

Abstract
Within the developing vertebrate spinal cord, motor neuron subtypes are distinguished by the settling positions of their cell bodies, patterns of gene expression, and the paths their axons follow to exit the CNS. The inclusive set of cues required to guide a given motor axon subtype from cell body to target has yet to be identified, in any species. This is attributable, in part, to the unavailability of markers that demarcate the complete trajectory followed by a specific class of spinal motor axons. Most spinal motor neurons extend axons out of the CNS through ventral exit points. In contrast, spinal accessory motor neurons (SACMNs) project dorsally directed axons through lateral exit points (LEPs), and these axons assemble into the spinal accessory nerve (SAN). Here we show that an antibody against BEN/ALCAM/SC1/DM-GRASP/MuSC selectively labels mouse SACMNs and can be used to trace the pathfinding of SACMN axons. We use this marker, together with a battery of transcription factor-deficient or guidance cue/receptor-deficient mice to identify molecules required for distinct stages of SACMN development. Specifically, we find that Gli2 is required for the initial extension of axons from SACMN cell bodies, and that netrin-1 and its receptor Dcc are required for the proper dorsal migration of these cells and the dorsally directed extension of SACMN axons toward the LEPs. Furthermore, in the absence of the transcription factor Nkx2.9, SACMN axons fail to exit the CNS. Together, these findings suggest molecular mechanisms that are likely to regulate key steps in SACMN development.
Keywords: spinal accessory motor neurons, BEN/ALCAM/SC1/DM-GRASP/MuSC, Gli2, netrin, DCC, Nkx2.9, exit point
Introduction
In the vertebrate CNS, specific subclasses of motor neurons compose distinct neural circuits (Jessell, 2000; Landmesser, 2001; Price and Briscoe, 2004). All motor neurons arise from a ventral progenitor domain defined by a set of homeodomain transcription factors that are induced by and interpret a gradient of Sonic hedgehog (Shh) expression (Briscoe et al., 2000). Subsequently, repressive interactions involving homeodomain and basic helix-loop-helix transcription factors give rise to several classes of postmitotic motor neurons (Shirasaki and Pfaff, 2002; Price and Briscoe, 2004). Ultimately, the combinatorial expression of LIM homeodomain transcription factors defines motor neuron subtypes that occupy specific settling positions, select particular axon pathways, and innervate distinct targets (Tsuchida et al., 1994; Sharma et al., 1998; Shirasaki and Pfaff, 2002).
Most motor axons emerge through nearby ventral exit points. However, a subset grow for some distance toward and extend through more dorsally located exit points (Shirasaki and Pfaff, 2002). Consistent with their initial migration away from the ventral midline, both ventrally and dorsally exiting motor axons are repelled by floor plate tissue, in vitro (Guthrie and Pini, 1995). Whereas the ventral midline-associated chemorepellent netrin-1 selectively repels dorsally exiting trochlear axons in vitro (Colamarino and Tessier-Lavigne, 1995a; Guthrie and Pini, 1995), but is apparently not required for repulsion from the ventral midline in vivo (Serafini et al., 1996), ventral spinal cord-associated semaphorins are capable of repelling these axons both in vitro (Varela-Echavarria and Guthrie, 1997; Varela-Echavarria et al., 1997; Giger et al., 2000) and in vivo (Chen et al., 2000; Giger et al., 2000; Sahay et al., 2003). Notably, the molecular mechanisms that facilitate the exit of either type of motor axon from the CNS are not known.
Spinal accessory motor neurons (SACMNs) are located within cervical regions of the spinal cord and selectively innervate the sternocleidomastoid and trapezius muscles in the neck and back (Greene, 1935; Wentworth and Hinds, 1978; Brichta et al., 1987; Snider and Palavali, 1990). Unlike most spinal motor neurons, SACMNs extend dorsally projecting axons along the lateral margins of the spinal cord that extend out of the CNS through the lateral exit point (LEP) (Snider and Palavali, 1990). Subsequently, SACMN axons execute a rostral turn and assemble into the longitudinally oriented spinal accessory nerve (SAN) (Snider and Palavali, 1990). Although the molecular mechanisms that regulate SACMN development have not been directly investigated, all spinal motor neurons acquire SACMN-like properties in mice lacking Lhx3 and Lhx4 (Sharma et al., 1998), and Nkx2.9 null mice exhibit a truncated SAN (Pabst et al., 2003).
Here, we use anti-BEN/ALCAM/SC1/DM-GRASP/MuSC (herein referred to as BEN) as a specific marker of SACMN to outline the steps their axons take to navigate through and out of the CNS. Moreover, we use this marker to analyze mice deficient in Gli2, netrin-1, Dcc, Nkx2.9, or BEN. Our findings identify molecules that are required for the initial extension of SACMN axons, the dorsally directed migration of SACMN cell bodies, the projection of their axons toward the LEP, and the exit of SACMN axons from the CNS.
Materials and Methods
Mice
For the analyses aimed at examining BEN expression in wild-type animals, timed pregnant CD1 mice were obtained from Charles River Laboratories (Wilmington, MA). For the analyses of the various aged Gli2 (Matise et al., 1998), netrin-1 (Serafini et al., 1996), Dcc (Fazeli et al., 1997), Nkx2.9 (Pabst et al., 2003), and BEN (Weiner et al., 2004) mutant mouse embryos, pregnant mice harboring litters containing homozygous, heterozygous, and wild-type animals were generated as described previously. Briefly, Gli2 heterozygotes were maintained on outbred Swiss Webster or CD1 backgrounds. Litters containing homozygous mutants were generated by intercrossing adults and were identified by PCR using yolk sac DNA as described previously (Ding et al., 1998; Matise et al., 1998). Chimeric netrin-1 mutant males were bred to CD1 females (Serafini et al., 1996), and chimeric Dcc mutant males were bred to 129/Sv females (Fazeli et al., 1997). The Nkx2.9 and BEN/ALCAM-GFP lines were maintained on a C57BL/6 background (Pabst et al., 2003; Weiner et al., 2004). The BEN/ALCAM-GFP line was made as described previously (Weiner et al., 2004). In all cases, pregnant dams were killed by exposure to compressed carbon dioxide. Embryos were removed by cesarean section and immersed in PBS (150 mm Na2HPO4, 20 mm NaH2PO4, and 150 mm NaCl, pH 7.4). The genotypes of the various embryos were determined as previously described for Gli2 (Matise et al., 1998), netrin-1 (Serafini et al., 1996), Dcc (Fazeli et al., 1997), Nkx2.9 (Pabst et al., 2003), and BEN (Weiner et al., 2004) mutant mice.
Antibodies
The following monoclonal antibodies were used to label cryosections and/or whole embryos: monoclonal antibody (mAb) 802C11 (Sekine-Aizawa et al., 1998), mAb 33.1 [anti-ALCAM; kind gift of Dr. Michael Bowen, Bristol-Myers Squibb (Wallingford, CT), now at Human Genome Sciences (Rockville, MD)], mAb 2H3 (Dodd et al., 1988), mAb 5272 (anti-mouse L1; rat IgG; Chemicon, Temecula, CA), mAb 347 (anti-GAP43; mouse IgG; Chemicon), and anti-DCC intracellular domain (G97-449; anti-human DCC; mouse IgG; PharMingen, San Diego, CA).
Immunohistochemistry
Cryosection preparation and immunolabeling. Whole mouse embryos were fixed in 4% paraformaldehyde (PFA) for 12 h at 4°C, followed by cryoprotection in 30% sucrose for 6 h at 4°C, and embedded at -20°C in optimal cutting temperature compound (Tissue Tek; Sakura Finetek, Torrance, CA). Cryosections (16 μm) were cut using a Leica (Nussloch, Germany) cryostat (model CM3050 S) and mounted onto glass slides (Superfrost Plus; Fisher Scientific, Houston, TX). The sections were rinsed in PBS, postfixed in 4% PFA for 20 min at room temperature (RT), and blocked with 10% goat serum (HyClone, Logan, UT) in PBS for 30 min at RT. For mAb 802C11 and mAb 5272 labeling, sections were incubated in blocking solution containing 1% Triton X-100 (Fisher Scientific). Sections were then incubated with the primary antibody diluted in blocking solution (or undiluted supernatant) for 90 min at RT and then rinsed in PBS. Primary antibodies were used as follows: mAb 802C11 (20 μg/ml), mAb 33.1 (1:10), mAb 2H3 (hybridoma supernatant), mAb 5272 (10 μg/ml), mAb 347 (0.5 μg/ml), and anti-DCC intracellular domain (2.5 μg/ml). Sections were then incubated with the following concentrations of isotype-appropriate cyanine 2 (Cy2) or Cy3-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA): mAb 802C11 (goat anti-Armenian hamster IgG; 1:100), mAb 33.1 (goat anti-rat IgG; 1:200) mAb 5272 (goat anti-rat IgG; 1:200), mAb 2H3 (goat anti-mouse IgG; 1:200), mAb 347 (goat anti-mouse IgG; 1:200), and anti-DCC intracellular domain (goat anti-mouse IgG; 1:200) for 60 min at RT. Slides were then mounted in a 1:1 mixture of glycerol and PBS. Double labeling of anti-DCC and anti-BEN on the same section was performed by first performing the anti-DCC staining followed by anti-BEN labeling according to the protocols described above, because anti-BEN, but not anti-DCC, labeling requires Triton X-100 (Fisher Scientific) in the blocking solution.
Whole-mount immunohistochemistry. Immunolabeling of whole embryos was performed using a modified version of a previously described protocol (Mukouyama et al., 2002). More specifically, mouse embryos were fixed in 4% PFA for 12 h at 4°C and dehydrated through a series of methanol steps (25% MeOH/PBS, 50% MeOH/PBS, and 75% MeOH/PBS) for 10 min each at RT. Embryos were then stored in 100% MeOH at -20°C. Embryos were rehydrated from 100% MeOH to 100% PBT (PBS, 0.2% BSA, and 0.1% Triton X-100) through a series of steps in MeOH/PBT (75, 50, and 25%) each for 10 min at RT, followed by two 20 min washes in 100% PBT at RT. Whole embryos were then immersed in blocking solution containing 10% heat-inactivated goat serum and 1% Triton X-100 in PBS for 2 h at RT and then incubated with anti-GAP43 (0.5 μl/ml), mAb 2H3 (hybridoma supernatant), or mAb 802C11 (10 μg/ml) diluted in blocking solution containing 10% heat-inactivated goat serum and 1% Triton X-100 overnight at 4°C. After eight 30 min washes at room temperature in PBS containing 2% heat-inactivated goat serum and 0.2% Triton X-100, whole embryos were incubated in PBS containing 10% heat-inactivated goat serum and 1% Triton X-100 for 1 h at RT. After 12 30 min washes at room temperature in 2% heat-inactivated goat serum and 0.2% Triton X-100 in PBS, embryos were treated with ABC solution (Vector Laboratories, Burlingame, CA) for 2 h at RT, followed by visualization with a DAB Substrate kit (Vector Laboratories). Stained whole embryos were then fixed in 4% PFA for 1-2 h at RT, rinsed with PBS, and cleared in a glycerol series to 100% glycerol. For β-galactosidase staining, embryos were fixed in 0.2% glutaraldehyde dissolved in PBS containing 5 mm EGTA and 2 mm MgCl2 for 15 min and washed three times for 15 min each in PBS containing 5 mm EGTA, 2 mm MgCl2, 0.01% NP-40, and 0.1% sodium deoxycholate. Staining was performed in the same buffer with 10 mm K3Fe(CN)6, 10 mm K4Fe(CN)6 and 0.5 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside at 37°C. Embryos were then sectioned using a vibratome and stained for BEN after the whole-mount immunohistochemistry protocol described above.
In situ hybridization
Dcc (Imondi et al., 2000) and Nkx2.9 (Briscoe et al., 1999) cDNAs were generated as described previously. For the BEN probe, the PCR was used to generate ∼1 kb cDNA fragments from embryonic day 11 (E11) mouse cDNA templates. Each cDNA was linearized with the appropriate restriction enzymes and transcribed using the Megascript kit (Ambion, Austin, TX) to generate antisense and sense digoxigenin-labeled riboprobes, and nonradioactive in situ hybridization was performed, all as described previously (Kaprielian et al., 1995), with the following exceptions: cryosections were prehybridized and hybridized at 60°C, and anti-digoxigenin Fab fragments were preadsorbed with extracts derived from E10-E11 mouse embryos. A color reaction was used to detect mRNA expression using 5-bromo-4-chloro-3-indolyl phosphate and nitroblue tetrazolium. Sections incubated with the antisense BEN mRNA probe were developed for 24 h, and Dcc probed sections were developed for 6 h. Subsequently, the sections were fixed in 4% PFA for 2 h, rinsed in PBS, and mounted in a 1:1 mixture of glycerol and PBS. For each gene, in situ hybridization performed with the sense probe yielded no specific signal (data not shown).
DiI injections
To directly visualize the trajectory of SACMNs, DiI (Invitrogen, Eugene, OR) dissolved in dimethylformamide (Sigma, St. Louis, MO) at a concentration of 3 mg/ml was iontophoretically applied into the SAN in thick (150 μm) vibratome sections of E12 mouse embryos through the use of glass electrodes with broken tips (50 μm in diameter) and a nanoject II automatic nanoliter injector (Drummond Scientific, Broomall, PA). The sections were then placed in fixative and stored at room temperature for 24 h to allow for retrograde labeling of SACMNs. Subsequently, DiI labeling was visualized with epifluorescence optics (Nikon, Tokyo, Japan) using a Cy3/DiI optical filter (Chroma Technology, Brattleboro, VT).
Photodocumentation
Primary antibody binding to transverse cryosections was visualized under epifluorescence optics (Nikon Eclipse TE300) using Cy2 or Cy3 optical filters (Chroma Technology). Black and white images were captured with a digital camera manufactured by Optronics (Bolton, MA) and compatible Magnafire software. Photomicrographs of whole mount-immunolabeled embryos were taken using an Olympus Optical (Tokyo, Japan) SZX-ZB12 stereomicroscope that was fitted onto a dark-field base and equipped for use as a macroscope and Eastman Kodak (Rochester, NY) 64T Ektachrome color film. Color slides were digitally captured using an Agfa Gevaert (Leverkusen, Germany) Duoscan flatbed scanner. In all cases, composites were assembled and annotated using Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA). For color images, the brightness, contrast, and/or color balance was adjusted in Photoshop for increased visibility of nerves/axons and for color matching. For black and white images, only the brightness and contrast were adjusted for increased visibility of cell bodies and axons. Sections subjected to in situ hybridization were visualized using an Olympus Optical AX70 microscope. Images of these sections were captured using a digital camera and compatible Magnafire software.
Quantification
To quantify the cell body/axon migration phenotype observed in the Dcc and netrin-1 mutant embryos, 10× micrographs of cervical sections derived from mutant or wild-type embryos were first opened in Photoshop 7.0. A grid was then created in Photoshop that covered the ventral half of the spinal cord, which contains SACMNs and the LEP. The width and height of each box in the grid is 20 μm, and each box represents one graphed “unit” (see Fig. 5D-F). The grid was placed as a new layer on top of each micrograph, and the region containing BEN-positive SACMNs was counted in boxes (units) along both the mediolateral (M-L) (medial being closest to the LEP) and the dorsoventral (D-V) axis (dorsal being closest to the LEP; see Fig. 5D). The average distance of BEN-positive SACMNs from the LEP, along both of these axes, was graphed (Fig. 5E) for netrin-1 or Dcc mutant embryos and their respective wild-type littermate controls. The area spread of BEN-positive SACMNs, defined as the product of the M-L and D-V distances, was also determined for each of the mutants and their wild-type controls and represented graphically (Fig. 5F).
Figure 5.

SACMN cell bodies fail to migrate dorsally, and some of their axons inappropriately extend toward the floor plate, in both Dcc and netrin-1 null embryos. A-C, Anti-BEN labeling of transverse, cervical spinal cord-containing cryosections derived from E11.5 wild-type and Dcc or netrin-1 null embryos. Anti-BEN selectively labels small clusters of SACMNs (arrowheads), which are appropriately positioned just beneath the LEP, as well as the SAN (arrows) in an E11.5 wild-type embryo (A). In a Dcc null littermate (B), anti-BEN labels disorganized clusters of SACMN cell bodies, many of which have failed to migrate to, and settle within, the vicinity of the LEP (arrowheads). Similarly disorganized anti-BEN-positive SACMN cell bodies, as well as a SACMN axon that appears to be inappropriately projecting toward the floor (arrowheads), are present in a netrin-1 null E11.5 embryo (C). D-F, Analysis and quantification of the defects observed in Dcc and netrin-1 mutant embryos. D, A schematic representation of the spinal cord indicating that SACMN/axon migration defects were scored along the D-V axis between the LEP and the ventral margin of the spinal cord, as well as along the M-L axis, between the LEP and the central canal. E, BEN-positive SACMNs were detected at significantly greater distances from the LEP in Dcc and netrin-1 mutants compared with their wild-type littermates, along both the dorsoventral and mediolateral axes. F, The area spread (product of average dorsoventral and mediolateral distances) of BEN-positive SACMNs was significantly greater in Dcc and netrin-1 mutants than in their wild-type littermates. Values shown are means; *p < 0.05, **p < 0.005. 1 unit is equivalent to 20 μm. B, n = 11; C, n = 4. Scale bar, 100 μm.
Statistical analyses
Linear mixed models were fitted using SAS PROC MIXED (version 9.1) to analyze the data. The number of units in the M-L direction, D-V direction, and area spread of BEN-positive SACMNs were evaluated. In all cases, these cells spread significantly more in the mutants than those in wild-type littermates.
Results
Anti-BEN is a marker of spinal accessory motor neurons in mouse embryos
We recently demonstrated that mAb SAC1 selectively labels SACMN in the embryonic rat spinal cord (Schubert and Kaprielian, 2001) and recognizes the rat ortholog of BEN/ALCAM/SC1/DM-GRASP/MuSC, a homophilic adhesion molecule that regulates axon outgrowth/fasciculation (Leppert et al., 1999; Schubert and Kaprielian, 2001; Avci et al., 2004; Weiner et al., 2004). Here we show that anti-BEN (Sekine-Aizawa et al., 1998) labels mouse SACMNs, their axons, and the SAN (Fig. 1A) and use this marker to selectively examine SACMN development in a variety of mutant mice.
Figure 1.

Anti-BEN is a marker of SACMNs, their axons, and the SAN in the developing mouse spinal cord. A, Schematic representation of SACMN cell bodies, the trajectory of SACMN axons, and the ascending SAN. At cervical levels of embryonic mouse spinal cord, SACMN cell bodies and their dorsally projecting axons selectively express the murine SC1-related protein (BEN). BEN continues to be expressed on these axons as they emerge from the spinal cord through the LEP, execute a rostral turn, and assemble into the longitudinally oriented SAN. A, Anterior; P, posterior; D, dorsal; V, ventral. B-D, Anti-BEN labeling of transverse cryosections derived from cervical spinal cord levels of various aged mouse embryos. At E9.25 (B), anti-BEN specifically labels SACMN cell bodies that line the lateral edges of the ventral and intermediate spinal cord (arrowheads) and their dorsally directed axons, which have just begun to exit the spinal cord through the LEP (arrows). One day later, at E10.5 (C), anti-BEN intensely and selectively labels ventrolaterally and intermediolaterally positioned SACMN cell bodies and their dorsally projecting axons, which have extended out of the spinal cord and assembled into the longitudinally oriented SAN (arrows). At E11.5 (D), anti-BEN labels SACMN cell bodies that have migrated to a more dorsal position just below the LEP (arrowheads), as well as the SAN (arrows). Anti-BEN immunoreactivity is also associated with dorsal root ganglia located adjacent to the LEP on either side of the spinal cord, as well as the neuroepithelium that lines the dorsal surface of the spinal cord. At each of these ages, anti-BEN also labels the floor plate and notochord. E, At E11.5, anti-ALCAM, a different anti-BEN mAb, also labels SACMN cell bodies that have migrated to a more dorsal position just below the LEP (arrowheads) and the SAN (arrows), as well as the floor plate (FP). Scale bars, 100 μm. FP, Floor plate; NC, notochord.
To determine whether BEN is selectively expressed by SACMNs in mice, we labeled spinal cord-containing transverse cryosections derived from cervical regions of various aged mouse embryos with an anti-BEN mAb (Sekine-Aizawa et al., 1998). Consistent with our previous findings in rat (Schubert and Kaprielian, 2001), BEN is transiently expressed by SACMNs in the developing mouse spinal cord. As early as E9.25, anti-BEN labeled SACMN cell bodies and their axons, which project dorsally to the LEP (Fig. 1B), as well as the notochord and floor plate. At E10.5, anti-BEN-positive SACMN cell bodies lined the lateral edge of the spinal cord from a ventral position near the floor plate to a more dorsal position adjacent to the LEP (Fig. 1C, arrowheads). Anti-BEN labeling was also observed on dorsally directed SACMN axons, including those segments that had extended out of the LEP, turned into the longitudinal plane, and assembled into the SAN (Fig. 1C, arrows). A day later, at E11.5, BEN-expressing SACMN cell bodies had migrated dorsally and were clustered in close proximity to the LEP (Fig. 1D, arrowheads). To further confirm that mouse SACMNs express BEN during this period of development, we labeled transverse cryosections derived from cervical levels of an E11.5 mouse embryo with anti-ALCAM, an independently generated mAb against BEN (Weiner et al., 2004). Consistent with anti-BEN labeling at E11.5 (Fig. 1D), anti-ALCAM also labels SACMNs and the SAN (Fig. 1E, arrowheads and arrows), as well as the floor plate. The close similarity between anti-BEN and anti-ALCAM labeling supports our finding that BEN expression marks SACMNs. Consistent with SACMNs being confined to C1-C4 levels of the rodent spinal cord (Greene, 1935; Wentworth and Hinds, 1978; Gottschall et al., 1980; Brichta et al., 1987), anti-BEN-positive cell bodies and axons were not detected caudal to the cervical spinal cord in mouse embryos (data not shown). To further support our identification of anti-BEN as a marker of SACMN, we retrogradely labeled SACMN by introducing small crystals of DiI into the SAN in E12 wild-type mouse embryos. This resulted in the labeling of SACMN cell bodies and dorsally directed axons (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). By E12, BEN expression was extinguished on the SAN and SACMN axons and was only present on SACMN cell bodies. By E12.5, BEN expression was no longer specific to SACMN and was also observed on subsets of ventrally positioned motor neurons that project axons out of the spinal cord through the ventral roots (data not shown).
As described above, BEN is expressed on SACMN axons as they exit the spinal cord and assemble into the SAN. To determine the rostrocaudal extent of BEN expression on the SAN, we labeled whole E10 mouse embryos with anti-BEN. Compared with the distribution of the pan-neuronal markers anti-neurofilament (NF) (Fig. 2A,B) and anti-GAP43 (Fig. 2E,F), which also label dorsal root ganglia, ventral roots, and cranial ganglia (Fig. 2), BEN is selectively expressed all along the hook-shaped SAN (Fig. 2C,D, arrow). The hook-shaped axon bundle appears thicker in the anti-NF- and anti-GAP43-labeled embryos, because these markers also label other nerves, which travel alongside the SAN (e.g., vagus nerve). It is important to note that the spinal accessory nerve, or cranial nerve XI, is generally considered to be composed of a cranial root and a spinal root. The cranial root originates from SACMN cell bodies in the medulla and innervates the soft palate and larynx muscles, whereas the spinal root originates from cell bodies found between C1 and C4 levels of the spinal cord and innervates the sternocleidomastoid and trapezius muscles (Wilson-Pauwels et al., 1988). Thus, BEN expression on the SAN extends from the hindbrain (the cranial root of the SAN) through the C4 level, which represents the caudal limit of the SAN in the spinal cord (spinal root).
Figure 2.

Anti-BEN labels the entire rostrocaudal extent of the SAN. A-F, Whole mouse embryos were labeled with anti-NF, anti-BEN, or anti-GAP43. The higher-magnification images in B, D, and F represent cervical spinal cord levels of the labeled embryos shown in A, C, and E, respectively. Anti-NF (A, B) brightly labels the hook-shaped SAN (arrow), as well as a variety of axons and cranial nerves/ganglia in an E10.5 mouse embryo. In a whole E10 mouse embryo, anti-BEN (C, D) rather selectively labels the SAN (arrow) and a small subset of cranial nerves/ganglia. Anti-GAP43 (E, F) labels the SAN (arrow) and a significant number of axons/nerves in a whole E10.5 mouse embryo. The thicker appearance of the hook-shaped axon bundle in the anti-NF and anti-GAP43 panels is likely attributable to these more general axonal markers, but not anti-BEN, also labeling the vagus nerve, which runs alongside the SAN. Scale bar, 0.5 mm.
Together with the results of previous anatomical (Greene, 1935; Wentworth and Hinds, 1978; Gottschall et al., 1980; Brichta et al., 1987; Snider and Palavali, 1990) and immunohistochemical (Schubert and Kaprielian, 2001) studies, the distribution of BEN suggests that SACMN/SAN development involves the following sequence of events: (1) ventrolaterally positioned SACMN cell bodies extend dorsally projecting axons away from the ventral midline, (2) SACMN cell bodies migrate dorsally and settle in the vicinity of the LEP, (3) SACMN axons extend toward, and exit through, the LEP, and (4) SACMN axons assemble into the longitudinally projecting SAN (Fig. 1A). In an attempt to identify molecules required for each of these steps, we examined the development of anti-BEN-positive SACMNs and their axons in a variety of mutant mice.
The spinal accessory nerve fails to form in Gli2-deficient mice
Based on their demonstrated ability to repel a variety of motor axons in vitro (Colamarino and Tessier-Lavigne, 1995a; Guthrie and Pini, 1995; Varela-Echavarria and Guthrie, 1997; Varela-Echavarria et al., 1997; Giger et al., 2000), floor plate-associated chemorepellents are good candidates for directing SACMN axons along a dorsally directed trajectory away from the ventral midline and toward the LEP. The floor plate fails to form in the spinal cords of mice that lack the Gli2 zinc-finger domain containing transcription factor (Matise et al., 1998). Thus, the Gli2 null mutant represents an in vivo system for assessing the role of the floor plate on SACMN development. However, the lack of a floor plate in this mutant is likely attributable to the loss of Gli2 itself and the subsequent disruption of Shh signaling. Therefore, the reduced expression of transcription factors and molecules normally found in the ventral spinal cord must be considered when interpreting the Gli2 mutant phenotype. That said, patterning in the ventral spinal cord of Gli2 mutants has been shown to be wild type-like, except for the absence of floor plate cells and the immediately adjacent V3 interneurons (Matise et al., 1998), and ventrally exiting motor neurons develop normally (Ding et al., 1998; Matise et al., 1999). Collectively, these observations support the use of the Gli2 mutant as an in vivo model for assessing the impact of floor plate-derived factors on SACMN.
To determine whether Gli2 is required for SACMN/SAN development, we labeled transverse cryosections derived from cervical and limb spinal cord levels of Gli2-null mice with anti-BEN. As described above, in wild-type mouse embryos, BEN is expressed by SACMNs and their axons as early as E9.25 (Fig. 1B) exclusively at cervical levels of the spinal cord (data not shown). In contrast, BEN-positive SACMN cell bodies were not detected between E9 and E11.5 in Gli2 null embryos. However, by E12, anti-BEN labeled SACMN cell bodies in homozygous embryos. These BEN-positive cell bodies were restricted to cervical levels of the spinal cord, suggesting that these late-developing motor neurons were, in fact, SACMNs. Consistent with the inappropriate medial positioning of ventrally exiting motor neurons in the absence of a floor plate (Ding et al., 1998; Matise et al., 1998), these SACMN cell bodies also converged on the ventral midline in Gli2 mutant embryos (Fig. 3E,F). However, in contrast to the finding that ventral roots form on schedule in animals lacking Gli2 (Ding et al., 1998; Matise et al., 1999), BEN-positive cell bodies located in cervical regions of the E12 Gli2 mutant spinal cord failed to extend axons. Antibodies that label SACMN axons as they extend toward the LEP, in wild-type embryos, failed to detect any dorsally projecting axons in the Gli2 null spinal cord. In addition, neither anti-BEN nor anti-neurofilament labeled the SAN in transverse cryosections. Whole-mount immunohistochemistry performed with anti-neurofilament and anti-BEN confirmed the absence of the entire SAN, both cranial and spinal portions, in E11Gli2 null embryos (Fig. 3B,D) compared with their wild-type littermates (Fig. 3A,C). To determine whether the absence of a floor plate simply delays SAN development, we labeled transverse cryosections derived from E13.5 Gli2-/- embryos with an antibody specific for the Ig domain-containing cell adhesion molecule L1. In E13.5 wild-type embryos, at a developmental stage when BEN is no longer expressed on SACMN, their axons, and the SAN, L1 was selectively expressed on those segments of SACMN axons that had exited the spinal cord and assembled into the longitudinally projecting SAN (Fig. 3G, arrows). In contrast, anti-L1 labeling clearly revealed a lack of the SAN in E13.5 Gli2 mutant littermates (Fig. 3H, arrows). As noted above, it is difficult to discern whether the observed SACMN defects are attributable to the absence of Gli2 itself or result from the loss of floor plate-derived factors. To circumvent this issue, we opted to take a candidate approach to identify genes that may have more selective roles in SACMN development.
Figure 3.

The SAN fails to develop, and late-developing anti-BEN-positive cell bodies do not extend axons, in floor plate-lacking Gli2 null mice. A-D, Whole-mount immunohistochemistry was used to examine the distribution of NF and BEN in E11 Gli2-deficient mouse embryos and their wild-type littermates. In E11 wild-type embryos, anti-NF (A) labels the SAN (arrow) and segmentally arranged dorsal root ganglia, whereas anti-BEN (C) immunoreactivity is rather selectively associated with the SAN (arrow). Anti-NF (B) and anti-BEN (D) do not label the SAN (arrows) in E11 Gli2 null embryos. E, F, Anti-BEN labeling of transverse, cervical spinal cord-containing cryosections derived from E12 Gli2-deficient and wild-type littermates. In E12 wild-type embryos (E), BEN is expressed by dorsomedially positioned SACMN cell bodies on either side of the ventral midline (arrows), as well as by the floor plate. Anti-BEN selectively labels cell bodies that are presumably SACMNs and have aberrantly converged on the ventral midline (arrows) in floor plate-lacking Gli2-deficient littermates (F). G, H, Anti-L1 labeling of transverse, cervical spinal cord-containing cryosections derived from E13.5 wild-type and Gli2 null embryos. Anti-L1 labels the longitudinally oriented SAN (arrows) in an E13.5 wild-type embryo (G) but reveals the selective absence of the SAN (arrows) in a Gli2-deficient littermate (H). B, D, n = 11; F, n = 3; H, n = 4. Scale bars: A-D, 1 mm; G, H, 100 μm.
Coexpression of DCC and BEN in SACMN
Netrin-1 functions as a chemoattractant for spinal commissural axons in rodents (Placzek et al., 1990; Kennedy et al., 1994; Serafini et al., 1994, 1996). Netrin-1 has also been shown to mediate both attractive and repulsive migration/guidance events through the DCC receptor (Colamarino and Tessier-Lavigne, 1995a; Keino-Masu et al., 1996; Varela-Echavarria et al., 1997; Schwarting et al., 2001; Jarjour et al., 2003). As a first step toward determining whether netrin-1 regulates the development of SACMN and their axons, we used in situ hybridization to compare the distribution of Dcc mRNA with the positions of BEN-expressing SACMN cell bodies in serial transverse cryosections derived from the cervical level of an E10.5 mouse embryo. Consistent with the distribution of anti-BEN-positive cell bodies at this developmental stage (Fig. 1C), BEN mRNA is expressed by small clusters of cell bodies located in ventrolateral and intermediolateral regions of the spinal cord, as well as by the floor plate and notochord (Fig. 4A). Dcc mRNA is rather broadly expressed throughout a region that contains ventrally located motor neurons, including BEN-expressing SACMNs, as well as more dorsally positioned interneurons (Fig. 4B). This overlapping expression pattern raised the possibility that netrin-1 via DCC regulates the development of SACMNs and their axons.
Figure 4.

Coexpression of Dcc and BEN on SACMNs. A, B, In situ hybridization was performed on transverse cryosections derived from cervical levels of an E10 mouse embryo using digoxigenin-labeled BEN or Dcc riboprobes. BEN mRNA (A) is expressed by small clusters of ventrolaterally and intermediolaterally positioned SACMN cell bodies (arrowheads), as well as in the notochord and floor plate. Dcc mRNA (B) is expressed in a broad lateral domain that is likely to contain ventrally located motor neurons and several populations of dorsal interneurons, as well as BEN-positive SACMN cell bodies. C-E, Double-antibody labeling was performed on transverse cryosections derived from cervical levels of an E10.5 mouse embryo using anti-BEN and anti-DCC. In the same section, anti-BEN (C, arrows) and anti-DCC (D, arrows) label SACMN cell bodies and their axons, which are positioned along the lateral margins of the spinal cord. A merged image reveals significant overlap (yellow) between anti-DCC and anti-BEN labeling (E, arrows). Scale bars, 100 μm.
To more directly address the possibility that SACMNs coexpress BEN and DCC, we double labeled transverse cryosections derived from cervical regions of E10.5 embryos with anti-BEN and anti-DCC. As described above, at E10.5, anti-BEN-positive SACMNs were detected along the ventrolateral edge of the spinal cord as their axons extend toward the LEP (Fig. 4C, arrows). In the same section, anti-DCC also labeled cell bodies that lined the ventrolateral edges of the spinal cord (Fig. 4D, arrows). A merged image (Fig. 4E, arrows) reveals colocalization of DCC and BEN in SACMNs (yellow, arrows).
Aberrant migration of SACMN cell bodies/axons in mice lacking netrin-1 or Dcc
To directly determine whether netrin-1 and/or DCC are required for the proper development of SACMNs, we examined the distribution of BEN in mice lacking functional netrin-1 (Serafini et al., 1996) or DCC (Fazeli et al., 1997). Specifically, we labeled transverse cryosections derived from the cervical level of netrin-1 or Dcc homozygous E11.5 embryos and their heterozygous and wild-type littermates with anti-BEN. By E11.5, in wild-type mouse embryos, anti-BEN-positive SACMN cell bodies are clustered near the LEP (Fig. 5A, arrowheads). In embryos lacking Dcc or netrin-1, however, SACMN cell bodies were scattered throughout the ventral spinal cord and failed to settle in close proximity to the LEP (Fig. 5 B, C, arrowheads). In addition, some anti-BEN-labeled SACMN axons aberrantly extended along a ventrally directed trajectory toward the floor plate (Fig. 5C, bottom left arrowhead). To quantify the migration defects observed in the netrin-1 and Dcc mutants, we scored the distance at which BEN-positive SACMNs were found from the LEP along the M-L and D-V axes (Fig. 5 D, E). In addition, we determined the area occupied by BEN-positive SACMNs, defined by the product of the M-L and D-V distance measurements (Fig. 5F). Using each of these criteria, anti-BEN-positive SACMNs were observed to spread more significantly in the mutant embryos compared with their wild-type littermate controls (Fig. 5). We predicted that the defective migration of a subset of SACMN cell bodies and the inappropriate projection of their axons toward the floor plate in Dcc and netrin-1 mutants would reduce the number of SACMN axons that ultimately exit the spinal cord. Consistent with this possibility, anti-BEN-labeled E10.5 Dcc null embryos displayed what appeared to be a thinner SAN than their heterozygous and wild-type littermates (data not shown). Together, these results suggest that netrin-1 is likely to function through DCC to repel SACMN cell bodies and their axons away from the ventral midline.
Nkx2.9 is downregulated in Gli2 null mice and is likely to be expressed in SACMN progenitors
Additional candidates for genes that may regulate specific aspects of SACMN development include those that are downregulated in Gli2 null embryos (Ding et al., 1998; Matise et al., 1998; Matise et al., 1999) and/or that are expressed by SACMNs. The Nkx family of homeodomain-containing transcription factors control patterning and cell fate determination in ventral regions of the developing vertebrate spinal cord in which their progenitor cell-associated expression is regulated by Sonic hedgehog signaling (Ericson et al., 1997). Transcription factors that are expressed in the ventral spinal cord, such as Gli1 and Nkx2.2, and that represent downstream effectors of the Sonic hedgehog signaling path-way are downregulated in Gli2-deficient embryos (Ding et al., 1998; Matise et al., 1998).
Consistent with a role for Nkx2.9 in SACMN development, we recently reported that the SAN fails to form properly in Nkx2.9 null mice (Pabst et al., 2003). To determine whether Nkx2.9 expression is dysregulated in the absence of Gli2 in cervical regions of the spinal cord that normally contain SACMNs, we performed in situ hybridization on transverse cryosections derived from E10 Gli2 embryos. In E10 wild-type embryos, at SACMN-containing cervical spinal cord levels, Nkx2.9 mRNA is expressed in a region that is just dorsal to the floor plate (Fig. 6A). However, in E10 Gli2 null mice, which lack BEN-positive SACMNs, Nkx2.9 mRNA expression is downregulated to essentially undetectable levels (Fig. 6B), consistent with the previously observed downregulation of Nkx2.9 protein expression at thoracic levels of the spinal cord (Lei et al., 2004). To determine whether Nkx2.9 is expressed by SACMN progenitors, we examined the distribution of SACMNs in mutant mice in which the first and most of the second exon of the Nkx2.9 gene had been replaced by the lacZ reporter gene. Specifically, we labeled transverse cryosections derived from E10.5 β-galactosidase-stained Nkx 2.9/lacZ embryos with anti-BEN (Fig. 6C). Because of the perdurance of lacZ expression in these embryos, we were able to compare the distribution of cells that arise from Nkx2.9-expressing progenitors with the expression of BEN in a single section. This analysis revealed that Nkx2.9 is likely to be expressed by progenitors that give rise to BEN-positive SACMNs. Together, the significant downregulation of Nkx2.9 mRNA in the Gli2 mutant spinal cord and the likely expression of Nkx2.9 in SACMN progenitors suggest that, in addition to the loss of guidance cues in the Gli2 mutant, a change in transcriptional regulation may account for the severe SACMN phenotype observed in these animals.
Figure 6.

Nkx2.9 mRNA expression is downregulated in Gli2 null mice; SACMN progenitors are likely to express Nkx2.9, and SACMN axons fail to exit the spinal cord in mice lacking Nkx2.9. A-C, In situ hybridization with a digoxigenin-labeled Nkx2.9 riboprobe was performed on transverse cryosections derived from cervical levels of an E10 wild-type embryo (A) and a Gli2 null littermate (B). Nkx2.9 mRNA expression is restricted to a narrow domain within the ventral ventricular zone that is immediately dorsal to the floor plate in E10 wild-type embryos (A, arrows) but is completely absent in an E10 Gli2 null littermate (B, arrows). C, A transverse vibratome section derived from an E10.5 Nkx2.9 homozygous embryo, in which most of the exons were replaced with the lacZ reporter gene, was processed for β-galactosidase staining and anti-BEN immunolabeling. The perdurance of lacZ allowed us to compare the distribution of cells/axons that arise from Nkx2.9 progenitors with BEN expression. In the ventralmost portion of the spinal cord, there exists significant overlap between Nkx2.9/lacZ reporter gene (blue) and BEN (purple) expression, suggesting that Nkx2.9 progenitors give rise to SACMNs. D, E, Anti-NF whole-mount immunohistochemistry was performed on E11 Nkx2.9 null mouse embryos and their wild-type littermates to visualize the SAN. The anti-NF-positive SAN appears foreshortened in an E11 Nkx2.9 null embryo (E) compared with the length of the SAN in a wild-type embryo (D). F-K, Anti-NF, anti-L1, and anti-BEN labeling of transverse, cervical spinal cord-containing cryosections derived from E10.5 wild-type and Nkx2.9 null embryos. The cryosections in F, H, J and G, I, K were derived from anteroposterior levels similar to those indicated by the dashed lines in D and E, respectively. Anti-NF (F, G), anti-L1 (H, I), and anti-BEN (J, K) reveal the presence of a longitudinally projecting SAN located outside the spinal cord and adjacent to the LEP (arrows) in a wild-type embryo (F, H, J) but not in a Nkx2.9 null litter mate (G, I, K). In the Nkx2.9 null embryo, anti-NF (G), anti-L1 (I), and anti-BEN (K) label groups of SACMN axons that possess what appear to be elaborated growth cones, which have reached the LEP but failed to exit the spinal cord (see concentrated immunoreactivity located inside the spinal cord and adjacent to the arrows). B, n = 2; C, n = 3; E, n = 2; G, I, K, n = 5. Scale bars: A, B, F-K, 100 μm; C, 1 mm; D, E, 0.5 mm.
SACMN axons fail to exit the spinal cord in mice lacking Nkx2.9
As alluded to above, we recently reported that anti-NF whole-mount immunohistochemistry revealed what appeared to be a shorter and thinner SAN in Nkx2.9 null mice (Pabst et al., 2003). Given the unique anatomical arrangement of the SAN, it seemed possible that the malformed SAN in the Nkx2.9 mutant might reflect the selective loss of the spinal root as opposed to the loss of both hindbrain and spinal roots (i.e., the complete SAN) we observed in Gli2 mutants (see above). Accordingly, the absence of the spinal root of the SAN could result from the complete loss of spinal accessory motor neurons in cervical regions of the spinal cord or from the inability of SACMN axons to properly exit the CNS. To distinguish between these possibilities, we labeled transverse cryosections of E11 Nkx2.9 null embryos with anti-BEN, anti-neurofilament, and anti-L1. Consistent with the results of our previous whole-mount analyses, anti-neurofilament (Fig. 6E,G, arrows), anti-L1 (Fig. 6I, arrows), and anti-BEN (Fig. 6K, arrows) each revealed the absence of the SAN at cervical spinal cord levels in Nk2.9 null embryos but the presence of the SAN at hindbrain levels (Fig. 6E and data not shown). In contrast to the phenotype we observed in Gli2 null embryos (Fig. 3), SACMN cell bodies developed on schedule and projected their axons toward the LEP. Strikingly, at cervical levels, SACMN axons apparently stalled in the immediate vicinity of the LEP because they failed to extend out of the spinal cord in all Nkx2.9 null embryos examined (Fig. 6G,I,K) (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). However, SACMN axons appropriately projected to, and exited from, the LEP at hind-brain levels, supporting the appearance of the wild type-like cranial root and absence of the spinal root in our whole-mount analysis (Fig. 6E, and data not shown). Anti-neurofilament (Fig. 6G) and anti-L1 (Fig. 6I) labeling demonstrated that ventrally exiting motor axons appear to follow wild type-like trajectories out of the spinal cord in Nkx2.9 mutant embryos. In E13.5 wild-type embryos, L1 was selectively expressed on those segments of SACMN axons that had exited the spinal cord and assembled into the longitudinally projecting SAN (supplemental Fig. 2A, arrows, available at www.jneurosci.org as supplemental material). However, anti-L1 labeling clearly revealed a lack of the SAN at spinal cord levels of E13.5 Nkx2.9 mutant littermates (supplemental Fig. 2B, arrows, available at www.jneurosci.org as supplemental material). Thus, these results suggest that Nkx2.9 is selectively required for the exit of dorsally directed SACMN axons through the spinal cord-associated LEP.
SACMN and their axons develop normally in BEN-/- mice
As we have described above, BEN is a rather selective marker of SACMNs, their axons, and the SAN during an early and transient phase of mouse spinal cord development. Together with the well established roles for BEN in regulating the outgrowth/guidance of a variety of CNS axons (Ott et al., 2001; Avci et al., 2004; Weiner et al., 2004), this unique expression profile suggests that BEN is a good candidate for mediating SACMN/SAN development. In particular, the recently reported defasciculation of motor and retinal ganglion cell (RGC) axons in BEN-deficient mice raised the possibility that BEN may be required for the assembly of SACMN axons into the SAN. To test this hypothesis, we examined SACMN and SAN development in a mutant mouse line in which green fluorescent protein (GFP) was knocked into the BEN locus (Weiner et al., 2004). In E11.5 transverse cryosections derived from BEN mutants, wild type-like SACMN that were appropriately restricted to cervical levels of the spinal cord expressed GFP (Fig. 7A, arrowheads). Anti-NF immunohistochemistry performed on transverse cryosections (data not shown) and whole-mutant embryos revealed that SACMN axons and the SAN appear to develop normally in the absence of BEN (Fig. 7C,E). Thus, despite our finding that BEN is quite selectively expressed by SACMN, BEN does not seem to be required for their development.
Figure 7.
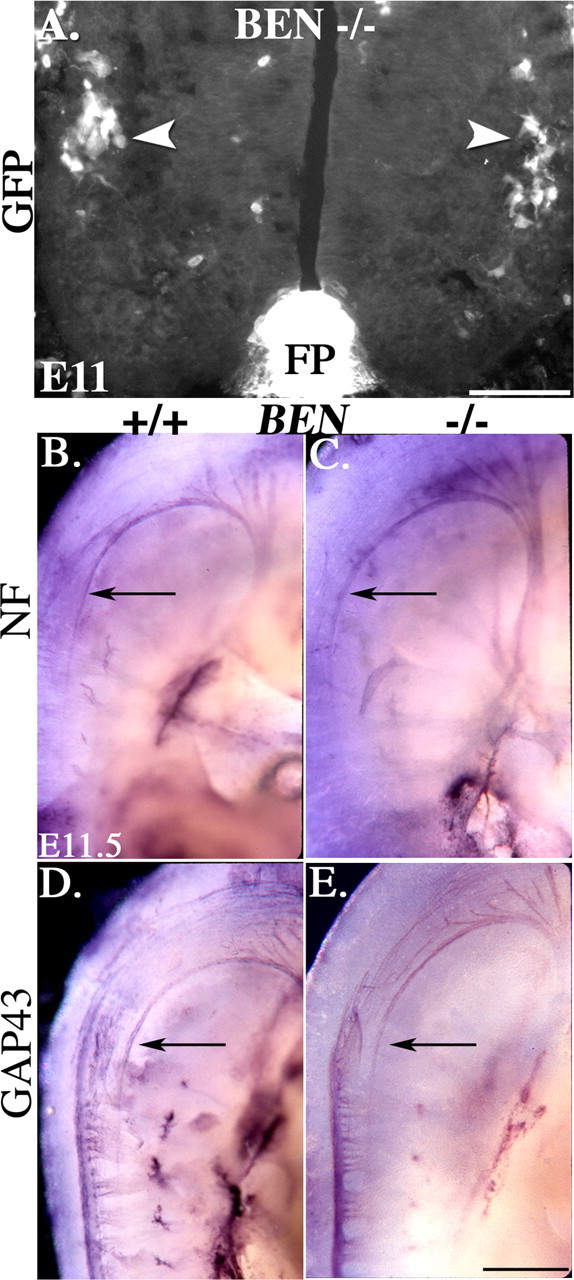
SACMNs and the SAN appear normal in BEN null mice. A, In a transverse cryosection derived from the cervical spinal cord level of an E11.5 BEN knock-in mouse embryo, GFP is expressed in wild type-like SACMN cell bodies. B-E, Whole E11.5 BEN-deficient mouse embryos (C, E) and their wild-type littermates (B, D) were labeled with anti-NF and anti-GAP43. Both anti-NF (B, C) and anti-GAP43 (D, E) labeling revealed a wild type-like SAN in embryos lacking BEN. A, n = 3; C, E, n = 5. Scale bars: A, 100 μm; E, 0.5 mm.
Discussion
Spinal motor neurons/axons represent a well studied model system for elucidating the molecular mechanisms that pattern neuronal connectivity. However, the full set of cues required to guide any motor axon subtype from cell body to target has yet to be defined. We previously identified mAb SAC1 as a marker of SACMNs and their axons in the developing rat spinal cord (Schubert and Kaprielian, 2001). Here we demonstrate that a mAb specific for the mouse homolog of the SAC1 antigen, BEN (Sekine-Aizawa et al., 1998), recognizes mouse SACMNs and used this marker to assess SACMN development in a variety of mutant mice. These analyses show that Gli2 is required for SACMN axon outgrowth, netrin-1 and Dcc are necessary for the dorsal migration of SACMN cell bodies/axons toward the LEP, and Nkx2.9 is required for the exit of SACMN axons through the LEP (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Despite its established role as a mediator of axon outgrowth/fasciculation (Weiner et al., 2004), we also show that BEN does not appear to be required for the assembly of SACMN axons into the SAN.
Several observations support our conclusion that SACMN selectively express BEN. First, anti-BEN labels a bilaterally symmetric population of cell bodies located at cervical levels of the embryonic mouse spinal cord. Second, anti-BEN-positive cell bodies project dorsally directed axons to the LEP. These axons then emerge from the spinal cord, turn into the longitudinal plane, and fasciculate into a longitudinally projecting nerve. Anti-BEN labeling of whole mouse embryos reveals the hook-like shape of this nerve, consistent with its identification as the SAN (Pabst et al., 2003). Third, application of DiI to the SAN retrogradely labels SACMNs and dorsally projecting axons that resemble anti-BEN-positive SACMNs and their axons. Fourth, in situ hybridization reveals that the distribution of BEN mRNA closely overlaps with the positions of anti-BEN-positive cell bodies. Furthermore, a different anti-BEN antibody, anti-ALCAM, selectively labels SACMN cell bodies, and SACMNs express GFP in BEN knock-in mice. Notably, anti-BEN failed to label these embryos, confirming the specificity of this antibody for BEN (data not shown). Together with our recent finding that anti-SC1, which recognizes chick BEN (Tanaka et al., 1991), labels SACMN and their axons in chick embryos (A. Dillon and Z. Kaprielian, unpublished observations), suggesting that BEN may be expressed by SACMNs across vertebrates.
The floor plate is a key source of factors that facilitate the growth/guidance of axons in the developing vertebrate CNS. Netrin-1 promotes the outgrowth of commissural axons in vitro (Kennedy and Tessier-Lavigne, 1995), and both netrin-1 (Serafini et al., 1996) and Sonic hedgehog (Charron et al., 2003) function as chemoattractants for these axons. In the spinal cord of floor plate-lacking Gli2 mutant embryos, anti-BEN reveals the absence of the SAN and dorsally projecting axons but the presence of late-developing BEN-positive cell bodies. The cervically restricted location of these cell bodies and their resemblance to retrogradely labeled SACMNs is consistent with their identification as anti-BEN-positive SACMNs. Given that these SACMN cell bodies ultimately develop in Gli2 mutant embryos, the missing-SAN phenotype likely reflects a defect in the initiation of SACMN axon outgrowth, presumably attributable to the loss of floor plate-derived factors. The absence of the trochlear nerve (Matise et al., 1999) and the wild type-like appearance of ventrally exiting motor axons in Gli2 mutants (Ding et al., 1998) raise the possibility that this factor(s) is selective for dorsally directed motor axons. Notably, netrin-1 cannot account for this activity because SACMN cell bodies extend axons in netrin-1-deficient mice. Alternatively, the absence of Gli2 or defects in Shh signaling may lead to a respecification of SACMNs in Gli2 mutant embryos. However, this scenario seems inconsistent with the finding that, except for a selective loss of V3 interneurons, patterning of the ventral spinal cord appears wild type-like in Gli2 mutants (Matise et al., 1998).
In wild-type embryos, anti-BEN-positive SACMN cell bodies and their axons migrate along a dorsally directed trajectory toward the LEP. Consistent with the ability of the floor plate (Colamarino and Tessier-Lavigne, 1995b; Guthrie and Pini, 1995; Tucker et al., 1996) and netrin-1 (Varela-Echavarria et al., 1997; Alcantara et al., 2000; Hamasaki et al., 2001) to repel hindbrain or spinal motor axons/cell bodies, a subset of SACMN cell bodies/axons fails to migrate dorsally in both netrin-1 and Dcc mutants. The observation that BEN-positive SACMNs are likely to express Dcc mRNA supports a cell-autonomous role for DCC in mediating the presumed repulsion of SACMNs and their axons. The in vivo SACMN/axon migration defects we describe here are consistent with the failure of oligodendrocyte precursors to migrate dorsally in Dcc mutants (Jarjour et al., 2003), the abnormal migration of leutinizing hormone-releasing hormone neurons, aberrant projections of the caudal vomeronasal nerve in both netrin-1 mutants and Dcc null embryos (Schwarting et al., 2001, 2004), and the finding that netrin-1 selectively repels dorsally projecting motor axons in vitro (Colamarino and Tessier-Lavigne, 1995a; Varela-Echavarria et al., 1997).
Because a subset of anti-BEN-positive SACMN cell bodies/axons migrate appropriately toward the LEP in both netrin-1 and Dcc mutants, other SACMN/axon-associated netrin-1 receptors and/or ventral spinal cord-associated chemorepellents, such as Slits and semaphorins (Varela-Echavarria et al., 1997; Brose et al., 1999; Patel et al., 2001), may also have roles in these guidance events. Previous studies in Caenorhabditis elegans, Drosophila, and Xenopus have shown that UNC-40/DCC binding to UNC-6/netrin mediates ventrally directed cell body/axon migration events, whereas UNC-40/DCC operates in concert with UNC5 receptors to facilitate dorsally directed cell body/axon migration in response to netrin (Hedgecock et al., 1990; Hamelin et al., 1993; Hong et al., 1999; Keleman and Dickson, 2001; Dickson, 2002; Araujo and Tear, 2003). UNC5H2, H3, and H4 (Engelkamp, 2002) are expressed in the developing rodent CNS during neuronal migration and axogenesis (Ackerman et al., 1997; Leonardo et al., 1997; Mattar et al., 2004). Furthermore, UNC5H3 has been implicated in the migration of cranial branchiomotor neurons and Purkinje cell progenitors (Przyborski et al., 1998) and corticospinal tract development (Finger et al., 2002). Thus, SACMNs may express a mouse UNC5H receptor that cooperates with DCC to drive SACMN cell bodies and axons along a dorsally directed trajectory to the LEP.
Motor axon exit from the CNS is a poorly understood phenomenon, and the underlying molecular mechanisms remain obscure. A specialized group of neural crest derivatives, referred to as boundary cap cells, prefigure motor axon exit points (Vermeren et al., 2003). Although boundary cap cells confine motor neuron cell bodies to the spinal cord, they are not required for motor axon exit through the ventral roots (Vermeren et al., 2003). We demonstrate that SACMN axons are incapable of exiting the spinal cord in Nkx2.9 null embryos. Our finding that SACMNs are likely to arise from Nkx2.9-expressing progenitors (Fig. 6C) suggests that Nkx2.9 functions cell autonomously. Mechanistically, we propose that Nkx2.9 regulates the expression of downstream targets that mediate the exit of SACMN axons from the spinal cord. Good candidates include receptors on SACMN axon/growth cones that respond to long- or short-range attractive guidance cues, which may be expressed in the immediate vicinity of the LEP (Guthrie and Lumsden, 1992; Niederlander and Lumsden, 1996; Irving et al., 2002; Vermeren et al., 2003). In contrast to the inability of axons to emerge from the spinal cord, hindbrain-associated SACMN axons exit the CNS normally in Nkx2.9 null embryos (data not shown), raising the possibility that another Nkx family member compensates for the loss of Nkx2.9 in the hindbrain. Notably, Nkx2.2 and Nkx2.9 are coexpressed for a longer period of time in the hindbrain compared with the spinal cord (Briscoe et al., 1999), and Nkx6 genes have evolutionarily conserved roles in motor neuron/axon development (Muller et al., 2003; Broihier et al., 2004).
BEN has been shown to mediate axon outgrowth and fasciculation (Weiner et al., 2004). The zebrafish homolog of BEN/DM-GRASP, Neurolin, is selectively expressed by secondary motoneurons (Fashena and Westerfield, 1999), and Neurolin-blocking antibodies promote pathfinding errors and fasciculation defects in the corresponding axons (Ott et al., 2001). DM-GRASP has also been shown to enhance RGC axon extension in vitro, and inhibition of DM-GRASP perturbs the fidelity of RGC axon projections in chick embryos (Avci et al., 2004). In addition, RGC axons and motor axons that compose the intercostal nerves are defasciculated in BEN null mouse embryos (Weiner et al., 2004). In contrast, our analyses failed to reveal a requirement for BEN in the development of SACMNs.
In summary, we have exploited the specificity of anti-BEN to identify molecules that regulate several phases of SACMN development. Specifically, we have shown that Gli2 is required for SACMN axon outgrowth (supplemental Fig. 3, red, available at www.jneurosci.org as supplemental material) and demonstrate a role for netrin-1/Dcc in the dorsal migration of SACMN cell bodies and their axons toward the LEP (supplemental Fig. 3, orange, available at www.jneurosci.org as supplemental material). We also shown that Nkx2.9 is essential for the exit of SACMN axons from the spinal cord (supplemental Fig. 3, blue, available at www.jneurosci.org as supplemental material). To our knowledge, this is the first identification of a gene that is required for motor axon exit from the vertebrate CNS.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Ruth L. Kirschstein Predoctoral Fellowship F31 NS43852-03 (A.K.D.), grants from the Canadian Institutes of Health Research and the Multiple Sclerosis Society of Canada (T.E.K.), and Muscular Dystrophy Association Research Grant 3043 (Z.K.). We thank Carol Mason, Todd Evans, and Scott Emmons for helpful suggestions and technical advice, as well as Carol Mason, Stephanie Kadison, Stacey Reeber, Cristina Aguirre-Chen, and Nozomi Sakai for critical reading of this manuscript. We thank Cuiling Wang in the Biostatistical Facility of the Albert Einstein College of Medicine for statistical assistance. We thank Michael Bowen for mAb 33.1. mAb 2H3 was developed by Thomas M. Jessell and Jane Dodd and was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by the Department of Biological Sciences, University of Iowa.
Correspondence should be addressed to Dr. Zaven Kaprielian, Departments of Neuroscience and Pathology, Albert Einstein College of Medicine, Kennedy Center, Room 624, 1410 Pelham Parkway South, Bronx, NY 10461. E-mail: kapriel@aecom.yu.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510119-12$15.00/0
References
- Ackerman SL, Kozak LP, Pryzborski SA, Rund LA, Boyer BB, Knowles BB (1997) The mouse rostral cerebellar malformation gene encodes an UNC-5-like protein. Nature 386: 838-842. [DOI] [PubMed] [Google Scholar]
- Alcantara S, Rulz M, De Castro F, Soriano E, Sotelo C (2000) Netrin 1 acts as an attractive or as a repulsive cue for distinct migrating neurons during the development of the cerebellar system. Development 127: 1359-1372. [DOI] [PubMed] [Google Scholar]
- Araujo S, Tear G (2003) Axon guidance mechanisms and molecules: lessons from invertebrates. Nat Rev Neurosci 4: 910-922. [DOI] [PubMed] [Google Scholar]
- Avci H, Zelina P, Thelen K, Pollerberg G (2004) Role of cell adhesion molecule DM-GRASP in growth and orientation of retinal ganglion cell axons. Dev Biol 271: 291-305. [DOI] [PubMed] [Google Scholar]
- Brichta AM, Callister RJ, Peterson EH (1987) Quantitative analysis of cervical musculature in rats: histochemical composition and motor pool organization. I. Muscles of the spinal accessory complex. J Comp Neurol 255: 351-368. [DOI] [PubMed] [Google Scholar]
- Briscoe J, Sussel L, Serup P, Hartigan-O'Connor D, Jessell TM, Rubinstein JL, Ericson J (1999) Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature 398: 622-627. [DOI] [PubMed] [Google Scholar]
- Briscoe J, Pierani A, Jessell TM, Ericson J (2000) A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell 101: 435-445. [DOI] [PubMed] [Google Scholar]
- Broihier H, Kuzin A, Zhu Y, Odenwald W, Skeath J (2004) Drosophila homeodomain protein Nkx6 coordinates motoneuron subtype identity and axogenesis. Development 131: 5233-5242. [DOI] [PubMed] [Google Scholar]
- Brose K, Bland KS, Wang KH, Arnott D, Henzel W, Goodman CS, Tessier-Lavigne M, Kidd T (1999) Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell 96: 795-806. [DOI] [PubMed] [Google Scholar]
- Charron F, Stein E, Jeong J, McMahon AP, Tessier-Lavigne M (2003) The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell 113: 11-23. [DOI] [PubMed] [Google Scholar]
- Chen H, Bagri A, Zupicich JA, Zou Y, Stoeckli E, Pleasure SJ, Lowenstein DH, Skarnes WC, Chedotal A, Tessier-Lavigne M (2000) Neuropilin-2 regulates the development of select cranial and sensory nerves and hippocampal mossy fiber projections. Neuron 25: 43-56. [DOI] [PubMed] [Google Scholar]
- Colamarino SA, Tessier-Lavigne M (1995a) The axonal chemoattractant netrin-1 is also a chemorepellent for trochlear motor axons. Cell 81: 621-629. [DOI] [PubMed] [Google Scholar]
- Colamarino SA, Tessier-Lavigne M (1995b) The role of the floor plate in axon guidance. Annu Rev Neurosci 18: 497-529. [DOI] [PubMed] [Google Scholar]
- Dickson BJ (2002) Molecular mechanisms of axon guidance. Science 298: 1959-1964. [DOI] [PubMed] [Google Scholar]
- Ding Q, Motoyama J, Gasca S, Rong M, Sasaki H, Rossant J, Hul C (1998) Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 125: 2533-2543. [DOI] [PubMed] [Google Scholar]
- Dodd J, Morton SB, Karagogeos D, Yamamoto M, Jessell TM (1988) Spatial regulation of axonal glycoprotein expression on subsets of embryonic spinal neurons. Neuron 1: 105-116. [DOI] [PubMed] [Google Scholar]
- Engelkamp D (2002) Cloning of three mouse Unc5 genes and their expression patterns at mid-gestation. Mech Dev 118: 191-197. [DOI] [PubMed] [Google Scholar]
- Ericson J, Rashbass P, Schedl A, Brenner-Morton S, Kawakami A, Herrington Vv, Jessell T, Briscoe J (1997) Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell 90: 169-180. [DOI] [PubMed] [Google Scholar]
- Fashena D, Westerfield M (1999) Secondary motoneuron axons localize DM-GRASP on their fasciculated segments. J Comp Neurol 406: 415-424. [DOI] [PubMed] [Google Scholar]
- Fazeli A, Dickinson SL, Hermiston ML, Tighe RV, Steen RG, Small CG, Stoeckli ET, Keino-Masu K, Masu M, Rayburn H, Simons J, Bronson RT, Gordon JI, Tessier-Lavigne M, Weinberg RA (1997) Phenotype of mice lacking functional deleted in colorectal carcinoma (DCC) gene. Nature 386: 796-804. [DOI] [PubMed] [Google Scholar]
- Finger J, Bronson R, Harris B, Johnson K, Przyborski S, Ackerman S (2002) The netrin 1 receptors Unc5h3 and Dcc are necessary at multiple choice points for the guidance of corticospinal tract axons. J Neurosci 22: 10346-10356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giger RJ, Cloutier JF, Sahay A, Prinjha RK, Levengood DV, Moore SE, Pickering S, Simmons D, Rastan S, Walsh FS, Kolodkin AL, Ginty DD, Geppert M (2000) Neuropilin-2 is required in vivo for selective axon guidance responses to secreted semaphorins. Neuron 25: 29-41. [DOI] [PubMed] [Google Scholar]
- Gottschall J, Zenker W, Neuhuber W, Mysicka A, Muntener M (1980) The sternomastoid muscle of the rat and its innervation. Anat Embryol (Berl) 160: 285-300. [DOI] [PubMed] [Google Scholar]
- Greene E (1935) Anatomy of the rat. New York: Hatner.
- Guthrie S, Lumsden A (1992) Motor neuron pathfinding following rhombomere reversals in the chick embryo hindbrain. Development 114: 663-673. [DOI] [PubMed] [Google Scholar]
- Guthrie S, Pini A (1995) Chemorepulsion of developing motor axons by the floor plate. Neuron 14: 1117-1130. [DOI] [PubMed] [Google Scholar]
- Hamasaki T, Satoshi G, Shigeyuki N, Yukitaka U (2001) A role of netrin-1 in the formation of the subcortical structure striatum: repulsive action on the migration of late-born striatal neurons. J Neurosci 21: 4272-4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelin M, Zhou Y, Su M-W, Scott IM, Culotti JG (1993) Expression of the UNC-5 guidance receptor in the touch neurons of C. elegans steers their axons dorsally. Nature 364: 327-330. [DOI] [PubMed] [Google Scholar]
- Hedgecock EM, Culotti JG, Hall DH (1990) The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans Neuron 2: 61-85. [DOI] [PubMed] [Google Scholar]
- Hong K, Hinck L, Nishiyama M, Poo M-M, Tessier-Lavigne M, Stein E (1999) A ligand-gated association between cytoplasmic domains of UNC5 and DCC family receptors converts netrin-induced growth cone attraction to repulsion. Cell 97: 927-941. [DOI] [PubMed] [Google Scholar]
- Imondi R, Wideman C, Kaprielian Z (2000) Complementary expression of transmembrane ephrins and their receptors in the mouse spinal cord: a possible role in constraining the orientation of longitudinally projecting axons. Development 127: 1397-1410. [DOI] [PubMed] [Google Scholar]
- Irving C, Malhas A, Guthrie S, Mason I (2002) Establishing the trochlear motor axon trajectory: role of the isthmic organiser and Fgf8. Development 129: 5389-5398. [DOI] [PubMed] [Google Scholar]
- Jarjour A, Manitt C, Moore S, Thompson K, Yuh S, Kennedy T (2003) Netrin-1 is a chemorepellent for oligodendrocyte precursor cells in the embryonic spinal cord. J Neurosci 23: 3735-3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessell TM (2000) Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat Rev Genet 1: 20-29. [DOI] [PubMed] [Google Scholar]
- Kaprielian Z, Cho K-O, Hadjiargyrou M, Patterson PH (1995) CD9, a major platelet cell surface glycoprotein, is a ROCA antigen and is expressed in the nervous system. J Neurosci 15: 562-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keino-Masu K, Masu M, Hinck L, Leonardo ED, Chan SS-Y, Culotti JG, Tessier-Lavigne M (1996) Deleted in colorectal carcinoma (DCC) encodes a netrin receptor. Cell 87: 175-185. [DOI] [PubMed] [Google Scholar]
- Keleman K, Dickson BJ (2001) Short- and long-range repulsion by the Drosophila Unc5 netrin receptor. Neuron 32: 605-617. [DOI] [PubMed] [Google Scholar]
- Kennedy TE, Tessier-Lavigne M (1995) Guidance and induction of branch formation in developing axons by target-derived diffusible factors. Curr Biol 5: 83-90. [DOI] [PubMed] [Google Scholar]
- Kennedy TE, Serafini T, delaTorre JR, Tessier-Lavigne M (1994) Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell 78: 425-435. [DOI] [PubMed] [Google Scholar]
- Landmesser LT (2001) The acquisition of motoneuron subtype identity and motor circuit formation. Int J Dev Neurosci 19: 175-182. [DOI] [PubMed] [Google Scholar]
- Lei Q, Zelman AK, Kuang E, Li S, Matise MP (2004) Transduction of graded Hedgehog signaling by a combination of Gli2 and Gli3 activator functions in the developing spinal cord. Development 131: 3593-3604. [DOI] [PubMed] [Google Scholar]
- Leonardo ED, Hinck L, Masu M, Keino-Masu K, Ackerman SL, Tessier-Lavigne M (1997) Vertebrate homologues of C. elegans UNC-5 are candidate netrin receptors. Nature 386: 833-838. [DOI] [PubMed] [Google Scholar]
- Leppert C, Diekmann H, Paul C, Laessing U, Marx M, Bastmeyer M, Stuermer CA (1999) Neurolin Ig domain 2 participates in retinal axon guidance and Ig domains 1 and 3 in fasciculation. J Cell Biol 144: 339-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise MP, Epstein DJ, Park HL, Platt KA, Joyner AL (1998) Gli 2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development 125: 2759-2770. [DOI] [PubMed] [Google Scholar]
- Matise MP, Lustig M, Sakurai T, Grumet M, Joyner AL (1999) Ventral midline cells are required for the local control of commissural axon guidance in the mouse spinal cord. Development 126: 3649-3659. [DOI] [PubMed] [Google Scholar]
- Mattar P, Britz O, Johannes C, Nieto M, Ma L, Rebeyka A, Klenin N, Polleux F, Guillemot F, Schuurmans C (2004) A screen for downstream effectors of Neurogenin2 in the embryonic neocortex. Dev Biol 273: 373-389. [DOI] [PubMed] [Google Scholar]
- Mukouyama Y, Donghun S, Britsch A, Masahiko T, Anderson D (2002) Sensory nerves determine the pattern of arterial differentiation and blood vessel branching in the skin. Cell 109: 693-705. [DOI] [PubMed] [Google Scholar]
- Muller M, Jabs N, Lorke D, Fritzsch B, Sander M (2003) Nkx6.1 controls migration and axon pathfinding of cranial branchio-motoneurons. Development 130: 5815-5826. [DOI] [PubMed] [Google Scholar]
- Niederlander C, Lumsden A (1996) Late emigrating neural crest cells migrate specifically to exit points of cranial branchiomotor nerves. Development 122: 2367-2374. [DOI] [PubMed] [Google Scholar]
- Ott H, Diekmann H, Stuermer CA, Bastmeyer M (2001) Function of neurolin (DM-GRASP/SC-1) in guidance of motor axons during zebrafish development. Dev Biol 235: 86-97. [DOI] [PubMed] [Google Scholar]
- Pabst O, Rummelies J, Winter B, Arnold H-H (2003) Targeted disruption of the homeobox gene Nkx2.9 reveals a role in development of the spinal accessory nerve. Development 130: 1193-1202. [DOI] [PubMed] [Google Scholar]
- Patel K, Nash JA, Itoh A, Liu Z, Sundaresan V, Pini A (2001) Slit proteins are not dominant chemorepellents for olfactory tract and spinal motor axons. Development 128: 5031-5037. [DOI] [PubMed] [Google Scholar]
- Placzek M, Tessier-Lavigne M, Jessell TM, Dodd J (1990) Orientation of commissural axons in vitro in response to a floor plate-derived chemoattractant. Development 110: 19-30. [DOI] [PubMed] [Google Scholar]
- Price S, Briscoe J (2004) The generation and diversification of spinal motor neurons: signals and responses. Mech Dev 121: 1103-1115. [DOI] [PubMed] [Google Scholar]
- Przyborski SA, Knowles BB, Ackerman SL (1998) Embryonic phenotype of unc5h3 mutant mice suggests chemorepulsion during the formation of the rostral cerebellar boundary. Development 125: 41-50. [DOI] [PubMed] [Google Scholar]
- Sahay A, Molliver ME, Ginty DD, Kolodkin AL (2003) Sempahorin 3F is critical for the developmentl of limbic system circuitry and is required in neurons for selective CNS axon guidance events. J Neurosci 23: 6671-6680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert W, Kaprielian Z (2001) Identification and characterization of a cell surface marker for embryonic rat spinal accessory motor neurons. J Comp Neurol 439: 368-383. [DOI] [PubMed] [Google Scholar]
- Schwarting G, Kostek C, Bless E, Ahmad N, Tobet S (2001) Deleted in colorectal cancer (DCC) regulates the migration of luteinizing hormone-releasing hormone neurons to the basal forebrain. J Neurosci 21: 911-919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarting G, Raitcheva D, Bless E, Ackerman S, Tobet S (2004) Netrin 1-mediated chemoattraction regulates the migratory pathway of LHRH neurons. Eur J Neurosci 19: 11-20. [DOI] [PubMed] [Google Scholar]
- Sekine-Aizawa Y, Omori A, Fujita SC (1998) MuSC, a novel member of the immunoglobulin superfamily, is expressed in neurons of a subset of cranial sensory ganglia in the mouse embryo. Eur J Neurosci 10: 2810-2824. [DOI] [PubMed] [Google Scholar]
- Serafini T, Kennedy TE, Galko MJ, Mirzayan C, Jessell TM, Tessier-Lavigne M (1994) The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell 78: 409-424. [DOI] [PubMed] [Google Scholar]
- Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, Tessier-Lavigne M (1996) Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell 87: 1001-1014. [DOI] [PubMed] [Google Scholar]
- Sharma K, Sheng HZ, Lettieri K, Li H, Karavanov A, Potter S, Westphal H, Pfaff SL (1998) LIM homeodomain factors Lhx3 and Lhx4 assign subtype identities for motor neurons. Cell 95: 817-828. [DOI] [PubMed] [Google Scholar]
- Shirasaki R, Pfaff SL (2002) Transcriptional codes and the control of neuronal identity. Annu Rev Neurosci 25: 251-281. [DOI] [PubMed] [Google Scholar]
- Snider WD, Palavali V (1990) Early axon and dendritic outgrowth of spinal accessory motor neurons studied with DiI in fixed tissues. J Comp Neurol 297: 227-238. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Matsui T, Agata A, Tomura M, Kubota I, McFarland KC, Kohr B, Lee A, Philips HS, Shelton DL (1991) Molecular cloning and expression of a novel adhesion molecule, SC1. Neuron 7: 535-545. [DOI] [PubMed] [Google Scholar]
- Tsuchida T, Ensini M, Morton SB, Baldassare M, Edlund T, Jessell TM, Pfaff SL (1994) Topographic organization of embryonic motor neurons defined by the expression of LIM homeobox genes. Cell 79: 957-970. [DOI] [PubMed] [Google Scholar]
- Tucker A, Lumsden A, Guthrie S (1996) Cranial motor axons respond differentially to the floor plate and sensory ganglia in collagen gel cocultures. Eur J Neurosci 8: 906-916. [DOI] [PubMed] [Google Scholar]
- Varela-Echavarria A, Guthrie S (1997) Molecules making waves in axon guidance. Genes Dev 1: 545-557. [DOI] [PubMed] [Google Scholar]
- Varela-Echavarria A, Tucker A, Puschel AW, Guthrie S (1997) Motor axon subpopulations respond differentially to the chemorepellents netrin-1 and semaphorin D. Neuron 18: 193-207. [DOI] [PubMed] [Google Scholar]
- Vermeren M, Maro GS, Bron R, McGonnell IM, Charnay P, Topilko P, Cohen J (2003) Integrity of developing spinal motor columns is regulated by neural crest derivatives at motor exit points. Neuron 37: 403-415. [DOI] [PubMed] [Google Scholar]
- Weiner JA, Koo SJ, Nicolas S, Fraboulet S, Pfaff SL, Pourquie O, Sanes JR (2004) Axon fasciculation defects and retinal dysplasias in mice lacking the immunoglobulin superfamily adhesion molecule BEN/ALCAM/SC1. Mol Cell Neurosci 27: 59-69. [DOI] [PubMed] [Google Scholar]
- Wentworth LE, Hinds JW (1978) Early motor neuron formation in the cervical spinal cord of the mouse: an electron microscopic, serial section analysis. J Comp Neurol 177: 611-634. [DOI] [PubMed] [Google Scholar]
- Wilson-Pauwels L, Akesson E, Stewart P (1988) Cranial nerves: anatomy and clinical comments. Philadelphia: Decker.