 2871 and 2565 L4 DRG neurons were measured in nine control and nine CFA rats, respectively). When CGRP-IR, tracer-labeled neurons were measured, the majority of these neurons were small with some medium and large cells, and the mean size increased after inflammation, from 648.1 ± 8.7 to 710.6 ± 8.8 μm2 (Fig. 4B)(p < 0.0001; 2068 and 2102 neurons were measured in nine control and nine CFA rats, respectively). In contrast, RT97-IR neurons showed a broad size spectrum, including small, medium, and large cells, and the mean size of RT97-IR tracer-labeled sensory neurons did not change after inflammation, remaining at ∼1090 μm2 (Fig. 4C)(p > 0.9999; 876 and 764 neurons were measured in five control and five CFA rats, respectively). Thus, 2 d after inflammation, the CGRP-IR, tracer-labeled neurons had a larger soma size, whereas RT97-IR neurons remained unchanged.
Figure 3.

CGRP expression in different subpopulations of L4 DRG neurons. A, C, E, L4 sensory neurons that innervated the ankle skin contained tracer. B, IB4 binding (green) and CGRP immunoreactivity (red) were sometimes coexpressed in small-sized neurons. D, TrkA (green) and CGRP (red) immunoreactivities were often coexpressed in neurons. F, RT97 (green) and CGRP (red) immunoreactivities were sometimes coexpressed, especially in medium- to largesized neurons. Where red and green labels were coexpressed, the cells appear yellow in the merged image. Arrows represent double-labeled neurons, and arrowheads represent neurons with a single label but not CGRP. Scale bar, 100 μm.
Figure 4.
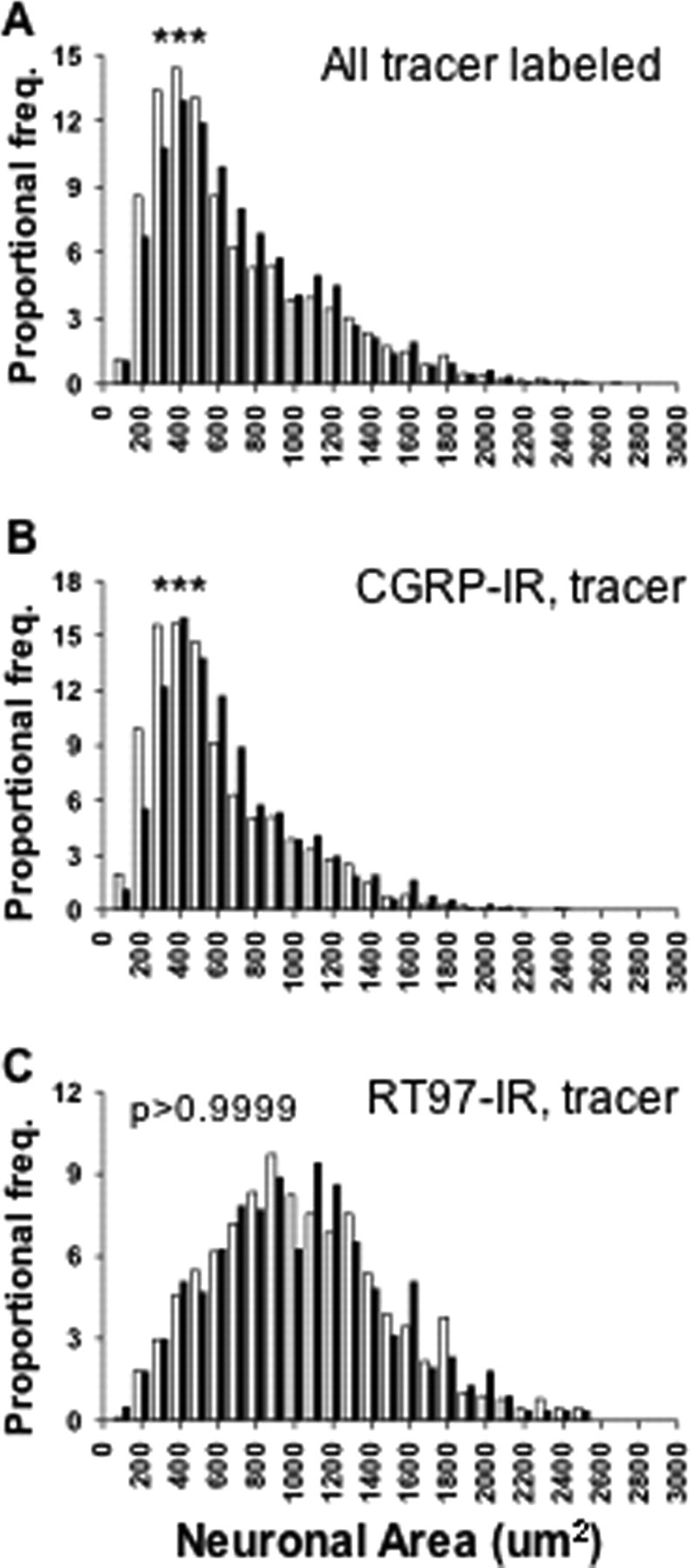
The effect of inflammation on the size distribution of sensory neurons innervating ankle skin. A, CFA increased the size of the total labeled neurons (2871 and 2565 L4 DRG neurons were measured in 9 control and 9 CFA rats, respectively). B, CFA increased the size of CGRP-IR tracer-labeled neurons (2068 and 2102 neurons were measured in 9 control and 9 CFA rats, respectively). C, The size of RT97-IR neurons was not changed after CFA (876 and 764 neurons were measured in 5 control and 5 CFA rats, respectively). All of the population comparisons were done using a Kolmogorov-Smirnov test. ***p < 0.0001; **p < 0.001. Open bar, Control L4 DRG neurons; filled bar, CFA L4 DRG neurons.
To test which population of DRG neurons accounted for the increased CGRP-IR neurons observed with inflammation, double-label immunohistochemistry of the tracer-labeled neurons was performed. Both in the control and after inflammation, ∼34% of tracer-labeled neurons in the L4 DRG showed IB4 binding, which suggests that IB4 binding was not affected by inflammation, and this marker was used as a stable marker. CGRP-IR and IB4 populations in the DRG were generally two distinct populations with only a minor overlap (Fig. 3A,B). In the control L4 DRG, 14.9 ± 0.7% tracer-labeled neurons expressed both IB4 and CGRP, whereas in the CFA L4 DRG, 19.1 ± 1.1% tracer-labeled neurons expressed both IB4 and CGRP (p < 0.05; 2356 and 1952 tracer-labeled neurons from five control and five CFA rats were counted, respectively). These data suggest that more IB4 neurons have detectable CGRP expression after inflammation. Indeed, the increase in CGRP-IR/IB4 double-labeled neurons accounts for one-third of the increase in CGRP-IR neurons seen after inflammation.
In our experiments, trkA-IR and CGRP-IR also showed considerable coexpression (Fig. 3C,D), and trkA-IR neurons represented a stable 59.0% of tracer-labeled L4 DRG neurons both in control and after inflammation. TrkA is the high-affinity neurotrophin receptor for NGF, and previous studies illustrated that approximately one-half of the L3 cutaneous DRG neurons were trkA-IR (McMahon et al., 1994), and 92% of trkA-IR neurons express CGRP (Averill et al., 1995). When the proportion of CGRP-IR, trkA-IR tracer-labeled neurons was quantified, this group increased from 41.7 ± 3.4 to 48.6 ± 4.9% after inflammation (p < 0.05; 2829 and 2522 tracer-labeled neurons from five control and five CFA rats were counted, respectively). Thus, these data suggest that ∼7% more CGRP/trkA neurons are detected after inflammation, and more trkA neurons have detectable CGRP expression after inflammation. In contrast, RT97-IR neurons and CGRP-IR neurons are two distinct populations with small overlap (Fig. 3E,F) and ∼46% of tracer-labeled neurons both in the control and CFA L4 DRG showed RT97-IR. Although these markers are occasionally present in the same sensory neuron, there was no significant difference found in the CGRP/RT97 doubly IR tracer-labeled population after inflammation (1914 and 1596 tracer-labeled neurons from five control and five CFA rats were counted, respectively). These data suggest that the increase in CGRP-IR neurons after inflammation did not arise from an RT97-IR pool. In summary, the increase in CGRP-IR neurons observed after inflammation is accounted for by summing the changes in CGRP/IB4 and CGRP/trkA neurons.
Central projection of sensory afferents may change after inflammation
To test whether the increase in CGRP-IR neurons after inflammation was associated with changes in their central projections in the superficial laminas of the dorsal horn, CGRP immunoreactivity in the spinal cord was assayed. Nociceptive afferent fibers terminate predominantly in lamina I and II of the dorsal horn of the spinal cord (Scott, 1992). In the control spinal cord section, CGRP staining occupied laminas I-IIo (outer) as a curved zone (Gibson et al., 1984) (Fig. 5A,B). IB4 staining was seen throughout laminas I and II but primarily in lamina IIi (inner) (Bennett et al., 1998), and some overlap between CGRP and IB4 staining could be found (Fig. 5B). Because the total proportion of IB4-labeled neurons did not change after inflammation in the DRG, it is reasonable to believe their central projections would also remain stable. Compared with the control spinal cord, the CGRP-IR zone in the CFA spinal cord became broader, and more CGRP-IR fibers projected deeply into the IB4-binding zone (Fig. 5C). The strong CGRP fluorescence in the IB4-binding zone and their broad overlap make fluorescence measurements very difficult to accomplish. For this reason, we used another more restricted lamina IIi marker, PKCγ. PKCγ staining occupied the inner layer of lamina II (Mori et al., 1990; Malmberg et al., 1997), and only a minor overlap between CGRP and PKCγ staining could be found in control spinal cord (Fig. 5D-F). To measure CGRP-IR fluorescence, sections of L4 spinal cord were doubly labeled with PKCγ and CGRP. In each case, the slides were processed together and were blinded as to treatment. The PKCγ area is ∼5.0 × 104 μm2, and total fluorescence of PKCγ is ∼2.4 × 107 U in both control and experimental CFA spinal cord (38 sections from three control rats and 60 sections from five CFA rats were included). We then identified the area of PKCγ reactivity and quantified the second marker, CGRP, in the same domain on the same sections. Compared with control spinal cord, there was a significant increase in CGRP immunofluorescence in the inner lamina II PKCγ domain from 1.46 ± 0.04 × 107 to 1.57 ± 0.03 × 107 after inflammation (p < 0.05).
Figure 5.

Central projections of CGRP-IR fibers after localized inflammation. A, Schematic figure of lumbar spinal cord dorsal horn lamina. B, L4 spinal cord section from the control rats was stained to show IB4 (green) and CGRP (red). The zone of CGRP labeling is in lamina I and the outer layer of lamina II. The zone of IB4 staining is in lamina II. C, L4 spinal cord section from CFA rats was stained to show IB4 and CGRP. The zone of CGRP labeling in the dorsal horn is broader and projects deeply into the IB4 zone. D, L4 spinal cord section reacted with PKCγ antibody. PKCγ staining was in lamina IIi. E, The same section reacted with CGRP antibodies. CGRP immunoreactivity is usually found in lamina I and the outer layer of lamina II. The white dotted line in D and E indicates the PKCγ-IR area and is used for CGRP fluorescence measurement in each section. The overlapping of D and E is shown in F. Scale bar, 100 μm.
Activin injection results in tactile allodynia and increases the proportion of CGRP-IR cutaneous neurons in innervating DRG
Systemic application of NGF (Lewin et al., 1993) as well as intraplantar NGF injections (Woolf et al., 1994) have been shown to produce thermal and mechanical hyperalgesia. Following a similar approach, rats were injected with 300 ng of NGF or 200 ng of activin A in the ankle and assessed for tactile allodynia responses. In each case, some animals did not respond behaviorally to injection (“failures”) (Fig. 6A). At 300 ng of NGF, ∼60% of the rats developed allodynia after 1 h with a mean response 4.4 ± 3.0 g, and this response was still present at 24 h with a mean response of 7.9 g. At low doses (200 ng) of activin A, 60% of the animals responded with mean response of 9.0 g at 6 h, and 80% of rats responded with a mean response of 10 g at 24 h (n = 4; data not shown). When the dose of activin A was increased to 1 μg, animal responses were more reliable, such that 43% of rats responded at 1 h with a 2.7 ± 1.3 g mean response. All rats responded 12 h after 1 μg of activin A injection with a 5.2 ± 1.6 g mean response. This allodynic response following activin A injection lasted at least 48 h (Fig. 6B). In contrast, control injections of 20 μg or 1 μg of BSA and saline did not induce any response (Fig. 6A). These data indicate that activin A injection alone is sufficient to cause tactile allodynia.
Figure 6.

Activin A or NGF injection induces tactile allodynia. Rats received a 20 μl injection of activin A, NGF, saline, or BSA solution under the skin of the left ankle. A, Leg withdrawal was assessed with von Frey filaments at 1-48 h after injection, and the number of animals with a response was scored in the table. B, The withdrawal threshold among responding animals was averaged and plotted in the bottom graph. One microgram of activin A induced tactile allodynia at 1 h that was still present at 48 h (*), and 300 ng of NGF induced allodynia and lasted at least 12 h after injection (#). Data from eight activin A, six NGF, six saline, or six BSA animals were analyzed by ANOVA followed by Fisher's PLSD. *,#p < 0.05 denotes the significance level.
To test whether injected activin can induce CGRP expression, cutaneous sensory neurons innervating the outer lateral left ankle were labeled by injection of retrograde tracer. Activin A or BSA injection was performed, and behavioral and immunohistochemical analyses were performed at 48 h. In BSA-injected animals, 57.8 ± 1.8% of the tracer-labeled neurons were CGRP-IR, whereas in activin-injected animals, 65.9 ± 1.4% of the tracer-labeled neurons were CGRP-IR (*p < 0.05; 3255 and 7259 tracer-labeled neurons in L4 and L5 DRG were counted in five BSA and eight activin A-injected animals, respectively, and one BSA-injected rat was not included because of failure to show any tracer labeling).
Discussion
Sensory neuropeptides such as CGRP are markers of nociceptive neurons and essential mediators of specific types of pain; therefore, the regulation of neuropeptides after injury is important for our understanding of pain. CGRP expression is regulated after injury or inflammation, but the factors that increase CGRP expression are not clear. This study highlights activin A as a candidate to mediate changes in CGRP expression and pain responses after injury.
Inflammation increases sensory neuropeptides and pain
Localized inflammation increases CGRP expression in innervating DRG neurons (Smith et al., 1992; Nahin and Byers, 1994; Hanesch and Schaible, 1995; Neumann et al., 1996; Bulling et al., 2001). In this study, a retrogradely transported tracer was used to label cutaneous neurons in the ankle. This approach allowed us to study the same population of DRG neurons with and without peripheral inflammation. The proportion of tracer labeled, CGRP-IR DRG neurons increased after inflammation, suggesting that additional neurons increased CGRP expression. We acknowledge several potential difficulties with this interpretation. These studies used antibody detection of CGRP, and neurons were subjectively scored as positive or negative in slides blinded as to experimental variable. Antibody specificity has been established (Hall et al., 1997). It remains possible that the present results reflect an increase in CGRP to detectable levels, rather than de novo induction in neurons. Despite this concern, the proportion of cutaneous CGRP-IR neurons in the present study is similar to that seen by others (Kuraishi et al., 1989; Donaldson et al., 1992; Bennett et al., 1996).
The inflammation model used causes primary and secondary hyperalgesia and tactile allodynia and involves CGRP. Peripheral nerves release transmitters and peptides, including CGRP (Lam and Ferrell, 1991; Mulder et al., 1997; Lawand et al., 2000). Unilateral inflammation causes alterations in nociceptive thresholds (Stein et al., 1988), and notably, mice deficient in the α-CGRP gene fail to develop secondary hyperalgesia after inflammation (Zhang et al., 2001). Given the central role of CGRP in inflammatory pain, this study focused on molecules known to modulate CGRP expression, namely NGF and activin A.
NGF administration, systemically or by injection into the skin, results in pain. Increased NGF contributes to hyperalgesia seen after inflammation (Lembeck et al., 1981; Taiwo et al., 1991; Donaldson et al., 1992; Davis et al., 1993; Lewin et al., 1993; Lewin and Mendell, 1994). For example, systemic NGF administration increases neuropeptides in sensory ganglia (Otten and Lorez, 1982) as well as mechanical and thermal hyperalgesia (Lewin et al., 1993). Subcutaneous NGF injection of 20-2000 ng into the footpad produced thermal and mechanical hyperalgesia at 6 h and increases in substance P/CGRP levels in the nerve at 24 and 48 h after injection (Woolf et al., 1994). Systemic anti-NGF antibody reduced hypersensitivity and CGRP increases induced by experimental inflammation but did not totally eliminate the increase (Woolf et al., 1994). Anti-NGF treatment did not reduce edema and reddening early in inflammation (Lewin et al., 1993). Thus, the behavioral and peptide expression changes depend, to some extent, on NGF, but NGF alone cannot explain all of the changes.
Increased CGRP-IR neurons and deeper central projections
Size was used as the first criteria to distinguish among different sensory neurons after inflammation. Indeed, the size of tracer-labeled neurons increased after inflammation, but these changes occurred only among the RT97-negative neurons. Two possibilities may underlie this observation: RT97-negative, tracer-labeled neurons may undergo hypertrophy resulting from growth factor availability after inflammation (Albers et al., 1994; Bradbury et al., 1998), or newly CGRP-IR neurons may derive from larger neurons. Markers of DRG subtypes were then used to learn which neurons gave rise to the increased CGRP-IR neurons after inflammation. Three populations were tested, IB4+ trkA-neurons that represent primarily light touch mechanoreceptors, trkA-IR neurons that are mostly cutaneous sensory neurons, and RT97-IR neurons that are primarily proprioceptors. One-third of the CGRP-IR neurons newly detectable after inflammation also bound IB4, suggesting that they were light-touch mechanoreceptors. The behavioral assays also support the notion that light touch applied induced pain after inflammation. Other newly CGRP-IR neurons were derived from trkA-IR neurons. In contrast, there was no increase in CGRP+RT97+ tracer-labeled neurons after inflammation, suggesting that proprioceptive and other large neurons were not responsible for the increase. A previous report (Neumann et al., 1996) used cholera toxin B to label neurons and suggested that some Aβ fibers contribute to inflammatory hypersensitivity by switching their phenotype to deliver pain sensation. However, following axotomy, cholera toxin B labeling may not identify the same group of nerves as in the control (Tong et al., 1999; Ma and Tian, 2001). Although some mechanoreceptors increase CGRP expression, it is not clear whether this phenotypic development is permanent or reversible after the inflammatory response has abated.
CGRP immunoreactivity increased in deeper spinal cord laminas after inflammation, suggesting that neurons in addition to primary nociceptors had become CGRP-IR. The expression of PKCγ is quite restricted (Mori et al., 1990; Malmberg et al., 1997) and serves as a marker for lamina IIi. Neurons in lamina IIi respond preferentially to non-noxious inputs (Light et al., 1979; Woolf and Fitzgerald, 1983; Malmberg et al., 1997) that may be important for neuropathic pain. It is not clear from this assay, however, whether new axons express detectable CGRP or whether the increased fluorescence reflects new branches deeper in the spinal cord.
Activin expression after inflammation and contribution to pain
Increased activin has been detected following wounds and inflammation (Hübner et al., 1997; Yu et al., 1998; Cruise et al., 2004). After wounding, activin increases in skin have been proposed to affect gene expression in neurons, possibly by retrograde signaling (Cruise et al., 2004). NGF and GDNF proteins also increase in the periphery after inflammation (Amaya et al., 2004), leading to the suggestion that NGF-dependent neurons convey inflammatory pain and GDNF-dependent neurons convey neuropathic pain (Snider and McMahon, 1998) (but see Nagano et al., 2003). Our data showed that in contrast to protein levels, only activin A mRNA was increased in skin at 2 d, suggesting that activin A might play a prolonged role in inflammation.
Pain following inflammation is thought to result from agents released from injured cells, immune cells, and sensory terminals including K+ and H+ ions, histamine, bradykinin, and NGF as well as cytokines (Richardson and Vasko, 2002). Immune cells may be one source of factors like NGF and activin A after inflammation. NGF is released by inflammatory cells in response to cytokines, including tumor necrosis factor-α (Safieh-Garabedian et al., 1995). NGF may produce hyperalgesia indirectly through mast cell degranulation (Mazurek et al., 1986). Activin A is released early during acute systemic inflammation, suggesting that proinflammatory cytokines modulate activin A (Erämaa et al., 1992; Shao et al., 1992; Yu and Dolter, 1997; Jones et al., 2004). However, CGRP can initiate the release of proinflammatory cytokines from mast cells (Preibisz, 1993). It remains critically important to clarify the cellular source of activin A and its target in inflammation so that the cascade of events leading to pain is understood.
Activin A induces tactile allodynia and increases CGRP-IR neurons
In this report, activin A or NGF was injected into ankle skin, and tactile allodynia was detected over 48 h. The timing of CGRP and pain responses following inflammation or activin injection suggests particular cellular mechanisms. Allodynia observed 24-48 h after injections is consistent with new CGRP synthesis by neurons [see this role for NGF in the study by Lewin and Mendell (1994)]. Our data demonstrating an early onset of allodynia also suggest a peripheral mechanism. Activin may directly alter the sensitivity of sensory neurons. NGF increases evoked release of neuropeptides (Richardson and Vasko, 2002), and bradykinin sensitizes sensory neurons through a PKC pathway (Vellani et al., 2001); but how activin A might sensitize neurons remains to be elucidated. Tactile allodynia is one measure of altered pain responses, in which benign stimulus produces pain behavior; in contrast, primary hyperalgesia is an augmented response to a painful stimulus and can involve both sensitization and central mechanisms. It is not yet clear whether activin will produce hyperalgesia or whether additional pain pathways involve this cytokine.
These data demonstrate that activin is sufficient to induce behavioral changes and increase CGRP-IR neurons. Thus, activin administration initiated a pathway that eventually regulated CGRP expression in neuronal cell bodies. Although activin is sufficient to initiate CGRP expression, the increased CGRP-IR neurons with activin injection were fewer than those observed with inflammation. It is not clear whether sufficient activin was injected, or whether additional molecules present with inflammation also raise CGRP levels. It will be interesting to learn whether activin inhibition during inflammation can abrogate the increase in CGRP-IR neurons, but because inflammation is complex, inhibition of one factor is likely to give only a partial result that may be difficult to resolve.
In summary, these data suggest that inflammation involves activin A upregulation in the skin resulting in increased sensory neuron expression of neuropeptides essential for pain. Additionally, activin A itself confers behavioral correlates of pain in addition to CGRP increases. Future studies using antibody perturbation or inhibitors to block activin A (Sulyok et al., 2004) during inflammation may clarify the role of activin A in pain responses.
Footnotes
This work was supported by National Institutes of Health Grant NS39316 (A.K.H.). We thank Dr. William D. Snider for his suggestion on PKCγ.
Correspondence should be addressed to Dr. Alison K. Hall, Department of Neuroscience, Case Western Reserve University School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106. E-mail: axh8@case.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259227-09$15.00/0
References
- Ai X, Cappuzzello J, Hall AK (1999) Activin and bone morphogenetic proteins induce calcitonin gene-related peptide in embryonic sensory neurons in vitro. Mol Cell Neurosci 14: 506-518. [DOI] [PubMed] [Google Scholar]
- Albers KM, Wright DE, Davis BM (1994) Overexpression of nerve growth factor in epidermis of transgenic mice causes hypertrophy of the peripheral nervous system. J Neurosci 14: 1422-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaya F, Shimosato G, Nagano M, Ueda M, Hashimoto S, Tanaka Y, Suzuki H, Tanaka M (2004) NGF and GDNF differentially regulate TRPV1 expression that contributes to development of inflammatory thermal hyperalgesia. Eur J Neurosci 20: 2303-2310. [DOI] [PubMed] [Google Scholar]
- Averill S, McMahon SB, Clary DO, Reichardt LF, Priestley JV (1995) Immunocytochemical localization of trkA receptors in chemically identified subgroups of adult rat sensory neurons. Eur J Neurosci 7: 1484-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker JC, Hertel M, Markmann A, Shahin M, Werner S, Domschke W, Pohle T (2003) Dynamics and localization of activin A expression in rat gastric ulcers. Scand J Gastroenterol 38: 260-267. [DOI] [PubMed] [Google Scholar]
- Bennett DL, Dmietrieva N, Priestley JV, Clary D, McMahon SB (1996) trkA, CGRP and IB4 expression in retrogradely labelled cutaneous and visceral primary sensory neurones in the rat. Neurosci Lett 206: 33-36. [DOI] [PubMed] [Google Scholar]
- Bennett DLH, Michael GJ, Ramachandran N, Munson JB, Averill S, Yan Q, McMahon SB, Priestley JV (1998) A distinct subgroup of small DRG cells express GDNF receptor components and GDNF is protective for these neurons after nerve injury. J Neurosci 18: 3059-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury E, Burnstock G, McMahon S (1998) The expression of P2X3 purinoreceptors in sensory neurons: effects of axotomy and glial-derived neurotrophic factor. Mol Cell Neurosci 12: 256-268. [DOI] [PubMed] [Google Scholar]
- Bulling D, Kelly D, Bond S, McQueen D, Seckl J (2001) Adjuvant-induced joint inflammation causes very rapid transcription of beta-preprotachykinin and alpha-CGRP genes in innervating sensory ganglia. J Neurochem 77: 372-382. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53: 55-63. [DOI] [PubMed] [Google Scholar]
- Cruise BA, Xu P, Hall AK (2004) Wounds increase activin in skin and a vasoactive neuropeptide in sensory ganglia. Dev Biol 271: 1-10. [DOI] [PubMed] [Google Scholar]
- Davis B, Lewin GR, Mendell LM, Jones M, Albers KM (1993) Altered expression of nerve growth factor in the skin of transgenic mice leads to changes in response to mechanical stimuli. Neuroscience 56: 789-792. [DOI] [PubMed] [Google Scholar]
- Donaldson LF, Harmar AJ, McQueen DS, Seckl JR (1992) Increased expression of preprotachykinin, calcitonin gene-related peptide, but not vasoactive intestinal polypeptide messenger RNA in dorsal root ganglia during the development of adjuvant monoarthritis in the rat. Brain Res Mol Brain Res 16: 143-149. [DOI] [PubMed] [Google Scholar]
- Erämaa M, Hurme M, Stenman U-H, Ritvos O (1992) Activin A/erythroid differentiation factor is induced during human monocyte activation. J Exp Med 176: 1449-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Djouhri L, Black JA, Dib-Hajj SD, Waxman SG, Lawson SN (2002) The presence and role of the tetrodotoxin-resistant sodium channel Nav1.9 (NaN) in nociceptive primary afferent neurons. J Neurosci 22: 7425-7433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson SJ, Polak JM, Bloom SR, Sabate IM, Mulderry PM, Gahtei MA, McGregor GP, Morrison JFB, Kelly JS, Evans RM, Rosenfeld MG (1984) Calcitonin gene-related peptide immunoreactivity in the spinal cord of man and of eight other species. J Neurosci 4: 3101-3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb BD, Stiller RU, Schaible HG (1993) Dynamic changes in the receptive field properties of spinal cord neurons with ankle input in rats with chronic unilateral inflammation in the ankle region. Exp Brain Res 92: 441-452. [DOI] [PubMed] [Google Scholar]
- Hall A, Ai X, Hickman G, MacPhedran S, Nduaguda C, Robertson C (1997) The generation of neuronal heterogeneity in a rat sensory ganglion. J Neurosci 17: 2775-2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AK, Dinsio KJ, Cappuzzello J (2001) Skin cell induction of calcitonin gene related peptide in embryonic sensory neurons in vitro involves activin. Dev Biol 229: 263-270. [DOI] [PubMed] [Google Scholar]
- Hanesch U, Schaible H-G (1995) Effects of ankle joint inflammation on the proportion of calcitonin gene-related peptide (CGRP)-immunopositive perikarya in dorsal root ganglia. Prog Brain Res 104: 339-347. [DOI] [PubMed] [Google Scholar]
- Holzer P (1998) Neurogenic vasodilation and plasma leakage in the skin. Gen Pharmacol 30: 5-11. [DOI] [PubMed] [Google Scholar]
- Hübner G, Hu Q, Smola H, Werner S (1996) Strong induction of activin expression after injury suggests an important role of activin in wound repair. Dev Biol 173: 490-498. [DOI] [PubMed] [Google Scholar]
- Hübner G, Brauchle M, Gregor M, Werner S (1997) Activin A: a novel player and inflammatory marker in inflammatory bowel disease. Lab Invest 77: 311-318. [PubMed] [Google Scholar]
- Jones KL, Brauman JN, Groome NP, De Kretser DM, Phillips DJ (2000) Activin A release into the circulation is an early event in systemic inflammation and precedes the release of pollistatin. Endocrinology 144: 1905-1908. [DOI] [PubMed] [Google Scholar]
- Jones KL, Kretser DM, Patella S, Phillips DJ (2004) Activin A and follistatin in systemic inflammation. Mol Cell Endocrinol 225: 119-125. [DOI] [PubMed] [Google Scholar]
- Koh D, Armugam A, Jeyaseelan K (2004) Sputa nerve growth factor forms a preferable substitute to mouse 7S-beta nerve growth factor. Biochem J 383: 149-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraishi Y, Nanayama T, Ohno H, Fujii N, Otaka A, Yajima H, Satoh M (1989) Calcitonin gene related peptide increases in the dorsal root ganglion of adjuvant arthritic rat. Peptides 10: 447-452. [DOI] [PubMed] [Google Scholar]
- Lam F, Ferrell WR (1991) Specific neurokinin receptors mediate plasma extravasation in the rat knee joint. Br J Pharmacol 103: 1263-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawand NB, McNearney T, Westlund KN (2000) Amino acid release into the knee joint: key role in nociception and inflammation. Pain 86: 69-74. [DOI] [PubMed] [Google Scholar]
- Lawson SN, Harper AA, Harper EI, Garson JA, Anderton BH (1984) A monoclonal antibody against neurofilament protein specifically labels a subpopulation of rat sensory neurones. J Comp Neurol 228: 263-272. [DOI] [PubMed] [Google Scholar]
- Lembeck F, Folkers K, Donnerer J (1981) Analgesic effect of antagonists of substance P. Biochem Biophys Res Commun 103: 1318-1321. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Mendell LM (1994) Regulation of cutaneous C-fiber heat nociceptors by nerve growth factor in the developing rat. J Neurophysiol 71: 941-949. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Ritter AM, Mendell LM (1993) Nerve growth factor-induced hyperalgesia in the neonatal and adult rat. J Neurosci 13: 2136-2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light A, Trevino D, Perl E (1979) Morphological features of functionally defined neurons in the marginal zone and substantia gelatinosa of the spinal dorsal horn. J Comp Neurol 186: 151-171. [DOI] [PubMed] [Google Scholar]
- Lindsay RM, Harmar AJ (1989) Nerve growth factor regulates expression of neuropeptide genes in adult sensory neurons. Nature 337: 362-364. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25: 402-408. [DOI] [PubMed] [Google Scholar]
- Ma QP, Tian L (2001) A-fibres sprouting from lamina I into lamina II of spinal dorsal horn after peripheral nerve injury in rats. Brain Res 904: 137-140. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Chen C, Tonegawa S, Basbaum AI (1997) Preserved acute pain and reduced neuropathic pain in mice lacking PKCγ. Science 278: 279-283. [DOI] [PubMed] [Google Scholar]
- Manni L, Lundeberg T, Tirassa PLA (2002) Role of cholecystokinin-8 in nerve growth factor and nerve growth factor mRNA expression in carrageenan-induced joint inflammation in adult rats. Rheumatology 41: 787-792. [DOI] [PubMed] [Google Scholar]
- Mazurek N, Weskamp G, Erne P, Otten U (1986) Nerve growth factor induces mast cell degranulation without changing intracellular calcium levels. FEBS Lett 198: 315-320. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Armanini MP, Ling LH, Phillips HS (1994) Expression and coexpression of trk receptors in subpopulations of adult primary sensory neurons projecting to identified peripheral targets. Neuron 12: 1161-1171. [DOI] [PubMed] [Google Scholar]
- Molliver DC, Wright DE, Leitner ML, Parsadanian AS, Doster K, Wen D, Yan Q, Snider WD (1997) IB4-binding DRG neurons switch from NGF to GDNF dependence in early postnatal life. Neuron 19: 849-861. [DOI] [PubMed] [Google Scholar]
- Mori M, Kose A, Tsujino T, Tanaka C (1990) Immunocytochemical localization of protein kinase C subspecies in the rat spinal cord: light and electron microscopic study. J Comp Neurol 299: 167-177. [DOI] [PubMed] [Google Scholar]
- Mu X, Silos-Santiago I, Carroll SL, Snider WD (1993) Neurotrophin receptor genes are expressed in distinct patterns in developing dorsal root ganglia. J Neurosci 13: 4029-4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder H, Zhang Y, Danielsen N, Sundler F (1997) Islet amyloid polypeptide and calcitonin gene-related peptide expression are upregulated in lumbar dorsal root ganglia after unilateral adjuvant-induced inflammation in the rat paw. Mol Brain Res 50: 127-135. [DOI] [PubMed] [Google Scholar]
- Munz B, Hübner G, Tretter Y, Alzheimer C, Werner S (1999) A novel role of activin in inflammation and repair. J Endocrinol 161: 187-193. [DOI] [PubMed] [Google Scholar]
- Nagano M, Sakai A, Takahashi N, Umino M, Yoshioka K, Suzuki H (2003) Decreased expression of glial cell line-derived neurotrophic factor signaling in rat models of neuropathic pain. Br J Pharmacol 140: 1252-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahin RL, Byers MR (1994) Adjuvant induced inflammation of rat paw is associated with altered calcitonin gene related peptide immunoreactivity within cell bodies and peripheral endings of primary afferent neurons. J Comp Neurol 349: 475-485. [DOI] [PubMed] [Google Scholar]
- Neumann S, Doubell TP, Leslie T, Woolf CJ (1996) Inflammatory pain hypersensitivity mediated by phenotypic switch in myelinated primary sensory neurons. Nature 384: 360-364. [DOI] [PubMed] [Google Scholar]
- Otten U, Lorez HP (1982) Nerve growth factor increases substance P, cholecystokinin and vasoactive intestinal polypeptide immunoreactivities in primary sensory neurones of newborn rats. Neurosci Lett 34: 153-158. [DOI] [PubMed] [Google Scholar]
- Phillips DJ, Jones KL, Scheerlinck JY, Hedger MP, de Kretser DM (2001) Evidence for activin A and follistatin involvement in the systemic inflammatory response. Mol Cell Endocrinol 180: 155-162. [DOI] [PubMed] [Google Scholar]
- Preibisz J (1993) Calcitonin gene-related peptide and regulation of human cardiovascular homeostasis. Am J Hypertens 6: 434-450. [DOI] [PubMed] [Google Scholar]
- Priestley JV, Michael GJ, Averill S, Liu M, Willmott N (2002) Regulation of nociceptive neurons by nerve growth factor and glial cell line derived neurotrophic factor. Can J Physiol Pharmacol 80: 495-505. [DOI] [PubMed] [Google Scholar]
- Ramer M, Bradbury EJ, Michael GJ, Lever IJ, McMahon SB (2003) Glial cell line-derived neurotrophic factor increases calcitonin gene-related peptide immunoreactivity in sensory and motoneurons in vivo. Eur J Neurosci 18: 2713-2721. [DOI] [PubMed] [Google Scholar]
- Richardson JD, Vasko MR (2002) Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther 302: 839-845. [DOI] [PubMed] [Google Scholar]
- Safieh-Garabedian B, Poole S, Allchorne A, Winter J, Woolf CJ (1995) Contribution of interluekin-1b to the inflammation-induced increase in nerve growth factor levels and inflammatory hyperalgesia. Br J Pharmacol 115: 1265-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott S (1992) Sensory neurons: diversity, development, and plasticity. New York: Oxford UP.
- Shao L, Frigon NJ, Sehy D, Yu A, Lofgren J, Schwall R, Yu J (1992) Regulation of production of activin A in human marrow stromal cells and monocytes. Exp Hematol 20: 1235-1242. [PubMed] [Google Scholar]
- Silverman J, Kruger L (1990) Selective neuronal glycoconjugate expression in sensory and autonomic ganglia: relation of lectin reactivity to peptide and enzyme markers. J Neurocytol 19: 789-801. [DOI] [PubMed] [Google Scholar]
- Smith GD, Harmar AJ, McQueen DS, Seckl JR (1992) Increase in substance p and CGRP but not somatostatin content of innervating dorsal root ganglia in adjuvant monoarthritis in the rat. Neurosci Lett 137: 257-260. [DOI] [PubMed] [Google Scholar]
- Snider WD, McMahon SB (1998) Tackling pain at the source: new ideas about nociceptors. Neuron 20: 629-632. [DOI] [PubMed] [Google Scholar]
- Stein C, Millan MJ, Herz A (1988) Unilateral inflammation of the hindpaw in rats as a model of prolonged noxious stimulation: alterations in behavior and nociceptive thresholds. Pharmacol Biochem Behav 31: 445-451. [DOI] [PubMed] [Google Scholar]
- Sulyok S, Wankell M, Alzheimer C, Werner S (2004) Activin: an important regulator of wound repair, fibrosis, and neuroprotection. Mol Cell Endocrinol 225: 127-132. [DOI] [PubMed] [Google Scholar]
- Taiwo Y, Levine J, Burch R, Woo J, Mobley W (1991) Hyperalgesia induced in the rat by the amino-terminal octapeptide of nerve growth factor. Proc Natl Acad Sci USA 88: 5144-5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong YG, Wang HF, Ju G, Grant G, Hokfelt T, Zhang X (1999) Increased uptake and transport of cholera toxin B-subunit in dorsal root ganglion neurons after peripheral axotomy: possible implications for sensory sprouting. J Comp Neurol 404: 143-158. [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis J, McNaughton P (2001) Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol (Lond) 534: 813-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verge VMK, Grondin J-P, Ernfors P, Persson H, Riopelle RJ, Hokfelt T, Richardson PM (1992) Colocalization of NGF binding sites, trk mRNA, and low-affinity NGF receptor mRNA in primary sensory neurons: responses to injury and infusion of NGF. J Neurosci 12: 4011-4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallengren J, Hakanson R (1987) Effects of substance P, neurokinin A and calcitonin gene related peptide in human skin and their involvement in sensory nerve mediated responses. Eur J Pharmacol 143: 267-273. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Fitzgerald M (1983) The properties of neurons recorded in the superficial dorsal horn of the rat spinal cord. J Comp Neurol 221: 313-328. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J (1994) Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience 62: 327-331. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Tagami M, Ikeda K, Tsumagari S, Yamori Y, Nara Y (2002) Differential regulation of glial cell line-derived neurotrophic factor (GDNF) mRNA expression during hypoxia and reoxygenation in astrocytes isolated from stroke-prone spontaneously hypertensive rats. Glia 37: 1-7. [DOI] [PubMed] [Google Scholar]
- Yu E, Dolter K, Shao L, Yu J (1998) Suppression of IL-6 biological activities by activin A and implications for inflammatory arthropathies. Clin Exp Immunol 112: 126-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Dolter K (1997) Production of activin A and its roles in inflammation and hematopoiesis. Cytokines Cell Mol Ther 3: 169-177. [PubMed] [Google Scholar]
- Zhang L, Hoff A, Wimalawansa SJ, Cote G, Gagel RF, Westlund KN (2001) Arthritic calcitonin/alpha calcitonin gene related peptide knockout mice have reduced nociceptive hypersensitivity. Pain 89: 265-273. [DOI] [PubMed] [Google Scholar]