

Abstract
Alzheimer's disease (AD) is the most common cause of dementia. Humanin (HN) is a short bioactive peptide abolishing neuronal cell death induced by various familial AD (FAD)-causative genes and amyloid-β (Aβ) in vitro. It has been shown that HN suppresses memory impairment of mice induced by intracerebroventricular administration of Aβ. To potentiate the neuroprotective effect of HN, we synthesized a hybrid peptide named Colivelin composed of activity-dependent neurotrophic factor (ADNF) C-terminally fused to AGA-(C8R)HNG17, a potent HN derivative. Colivelin completely suppresses death induced by overexpressed FAD-causative genes and Aβ1-43 at a concentration of 100 fm, whereas AGA-(C8R)HNG17 does so at a concentration of 10 pm. Colivelin-induced neuroprotection has been confirmed to occur via two neuroprotective pathways: one mediated by Ca2+/calmodulin-dependent protein kinase IV, triggered by ADNF, and one mediated by signal transducer and activator of transcription 3, triggered by HN. In vivo animal studies have further indicated that intracerebroventricular administration of Colivelin not only completely suppresses impairment in spatial working memory induced by repetitive intracerebroventricular injection of Aβ25-35 or Aβ1-42, but also it antagonizes neuronal loss in the CA1 region of hippocampus induced by hippocampal injection of Aβ1-42. In addition, intraperitoneally administered Colivelin suppresses memory impairment caused by a muscarinic acetylcholine receptor antagonist, 3-quinuclidinyl benzilate, indicating that a substantial portion of intraperitoneally administered Colivelin passes through the blood-brain barrier and suppresses functional memory deficit. Thus, Colivelin might serve as a novel drug candidate for treatment of AD.
Keywords: Alzheimer's disease, APP, Aβ, ADNF, Humanin, Colivelin
Introduction
Currently, there is no curative therapy for Alzheimer's disease (AD). The acetylcholinesterase inhibitors, such as Donepezil, antioxidants such as vitamin E, and inhibitors to NMDA excitotoxicity are clinically used in AD therapy, because they marginally delay disease progression of AD (Sano et al., 1997; Reisberg et al., 2003; Lane et al., 2004). Because progressive neuronal loss is implicated in the development of AD-associated dementia (Mattson, 2004), reagents that suppress neuronal death can be promising candidates for AD therapy. However, the mechanism underlying neuronal loss still remains to be elucidated.
It has been generally accepted that increased levels of toxic amyloid-βs (Aβs) are closely related to AD pathogenesis, including neuronal cell death (for review, see Hardy and Selkoe, 2002). In agreement, it has been reported that superphysiological concentrations of toxic Aβs cause neuronal cell death in vitro (for review, see Kawasumi et al., 2002; Niikura et al., 2002). Moreover, several groups have reported that familial AD (FAD)-linked mutants of amyloid precursor protein (APP), presenilin 1 (PS1), and PS2 (Shastry and Giblin, 1999) cause death in multiple neuronal cell lines and primary neurons (for review, see Kawasumi et al., 2002; Niikura et al., 2002). In addition, some antibodies recognizing the extracellular domain of APP induce death in primary neurons by activating intracellular death pathways (Rohn et al., 2000; Sudo et al., 2000, 2001; Hashimoto et al., 2003).
Using an expression gene cloning technique called “death-trap” screening (D'Adamio et al., 1997), we identified a novel gene encoding a 24 amino acid peptide named Humanin (HN) that suppressed neuronal death induced by any type of FAD genes thus far examined, toxic Aβ peptides, and anti-APP antibody (Hashimoto et al., 2001a, 2003; Niikura et al., 2004; Nishimoto et al., 2004). From structure/function analysis (Hashimoto et al., 2001a,b; Terashita et al., 2003), we have obtained several HN derivatives with more potent neuroprotective activities (see Table 1). Among them, AGA-(C8R)HNG17, one of the most potent derivatives of HN, completely suppresses neuronal cell death by AD-relevant insults at a concentration of 10 pm in vitro.
Table 1.
List of HN derivatives
|
HN derivatives |
Sequence |
Effect (Aβ1-43) |
|---|---|---|
| HN | MAPRGFSCLLLLTSEIDLPVKRRA | 10 μm |
| S14G-HN (HNG) | MAPRGFSCLLLLTGEIDLPVKRRA | 10 nm |
| C8A-HN (HNA) | MAPRGFSALLLLTSEIDLPVKRRA | NE |
| d-Ser14 HN | MAPRGFSCLLLLT(dS)EIDLPVKRRA | 10 nm |
| AGA-HNG | MAPAGASCLLLLTGEIDLPVKRRA | 100-300 pm |
| AGA-(d-Ser14)HN | MAPAGASCLLLLT(dS)EIDLPVKRRA | 100-300 pm |
| AGA-(d-Ser14)HN17 | PAGASCLLLLT(dS)EIDLP | 100 pm |
| AGA-(C8R)HNG17 | PAGASRLLLLTGEIDLP | 10 pm |
| EF-HN | EFLIVIKSMAPRGFSCLLLLTSEIDLPVKRRA | 100 nm |
| EF-HNA | EFLIVIKSMAPRGFSALLLLTSEIDLPVKRRA | NE |
| EF-HNG | EFLIVIKSMAPRGFSCLLLLTGEIDLPVKRRA | 1 nm |
| EF-AGA-HNG | EFLIVIKSMAPAGASCLLLLTGEIDLPVKRRA | 10 pm |
| Colivelin |
SALLRSIPA-PAGASRLLLLTGEIDLP |
100 fm
|
The abbreviated names, sequences, and the potencies of the antagonizing effect of Aβ1-43-mediated neurotoxicity are indicated for each peptide. Effect indicates minimal concentration of each peptide that exerted full neuroprotection. NE, No protective effect.
Furthermore, Mamiya and Ukai (2001) reported that intracerebroventricular injection of S14G-HN (HNG) prevented scopolamine-induced amnesia in mice. We also found that intracerebroventricular injection of HNG reverses memory impairment of mice induced by Aβ25-35 (Tajima et al., 2005). Krejcova et al. (2004) showed recently that an HN derivative may pass through the blood-brain barrier and suppress experimentally induced behavioral abnormalities. These findings indicate that discovery of additional HN derivatives with stronger anti-AD activity both in vitro and in vivo that can pass through the blood-brain barrier will encourage trials for clinical applications of HN.
Activity-dependent neurotrophic factor (ADNF), a nine amino acid peptide, was originally purified from conditioned medium of astrocytes (Brenneman and Gozes, 1996). ADNF protects neurons from death induced by Aβ at extremely low concentrations such as 100 fm. In contrast to HN, however, ADNF completely loses its protective effect at or above a concentration of 1 nm (Brenneman et al., 1998).
By taking advantage of different action properties of HN and ADNF, we have herein generated a novel hybrid peptide named Colivelin, which is composed of ADNF C-terminally fused to AGA-(C8R)HNG17. In this study, we show that Colivelin completely suppresses AD-relevant toxicity at 100 fm concentrations and keeps its neuroprotective action at or above the levels of 1 nm. We further demonstrate that Colivelin efficiently prevents memory impairment in vivo by intraperitoneal as well as intracerebroventricular administration.
Materials and Methods
Genes, polypeptides, and materials. cDNAs encoding V642I-APP and M146L-PS1 in pcDNA vectors were described previously (Hashimoto et al., 2001a,b). Kinase-negative [dominant-negative (dn)] forms of Ca2+/calmodulin-dependent protein kinase IV (CaMKIV) and signal transducer and activator of transcription 3 (STAT3) cDNAs were described previously (Minami et al., 1996; Chiba et al., 2004). Colivelin (SALLRSIPAPAGASRLLLLTGEIDLP), ADNF (SALLRSIPA), and AGA-(C8R)HNG17 (PAGASRLLLLTGEIDLP) were synthesized by Asahi Techno Glass Corporation (Funabashi, Japan). Monoclonal antibody against APP (22C11), rat anti-PS1 antibody, and anti-cholineacetyl transferase (ChAT) antibody were from Chemicon (Temecula, CA). Aβ1-42, Aβ1-43, and Aβ25-35 were purchased from Peptide Institute (Osaka, Japan).
Cells, cell death, and viability experiments. F11 cells, a hybrid of a rat embryonic day 13 primary cultured neuron and a mouse neuroblastoma N18TG2, were grown in Ham's F-12 medium (Sigma, St Louis, MO) containing 18% fetal bovine serum (FBS) (Hyclone, Logan, UT) and antibiotics, as described previously (Platika et al., 1985; Hashimoto et al., 2000). F11 cells (7 × 104 cells per well in a six-well plate), cultured in Ham's F-12 plus 18% FBS (HF-18%) for 12-16 h, were transfected with V642I-APP or M146L-PS1 genes by lipofection [cDNAs, 1.0 μg; LipofectAMINE (Invitrogen, Carlsbad, CA), 2 μl; PLUS Reagent (Invitrogen), 4 μl unless otherwise mentioned] in the absence of serum for 3 h and were incubated with HF-18% for 2 h. Culture media were then changed to Ham's F-12 plus 10% FBS with or without rescue peptides (ADNF, AGA-(C8R)HNG17, and Colivelin), and cell mortality was measured at 72 h after transfection by Trypan blue exclusion assay, as described previously (Hashimoto et al., 2001a,b). The primary culture of mouse primary cortical neurons (PCNs) was performed in poly-l-lysine-coated 96-well plates (Sumitomo Bakelite, Akita, Japan) in the absence of serum and the presence of N2 supplement, as described previously (Hashimoto et al., 2001a,b). Prepared PCNs (5.0 × 104 cells per well in a 96-well plate) were preincubated with or without synthetic Colivelin or AGA-(C8R)HNG17 for 16 h and treated with 25 μm Aβ1-43 or 20 μm l-glutamate in the presence or absence of the factors for 72 h. Cell viability assays were performed as described previously (Hashimoto et al., 2001a,b) with calcein AM (Dojindo, Kumamoto, Japan) and water-soluble tetrazolium salt (WST-8; Cell Counting Kit 8; Dojindo).
Immunoblot analysis. Immunoblot analysis was performed as described previously (Hashimoto et al., 2001a,b). Lysates (20 μg per lane) from cells transfected with the V642I-APP or M146L-PS1 expression vectors were subject to SDS-PAGE and were then electrically blotted to a polyvinylidene difluoride sheet. The sheet was soaked with 22C11 or anti-PS1 antibody and then with 1:5000 of horseradish peroxidase-labeled anti-mouse or anti-rat IgG antibody (Bio-Rad, Hercules, CA). The antigenic bands were visualized by ECL (Amersham Biosciences, Uppsala, Sweden).
Animals and peptide treatments (in vivo AD models). This study was conducted in accordance with the Policies on the Use of Animals and Humans in Neuroscience Research, the Society for Neuroscience, and Guideline for the Care and Use of Laboratory Animals of Keio University School of Medicine. All experimental procedures were approved by the Institutional Animal Experiment Committee at Keio University. Seven-week-old CD-1 mice were purchased from Charles River Japan (Yokohama, Japan). Nine-week-old C57BL/6J (C57) mice were purchased from CLEA Japan (Tokyo, Japan). Animals were housed in a specific pathogen-free animal facility (23 ± 1°C, 50 ± 5% humidity) under a 12 h light/dark cycle (lights on 7:00 A.M. to 7:00 P.M.), as described previously (Kawasumi et al., 2004).
To develop an AD mouse model by Aβ repetitive intracerebroventricular injection, 8-week-old CD-1 mice were implanted with cannulas in the left lateral ventricle of brain, as described previously (Chiba et al., 2004; Yamada et al., 2005). Mice were placed in a stereotaxic surgery apparatus under anesthesia by 10% Nembutal. A C315GS-4 cannula system for mice (Plastics One, Roanoke, VA) was implanted into the left lateral ventricles under aseptic conditions through a hole drilled in the skull at the following coordinates: anteroposterior, +0.3 mm; lateral, 1.0 mm; horizontal, 3.0 mm from the bregma, according to the atlas by Paxinos and Franklin (2000). The cannula was secured with surgical glue. Ten days after cannula implantation, animals were assigned randomly to the control group, the Aβ-injection group, or the Aβ plus Colivelin-injection group, receiving intracerebroventricular injection of 3 μl of sterile deionized distilled water (ddw), 300 pmol of Aβ1-42, or 1 nmol of Aβ25-35 peptide in 3 μl of ddw, every other day for 3 weeks together with injection of 3 μl of sterile saline or 10 pmol of Colivelin in 3 μl of sterile saline once every 6 d as depicted schematically in Figure 5A. Behavioral tests were performed from 2 d after the last intracerebroventricular injection.
Figure 5.
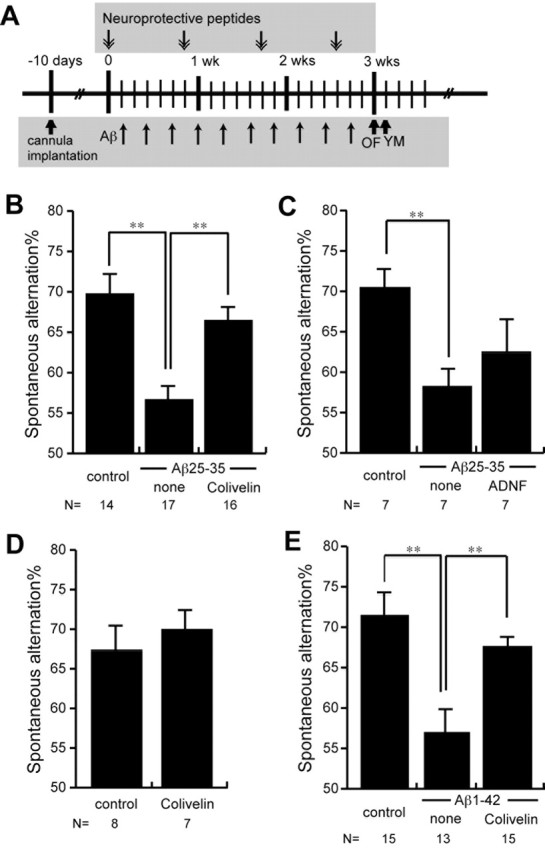
In vivo effect of Colivelin on spatial working memory deficits caused by repetitive administration of toxic Aβ peptides. A, The treatment schedule of repetitive Aβ injection. One nanomole of Aβ25-35 or 300 pmol of Aβ1-42 was injected 10 times every other day for 3 weeks (wks) together with or without Colivelin injection once in every 6 d (10 pmol each for Aβ25-35; 100 pmol each for Aβ1-42). B, C, SA% of mice repetitively injected with or without Aβ25-35 together with or without 10 pmol of Colivelin (B) or ADNF (C) were compared. D, Colivelin does not increase SA% of mice. SA% of mice injected with 10 pmol of Colivelin was compared with SA% of mice injected with saline. E, SA% of mice repetitively injected with Aβ1-42 together with 100 pmol of Colivelin were compared. Data are shown in means ± SEM. Statistical analyses were performed by one-way ANOVA followed by Fisher's PLSD (*p < 0.05; **p < 0.01).
To develop another AD mouse model with neuronal cell death by hippocampal injection of Aβ, CD-1 mice (8 weeks of age), intracerebroventricularly injected with a rescue factor into the left lateral ventricles on the previous day, were similarly placed in a stereotaxic apparatus under anesthesia by 10% Nembutal and received hippocampal injection of 3 μl of sterile ddw or 300 pmol of Aβ1-42 in 3 μl of sterile ddw by 31 gauge needles on the next day at following coordinates: anteroposterior, +1.8 mm; lateral, 1.0 mm; horizontal, 1.3 mm from the bregma. Seventy-two hours after Aβ injection, mice were killed for histological analysis of brain.
To establish a mouse model with cholinergic dysfunction, CD-1 mice (8 weeks of age) or C57 mice (10 weeks of age) were habituated in an open-field test (OF) 1 d before a Y-maze test (YM). On the YM day, the mice were injected intraperitoneally with 0.21 ml of sterile saline or the indicated amount (1.4, 7, or 35 nmol) of Colivelin in 0.21 ml of sterile saline at 30 min before YM. The mice were then injected with 0.2 ml of sterile ddw containing 20% methanol in the presence or absence of 3-quinuclidinyl benzilate (3-QNB) [17.5 ng (0.5 mg/kg body weight) for CD-1 mice; 70 ng (2.0 mg/kg body weight) for C57 mice], 15 min before YM (Krejcova et al., 2004).
Behavioral test. OF and YM were performed as described previously (Kawasumi et al., 2004; Yamada et al., 2005). In OF, mice were placed individually at the center of a 100 cm square gray-plastic field (with 20 cm interval black grids) and allowed to move freely for 3 min to habituate to the experimental environment. The apparatus for YM is made of three gray plastic arms (40 cm long, 12 cm high, 3 cm wide at the bottom, and 10 cm wide at the top) connected to each other at angles of 120°. Mice were placed individually at the end of an arm and allowed to explore the arms freely for 8 min. Examined parameters were (1) total arm entries and (2) spontaneous alternation percentage (SA%). SA% was defined as a ratio of the arm choices that differed from the previous two choices (“successful choices”) to total choices during the run (“total entry minus two,” because the first two entries could not be evaluated). For example, if a mouse made 10 entries, such as 1-2-3-2-3-1-2-3-2-1, there were five successful choices in eight total choices (10 entries minus two). Therefore, SA% in this case was 62.5%.
Immunohistochemistry. Immunohistochemical analysis was performed as described previously (Kawasumi et al., 2004; Yamada et al., 2005). After the behavioral experiments, mice were anesthetized, perfused transcardially with PBS, and fixed by ethanol containing 5% acetic acid. Brains were embedded in paraffin, and 10 μm coronal sections were prepared on New-Silane slide glasses (Muto Pure Chemicals, Tokyo, Japan). Samples were subsequently dewaxed and washed in ethanol and PBS. Immunohistochemical detection of cholinergic neurons was performed with anti-ChAT antibody (1:50 dilution; Chemicon) and visualized with the ABC method (Vectastain Elite kit; Vector Laboratories, Burlingame, CA). ChAT-immunoreactive neurons in the medial septum of five coronal sections, 10 μm thickness, with a 50 μm interval (∼0.6-0.9 mm anterior from the bregma), were counted, and the averages of the total numbers of ChAT-immunoreactive neurons in three mice per treatment group (n = 3) were compared.
Statistics. All of the experiments on neuronal cell lines described in this study were repeated at least three times with independent transfections and treatments. All values in the figures of the in vitro study indicate means ± SD, and the values of the in vivo study indicate means ± SEM. Statistical analysis for the in vivo experiment was performed with one-way ANOVA followed by a Fisher's PLSD, in which p < 0.05 was assessed as significant.
Results
Effect of Colivelin on neuronal death by Aβ1-43 in vitro
From detailed characterization of the structure-function relationship of Humanin (Hashimoto et al., 2001a,b; Terashita et al., 2003) (Table 1), we found that (1) a Gly substitution of Ser14 (HNG) potentiates neuroprotective activity of HN by 1000-fold; (2) Ala substitutions of both Arg4 and Phe6 (AGA-HNG), disrupting sites for trypsin and chymotrypsin digestion, further augment the potency of HNG by additional 10 times; (3) the core domain of HN that mediates neuroprotective activity is the 17 amino acid sequence from Pro3 to Pro19 (HN17); (4) modulation of the free thiol group in Cys8 negatively affects the neuroprotective activity of HN and a substitution of C8R or C8K potentiates HN activity. Based on these data, we assumed that AGA-(C8R)HNG17 might result in a more potent HN derivative than AGA-HNG, the IC50 against neurotoxicity by Aβ1-43 of which is ∼10-100 pm (Table 1) (Hashimoto et al., 2001b, Niikura et al., 2004). In accordance, our preliminary examination revealed that AGA-(C8R)HNG17 completely protected neurons from toxicity induced by Aβ1-43 at a concentration of 10 pm, suggesting that the Arg substitution of Cys8 (C8R) augmented the potency of AGA-HNG by 10-fold.
To further potentiate the rescue activity of AGA-(C8R)HNG17, we fused ADNF to it. ADNF was shown previously to suppress Aβ neurotoxicity (Brenneman and Gozes, 1996; Brenneman et al., 1998; Hashimoto et al., 2001b). It completely suppresses Aβ neurotoxicity at a concentration of 100 fm. Unfavorably, however, ADNF loses its protective effect at concentrations higher than 1 nm. We hypothesized that N-terminal attachment of ADNF to AGA-(C8R)HNG17, which gives rise to the 26 amino acid peptide named Colivelin, may enhance the neuroprotective effect without decreasing its neuroprotective potency at higher concentrations.
To test this hypothesis, we examined the effect of the synthetic Colivelin peptide on Aβ1-43-induced neuronal death, compared with that of AGA-(C8R)HNG17. We used Aβ1-43 as the toxicity inducer in vitro, because Aβ1-43 is the longest Aβ species detected in AD brain (Mori et al., 1992). There is evidence showing that treatment with Aβ1-43 induces death in primary cortical neurons (PCNs) with EC50 almost comparable with that of Aβ1-42 (Hashimoto et al., 2001b). Furthermore, HN or HNG suppresses neurotoxicity by Aβ1-43 with an approximate IC50 of 1-10 μm or 1-10 nm, respectively, suggesting that these IC50 values are almost comparable with those of HN- and HNG-mediated inhibition of neurotoxicity by Aβ1-42 (Hashimoto et al., 2001a,b). When PCNs were treated with 25 μm Aβ1-43, cell viability was reduced significantly within 72 h compared with nontreated PCNs (Fig. 1). When PCNs were treated with 25 μm Aβ1-43 in the presence of 10 fm Colivelin or 1 pm AGA-C8R-HNG17, about one-half of the neurons were rescued. Full neuronal protection against Aβ1-43 was obtained by treatment with either 100 fm Colivelin or 10 pm AGA-(C8R)HNG17, suggesting that Colivelin is 100 times more potent than AGA-(C8R)HNG17. Considering that HN or HNG exerts their full protection at 10 μm or 10 nm, respectively (Table 1), Colivelin is 100,000,000 or 100,000 times more potent than HN or HNG, respectively. Accordingly, we have concluded that Colivelin is a femtomolar-acting bioactive peptide at least as potent as ADNF in vitro. Furthermore, Colivelin does not lose its neuroprotective activity even at higher concentrations.
Figure 1.

Effect of Colivelin on neuronal death induced by Aβ1-43. PCNs were treated with 25 μm Aβ1-43 in the presence or absence of Colivelin or AGA-(C8R)HNG17 (100 am, 1 fm, 10 fm, 100 fm, 1 pm, 10 pm, 100 pm, 1 nm, 10 nm, or 100 nm). Indicated concentrations of neuroprotective peptides were added at 16 h before the onset of Aβ treatment. Cell viability was measured 72 h after Aβ1-43 treatment by WST-8 assay (left) and calcein assay (right). noT, Not transfected. All values in this study indicate means ± SD.
Effect of Colivelin on neuronal cell death caused by V642I-APP in vitro
In our previous study (Hashimoto et al., 2001b), HNG completely suppressed neuronal cell death induced by V642I-APP at 100 nm, whereas ADNF partially suppressed death caused by V642I-APP at concentrations from 100 fm to 10 pm. To examine the effect of Colivelin against neurotoxicity by V642I-APP, F11 cells were transfected with a V642I-APP gene-encoding vector and then treated with various concentrations of Colivelin or AGA-(C8R)HNG17 (Fig. 2A). Ectopic expression of V642I-APP induced death in ∼55% of F11 neurohybrid cells, whereas no transfection or transfection with the empty vector induced death in only 10% of cells. AGA-(C8R)HNG17 treatment suppressed V642I-APP-induced neuronal cell death in a dose-dependent manner with complete neuroprotection observed at a concentration of 10 pm as it did against Aβ1-43. Colivelin exhibited more potent neuroprotective effect on neuronal cell death caused by V642I-APP. Colivelin elicited full neuroprotection at a concentration of 100 fm, indicating that Colivelin has 100-fold more potent neuroprotective activity against toxicity caused by V642I-APP than AGA-(C8R)HNG17. It has been also shown that neither Colivelin nor AGA-(C8R)HNG17 loses their neuroprotective effect against V642I-induced neuronal cell death even at 100 nm, whereas ADNF has been reported to completely lose its neuroprotective effect at concentrations of 1-10 nm or more (Hashimoto et al., 2001b). The expression levels of V642I-APP were confirmed to be unaltered by treatment with rescue factors (Fig. 2B).
Figure 2.
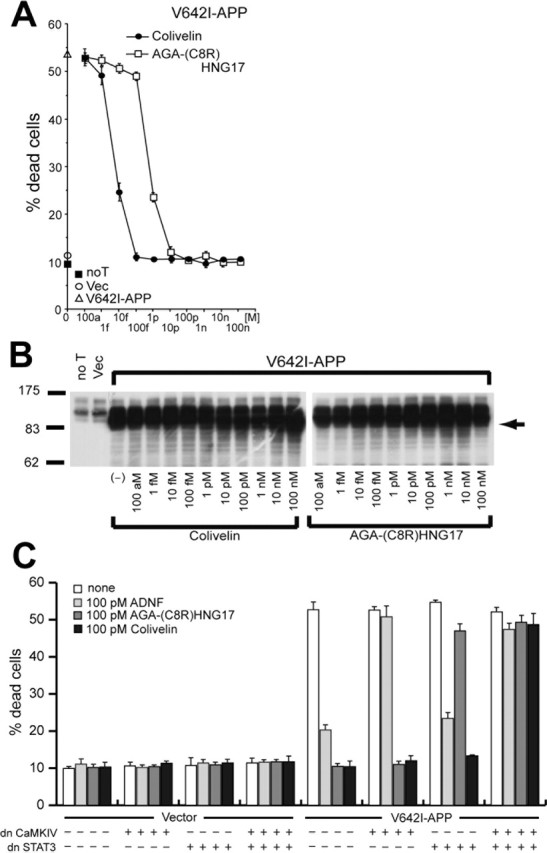
Effect of Colivelin on neuronal cell death induced by V642I-APP. A, F11 cells were transfected with the V642I-APP expression vector and then treated with various concentrations of Colivelin or AGA-(C8R)HNG17 (100 am, 1 fm, 10 fm, 100 fm, 1 pm, 10 pm, 100 pm, 1 nm, 10 nm, or 100 nm). Seventy-two hours after transfection, cell mortality was measured by Trypan blue exclusion assay. As controls, mortality of cells not transfected or transfected with the pcDNA vector was measured. B, Protein expression of V642I-APP was determined by immunoblot analysis with antibody to PS1. C, Roles of CaMKIV and STAT3 in Colivelin-mediated neuroprotection against V642I-APP-induced neurotoxicity. F11 cells, transfected with or without the V642I-APP expression vector together with dnCaMKIV or dnSTAT3 expression vectors, were incubated with or without 100 pm ADNF, AGA-(C8R)HNG17, or Colivelin. Seventy-two hours after transfection, cell mortality was measured by Trypan blue exclusion assay. All experiments performed with n = 3. noT, Not transfected; Vec, vector. Error bars represent SD.
Colivelin-mediated neuroprotection against V642I-APP through both CaMKIV and STAT3
We then asked whether Colivelin suppresses neurotoxicity induced by V642I-APP via both ADNF- and HN-mediated prosurvival pathways. We actually examined how expression of a dn form of CaMKIV or STAT3, the downstream effectors of ADNF or HN, modified the rescue function of Colivelin. To this end, we cotransfected F11 cells with the V642I-APP vector together with an expression vector encoding dnCaMKIV or dnSTAT3, or together with both vectors, and then treated cells with 100 pm ADNF, AGA-(C8R)HNG17, or Colivelin. As shown in Figure 2C, death induced by V642I-APP was suppressed completely by 100 pm AGA-(C8R)HNG17 or Colivelin and was considerably suppressed (∼80% of the control) by 100 pm ADNF. The neuroprotective effect of ADNF was antagonized by coexpression of dnCaMKIV (Chiba et al., 2004) but not of dnSTAT3, whereas the neuroprotective effect of AGA-(C8R)HNG17 was antagonized by coexpression of dnSTAT3 (Hashimoto et al., 2005) but not of dnCaMKIV. The neuroprotective effect of Colivelin was diminished only when both dnCaMKIV and dnSTAT3 were coexpressed simultaneously, indicating that Colivelin protects neuronal cells from death induced by V642I-APP via both ADNF- and HN-mediated prosurvival pathways.
Effect of Colivelin on neuronal cell death caused by M146L-PS1
We have reported previously that HNG completely suppresses neuronal cell death induced by ectopic overexpression of M146L-PS1 at 100 nm, whereas ADNF does not show the neuroprotective effect against M146L-PS1 even at 100 nm (Hashimoto et al., 2001b). To examine the effect of Colivelin on neurotoxicity caused by M146L-PS1, we transfected F11 cells with a M146L-PS1 gene-encoding vector and then treated cells with various concentrations of Colivelin or AGA-(C8R)HNG17 (Fig. 3A). Trypan blue exclusion assay revealed that ∼50% of neuronal cells died after transfection with the M146L-PS1 gene, whereas only 10% of cells without transfection or cells transfected with the empty vector died. Colivelin suppressed M146L-PS1-induced neuronal cell death in a dose-dependent manner with complete neuroprotection observed at a concentration of 100 fm. Although AGA-(C8R)HNG17 similarly suppressed neuronal cell death induced by M146L-PS1, 10 pm or more AGA-(C8R)HNG17 is necessary to completely suppress M146L-PS1-mediated neurotoxicity, indicating that Colivelin has 100-fold more potent neuroprotective effect than AGA-(C8R)HNG17 to suppress M146L-PS1-mediated neurotoxicity. The expression levels of M146L-PS1 were confirmed to be unaltered in the presence of neuroprotective peptides (Fig. 3B)
Figure 3.
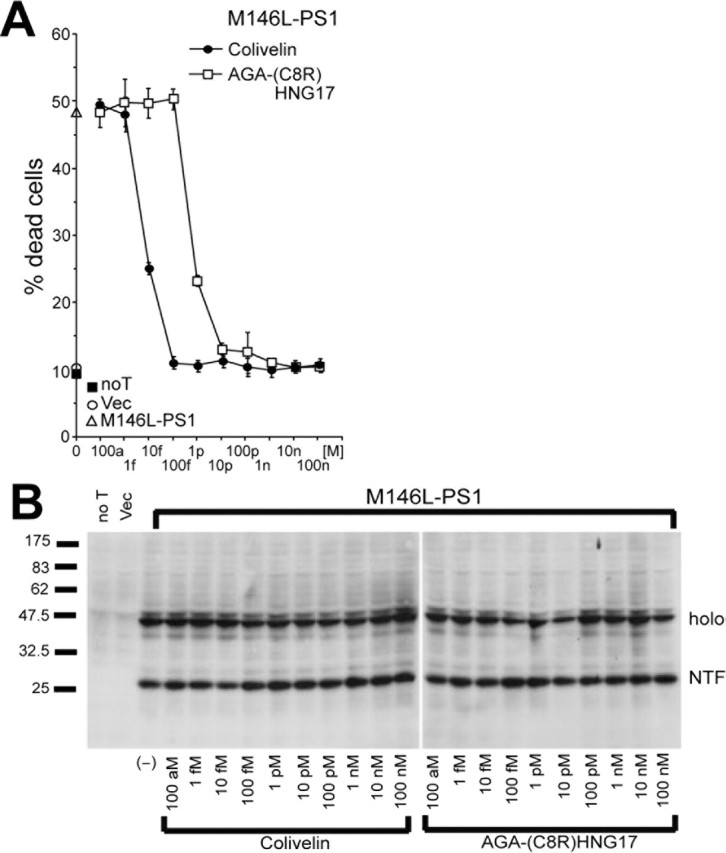
Effect of Colivelin on neuronal cell death induced by M146L-PS1. A, B, Effect of Colivelin on neuronal cell death induced by M146L-PS1. F11 cells were transfected with the M146L-PS1 expression vector and then treated with various concentrations of Colivelin or AGA-(C8R)HNG17 (100 am, 1 fm, 10 fm, 100 fm, 1 pm, 10 pm, 100 pm, 1 nm, 10 nm, or 100 nm). As controls, mortality of cells not transfected or transfected with the pcDNA vector was measured. Seventy-two hours after transfection, cell mortality was measured by Trypan blue exclusion assay (A). Protein expression of M146L-PS1 was determined by immunoblot analysis with antibody to PS1 (B). All experiments were performed with n = 3. noT, Not transfected; Vec, vector; holo, holoprotein; NTF, N-terminal fragment. Error bars represent SD.
Given that ADNF does not suppress neuronal cell death by M146L-PS1, it is strongly suggested that neuroprotection by Colivelin is mediated only via the AGA-(C8R)HNG17 domain. If this is the case, it is highly likely that N-terminal attachment of ADNF enhances the neuroprotective effect of AGA-(C8R)HNG17. One possible mechanism underlying potentiation of HN-mediated neuroprotective activity by attachment of an N-terminal tag is that it enhances HN dimerization. Our previous study indicated that HN needs to be dimerized to be active, and N-terminal attachment of a dimerization tag (EFLIVIKS) facilitates neuroprotective activity of HN (Terashita et al., 2003). BIA-core binding analysis (BIAcore 2000; BIAcore AB, Uppsala, Sweden), which uses the natural phenomenon of surface plasmon resonance to monitor in real time the protein-protein interaction (association/dissociation), has indicated that Colivelin also got dimerized or polymerized (data not shown). Calculated KD for Colivelin dimerization was ∼500 times smaller than that for HN dimerization [9.41 μm vs 4.79 mm, fitted to a simple 1:1 (Langmuir) binding model], on the artificial dimerization condition that Colivelin (26 amino acids) or HN (24 amino acids), immobilized onto the BIAcore dextran matrix (CM5 sensor tip) through its internal Arg and Lys residues, formed a complex with free Colivelin or free HN. In this regard, our previous pull-down study (Terashita et al., 2003) showed that His-tagged HN immobilized onto the nickel bead through its His-tag portion made a complex with FLAG-tagged HN at their concentration of 2 μm, suggesting that our condition of BIAcore binding analysis under-estimated KD values of Colivelin and HN, possibly because immobilization of short peptides onto the matrix through internal Arg and Lys residues interfered their association with free peptides. In addition, calculated KD for Colivelin might be further underestimated compared with HN, because Colivelin has only two Arg residues, whereas HN contains three Arg and one Lys residues, which resulted in much smaller immobilized amount of Colivelin than that of HN. In any event, based on these binding finding, we speculate that attachment of ADNF may potentiate the neuroprotective activity by accelerating peptide dimerization, although there is no direct evidence supporting this notion.
Colivelin shows protection against excitotoxicity
ADNF has been originally reported to have protective activity against a variety of neurotoxic insults including excitotoxicity (NMDA), Aβ, and tetrodotoxin (electrical blockade) (Brenneman and Gozes, 1996; Brenneman et al., 1998). It is therefore highly possible that Colivelin also shows protective activity against these neurotoxic insults. To address this issue, we treated PCNs with 20 μm glutamate in the presence or absence of increasing amounts of Colivelin, ADNF, or AGA-(C8R)HNG17. As shown in Figure 4, treatment with 20 μm glutamate decreased viability in PCNs. We found that although ADNF showed suppressive activity at a concentration of 10 pm, it loses its activity at a concentration of 1 nm. This unique character of ADNF was already mentioned (Brenneman and Gozes, 1996). In contrast, AGA-(C8R)HNG17 did not attenuate neurotoxicity induced by glutamate at any concentration tested, as expected from our previous experiments (Hashimoto et al., 2001a,b). Interestingly, Colivelin began to exhibit anti-excitotoxicity activity at a concentration of 100 fm and kept its activity even at higher concentrations (10 pm-1 nm). We thus concluded that Colivelin has neuroprotective activity not only against AD-related neurotoxicity but also against non-AD-related toxicity, probably through its ADNF moiety. In addition, it is speculated from Figure 4 that the anti-excitotoxicity activity of Colivelin appears to be enhanced by attachment of AGA-(C8R)HNG17 to ADNF.
Figure 4.

Effect of Colivelin on death induced by treatment with 20 μm glutamate in PCNs. PCNs were treated with 20 μm glutamate in association with various concentrations of AGA-(C8R)HNG17, ADNF, or Colivelin (1 fm, 100 fm, 10 pm, or 1 nm). Seventy-two hours after the start of treatment, cells were measured with WST-8 assay (n = 3). Error bars represent SD. Statistical analysis was performed by one-way ANOVA followed by Fisher's PLSD (*p<0.05, **p<0.01).
In vivo effect of Colivelin on impairment in spatial working memory by toxic Aβs Intracerebroventricular injection of toxic Aβ peptides has been reported to impair spatial working memory in rodents (Flood et al., 1991; Delobette et al., 1997; Yamada et al., 1999; Yamaguchi and Kawashima, 2001; Stepanichev et al., 2003b). Although administration of Aβ into cerebral ventricles has usually been performed by a one-shot or bolus intracerebroventricular injection of a large dose of Aβ, rats with dementia have been constructed successfully by continuous infusion of Aβ via an osmotic pump connected to the cerebral ventricle (Nitta et al., 1997; Yamada et al., 1999). We have recently developed another toxic Aβ-induced mouse model of AD by using repetitive injection of Aβ25-35 via an implanted cannula (Yamada et al., 2005). Continuous administration and repetitive injection of a small dose of Aβ25-35 via an implanted cannula are superior to a bolus administration of toxic Aβ in the sense that the former procedure gives rise to a brain state more resembling AD than the latter procedure. We mainly used Aβ25-35, which shares with Aβ1-42 the ability to self-aggregate and to induce neurotoxicity both in vitro and in vivo (Mori et al., 1992; Delobette et al., 1997), in this animal experiment, because they are more easily solubilized in water and show neurotoxicity in vitro similar to that of Aβ1-42 or Aβ1-43, which are suppressed by treatment with Colivelin (data not shown).
Using this procedure, we examined the neuroprotective effect of Colivelin against Aβ toxicity in vivo. After 10 injections of 1 nmol of Aβ25-35 were performed (as schematically shown in Fig. 5A), spatial working memory of each mouse was examined in a continuous alternation version of the YM. SA%, which reflects spatial working memory, of control mice was 69.7 ± 2.5%, whereas that of Aβ25-35-administered mice was 56.6 ± 1.8%, indicating that repetitive administration of Aβ25-35 efficiently impaired spatial working memory (Fig. 5B). In contrast, SA% of Aβ25-35-administered mice treated with Colivelin at 10 pmol per 6 d was 66.4 ± 1.7%, indicating that Colivelin treatment almost completely suppressed Aβ25-35-mediated impairment in spatial working memory. As shown in Figure 5C, however, SA% of Aβ25-35-administered mice treated with ADNF at 10 pmol per 6 d was 62.6 ± 4.3%, whereas those of control and Aβ25-35-adiministered mice were 70.9 ± 2.3 and 58.3 ± 2.2%, respectively, suggesting that ADNF treatment resulted in a marginal but statistically insignificant tendency to improve memory impairment induced by repetitive administration of Aβ25-35. Colivelin treatment alone at 10 pmol per 6 d was deemed not to increase SA% in YM, as shown in Figure 5D (control, 67.3 ± 3.0%; Colivelin-treated mice, 69.9 ± 2.5%). We further examined the effect of Colivelin on neurotoxicity induced by repetitive administration of Aβ1-42, another toxic Aβ species detected in AD brain, which has been believed to play a critical role in the pathogenesis of AD. Ten repetitive administrations of 300 pmol of Aβ1-42 similarly impaired spatial working memory; SA% in YM of Aβ1-42-administered mice was 56.9 ± 3.1%, whereas that of control mice was 71.4 ± 2.8% (Fig. 5E). Colivelin treatment at 100 pmol per 6 d again almost completely antagonized Aβ1-42-mediated impairment in spatial working memory (SA%, 67.4 ± 1.4%).
As reported in our previous study (Tajima et al., 2005; Yamada et al., 2005), intracerebroventricular injection of Aβ downregulates the expression level of ChAT of neurons in the medial septum. We further recognized correlation between the extent of impairment in spatial working memory and the numbers of ChAT-immunoreactive neurons in the medial septum in Aβ-injected mice. To confirm the neuroprotective effect of Colivelin in vivo, we performed immunohistochemical analysis of medial septa of Aβ25-35-administered mice with or without Colivelin treatment. Brain sections of mice were immunohistochemically stained with anti-ChAT antibody, and the numbers of immunoreactive neurons in medial septa were compared (Fig. 6A-C). We observed a significant decrease in the numbers of ChAT-immunoreactive neurons in medial septa of Aβ25-35-injected mice (188.7 ± 40.2 ChAT-immunoreactive neurons) compared with the control mice (517.7 ± 17.7) (Fig. 6D). Consistent with the results of SA% in YM, Colivelin treatment significantly increased the number of immunoreactive neurons, which were downregulated by repetitive Aβ25-35 injection, to a comparable with that of control (445.7 ± 35.0).
Figure 6.

Immunohistochemical analysis of ChAT-immunoreactive neurons in the medial septum. A-C, Coronal sections of medial septa of mice repetitively infused with Aβ25-35 together with or without Colivelin treatment were immunohistochemically stained with anti-ChAT antibody. Scale bar, 100 μm. D, The average numbers of ChAT-immunoreactive [ChAT (+)] neurons in the medial septa of five coronal sections, 10 μm in thickness (total, 50 μm thickness), were compared (n = 3). Data are shown as means ± SEM. Statistical analyses were performed by one-way ANOVA followed by Fisher's PLSD (**p < 0.01).
In vivo effect of Colivelin on hippocampal neuronal death by Aβ1-42 Multiple groups have reported that intracerebral or hippocampal injection of Aβ causes neuronal degeneration in rodents (Frautschy et al., 1991; Kowall et al., 1992; Miguel-Hidalgo and Cacabelos, 1998; Stephan et al., 2001; Stepanichev et al., 2003a; Ryu et al., 2004). To examine the in vivo neuroprotective effect of Colivelin on neuronal death by Aβ1-42, we performed hippocampal injection of 300 pmol of Aβ1-42 into the CA1 region 1 d after intracerebroventricular injection of neuroprotective factors. Seventy-two hours after hippocampal injection, mice were killed to prepare paraffin-embedded hippocampal sections, which were subsequently stained with cresyl violet. As shown in Figure 7, hippocampal injection of Aβ1-42 resulted in robust neuronal loss in the CA1 region, whereas there was no significant neuronal loss in saline-injected control mice. Intracerebroventricular injection of 100 pmol of Colivelin before hippocampal Aβ injection completely protected neurons from toxicity induced by Aβ1-42 injection. On the other hand, neither administration of 100 pmol of AGA-(C8R)HNG17 nor administration of a mixture of ADNF and AGA-(C8R)HNG17 at the same dosage exhibited as potent a neuroprotective effect as Colivelin, suggesting that fusion of ADNF and AGA-(C8R)HNG17 gives rise to a much superior neuroprotective peptide over its either parent.
Figure 7.

Antagonistic effect of Colivelin on neuronal death in the CA1 induced by direct hippocampal injection of Aβ1-42. Coronal sections of the hippocampal CA1 regions of mice injected with 300 pmol of Aβ1-42 in the presence or absence of rescue factors (100 pmol each) were stained with cresyl violet (n = 2). Rescue factors were intracerebroventricularly injected 16 h before Aβ1-42 injection. Top left, Control; top middle, 300 pmol of Aβ1-42; top right, 300 pmol of Aβ1-42 plus 100 pmol of Colivelin; bottom left, 300 pmol of Aβ1-42 plus 100 pmol of AGA-(C8R)HNG17; bottom right, 300 pmol of Aβ1-42 plus 100 pmol of ADNF plus 100 pmol of AGA-(C8R)HNG17. Scale bars, 100 μm.
Protection against cholinotoxin-induced amnesia by intraperitoneal injection of Colivelin
Dysfunction of the cholinergic system in the CNS, including the medial septal nucleus and the nucleus basalis of Mynert, is implicated in the development of cognitive impairment of AD (Bartus et al., 1982; Coyle et al., 1983). In accordance, administration of several cholinotoxins, including scopolamine, 3-QNB, and ethylcholine aziridium (AF64A), was reported to induce memory impairment in rodents and humans (Fisher et al., 1989; Ebert and Kirch, 1998; Gozes et al., 2000; Krejcova et al., 2004). Consequently, mice treated with these cholinotoxins are also used widely as AD models, in addition to Aβ-injected or genetically engineered models. We examined the effect of intraperitoneal injection of 7 nmol of Colivelin on spatial working memory impairment caused by intraperitoneally administered 3-QNB (Fig. 8A). SA% in YM of control C57 mice was 60.9 ± 3.7%. In agreement with the previous findings (Krejcova et al., 2004), intraperitoneally administered 3-QNB at 2.0 mg/kg elicited potent neurotoxicity in C57 mice, and SA% of 3-QNB-treated mice was 38.3 ± 3.5%. Four mice of a total of eight were excluded from the test because they did not enter more than three arms in this particular experiment. In contrast, SA% of 3-QNB-administered mice preinjected intraperitoneally with 7 nmol of Colivelin was 52.4 ± 3.1%. Only two mice of a total of eleven were excluded from the test. This result suggests that intraperitoneally administered Colivelin passes through the blood-brain barrier and significantly attenuates cholinotoxin-induced amnesia. To see whether there is dose dependency in the neuroprotective effect by intraperitoneal injection of Colivelin, we intraperitoneally injected 3-QNB-treated mice with three different doses of Colivelin, using CD-1 mice, which may be less sensitive to the intraperitoneal procedure than C57 mice. As shown in Figure 8B, Colivelin showed anti-cholinotoxicity activity in a dose-response manner with maximal protection at a dose of 7 nmol. In this experiment, no mice were excluded from the analysis because of 3-QNB-induced immobility observed in C57 mice. We further examined the effect of intraperitoneally administered Colivelin in comparison with that of HNG. As shown in Figure 8C, CD-1 mice with 3-QNB treatment resulted in deterioration of SA% (50.6 ± 2.7%) compared with control mice (68.2 ± 1.6%). Intraperitoneal injection of Colivelin again significantly attenuated 3-QNB-induced spatial working memory deficits (59.5 ± 2.3%) to a greater extent than that of HNG (54.8 ± 2.1%), indicating that Colivelin is superior to HNG even in vivo.
Figure 8.

In vivo effect of intraperitoneal injection of Colivelin on 3-QNB-induced amnesia. A, SA% of C57 mice with 3-QNB treatment (70 ng) in the presence or absence of intraperitoneal preinjection of Colivelin (7 nmol) were compared. B, SA% of CD-1 mice with 3-QNB treatment (17.5 ng) in the presence of intraperitoneal preinjection of increasing doses of Colivelin (1.4, 7, and 35 nmol). C, SA% of CD-1 mice with 3-QNB treatment (17.5 ng) in the presence or absence of intraperitoneal injection of Colivelin or HNG (7 nmol) were compared. Data are shown as means ± SEM. Statistical analyses were performed by one-way ANOVA followed by Fisher's PLSD (*p < 0.05; **p < 0.01).
Discussion
We have herein shown that a hybrid peptide composed of AGA-(C8R)HNG17 C-terminally attached with ADNF is the most potent derivative of HN so far developed. Colivelin completely suppressed neuronal cell death induced by 25 μm Aβ1-43 and overexpression of V642I-APP or M146L-PS1 at a concentration as low as 100 fm in vitro. Furthermore, HN does not lose its anti-AD activity at its higher concentrations, in contrast to ADNF.
ADNF itself has a neuroprotective effect on multiple types of toxicities including Aβ neurotoxicity, electrical blockade with tetrodotoxin, excitotoxicity (NMDA), and oxidative stress (Brenneman and Gozes, 1996; Brenneman et al., 1998). ADNF completely suppresses Aβ neurotoxicity at a concentration as low as 100 fm in vitro. One of unfavorable characteristics of ADNF, however, is that it completely loses its protective effect at or above the nanomolar levels. On the other hand, AGA-(C8R)HNG17 displays its complete neuroprotective effect at concentrations of 10 pm or more but does not lose its neuroprotective activity at higher concentrations. As shown in Figure 2C, Colivelin simultaneously activates two distinct prosurvival pathways, which are triggered by ADNF and HN. Accordingly, it is likely that neuroprotection against Aβ at low concentrations (100 fm-10 pm) of Colivelin might be primarily mediated by ADNF, whereas neuroprotection against Aβ at higher concentrations (10 nm) of Colivelin might be mainly mediated by AGA-(C8R)HNG17. In addition, as shown in Figure 3, it is highly likely that N-terminal attachment of ADNF promotes the neuroprotective effect of AGA-(C8R)HNG17.
It is further suggested that via the ADNF-mediated pathway, Colivelin can suppress multiple types of toxicity other than neurotoxicity induced by AD-relevant insults. For instance, as shown in Figure 4, Colivelin attenuates excitotoxicity induced by glutamate, although HN is reported to be ineffective against glutamate-induced neurotoxicity (Hashimoto et al., 2001b). Furthermore, we have found recently that the anti-ALS effect in vivo of Colivelin is stronger than ADNF, possibly because attachment of AGA-(C8R)HNG17 stabilizes the peptide in vivo. Consequently, intracerebroventricular administration of Colivelin prolongs survival of ALS model mice (T. Chiba, M. Yamada, and M. Matsuoka, unpublished observation).
In the rescue experiments with PCNs treated with Aβs, cells were necessarily preincubated with HN or its derivative for 16 h before addition of Aβs. Such long preincubation appears to result in protein synthesis. This fact is in accordance with recently characterized signal transduction pathways mediated by the HN receptor on the cell membrane that is connected with activation of a transcription factor, STAT3 (Hashimoto et al., 2005).
We have reported that HN is secreted from cells, and from outside cells it binds to the membrane receptor and activates a prosurvival pathway linked to STAT 3 activation (Hashimoto et al., 2001a,b, 2005; Yamagishi et al., 2003). Independently, Guo et al. (2003) reported that HN intracellularly inhibits Bax-mediated apoptosis by binding to Bax. Together, it could be hypothesized that HN acts as an anti-cell-death factor by two ways of action. However, it remains to be addressed whether the latter mechanism is really involved in neuroprotective activity by HN or Colivelin.
We also confirmed the potent neuroprotective effect of Colivelin on AD-relevant insults in vivo. To examine this effect, we used three types of models: the repetitive Aβ intracerebroventricular model, the 3-QNB model, and the Aβ hippocampal injection model. We have recognized that repeated intracerebroventricular administration of toxic Aβ did not cause apparent death in hippocampal neurons including cholinergic neurons (Yamada et al., 2005). The first two models were used primarily to detect impairment in spatial working memory; the Aβ hippocampal injection model was used to histologically examine neuronal cell death in the CA1 region. Therefore, the first two recapitulate the functional abnormality of AD, and the third reproduces neuronal cell death, one of the most important pathological hallmarks of AD. Using repetitive Aβ intracerebroventricular model mice, we have demonstrated that Colivelin has a potent in vivo neuroprotective effect on Aβ-induced memory dysfunction. Based on the finding that ADNF in the same amount as Colivelin only showed a slight tendency to compromise Aβ neurotoxicity, we have concluded that Colivelin has a more potent neuroprotective effect than ADNF in vivo. We reported in our previous study that intracerebroventricular injection of HNG at 50 pmol or 1 nmol per week for 3 weeks attenuated toxicity induced by single intracerebroventricular injection of Aβ25-35 (Tajima et al., 2005). Mamiya and Ukai (2001) also reported that intracerebroventricular injection of 1 nmol of HNG attenuated spatial working memory impairment induced by scopolamine. Our present study has shown clearly that smaller amounts of Colivelin suppress impairment in spatial working memory induced by repetitive intracerebroventricular injection of Aβ25-35 and AD-relevant Aβ1-42, demonstrating a more potent neuroprotective action of Colivelin than HNG in vivo. Using Aβ hippocampal injection models, we further demonstrated that focal neuronal cell death was induced by injection of 300 pmol of Aβ1-42 around Aβ-injected areas in the CA1 region of hippocampus. Intracerebroventricular pretreatment of Colivelin at 100 pmol almost completely antagonized Aβ-mediated neuronal death in vivo, as observed in vitro, whereas neither the same amount of AGA-(C8R)HNG alone nor administration of a mixture of ADNF and AGA-(C8R)HNG17 completely suppressed neuronal death caused by Aβ1-42 (Fig. 7). It is notable that administration of a mixture of ADNF and AGA-(C8R)HNG17 did not exhibit as potent a neuroprotective effect in vivo as Colivelin, suggesting that fusion of two peptides gives rise to additional advantages, even in vivo.
Using the 3-QNB model, we further examined the effect of intraperitoneally administered Colivelin on impairment in spatial working memory induced by 3-QNB. In agreement with the previous report (Krejcova et al., 2004), we observed a suppressive effect of intraperitoneal injection of Colivelin on memory impairment by 3-QNB in both C57 and CD-1 mice. However, Colivelin did not completely suppress 3-QNB-induced neurotoxicity. Possible reasons are, first, that 3-QNB has additional toxic effects except for the antagonistic effect on muscarinic acetylcholine receptor that is not blocked by treatment with HN derivatives and/or second, that the intraperitoneally administered dose of Colivelin was not sufficient to suppress neuronal dysfunction by 3-QNB. In any event, it is apparent that Colivelin exhibited a more potent neuroprotective effect than HNG even when it was administered intraperitoneally.
In summary, this study has characterized the most potent HN derivative named Colivelin, composed of ADNF and AGA-(C8R)HNG17. Its potent anti-AD activity, both in vitro and in vivo, will encourage clinical application of Colivelin as an anti-AD candidate drug to suppress both AD-related neuronal cell death and dysfunction of the memory system.
Footnotes
This work was supported in part by a grant from Japan Science and Technology Agency, Noevir, Keio University Grant-in-Aid for Encouragement of Young Medical Scientists (T.C. and M.Y.), and the Japan Society for the Promotion of Science. We are indebted to Dr. Masaki Kitajima for essential help. We are especially grateful to Takako Hiraki for essential assistance throughout this study. We thank Dr. Mark C. Fishman for F11 neurohybrid cells; Dr. John T. Potts Jr, Dr. Etsuro Ogata, and Yoshiomi and Yumi Tamai for indispensable support; Shinano Manufacturing Company for expert technical support; H. Yanagisawa, M. Kimura, M. Nakajima, M. Akiyama, and Y. Nakano for indispensable assistance; Tomo Yoshida-Nishimoto for essential cooperation; Dr. Dovie Wylie for expert assistance; R. Kato, Y. Kamei, and K. Negishi for assistance in animal husbandry; Dr. Katsuaki Dan for technical support; and all members of the Departments of Pharmacology and Anatomy for essential cooperation.
Correspondence should be addressed to Masaaki Matsuoka, Department of Pharmacology, Keio University School of Medicine, 35 Shinanomachi, Shinjuku-ku, Tokyo 162-8582, Japan. E-mail: sakimatu@sc.itc.keio.ac.jp.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510252-10$15.00/0
T.C. and M.Y. contributed equally to this work.
References
- Bartus RT, Dean III RL, Beer B, Lippa AS (1982) The cholinergic hypothesis of geriatric memory dysfunction. Science 217: 408-417. [DOI] [PubMed] [Google Scholar]
- Brenneman DE, Gozes I (1996) A femtomolar-acting neuroprotective peptide. J Clin Invest 97: 2299-2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman DE, Hauser J, Neale E, Rubinraut S, Fridkin M, Davidson A, Gozes I (1998) Activity-dependent neurotrophic factor: structure-activity relationships of femtomolar-acting peptides. J Pharmacol Exp Ther 285: 619-627. [PubMed] [Google Scholar]
- Chiba T, Hashimoto Y, Tajima H, Yamada M, Kato R, Niikura T, Terashita K, Schulman H, Aiso S, Kita Y, Matsuoka M, Nishimoto I (2004) Neuroprotective effect of activity-dependent neurotrophic factor against toxicity from familial amyotrophic lateral sclerosis-linked mutant SOD1 in vitro and in vivo. J Neurosci Res 78: 542-552. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Price DL, DeLong MR (1983) Alzheimer's disease: a disorder of cortical cholinergic innervation. Science 216: 1184-1190. [DOI] [PubMed] [Google Scholar]
- D'Adamio L, Lacana E, Vito P (1997) Functional cloning of genes involved in T-cell receptor-induced programmed cell death [review]. Semin Immunol 9: 17-23. [DOI] [PubMed] [Google Scholar]
- Delobette S, Privat A, Maurice T (1997) In vitro aggregation facilities β-amyloid peptide-(25-35)-induced amnesia in the rat. Eur J Pharmacol 319: 1-4. [DOI] [PubMed] [Google Scholar]
- Ebert U, Kirch W (1998) Scopolamine model of dementia: electroencephalogram findings and cognitive performance. Eur J Clin Invest 28: 944-949 Rev. [DOI] [PubMed] [Google Scholar]
- Fisher A, Brandeis R, Pittel Z, Karton I, Sapir M, Dachir S, Levy A, Heldman E (1989) (+)-cis-2-Methyl-spiro(1,3-oxathiolane-5,3′) quinuclidine (AF102B): a new M1 agonist attenuates cognitive dysfunctions in AF64A-treated rats. Neurosci Lett 102: 325-331. [DOI] [PubMed] [Google Scholar]
- Flood JF, Morley JE, Roberts E (1991) Amnestic effects in mice of four synthetic peptides homologous to amyloid beta protein from patients with Alzheimer disease. Proc Natl Acad Sci USA 88: 3363-3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frautschy SA, Baird A, Cole GM (1991) Effects of injected Alzheimer beta-amyloid cores in rat brain. Proc Natl Acad Sci USA 88: 8362-8366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozes I, Giladi E, Pinhasov A, Bardea A, Brenneman DE (2000) Activity-dependent neurotrophic factor: intranasal administration of femtomolar-acting peptides improve performance in a water maze. J Pharmacol Exp Ther 293: 1091-1098. [PubMed] [Google Scholar]
- Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC (2003) Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 423: 456-461. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353-356. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Ito Y, Nishimoto I (2000) Multiple mechanisms underlie neurotoxicity by different types of Alzheimer's disease mutations of amyloid precursor protein. J Biol Chem 275: 34541-34551. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, Sobue G, Koide T, Tsuji S, Lang J, Kurokawa K, Nishimoto I (2001a) A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Aβ. Proc Natl Acad Sci USA 98: 6336-6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Ito Y, Sudo H, Hata M, Arakawa E, Abe Y, Kita Y, Nishimoto I (2001b) Detailed characterization of neuroprotection by a rescue factor humanin against various Alzheimer's disease-relevant insults. J Neurosci 21: 9235-9245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Tsuji O, Niikura T, Yamagishi Y, Ishizaka M, Kawasumi M, Chiba T, Kanekura K, Yamada M, Tsukamoto E, Kouyama K, Terashita K, Aiso S, Lin A, Nishimoto I (2003) Involvement of c-Jun N-terminal kinase in amyloid precursor protein-mediated neuronal cell death. J Neurochem 84: 864-877. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Suzuki H, Aiso S, Niikura T, Nishimoto I, Matsuoka M (2005) Involvement of tyrosine kinases and STAT3 in Humanin-mediated neuroprotection. Life Sci 77: 3092-3104. [DOI] [PubMed] [Google Scholar]
- Kawasumi M, Hashimoto Y, Chiba T, Kanekura K, Yamagishi Y, Ishizaka M, Tajima H, Niikura T, Nishimoto I (2002) Molecular mechanisms for neuronal cell death by Alzheimer's amyloid precursor protein-relevant insults. Neurosignals 11: 236-250. [DOI] [PubMed] [Google Scholar]
- Kawasumi M, Chiba T, Yamada M, Miyamae-Kaneko M, Matsuoka M, Nakahara J, Tomita T, Iwatsubo T, Kato S, Aiso S, Nishimoto I, Kouyama K (2004) Targeted introduction of V642I mutation in amyloid precursor protein gene causes functional abnormality resembling early stage of Alzheimer's disease in aged mice. Eur J Neurosci 19: 2826-2838. [DOI] [PubMed] [Google Scholar]
- Kowall NW, McKee AC, Yankner BA, Beal MF (1992) In vivo neurotoxicity of beta-amyloid [beta(1-40)] and the beta(25-35) fragment. Neurobiol Aging 13: 537-542. [DOI] [PubMed] [Google Scholar]
- Krejcova G, Patocka J, Slaninova J (2004) Effect of humanin analogues on experimentally induced impairment of spatial memory in rats. J Pept Sci 10: 636-639. [DOI] [PubMed] [Google Scholar]
- Lane RM, Kivipelto M, Greig NH (2004) Acetylcholinesterase and its inhibition in Alzheimer disease [review]. Clin Neuropharmacol 27: 141-149. [DOI] [PubMed] [Google Scholar]
- Mamiya T, Ukai M (2001) [Gly(14)]-Humanin improved the learning and memory impairment induced by scopolamine in vivo. Br J Pharmacol 134: 1597-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP (2004) Pathways towards and away from Alzheimer's disease. Nature 430: 631-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ, Cacabelos R (1998) Beta-amyloid(1-40)-induced neurodegeneration in the rat hippocampal neurons of the CA1 subfield. Acta Neuropathol 95: 455-465. [DOI] [PubMed] [Google Scholar]
- Minami M, Inoue M, Wei S, Takeda K, Matsumoto M, Kishimoto T, Akira S (1996) STAT3 activation is a critical step in gp130-mediated terminal differentiation and growth arrest of a myeloid cell line. Proc Natl Acad Sci USA 93: 3963-3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Takio K, Ogawara M, Selkoe DJ (1992) Mass spectrometry of purified amyloid beta protein in Alzheimer's disease. J Biol Chem 267: 17082-17086. [PubMed] [Google Scholar]
- Niikura T, Hashimoto Y, Tajima H, Nishimoto I (2002) Death and survival of neuronal cells exposed to Alzheimer's insults. J Neurosci Res 70: 380-391 Rev. [DOI] [PubMed] [Google Scholar]
- Niikura T, Chiba T, Aiso S, Matsuoka M, Nishimoto I (2004) Humanin: after the discovery. Mol Neurobiol 30: 327-340. [DOI] [PubMed] [Google Scholar]
- Nishimoto I, Matsuoka M, Niikura T (2004) Unravelling the role of Humanin. Trends Mol Med 10: 102-105. [DOI] [PubMed] [Google Scholar]
- Nitta A, Fukuta T, Hasegawa T, Nabeshima T (1997) Continuous infusion of β-amyloid protein into the rat cerebral ventricle induces learning impairment and neuronal and morphological degeneration. Jpn J Pharmacol 73: 51-57. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB (2000) The mouse brain in stereotaxic coordinates, Ed 2. New York: Academic.
- Platika D, Boulos MH, Baizer L, Fishman MC (1985) Neuronal traits of clonal cell lines derived by fusion of dorsal root ganglia neurons with neuroblastoma cells. Proc Natl Acad Sci USA 82: 3499-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ, Memantine Study Group (2003) Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med 348: 1333-1341. [DOI] [PubMed] [Google Scholar]
- Rohn TT, Ivins KJ, Bahr BA, Cotman CW, Cribbs DH (2000) A monoclonal antibody to amyloid precursor protein induces neuronal apoptosis. J Neurochem 74: 2331-2342. [DOI] [PubMed] [Google Scholar]
- Ryu JK, Franciosi S, Sattayaprasert P, Kim SU, McLarnon JG (2004) Minocycline inhibits neuronal death and glial activation induced by beta-amyloid peptide in rat hippocampus. Glia 48: 85-90. [DOI] [PubMed] [Google Scholar]
- Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, Schneider LS, Thal LJ (1997) A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's Disease Cooperative Study. N Engl J Med 336: 1216-1222. [DOI] [PubMed] [Google Scholar]
- Shastry BS, Giblin FJ (1999) Genes and susceptible loci of Alzheimer's disease. Brain Res Bull 48: 121-127. [DOI] [PubMed] [Google Scholar]
- Stepanichev MY, Zdobnova IM, Yakovlev AA, Onufriev MV, Lazareva NA, Zarubenko II, Gulyaeva NV (2003a) Effects of tumor necrosis factor-alpha central administration on hippocampal damage in rat induced by amyloid beta-peptide (25-35). J Neurosci Res 71: 110-120. [DOI] [PubMed] [Google Scholar]
- Stepanichev MY, Moiseeva YV, Lazareva NA, Onufriev MV, Gulyaeva NV (2003b) Single intracerebroventricular administration of amyloid-beta (25-35) peptide induces impairment in short-term rather than long-term memory in rats. Brain Res Bull 61: 197-205. [DOI] [PubMed] [Google Scholar]
- Stephan A, Laroche S, Davis S (2001) Generation of aggregated β-amyloid in the rat hippocampus impairs synaptic transmission and plasticity and causes memory deficits. J Neurosci 21: 5703-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo H, Jiang H, Yasukawa T, Hashimoto Y, Niikura T, Kawasumi M, Matsuda S, Takeuchi Y, Aiso S, Matsuoka M, Murayama Y, Nishimoto I (2000) Antibody-regulated neurotoxic function of cell-surface beta-amyloid precursor protein. Mol Cell Neurosci 16: 708-723. [DOI] [PubMed] [Google Scholar]
- Sudo H, Hashimoto Y, Niikura T, Shao Z, Yasukawa T, Ito Y, Yamada M, Hata M, Hiraki T, Kawasumi M, Kouyama K, Nishimoto I (2001) Secreted Abeta does not mediate neurotoxicity by antibody-stimulated amyloid precursor protein. Biochem Biophys Res Commun 282: 548-556. [DOI] [PubMed] [Google Scholar]
- Tajima H, Kawasumi M, Chiba T, Yamada M, Yamashita K, Nawa M, Kita Y, Kouyama K, Aiso S, Matsuoka M, Niikura T, Nishimoto I (2005) A Humanin derivative, S14G-HN, prevents amyloid-β-induced memory impairment in mice. J Neurosci Res 79: 714-723. [DOI] [PubMed] [Google Scholar]
- Terashita K, Hashimoto Y, Niikura T, Tajima H, Yamagishi Y, Ishizaka M, Kawasumi M, Chiba T, Kanekura K, Yamada M, Nawa M, Kita Y, Aiso S, Nishimoto I (2003) Two serine residues distinctly regulate the rescue function of Humanin, an inhibiting factor of Alzheimer's disease-related neurotoxicity: functional potentiation by isomerization and dimerization. J Neurochem 85: 1521-1538. [DOI] [PubMed] [Google Scholar]
- Yamada K, Tanaka T, Mamiya T, Shiotani T, Kameyama T, Nabeshima T (1999) Improvement by nefiracetam of β-amyloid-(1-42)-induced learning and memory impairments in rats. Br J Pharmacol 126: 235-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Chiba T, Sasabe J, Nawa M, Tajima H, Niikura T, Terashita K, Aiso S, Kita Y, Matsuoka M, Nishimoto I (2005) Implanted cannula-mediated repetitive administration of Aβ25-35 into the mouse cerebral ventricle effectively impairs spatial working memory. Behav Brain Res 164: 139-146. [DOI] [PubMed] [Google Scholar]
- Yamagishi Y, Hashimoto Y, Niikura T, Nishimoto I (2003) Identification of essential amino acids in Humanin, a neuroprotective factor against Alzheimer's disease-relevant insults. Peptides 24: 585-595. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Kawashima S (2001) Effects of amyloid-β-(25-35) on passive avoidance, radial-arm maze learning and choline acetyltransferase activity in the rat. Eur J Pharmacol 412: 265-272. [DOI] [PubMed] [Google Scholar]