

One of the most powerful means for studying the function of proteins, synapses, cells, and networks of cells is to control their activation in time and in space. Many commonly used approaches are inadequate for some experimental questions. Modulation of protein function by traditional genetic approaches depends on natural protein turnover, which occurs on a sufficiently slow timescale to permit homeostatic compensation by other proteins. Synapses and cells are usually activated electrically, with stimuli delivered by wire or glass electrodes, or chemically, by applying exogenous neurotransmitters and other modulatory substances. The spatial, temporal, and molecular resolution of these approaches is limited, however. Moreover, the precise anatomical location of the synapses activated by electrical stimuli are generally unknown. Application of receptor agonists for only brief periods in a manner that mimics naturally occurring transmitter release can only be performed with isolated cells and membrane patches but is difficult in more intact preparations. For cells within networks, inactivating them with traditional lesioning approaches is irreversible, difficult to localize to small or distinct cell populations, and often accompanied by damage to fibers of passage. Finally, it is difficult to control the function of individual cells in complex tissues or of individual proteins in complex signaling cascades.
The development of technology to observe changes in the intracellular concentration of Ca2+ and other ions with fluorescence microscopy has increased our knowledge of cellular and synaptic function enormously. Recent advances in microscopy, optical physics, laser instrumentation, and molecular probe development have now engendered methodology that permits not only the observation but also the simultaneous manipulation of neural function with unprecedented spatial and temporal resolution through the use of light. In this mini-symposium, some practitioners of this emergent technology will highlight the state of the art in the development and application of new techniques in this rapidly advancing field. In particular, improvements in microscopy, photonics, and laser “hardware,” as well as results obtained using newly developed “software,” such as novel chemically synthesized and genetically encoded light-sensitive probes, are illustrated.
Elucidating neuronal function with caged molecules
A caged molecule is an inert but photosensitive molecule that has latent biological activity. Light absorption transforms a caged molecule into a fully bioactive molecule. The photochemically generated molecules can be agonists or antagonists; therefore, photolysis of caged molecules is a convenient method for using light to switch biological processes on or off. Caged molecules are most often made by chemically modifying the structure of a bioactive molecule to mask its activity. Absorption of light then leads to photochemical alteration and/or cleavage of the activity-blocking modification to restore the activity of the biomolecule. Metaphorically, the biological activity of the molecule is “caged,” and light is said to “uncage” or “photorelease” that activity.
Photolysis of caged molecules offers three advantages over conventional techniques of reagent delivery: temporal resolution, spatial resolution, and control over the chemical identity of the reagent being delivered. First, because they are biologically inactive, caged molecules can be preloaded into cells or preequilibrated with tissues before experimentation. A flash of light rapidly generates bioactive molecules in situ, which can immediately interact with their receptors. Problems with mixing, diffusion, or access to anatomically complex tissue are thus minimized. Photorelease typically occurs within a few microseconds to ∼1 ms, so neurophysiology can be manipulated with comparable time resolution. Second, a light beam is easy to manipulate and steer and can be focused to a diffraction-limited spot with diameter d = 1.22λ/NA, where λ is the wavelength of light used, and NA is the numerical aperture of the focusing lens. In typical experiments, photolysis is achieved by focusing UV light near 350 nm through a microscope objective with NA of >1.0, which means spatial resolution better than 0.5 μm is possible. Third, many biochemical messengers are chemically or enzymatically labile. Two examples are dopamine and anandamide; the former air-oxidizes readily, and the latter is cleaved rapidly by endogenous lipases. That the degradation products themselves can have independent biological action is problematic. Judicious caging converts labile molecules to stable species that resist chemical and biological breakdown. Focal photolysis then generates the authentic bioactive molecule in situ, thus affording precise control over the chemical identity of the applied reagent. Below, we describe novel caged compounds and technologies that illustrate the advantages of focal photolysis.
Exploring the functional properties and plasticity of synaptic receptors with caged glutamate
Relatively little is known about the properties of glutamate receptors within the synapse, as opposed to extrasynaptic receptors, because of their inaccessibility to conventional methods such as outside-out patch-clamp recording. Presynaptic nerve terminals, the conventional source of neurotransmitter for activating synaptic receptors, have variable release probabilities, making them a poor source of glutamate for many types of experiments. The properties of the synaptic receptors are important because they are thought to be the major determinant of the EPSC amplitude and time course (Jonas and Spruston, 1994), which determines the reliability and the precision of synaptic signaling (Galarreta and Hestrin, 2001; Cathala et al., 2003). Moreover, their properties are likely to be different from nonsynaptic receptors because other proteins to which they are bound can influence AMPA receptor (AMPAR) behavior and may be different at the two sites (Priel et al., 2005; Tomita et al., 2005). Silver, Thompson, Wang, and colleagues have designed systems for local photorelease of caged glutamate to examine the properties of glutamate receptor channels at single synapses and dendritic spines.
Silver's group has built, in collaboration with Prairie Technologies (Middleton, WI), a photolysis system that comprises a continuous-wave (CW) argon ion UV laser, an acousto-optical tunable filter (AOTF), and a single-mode fiber to deliver the light to the microscope. By filling the back aperture of the objective with the UV light, they can achieve a diffraction-limited illumination volume that has lateral dimensions approaching 220 nm (full-width at half-maximum), comparable with the size of a single postsynaptic density (PSD) and smaller than the lateral resolution of a two-photon microscope (Matsuzaki et al., 2001; Zipfel et al., 2003). The position of the uncaging spot is computer controlled with stepper motors and can be placed at the desired location with high precision using a CCD image of the preparation as a reference.
Thompson and colleagues have modified an upright microscope, equipped for whole-cell recording, to allow the UV light needed for photolysis to be combined with conventional wide-field illumination via a dichroic mirror in the excitation light path (Fig. 1A) (Wei et al., 2001). UV light from an argon ion or diode pumped solid-state laser is launched into a thin (10-25 μm) multimode quartz fiber and then focused in a plane conjugate to the preparation via a lens assembly above the microscope.
Figure 1.

Potentiation of exogenous glutamate responses at single dendritic spines. A, Diagram of the experimental apparatus. UV light is launched into a quartz multimode fiber and focused in a conjugate focal plane using a lens assembly. The intensity of the light is controlled by a neutral density filter wheel. A dichroic mirror (DM) allows the UV light to be combined with wide-field illumination from a conventional mercury arc (HBO) lamp, after passing through an excitation filter (EX). The normal filter cube contains a second dichroic mirror and the desired emission filter (EM). Mechanical shutters gate the UV and visible light pulses. A CCD camera captures images of the cells. Inset, With this apparatus, UV light for microphotolysis can be directed to a spot (gray circle) near the head of a single dendritic spine, visualized with fluorescent dye loaded into the cell from the whole-cell patch pipette. These spots have a full-width at half-maximal height of <1 μm. B, Pooled data showing the mean ± SEM potentiation of photolytic EPSCs in 13 cells. Averaged photolytic EPSCs 3 min before and 20 min after the pairing are shown above and were elicited from the spine illustrated in A. Modified from Bagal et al. (2005).
There are a number of advantages to these arrangements. First, the use of a CW or quasi-CW laser, particularly in combination with an AOTF, allows flexible uncaging protocols, including variable duration pulses, ramps, paired pulse, and trains. Second, UV laser excitation produces highly efficient uncaging of 4-methoxy-7-nitroindolinyl-caged l-glutamate (MNI-glutamate), allowing receptor activation with brief illumination pulses (∼20 μs) that mimic synaptic activation and produce minimal photodamage. In contrast, the two-photon cross-section of MNI-glutamate is low (Kiskin et al., 2002), so that it requires high light intensities (Matsuzaki et al., 2001; Kiskin et al., 2002) or extended illumination durations (Carter and Sabatini, 2004) to achieve millimolar concentrations of glutamate. The power levels necessary can induce photodamage (Kiskin et al., 2002), which increases nonlinearly with intensity for two-photon excitation (Koester et al., 1999; Hopt and Neher, 2001). Third, diffraction-limited illumination permits highly localized activation, allowing the investigation of spatial variations in receptor activation on the micrometer scale. It also permits paired-pulse activation protocols because the small photolysis volume can be replenished extremely rapidly by diffusion of unphotolysed caged molecules from outside the focal volume. Fourth, by having one fewer nonlinearity than two-photon excitation, calibration of the glutamate concentration within the UV illumination volume is potentially simpler. The limitations of using UV excitation include substantially larger scattering by tissue than with the long-wavelength light used for two-photon excitation and the fact that the defocused UV light can still cause photolysis above and below the focal illumination volume, albeit at a much lower rate. Unlike the case for two-photon uncaging, these properties will restrict the application of diffraction-limited UV uncaging to the surface of slices or to tissue culture preparations. For example, Thompson's group uses cultured hippocampal slices prepared with the roller tube method so that individual spines on cells loaded with fluorescent dye from the patch pipette may be targeted near the upper surface of the culture, thus minimizing the depth of tissue the light must traverse.
Synaptic AMPAR properties
Preliminary experiments from Silver's group show that a combination of diffraction-limited UV photolysis and whole-cell patch clamping can be used to study synaptic AMPA receptors in acute brain slices at room temperature (DiGregorio et al., 2004). The cerebellar granule cell is particularly suitable for studying synaptic receptors because their previous studies show that AMPA receptors are located exclusively in PSDs (DiGregorio et al., 2002). The initial findings suggest that they will be able to quantify the kinetic properties of synaptic channels by using various illumination protocols that can be generated with the AOTF. For example, Figure 2A shows AMPA receptor-mediated currents from a cerebellar granule cell under voltage clamp evoked with 20 μs UV pulses using MNI-glutamate. Application of a paired-pulse UV protocol, in which the illumination intensity of the first pulse is varied and that of the second is fixed, has allowed the investigation of the activation dependence of AMPAR desensitization (Fig. 2A). As the fraction of synaptic receptors activated increased, the size of their maximal response 10 ms later decreased attributable to an increased level of desensitization. Using their recently developed glutamate diffusion model of the synapse (Nielsen et al., 2004) and an experimental estimate of the uncaging efficiency, it is possible to predict both the concentration of the caged compound during the illumination pulse and the glutamate concentration generated (Fig. 2B). By combining these approaches, it should be possible to quantify the functional properties of receptors within the synaptic environment and build a kinetic model of the synaptic channels. This new approach should provide a better understanding of the role of receptor properties in determining the input-output function of single synaptic contacts.
Figure 2.
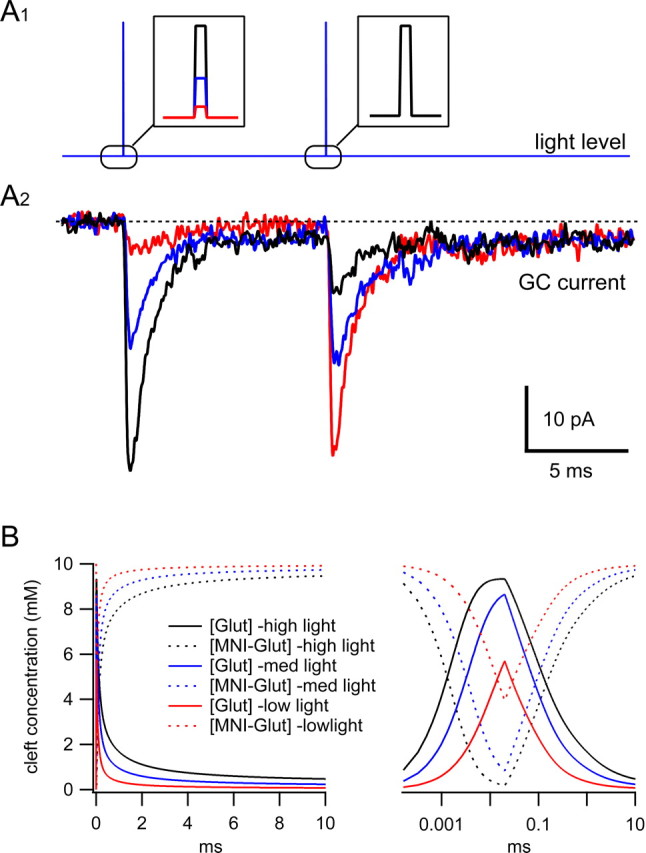
Glutamate uncaging at the cerebellar mossy fiber-granule cell synapse using diffraction-limited UV spot. A1, Paired-pulse illumination protocol (each 20 μs duration, insets) achieved by shuttering the UV light from an I328 Coherent laser (Coherent, Santa Clara, CA) with an AOTF. The intensity of light, and thus the amount of glutamate uncaged, was varied on pulse 1 by altering the voltage applied to the AOTF. For each trace, the second pulse amplitude was set to match the largest of pulse 1. A2, AMPA receptor-mediated currents (averages of 3 trials) recorded under voltage clamp from the granule cell (GC) soma evoked by uncaging locally perfused MNI-glutamate (10 mm) using a diffraction-limited UV spot using the protocol in A1. The colors of the current traces correspond to those for pulse 1 (A1 inset). B, Simulation of the concentration of MNI-glutamate and free glutamate within the diffraction-limited illumination volume for pulse 1 as in A on a linear (left) and logarithmic (right) timescale. The concentrations were calculated using a three-dimensional diffusion model of the mossy fiber-granule cells synapse (Nielsen etal., 2004) and the measured UV point-spread function. The MNI-glutamate concentration recovered to 95% in the 10 ms interpulse interval (D. Digregorio, T. Nielsen, J. Rothman, and R. A. Silver, unpublished observation).
Long-term potentiation
NMDA receptor (NMDAR)-dependent long-term plasticity of excitatory synaptic transmission probably underlies learning and memory. Persistent uncertainty and controversy about the mechanisms of long-term potentiation (LTP) induction and expression remain, at least in part, because of the unreliability of transmitter release from presynaptic nerve terminals. For example, one of the stronger pieces of evidence for a presynaptic expression mechanism in LTP is the decrease in paired-pulse ratio that has been reported in some studies (Schulz et al., 1994). Matsuzaki et al. (2004) have presented preliminary evidence that potentiation of glutamate responses can be achieved with photorelease from caged glutamate targeted to dendritic spines, offering a powerful tool to answer fundamental questions about the kinetics and mechanisms of LTP expression.
Using microphotolysis of caged N-(6-nitro-7-coumarylmethyl)-l-glutamate (Ncm-Glu) (Cai et al., 2004) to activate receptors at single dendritic spines in CA1 cells in hippocampal slice cultures, Thompson's group can induce a long-lasting potentiation of the photolysis-induced AMPAR-mediated currents with a single pairing of photoreleased glutamate and brief postsynaptic depolarization (Bagal et al., 2005). The potentiation has a magnitude of 80% over the control amplitude, is stable for >30 min after induction (Fig. 1 B), and cannot be induced at extrasynaptic sites on the dendritic shaft. Like conventional synaptic LTP, this potentiation is blocked by NMDAR antagonists and is reversed with low-frequency photostimulation. Because they can induce the potentiation with a single pairing, they discovered that potentiation of photolytic responses develops at single spines in a rapid, stepwise manner after a ∼40 s delay. The potentiation is accompanied by a decrease in postsynaptic paired-pulse facilitation of the photolytic currents, indicating that a decrease in the proportion of glutamate receptor sub-type 2-lacking AMPARs at the spine head contributes to the potentiation. These results establish that activation of postsynaptic glutamate receptors by glutamate is not only necessary, but sufficient, for the induction of NMDA receptor-dependent LTP. Furthermore, the ability to stimulate single dendritic spines with photoreleased glutamate provides a powerful new means to study LTP expression.
Patterned stimulation
Of course, learning does not involve single presynaptic and postsynaptic elements, however, but ensembles of cells and synapses acting in concert. Wang's laboratory is interested in synaptic learning rules and the cellular mechanisms that govern them. In brain slices, they are using photolysis of caged molecules as a means of rapidly and non-invasively manipulating biochemical signals with submicrometer spatial resolution. They have developed an optical system for rapid uncaging in arbitrary spatiotemporal patterns to emulate complex neural activity (Shoham et al., 2005) (Fig. 3A). They use patch-clamp recording, two-photon fluorescence microscopy and patterned single-photon uncaging of neurotransmitters and second messengers to probe network activity in functioning neural tissue. Uncaging is done using a frequency tripled Nd:YVO4 laser (DPSS, San Jose, CA) (pulse width, 50-60 nsec;λ = 355 nm; 100 kHz repetition rate) as the ultraviolet light source. The system uses TeO2 acousto-optical deflectors to steer the UV beam and can uncage at more than 20,000 locations per second. The uncaging beam is projected into the focal plane of a two-photon microscope, allowing patterned uncaging to be performed concurrently with imaging and electrophysiology. This technology has been used to generate precise, complex activity patterns of glutamate, other neurotransmitters, and second messengers. The spatial resolution of this system is well suited for studying integration in whole dendrites and neurons, in which signals are integrated on length scales of tens to hundreds of micrometers.
Figure 3.
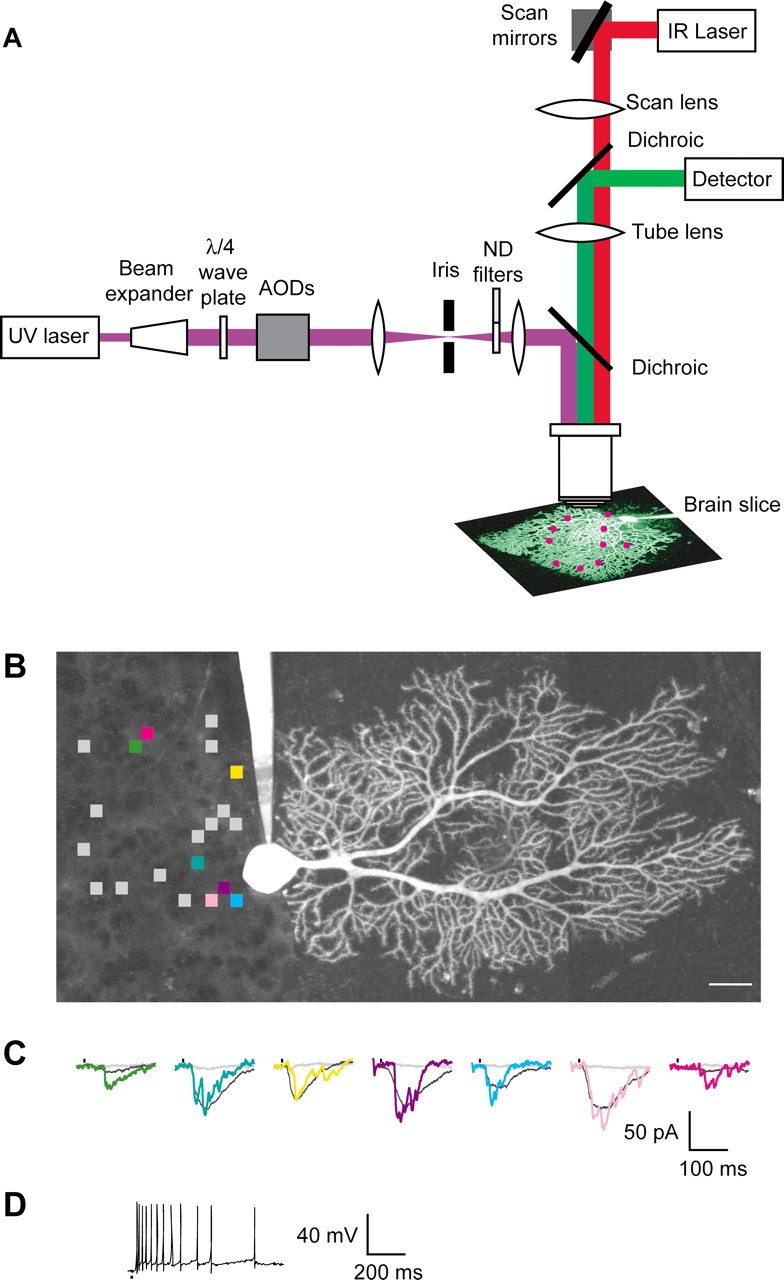
A system for patterned uncaging and two-photon imaging. A, An ultraviolet uncaging laser beam is projected into the focal plane of a two-photon microscope. IR, Infrared; AOD, acousto-optical deflector; ND, neutral density. B, Patterned activation of cells synaptically connected to a cerebellar Purkinje neuron filled via patch-clamp recording electrode with fluorescent dye. The squares indicate the positions of 20 uncaging locations in the presynaptic granule cell layer. Scale bar, 25 μm. C, Postsynaptic voltage-clamp responses to photolysis of caged glutamate at the colored locations shown in B. Vertical marks indicate times of uncaging. Shown in gray at each location are responses after addition of APV (400 μm) and lidocaine (500 μm). Also shown (colored traces) are single example traces. D, The same uncaging locations as C, activated in current clamp with interlocation intervals of 50 μs. Modified from Shoham et al. (2005).
This technology promises to be a powerful technology for the study of dendritic integration or for applications that require the activation of many presynaptic neurons at once (Fig. 3B-D). In preliminary results, they have assayed local amplification by uncaging glutamate at multiple locations at once on a basal dendrite, each producing a response reminiscent of those measured with single-site stimulation (Polsky et al., 2004). They also find that uncaging sequences at dozens of locations distributed over a dendritic arbor give reproducible responses with multiple repetitions of the same sequence in either voltage clamp or current clamp. In current clamp, patterned activation evokes a sequence of action potentials with similar timing from trial to trial. These preliminary results demonstrate the utility of rapid patterned uncaging to act as a high-throughput assay for dendritic physiology and synaptic integration.
Caged lipid messengers
Endocannabinoids (eCBs) have emerged as putative retrograde intercellular messengers in the mammalian CNS (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). Two major eCBs have been characterized; both are derivatives of arachidonic acid [arachidonoyl ethanolamide (AEA or anandamide) and 2-arachidonoylglycerol] that act at the G-protein-coupled receptor CB1 (for review, see Freund et al., 2003; Iversen, 2003). Mobilization of eCBs can occur through Ca2+-dependent or relatively Ca2+-independent path-ways (Maejima et al., 2001; Kim et al., 2002), with different down-stream effects. Ca2+-dependent eCB mobilization causes a reversible, short-term depression of synaptic transmission, a process termed depolarization-induced suppression of inhibition (DSI), in which excitation of a hippocampal CA1 pyramidal neuron (Pitler and Alger, 1992) or cerebellar Purkinje cell (Llano et al., 1991) leads to a transient reduction of GABA release from presynaptic terminals of inhibitory interneurons. Hitherto, insights into eCB action have been derived primarily from pharmacological experiments (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001), because intrinsic properties of eCBs have hampered direct physiological study of eCB signaling at CNS synapses. The hydrophobicity of eCBs severely limits their penetration into brain tissue, and eCBs are rapidly degraded by abundant endogenous lipases.
To circumvent these obstacles, Kao and colleagues designed a highly water-soluble caged AEA that is inert to lipases. When perfused into hippocampal slice preparations, the caged AEA serves as a latent eCB pool, and focal photolysis rapidly liberates highly hydrophobic AEA in situ to activate CB1 receptors. They have used whole-cell voltage-clamp recording and intracellular Ca2+ measurement in combination with photorelease of caged AEA and Ncm-Glu (Cai et al., 2004) to probe the dynamics of eCB signaling in the hippocampal CA1 region (Heinbockel et al., 2005). Photorelease of AEA transiently suppressed spontaneous IPSCs with a time course comparable with that of DSI (Fig. 4), supporting the role of eCB as mediator of the process. These experiments also allowed them to determine the time for synthesis and release of eCB from the postsynaptic neuron, which was estimated to be 75-190 ms at room temperature, comparable with the timescale of metabotropic signaling and at least an order of magnitude faster than previously supposed. These findings suggest that, far from simply serving long-term neuromodulatory functions, eCB signaling is sufficiently fast to exert moment-by-moment control of synaptic transmission.
Figure 4.

Dynamics of spontaneous IPSC suppression by photorelease of AEA in cultured hippocampal slices. Left, Chemical scheme showing photorelease of caged AEA. Center, Photorelease of caged AEA, present at 200 μm, by a 100 ms UV laser flash (jagged arrow) transiently suppresses spontaneous IPSCs (downward deflections) recorded under whole-cell voltage clamp from a pyramidal cell at room temperature with ionotropic glutamate receptors blocked. Right, Grouped data showing time course of suppression of inhibition induced by AEA photorelease (17 cells). Modified from Heinbockel et al. (2005).
Whereas neuronal processes in the CNS and their integrative functions have been extensively studied by both optical and electrophysiological methods, most peripheral sensory nerve terminals, being embedded in opaque tissue, are inaccessible to experimental manipulation. A notable exception is the cornea, which has the highest density of peripheral sensory nerve innervation of any tissue and which is entirely transparent and thus ideal for study by optical methods. Corneal nerve terminals reside in the epithelial (outer-most) layer of the cornea (Zander and Weddell, 1951; MacIver and Tanelian, 1993), are almost exclusively nociceptive Aδ and C fibers bearing the vanilloid receptor (TRPV1), and originate from the ophthalmic branch of the trigeminal ganglion (Marfurt, 2000). Kao and colleagues have developed a novel preparation that permits fluorescence imaging of [Ca2+]i in corneal nerve terminals (Gover et al., 2003). They have also developed methods to stimulate individual nerve terminals by photorelease of N-(2-nitroveratryl)-N-vanillyl-nonanoylamide (Nv-VNA), a caged vanilloid. Because of the high density of nerve endings in the 50-μm-thick corneal epithelium (>106 in ∼0.5 cm2 area), selective stimulation of individual nerve terminals is extremely difficult even with standard uncaging techniques. This is because above and below the focal plane, the defocused UV light beam can still photorelease some vanilloids, which can inadvertently excite nearby nerve terminals. Therefore, they resorted to two-photon photolysis, in which ∼120 fs pulses of 720 nm (near-infrared) light from a titanium:sapphire laser are focused onto the target nerve terminal. The extremely high light intensity thus achieved within the subfemtoliter focal volume permits Nv-VNA to capture two 720 nm photons “simultaneously.” This is equivalent to absorption of a single 360 nm (UV) photon and leads to uncaging. Light intensity outside the focal volume is too low to permit two-photon absorption; therefore, photorelease can be achieved with very high spatial resolution. Selective stimulation of a corneal nerve terminal by two-photon uncaging of Nv-VNA is shown in Figure 5.
Figure 5.

Stimulation of corneal nerve terminals by two-photon photorelease of vanilloid agonist. Rat cornea was loaded in vivo with the fluorescent Ca2+ indicator Oregon Green 488 BAPTA-1 dextran (10 kDa), as described previously (Gover et al., 2003). Confocal fluorescence images of the dissected cornea were acquired with 488 nm excitation on a confocal microscope (LSM-510 NLO; Zeiss, Oberkochen, Germany) fitted with a titanium:sapphire laser (Mira 900-F; Coherent-AMT). The cornea was superfused with 5 μm Nv-VNA in Locke's solution. Three corneal nerve terminals are discernible in the field of view. For two-photon photolysis, the 720 nm output of the titanium:sapphire laser was scanned seven times over a ∼5 μm2 area centered at the site marked by the yellow arrow (pixel dwell time, 1.6 μs). Changes in Ca2+ -sensitive fluorescence from the two terminals marked by the red and green arrows are shown in the correspondingly colored traces at right; data are represented as fluorescence change relative to baseline intensity (ΔF/F0). The images shown were acquired immediately before (Pre) and 5.05 s after (Post) two-photon excitation and correspond to the points marked by filled symbols in the ΔF/F0 traces. The nerve terminal stimulated by vanilloid photorelease showed a marked increase in [Ca2+]i, whereas the adjacent, unstimulated terminal showed no change. Scale bar, 5 μm. (T. D. Gover, J. P.Y. Kao, and D. Weinreich, unpublished observation.)
Controlling gene expression with light
Although light has been used to manipulate physiology, it has rarely been used to control gene expression. Building on the inducible gene expression technology based on the ecdysone promoter system (No et al., 1996), Kao and colleagues have developed a method for using light to turn on transgene expression (Kao and Freilich, 2004). In this approach, components of the ecdysone-inducible expression system (the gene for the ecdysone steroid receptor and the transgene under control of the ecdysone-dependent promoter) are introduced into cells or tissues by standard transfection techniques or by virally mediated gene transfer. Application of steroid analogs of ecydysone (e.g., ponasterone) or nonsteroidal ecdysoids (NSEs) effectively activates transgene expression. The AM approach, widely used for loading fluorescent indicators into cells (Tsien, 1981), is used to load a caged NSE into transgenic cells at high concentrations (>0.1 mm) by passive incubation. Focal UV or two-photon photolysis generates active NSE intracellularly to activate the ecdysone receptor and turn on transgene expression. This is illustrated in Figure 6.
Figure 6.

Light-activated gene expression. A549 cells were infected with adenoviruses Ad-VgRXR (bearing constructs for the ecdysone and RXR coreceptors) and AdEGIKir2.1 [bearing genes for enhanced GFP (EGFP) and Kir2.1 K+ channel with an intervening internal ribosomal entry site, under ecdysone promoter control]. Twenty hours after infection, cultures were subjected to NSE uncaging, and fluorescence imaging and electrophysiological measurements were undertaken 24 h later. Cells stimulated by NSE photorelease expressed EGFP reporter abundantly (top right image), as evidenced by the bright green fluorescence. Expression of the exogenous K+ channel was also high, as shown by the massive outward currents measured under whole-cell voltage clamp. In contrast, control cells expressed neither EGFP reporter (top left image) nor exogenous K+ channels. A light image (bottom left) and merged light and EGFP image (bottom right) are also shown. From a holding potential of -45 mV, outward currents were evoked by a series of voltage steps to -15 to +25 mV in 5 mV increments (D. A. Freilich, J. Ni, D. C. Johns, T. H. V. Moreira, A. Garzino-Demo, and J. P. Y. Kao, unpublished observation).
Detection and dissection of synaptic vesicle endocytosis using light
Defining the molecular mechanisms that control synaptic vesicle endocytosis has been challenging, in part because endocytosis is a rapid, compensatory process that follows exocytosis. To identify and characterize the molecular players in compensatory endocytosis, it is therefore necessary to use techniques that perturb specific proteins only during endocytosis, but not exocytosis. Because these two processes are temporally and spatially coupled, it is technically difficult to inhibit protein function during only one of these phases of the synaptic vesicle cycle. Furthermore, teasing out the role of a protein that may be involved in both exocytosis and endocytosis is especially difficult, because disruption of a protein involved in exocytosis would likely obscure analysis of its potential endocytic function.
Important advances in the study of endocytosis have come from the examination of temperature-sensitive mutations, such as shibire (dynamin), which cause specific inactivation of protein function at high temperatures (Koenig et al., 1983). However, even temperature-sensitive mutations have a fairly slow temporal resolution, and most proteins and individual protein domains are not accessible to this approach. Protein photoinactivation has the potential to combine high spatial resolution of inactivation with even better temporal resolution than temperature-sensitive mutations. Using photoinactivation, it may be possible to dissect out the precise exocytic and endocytic functions of a protein, as well as the function of a protein at discrete stages in the multistep process of synaptic vesicle endocytosis.
FlAsH [4′,5′-bis(1,2,3-dithioarsolan-2-yl)fluorescein] is a membrane-permeant fluorescein derivative that fluoresces when bound to a short tetracysteine motif engineered into a protein of interest (Griffin et al., 1998; Gaietta et al., 2002). Graeme Davis and colleagues have shown that FlAsH can specifically label proteins in Drosophila in vivo and that a FlAsH-bound protein can be specifically photoinactivated in seconds in a dose-sensitive manner, with the spatial and temporal precision of light (Marek and Davis, 2002; Tour et al., 2003). Poskanzer et al. (2003) have combined this genetically encodable system of photoinactivation with use of the synapto-pHluorins, pH-sensitive green fluorescent protein (GFP)-tagged synaptic vesicle proteins that monitor synaptic vesicle recycling in real time (Miesenbock et al., 1998). In combination, these techniques enabled them to assay endocytosis while disrupting the function of specific proteins.
Synaptotagmin I (Syt I), a Ca2+ sensor for synaptic vesicle fusion, has been implicated previously in synaptic vesicle endocytosis. However, this hypothesis could not be tested directly, in part because neurotransmitter release is severely reduced in Syt I mutants. By FlAsH photoinactivating Syt I only during endocytosis and assaying vesicle recycling with the synapto-pHluorins, they have shown that this protein is necessary for synaptic vesicle endocytosis (Fig. 7) (Poskanzer et al., 2003). Thus, Syt I, during insertion in the synaptic plasma membrane during vesicle fusion, is poised to link the exocytosis of a synaptic vesicle with its subsequent internalization. They are using these techniques to define the function of Syt I in endocytosis more specifically and to explore how this protein coordinates different aspects of the synaptic vesicle cycle. These genetically encoded fluorescent tools should enable the analysis of other molecular interactions important in synaptic vesicle recycling, including those of other known exocytic proteins. In addition, this type of temporally and spatially specific light inactivation could be used in the future to dissect the mechanisms of other rapid cellular processes.
Figure 7.
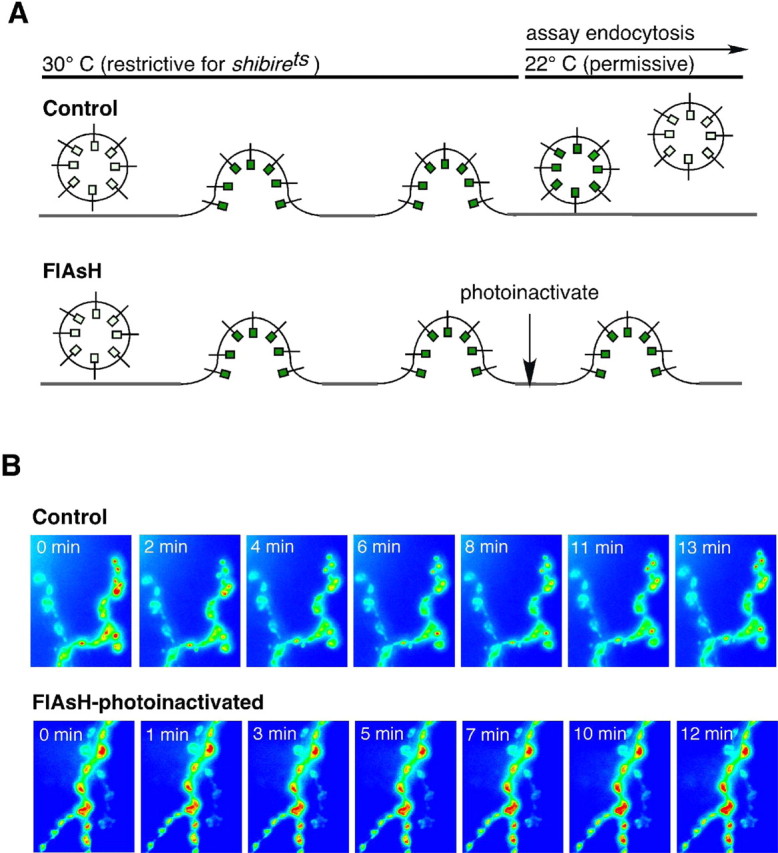
Photoinactivation of Syt I impairs endocytosis after normal vesicle fusion at the Drosophila neuromuscular junction. A, Schematic of experimental protocol and results. Nerve stimulation causes exocytosis, but no endocytosis, at the shibirets restrictive temperature in both conditions. Synaptic vesicle proteins and membrane are therefore trapped at the plasma membrane. When returned to the permissive temperature, control synapses endocytose synaptic vesicles, as indicated by synapto-pHluorin fluorescence decay (Control). At synapses in which Syt I has been photoinactivated (FlAsH), there is no synapto-pHluorin fluorescence decay, indicating that endocytosis is blocked. B, Images demonstrating synapto-pHluorin intensity changes at the neuromuscular junction after the shift from the restrictive to the permissive temperature. Time indicates minutes at the permissive temperature. Modified from Poskanzer et al. (2003).
Light-activated ion channels for remote control of neuronal firing
Neurons possess ion channels that are directly sensitive to voltage, ligands, temperature, or mechanical forces, but not to light. To allow optical regulation of neural activity, Kramer and colleagues have used a combination of organic chemistry and molecular biology to engineer ion channels that are directly opened and closed with different wavelengths of light (Banghart et al., 2004). These “hybrid” channels have two parts: a synthetic photoswitch molecule and a specially modified Shaker K+ channel protein. The photoswitch molecule (MAL-AZO-QA) contains a cysteine-reactive maleimide group (MAL), for covalent attachment to the channel protein, a photoisomerizable azobenzene group (AZO), which changes length in response to light, and a quaternary ammonium ion (QA), with blocks the pore of K+ channels (Fig. 8A). Various engineered mutations in the Shaker K+ channel minimize inactivation and shift the voltage dependence of activation, rendering the channel constitutively open in the absence of the photoswitch. The channel also has a cysteine engineered into a key extracellular location, at which the photo-switch can covalently attach to the channel, thus tethering the QA group near the pore of the channel. In the dark or in visible light (>450 nm), the AZO group is in its extended trans configuration, and the QA can reach the pore and block conduction of K+ ions. Exposure to near-UV light (380 nm) photoisomerizes the AZO to its bent cis-configuration, thereby shortening the molecule so that the QA cannot reach the pore, which then permits conduction of K+ ions. Thus, different wavelengths of light open or close the channel by advancing or retracting the blocking group from the pore.
Figure 8.

Design and use of light-activated SPARK channels. A, The channels contain a synthetic photoswitch molecule (MAL-AZO-QA; top) that changes length from 17Å (trans) to 10Å (cis) during exposure to light. Covalent attachment of the molecule to the outside of a Shaker K+ channel (bottom) allows the pore-blocking QA group (blue ball) to be retracted or advanced into the pore with different wavelengths of light. B, Rapid control of SPARK channels with light. Activation of the channels with 380 nm light (violet bars) and deactivation with 500 nm light (green bars). Data are from a membrane patch excised from a Xenopus oocyte expressing the H-SPARK channel. C, Expression of the H-SPARK channel in cultured hippocampal pyramidal neurons allows 380 nm light (violet bars) to suppress spontaneous action potential firing and 500 nm light (green bars) to restore activity. Modified from Banghart et al. (2004).
This synthetic photoisomerizable azobenzene-regulated K+ channel (called the “SPARK” channel) can be exogenously expressed in various cell types with standard gene transfection methods. Light opens and closes SPARK channels within seconds, and it can be used repeatedly with little or no decrement in the response (Fig. 8B). In hippocampal neurons, expression of the modified Shaker channel, followed by treatment with MAL-AZO-QA, generates SPARK channels that can be used for photic activation of the K+ conductance and thus can reversibly suppress action potential firing (Fig. 8C). Remarkably, MAL-AZO-QA has no detectable effect on neurons that fail to express the Shaker channel. This is because successful coupling of MAL-AZO-QA with the channel requires that the distance between the cysteine attachment site and the QA binding site in the pore to closely match the length of the photoswitch molecule. The specificity of the photoswitch for the exogenously expressed Shaker channel implies that SPARK channels can be targeted to specific types of neurons simply by targeting Shaker channel expression with cell-type-specific promoters.
Because the photoswitch is covalently attached to an ion channel, which is integral to the plasma membrane of the targeted neuron, this approach avoids the use of diffusible chemical activators that can limit the spatial and temporal accuracy of photic control of neuronal activity. The structural basis of channel function is perhaps better understood for Shaker than for any other voltage-gated channel (Jan and Jan, 1997), making it easy to customize SPARK for particular uses by altering the channel protein. Thus, mutations can be made in the pore-lining domain to convert the K+-selective Shaker channel into a nonselective cation channel (Heginbotham et al., 1994), changing the channel from one that hyperpolarizes cells when it is opened with light (H-SPARK) into a channel that depolarizes (D-SPARK). Introduction of a specific dendritic targeting sequence (Rivera et al., 2003) directs SPARK channels to the dendritic tree, allowing optical regulation of dendritic spiking. The photoswitch molecule can also be modified, for example, to change the stability of the cis- configuration, resulting in persistent SPARK channel activation triggered by a brief flash of light.
The creation of SPARK channels provides a precise and non-invasive way to control neural activity, with experimental and, perhaps eventually, medical applications. Acute activation of SPARK channels, either in single neurons or many neurons of a given type, can be used to assess the role of neurons in complex neural circuits. Sustained activation of SPARK channels, for example, in the dendritic tree, can be used to investigate homeostasis of neuronal excitability. Farther down the road is the possibility of introducing SPARK channels in vivo by transgenesis or viral transduction, to allow light to control activity in intact neural structures. The most intriguing possibility is the retina, the one part of the nervous system normally accessible to light. By expressing D-SPARK and H-SPARK channels in retinal ganglion cells, it may be possible to generate virtual “On” and “Off” cells that are directly activated or inhibited by light. This could enable light to directly control spiking in these cells, even if the natural photoreceptors (the rods and cones) have degenerated because of experimental manipulations or disease.
Summary
In conclusion, the data discussed here demonstrate that light is not merely a medium for passively visualizing structures and events but rather a tool to be used actively for manipulating those structures and events. The approaches described above represent enabling technology that has wide application in neurobiology. We are convinced that the introduction of this powerful new technology will invariably stimulate new, as well as new types of, experiments.
Footnotes
This work was supported by grants from the National Institutes of Health (MH65488 and NS40338 to S.M.T., GM56481 and GM64706 to J.P.Y.K., EY 12649 to R.H.K., MH072698 to Graeme Davis, and NS045193 to S.S.-H.W.) and the National Science Foundation (IOB 0347719 to S.S.-H.W.), by a Keck Foundation Young Investigator Award (S.S.-H.W.), and by a Wellcome Trust Senior Fellowship (R.A.S.).
Correspondence should be addressed to Scott Thompson, Department of Physiology, University of Maryland School of Medicine, 655 West Baltimore Street, Baltimore, MD 21201. E-mail: sthom003@umaryland.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510358-08$15.00/0
References
- Bagal AA, Kao JPY, Tang C-M, Thompson SM (2005) Long-term potentiation of exogenous glutamate responses at single dendritic spines. Proc Natl Acad Sci USA 102: 14434-14439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banghart M, Borges K, Isacoff E, Trauner D, Kramer RH (2004) Light-activated ion channels for remote control of neuronal firing. Nat Neurosci 7: 1381-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Liang CW, Muralidharan S, Kao JP, Tang CM, Thompson SM (2004) Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron 44: 351-364. [DOI] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL (2004) State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron 44: 483-493. [DOI] [PubMed] [Google Scholar]
- Cathala L, Brickley S, Cull-Candy S, Farrant M (2003) Maturation of EPSCs and intrinsic membrane properties enhances precision at a cerebellar synapse. J Neurosci 23: 6074-6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGregorio DA, Nusser Z, Silver RA (2002) Spillover of glutamate onto synaptic AMPA receptors enhances fast transmission at a cerebellar synapse. Neuron 35: 521-533. [DOI] [PubMed] [Google Scholar]
- DiGregorio DA, Nielsen TA, Silver RA (2004) Investigation of synaptic AMPA receptors with glutamate uncaging using a diffraction-limited UV spot. Soc Neurosci Abstr 30: 404.4. [Google Scholar]
- Freund TF, Katona I, Piomelli D (2003) Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83: 1017-1066. [DOI] [PubMed] [Google Scholar]
- Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH (2002) Multicolor and electron microscopic imaging of connexin trafficking. Science 296: 503-507. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S (2001) Spike transmission and synchrony detection in networks of GABAergic interneurons. Science 292: 2295-2299. [DOI] [PubMed] [Google Scholar]
- Gover T, Kao JPY, Weinreich D (2003) Calcium signaling in single peripheral sensory nerve terminals. J Neurosci 23: 4793-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin BA, Adams SR, Tsien RY (1998) Specific covalent labeling of recombinant protein molecules inside live cells. Science 281: 269-272. [DOI] [PubMed] [Google Scholar]
- Heginbotham L, Lu Z, Abramson T, MacKinnon R (1994) Mutations in the K+ channel signature sequence. Biophys J 66: 1061-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinbockel T, Brager DH, Reich CG, Zhao J, Muralidharan S, Alger BE, Kao JPY (2005) Endocannabinoid signaling dynamics probed with optical tools. J Neurosci 25: 9449-9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopt A, Neher E (2001) Highly nonlinear photodamage in two-photon fluorescence microscopy. Biophys J 80: 2029-2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen L (2003) Cannabis and the brain. Brain 126: 1252-1270. [DOI] [PubMed] [Google Scholar]
- Jan LY, Jan YN (1997) Cloned potassium channels from eukaryotes and prokaryotes. Annu Rev Neurosci 20: 91-123. [DOI] [PubMed] [Google Scholar]
- Jonas P, Spruston N (1994) Mechanisms shaping glutamate-mediated excitatory postsynaptic currents in the CNS. Curr Opin Neurobiol 4: 366-372. [DOI] [PubMed] [Google Scholar]
- Kao JPY, Freilich DA (2004) Reagents and method for spatio-temporal control of gene expression by illumination. U.S. Patent 6, 803: 479. [Google Scholar]
- Kim J, Isokawa M, Ledent C, Alger BE (2002) Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci 22: 10182-10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskin NI, Chillingworth R, McCray JA, Piston D, Ogden D (2002) The efficiency of two-photon photolysis of a “caged” fluorophore, o-1-(2-nitrophenyl)ethylpyranine, in relation to photodamage of synaptic terminals. Eur Biophys J 30: 588-604. [DOI] [PubMed] [Google Scholar]
- Koenig JH, Saito K, Ikeda K (1983) Reversible control of synaptic transmission in a single gene mutant of Drosophila melanogaster J Cell Biol 96: 1517-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester HJ, Baur D, Uhl R, Hell SW (1999) Ca2+ fluorescence imaging with pico- and femtosecond two-photon excitation: signal and photodamage. Biophys J 77: 2226-2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29: 717-727. [DOI] [PubMed] [Google Scholar]
- Llano I, Leresche N, Marty A (1991) Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron 6: 565-574. [DOI] [PubMed] [Google Scholar]
- MacIver MB, Tanelian DL (1993) Structural and functional specialization of Aδ and C fiber free nerve endings innvervating rabbit corneal epithelium. J Neurosci 13: 4511-4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M (2001) Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31: 463-475. [DOI] [PubMed] [Google Scholar]
- Marek KW, Davis GW (2002) Transgenically encoded protein photoinactivation (FlAsH-FALI): acute inactivation of synaptotagmin I. Neuron 36: 805-813. [DOI] [PubMed] [Google Scholar]
- Marfurt CF (2000) Nervous control of the cornea. In: Nervous control of the eye (Burnstock G, Sillito AM, eds), pp 41-92. Amsterdam: Harwood.
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H (2001) Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci 4: 1086-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429: 761-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE (1998) Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394: 192-195. [DOI] [PubMed] [Google Scholar]
- Nielsen TA, DiGregorio DA, Silver RA (2004) Modulation of glutamate mobility reveals the mechanism underlying slow-rising AMPAR EPSCs and the diffusion coefficient in the synaptic cleft. Neuron 42: 757-771. [DOI] [PubMed] [Google Scholar]
- No D, Yao TP, Evans RM (1996) Ecdysone-inducible gene expression in mammalian cells and transgenic mice. Proc Natl Acad Sci USA 93: 3346-3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M (2001) Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to pre-synaptic terminals. Neuron 29: 729-738. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE (1992) Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci 12: 4122-4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polsky A, Mel BW, Schiller J (2004) Computational subunits in thin dendrites of pyramidal cells. Nat Neurosci 7: 621-667. [DOI] [PubMed] [Google Scholar]
- Poskanzer KE, Marek KW, Sweeney ST, Davis GW (2003) Synaptotagmin I is necessary for compensatory synaptic vesicle endocytosis in vivo. Nature 426: 559-563. [DOI] [PubMed] [Google Scholar]
- Priel A, Kolleker A, Ayalon G, Gillor M, Osten P, Stern-Bach Y (2005) Stargazin reduces desensitization and slows deactivation of the AMPA-type glutamate receptors. J Neurosci 25: 2682-2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera JF, Ahmad S, Quick MW, Liman ER, Arnold DB (2003) An evolutionarily conserved dileucine motif in Shal K+ channels mediates dendritic targeting. Nat Neurosci 6: 243-250. [DOI] [PubMed] [Google Scholar]
- Schulz PE, Cook EP, Johnston D (1994) Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J Neurosci 14: 5325-5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoham S, O'Connor DH, Sarkisov DV, Wang SS-H (2005) Rapid neurotransmitter uncaging in spatially defined patterns. Nat Methods 2: 837-843. [DOI] [PubMed] [Google Scholar]
- Tomita S, Adesnik H, Sekiguchi M, Zhang W, Wada K, Howe JR, Nicoll RA, Bredt DS (2005) Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature 435: 1052-1058. [DOI] [PubMed] [Google Scholar]
- Tour O, Meijer RM, Zacharias DA, Adams SR, Tsien RY (2003) Genetically targeted chromophore-assisted light inactivation. Nat Biotechnol 21: 1505-1508. [DOI] [PubMed] [Google Scholar]
- Tsien RY (1981) A non-disruptive technique for loading calcium buffers and indicators into cells. Nature 290: 527-528. [DOI] [PubMed] [Google Scholar]
- Wei DS, Mei YA, Bagal A, Kao JP, Thompson SM, Tang CM (2001) Compartmentalized and binary behavior of terminal dendrites in hippocampal pyramidal neurons. Science 293: 2272-2275. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signaling at hippocampal synapses. Nature 410: 588-592. [DOI] [PubMed] [Google Scholar]
- Zander E, Weddell G (1951) Observations of the innervation of the cornea. J Anat 85: 68-69. [PMC free article] [PubMed] [Google Scholar]
- Zipfel WR, Williams RM, Webb WW (2003) Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol 21: 1369-1377. [DOI] [PubMed] [Google Scholar]