

Abstract
Peptide YY3-36 (PYY3-36) is released by endocrine cells of the gut and may serve as an important long-distance neuropeptide signal relating energy balance information to the brain to depress food intake. The postulated mechanism is the activation of anorexigenic proopiomelanocortin (POMC) neurons of the hypothalamic arcuate nucleus. In striking contrast, using voltage and current-clamp recording, we found that PYY3-36 consistently, dose dependently, and reversibly inhibited POMC cells by reducing action potentials, hyperpolarizing the membrane potential, decreasing input resistance and inward calcium currents, increasing G-protein-gated inwardly rectifying K+ channel currents, and presynaptically inhibiting release of excitatory glutamate. Importantly, we found PYY3-36 had similar inhibitory effects on identified orexigenic neuropeptide Y (NPY) neurons. In both cell types, these effects were blocked by BIIE0246, a Y2 receptor antagonist. Together, these data argue that anorexigenic actions of PYY3-36 are mediated more likely by inhibition of NPY neurons. Dual PYY3-36 inhibition of both NPY and POMC cells may temporarily reduce the contribution of arcuate cells to feeding circuits, enhancing the role of other CNS loci.
Keywords: arcuate, feeding, hypothalamus, glutamate, Y receptors, peptide YY
Introduction
The prevalence of obesity and its consequent health risks continue to escalate. Understanding how the brain controls energy homeostasis is therefore a critical factor in understanding the origins of body weight and food intake regulation. Neurons of the hypothalamic arcuate nucleus that synthesize and release proopiomelanocortin (POMC) peptides may play a key role in reducing food intake (Wisse and Schwartz, 2001). In contrast, the nearby neurons that synthesize neuropeptide Y (NPY) and agouti-related protein are thought to enhance food intake and positive caloric balance by orexigenic actions within the hypothalamus (Elmquist et al., 1999; Schwartz et al., 2000; Spiegelman and Flier, 2001; Saper et al., 2002; Seeley and Woods, 2003).
Peptide YY (PYY) is released by cells of the gut as a longdistance signal of energy state (Stanley et al., 2004). The primary circulating signal is PYY3-36, a cleavage product of PYY (Grandt et al., 1994). PYY3-36 is reported to signal the brain that energy stores are up, thereby reducing further food intake (Batterham et al., 2002). PYY3-36 in the circulatory system or brain was reported to reduce food intake in rodents and humans by acting on the neurons of the arcuate nucleus. This model has received considerable attention in that it reveals a possible inhibitory feedback system from the gut to the brain signaling energy state in parallel to other feedback systems that signal fat deposition (leptin), meals (glucose), or endocrine response to food intake (insulin or ghrelin) and has generated excitement both as a novel sensory feedback system regulating energy homeostasis and as a possible target for treatment of obesity (Saper et al., 2002; Schwartz and Morton, 2002; Batterham and Bloom, 2003; Cowley et al., 2003a; Jobst et al., 2004). PYY3-36 causes a reduction in food intake in obese humans, and basal levels of PYY3-36 are reduced in obese subjects, suggesting that this pathway may underlie some types of obesity (Batterham et al., 2003).
The mechanism of PYY3-36 action was reported to be an excitation of POMC neurons that activates anorexigenic circuits, based on a reduction in the inhibitory synaptic input to the POMC cells mediated by a Y2 receptor expressed by inhibitory axons.
In the present report, we confirmed that PYY3-36 attenuated the inhibitory input. However, we also found that the glutamatemediated excitatory input to the POMC cells was inhibited by PYY3-36, counteracting a decrease in synaptic inhibition. In addition, PYY3-36 exerted a robust direct inhibitory action on POMC cells. PYY3-36 also was strongly inhibitory on NPY cells, and this inhibition would more likely explain the reduction in feeding mediated by PYY3-36, consistent with recent work showing that PYY3-36 reduces food intake in POMC knock-out mice (Challis et al., 2004) and in mice lacking the MC4 receptor (Halatchev et al., 2004). Furthermore, as PYY3-36 inhibited both POMC and NPY neurons, this may temporarily reduce the contribution of the arcuate nucleus to energy homeostasis, potentially resulting in a greater sensitivity to environmental stressors, which might block the anorectic actions of PYY3-36 (Tschop et al., 2004).
Materials and Methods
Animals. All of the experiments were performed on hypothalamic slices obtained from transgenic mice that selectively express enhanced green fluorescent protein (GFP) in POMC-containing neurons, as reported previously (Cowley et al., 2001, 2003b; Batterham et al., 2002) (kindly provided by Dr. M. Low, Oregon Health and Science University, Portland, OR), or express GFP selectively in NPY neurons (Pinto et al., 2004; Roseberry et al., 2004) [kindly provided by Drs. T. Horvath (Yale University, New Haven, CT) and J. Friedman (The Rockefeller University, New York, NY)]. All experimental procedures involving animals were approved by the Yale University Committee on Animal Care and Use.
Hypothalamic slices. Slices containing the arcuate nucleus (250-360 μm) were obtained as described previously (Acuna-Goycolea et al., 2004). Briefly, 2- to 7-week-old animals were deeply anesthetized with sodium pentobarbital (100 mg/kg) during the active (dark) and inactive (light) periods, and then the brain was rapidly removed and placed in ice-cold oxygenated (95% O2, 5% CO2) solution containing the following (in mm): 220 sucrose, 2.5 KCl, 6 MgCl2, 1 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose. A block of tissue containing the mediobasal hypothalamus was dissected, and coronal slices were cut in sucrose solution with a vibratome. After 2 h of recovery from sectioning, slices were moved to the recording chamber mounted on an Olympus (Tokyo, Japan) BX51WI upright microscope equipped with video-enhanced infrared-differential interference contrast (IR-DIC) and fluorescence. Tissue was superfused with gassed artificial CSF (ACSF) (95% O2, 5% CO2) that contained the following (in mm): 124 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 1.23 NaH2PO4, 26 NaHCO3, and 10 glucose. The solutions were heated to 34-36°C using a TC-344B heater controller (Warner Instruments, Hamden, CT). Neurons were visualized with an Olympus 40× water-immersion lens.
Electrophysiology. Whole-cell current- and voltage-clamp recordings were performed using pipettes with 4-6 MΩ resistance after filling with internal solution. The pipettes were made of borosilicate glass (World Precision Instruments, Sarasota, FL) using a PP-83 vertical puller (Narishige, Tokyo, Japan). For most recordings, the composition of the internal solution was as follows (in mm): 130 KMeSO4 (or KCl for IPSCs), 1 MgCl2, 10 HEPES, 1.1 EGTA, 2 Mg-ATP, 0.5 Na2-GTP, and 10 Na2-phosphocreatine, pH 7.3, with KOH. The pipette solution for barium current (IBa) recording contained the following (in mm): 130 CsMeSO3, 1 MgCl2, 11 EGTA-Cs, 2 Mg-ATP, 0.5 Na2-GTP, and 10 HEPES, pH 7.3, with CsOH. The bath solution for IBa recording contained the following (in mm): 79.5 NaCl, 40 tetraethylammonium (TEA)-Cl, 3 KCl, 2 MgCl2, 5 BaCl2, 1.23 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4, after bubbling with 95% O2 and 5% CO2. POMC-GFP or NPY-GFP neurons were identified by green fluorescence, and by using video-enhanced differential interference contrast, the cells were approached with the recording pipette. After a GΩ seal was obtained, a gentle negative pressure was applied to break through to the whole-cell configuration. Seal resistance was ≥1 GΩ in all of the experiments. An EPC9 amplifier and Pulse software (HEKA Elektronik, Lambrecht/Pfalz, Germany) were used for data acquisition.
Slow and fast capacitance and access resistance were compensated in voltage clamp. In current clamp, access resistance was bridge adjusted through Pulse software. Access resistance of 15-20 mΩ was typically observed and compensated up to 75% throughout the experiments. Only those cells with stable access resistance (change <10%) were used for analysis, assuring adequate membrane potential control under voltageclamp conditions. For the experiments in which voltage-activated calcium channels were studied, automatic whole-cell capacitance and access resistance compensation was performed before each voltage command.
Drug delivery was generally performed via a 250-μm-diameter flow pipe placed on the arcuate nucleus near the recording neurons, allowing the reagent to reach recorded cells faster than after bath application. This flow pipe was connected to multibarreled tubing used to change the locally puffed solution throughout the experiment. All solutions in the puffer were continuously CO2/O2 bubbled. For Y receptor agonist/antagonist interaction studies (see Fig. 2), the whole hypothalamic slices were preincubated for ≥10 min with saturating concentrations of tested Y receptor subtype antagonist. Subsequently, the agonist (dissolved in ACSF-containing the antagonist) was locally applied through the flow pipe. Potassium concentration was altered in the bath in those experiments in which the effect of increased external potassium concentration on PYY-induced inwardly rectifier current reversal potential was studied. In these studies, potassium concentration was also accordingly altered in the puffer.
Figure 2.

The depressing actions of PYY3-36 are consistent with a postsynaptic activation of Y2 receptors in POMC neurons. A, PYY3-36 reduced the spike frequency in a dose-dependent manner. B, The graph shows the time course of PYY3-36 (open circles) and NPY13-36 (filled circles) inhibitory actions on the frequency of action potentials in identified arcuate POMC cells. C, The actions of PYY3-36 and NPY13-36, two agonists of Y2 receptors, on POMC neuron membrane potential were dose dependent. D, Both Y1 (Pro34NPY or d-Arg25NPY) and Y2 (PYY3-36 or NPY13-36) receptor agonists hyperpolarized POMC neurons. BIBP3226, a selective Y1 receptor antagonist, eliminated the Y1 receptor-mediated hyperpolarization but did not affect Y2 receptor-dependent changes in membrane potential.
Postsynaptic currents were detected in Axograph 4.8, using a double exponential function sensitive to the kinetics of rise and decay of the currents (Bekkers and Stevens, 1995). Only those events with amplitude ≥5 pA were considered for the analysis. This procedure has been described in detail previously (Gao and van den Pol, 1999). Data in the Results are expressed as mean ± SEM. Statistical analyses were performed using one-way ANOVA followed by a Bonferroni's post hoc procedure for pairwise between-group comparison (i.e., control vs treatment). The nonparametric Kolmogorov-Smirnov test was used for comparison of the cumulative fractions before and during drug applications. p < 0.05 was considered statistically significant.
Reagents. Some reagents were purchased from Sigma (St. Louis, MO) and included the following: dl-2-amino-5-phosphonovaleric acid (AP-5), bicuculline methiodide (BIC), and 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX). Neuropeptide Y (human, rat) and [D-Arg25-NPY] (human, rat) were obtained from Phoenix Pharmaceuticals (Belmont, CA). PYY3-36 (human), NPY13-36 (porcine), and [Pro34]-NPY (human) were purchased from American Peptide (Sunnydale, CA). BIBP3226 [N2-(diphenylacetyl)-N-[(4-hydroxyphenyl)methyl]-d-arginine-amide] was purchased from Bachem (San Carlos, CA), BIIE0246 was purchased from Tocris Cookson (Ellisville, MO), and tetrodotoxin (TTX) and Tertiapin-Q were purchased from Alomone Labs (Jerusalem, Israel).
Results
POMC neurons
POMC-containing cells (n > 120) in the arcuate nucleus were identified by selective GFP expression in transgenic mice (Cowley et al., 2001, 2003b; Batterham et al., 2002). A typical GFP-expressing cell is shown in Figure 1A (left; GFP expression is shown in white). The same cell (arrow) is presented on the right with the recording electrode (arrowhead) under IR-DIC. Brief negative current injections (-60 to -100 pA; 10 pA steps for 100-200 ms) revealed a membrane rectification characterized by a sag (data not shown). Although this membrane potential rectification has been associated with the presence of an Ih current in hypothalamic neurons (Eggermann et al., 2003, Ibrahim et al., 2003), its ionic bases in POMC-expressing cells merits additional characterization. Low-threshold spikes were also typically observed in arcuate POMC neurons at the end of hyperpolarizing current steps (Fig. 1B, black trace). Positive current steps delivered to the recorded cells through the recording pipette evoked a burst of action potentials as shown in Figure 1B (gray). Spike frequency adaptation, a decrease in spike frequency with continued stimulation, was consistently observed when current steps ≥40 pA were applied to the recorded cells (Fig. 1B, gray trace). In addition, a consistent reduction in the action potential amplitude was seen near the end of a 200 ms depolarizing current step (Fig. 1B).
Figure 1.
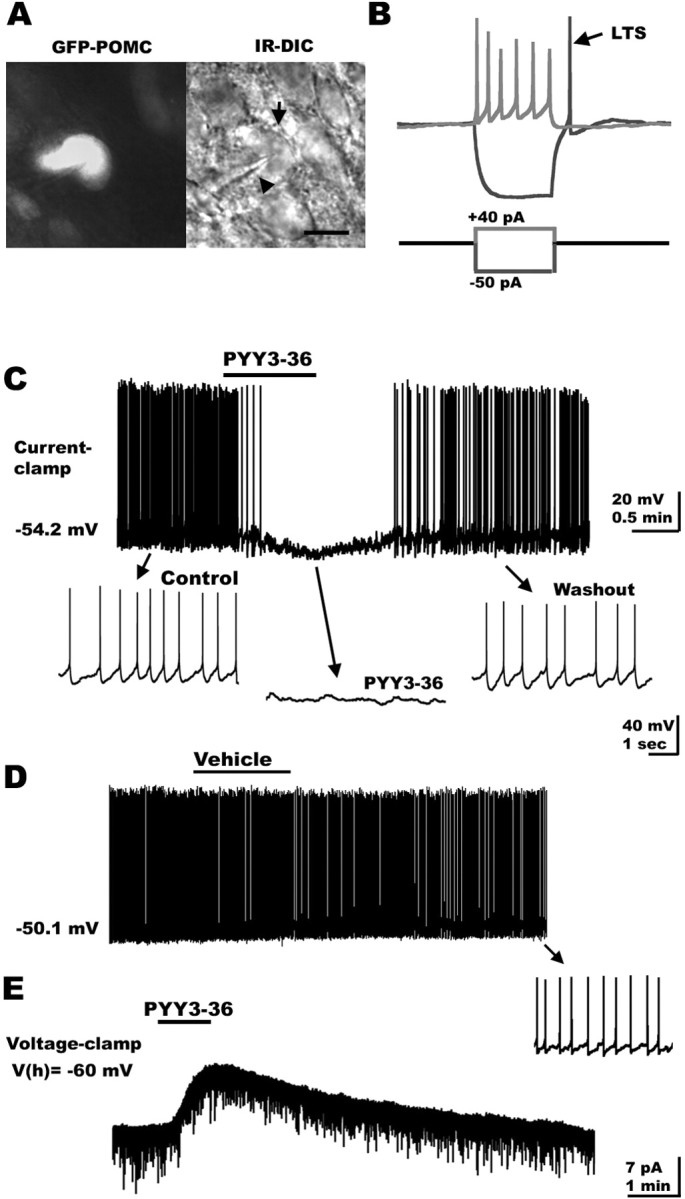
PYY3-36 inhibits GFP-expressing POMC neurons in the hypothalamus. A, Identification of hypothalamic POMC neurons in a slice from GFP-POMC transgenic mouse. Fluorescent (left, white) and IR-DIC (right) images of a POMC cell body (arrow) being approached by the recording pipette (arrowhead). B, When negative current steps (-50 to -100 pA for 50-200 ms; blue trace, bottom) were delivered to POMC neurons, low-threshold spikes (LTS) became evident (blue trace, top). Positive current steps (+20 to +30 pA; red trace, bottom) generated a burst of spikes. Spike-frequency adaptation was typically observed in POMC cells during extended current injection. C, Under current clamp, PYY3-36 blocked spike frequency and hyperpolarized POMC-expressing neurons. D, Flow pipe application of the vehicle (ACSF) did not change the spike frequency or membrane potential in POMC neurons. E, In voltage clamp, PYY3-36 induced a consistent outward current in these hypothalamic cells.
The PYY3-36 responses of GFP-expressing neurons were studied under whole-cell current clamp. Under resting conditions (when no current was delivered through the recording pipette), hypothalamic POMC neurons showed spontaneous discharge of action potentials (mean spontaneous firing, 1.6 ± 0.9 Hz; n = 8). PYY3-36 was locally applied through a 250-μm-diameter flow pipe placed on the arcuate nucleus and aimed at recorded cells, as described previously (Acuna-Goycolea et al., 2004). PYY3-36 (100 nm) induced a robust and relatively fast (20-60 s) hyperpolarization leading to a strong suppression of action potentials in six POMC neurons studied. The mean membrane potential was hyperpolarized by 5.8 ± 1.3 mV (p < 0.05) returning to the control pretreated levels 1.5-4 min after peptide washout (Fig. 1C). No change in POMC neuronal activity (spike frequency) was observed with application of ACSF (vehicle) alone, consistent with the lack of effects associated with either the vehicle or mechanical stimulation (pressure) with flow pipe delivery (Fig. 1D) (n = 6). Spike frequency was reduced by 79.7 ± 7.9% compared with control pretreated conditions (p < 0.05). Under voltage clamp, the application of 100 nm PYY3-36 resulted in a 10.1 ± 2.3 pA outward current at -60 mV holding potential (n = 7; p < 0.05) (Fig. 1E). No outward current was observed after vehicle application under the same experimental conditions (n = 4; data not shown).
Whole-cell patch-clamp recording can induce cellular rundown that might affect transmitter responses, particularly those mediated by peptidergic G-protein-coupled receptors; this can be a result of the diffusion of the pipette solution into the recorded neuron. To rule out this possibility, we evaluated PYY3-36 actions using patch pipettes placed near the GFP-expressing POMC neurons (extracellular or “loose cell-attached” configuration) PYY3-36 (100 nm) drastically decreased the firing rate by 89.1 ± 6.7% (n = 6; data not shown; p < 0.05). In two of these cells, whole-cell configuration was further achieved, and PYY3-36 actions were again tested under current clamp. In both cases, we found a consistent depression of spike frequency as well as hyperpolarization in GFP-containing cells (reduction in firing, 84.1 ± 3.2%; hyperpolarization, 5.1 ± 2.1 mV). Together, the results presented above are consistent with the view that PYY3-36 exerts robust depressing actions on hypothalamic POMC neurons and suggest that these inhibitory effects are relatively independent of the experimental conditions used in the present series of experiments.
PYY3-36 inhibited POMC cells in a dose-dependent manner (Fig. 2A). PYY3-36, at 10 nm, 100 nm, or 1 μm, significantly depressed the spike frequency by 26.6 ± 7.9 (n = 6), 78.6 ± 7.6 (n = 6), and 93.6 ± 3.6% (n = 7), respectively (p < 0.05). A lower dose of PYY3-36 (1 nm) did not significantly affect the spike frequency of GFP-POMC cells (change in spike frequency, 5.2 ± 3.6% compared with control levels; n = 7; p > 0.05, nonsignificant). The EC50 value for the PYY3-36 actions on POMC firing rate was near 51.2 nm. Previous studies have shown that PYY3-36 might selectively act at Y2 receptors (Keire et al., 2000; Batterham et al., 2002). To further study the role of Y2 receptors in POMC neurons, we used current clamp and evaluated the physiological actions of NPY13-36, a selective Y2 agonist (Guo et al., 2002). NPY13-36 (100 nm) strongly inhibited spiking of POMC neurons (mean reduction in spike frequency after NPY13-36, 84.1 ± 3.5%) and hyperpolarized these hypothalamic cells by 6.7 ± 2.4 mV (n = 6; p < 0.05). A time course graph comparing the PYY3-36 (n = 4) and NPY13-36 (n = 6) actions on the action potential frequency is shown in Figure 2B. Two additional cells showed no sign of recovery after PYY3-36 application, and those data were not included in the means. Recovery was achieved after 2-5 min of NPY13-36 washout. We further determined the direct postsynaptic Y2 agonist actions on the membrane potential using TTX (0.5 μm) in the extracellular solution to block spike-dependent synaptic activity and antagonists of ionotropic glutamate (50 μm AP-5 plus 10 μm CNQX) and GABA (BIC, 30 μm) receptors to eliminate spontaneous excitatory and inhibitory synaptic activity, respectively. Under these conditions, both PYY3-36 and NPY13-36 induced a dose-dependent shift in the POMC neuron membrane potential as shown in Figure 2C. PYY3-36 at 10 nm, 100 nm, and 1 μm hyperpolarized these hypothalamic cells by 1.2 ± 0.9 mV (n = 5), 6.9 ± 1.1 mV (n = 8), and 9.9 ± 2.6 mV (n = 6), whereas the same doses of NPY13-36 evoked a hyperpolarization of 0.9 ± 0.9 mV (n = 5), 7.1 ± 1.1 mV (n = 7), and 10.1 ± 1.7 mV (n = 5), respectively (Fig. 2C).
Functional Y1 receptors have been found previously in many brain regions, including the arcuate nucleus (Rhim et al., 1997; Roseberry et al., 2004). Consistent with this, when the selective Y1 agonists Pro34NPY (100 nm) (Potter et al., 1991) or d-Arg25NPY (100 nm) (Mullins et al., 2001) were applied to the hypothalamic slices, a robust and significant hyperpolarization was observed in POMC neurons (Fig. 2D). All of these experiments were done with 0.5 μm TTX, 50 μm AP-5, 10 μm CNQX, and 30 μm BIC in the external solution to prevent any indirect actions of Y1 analogs. Pro34NPY (100 nm) hyperpolarized POMC cells by 7.5 ± 2.3 mV (n = 6; p < 0.05) and d-Arg25NPY induced an 8.2 ± 2.2 mV (n = 5; p < 0.05) negative shift in the membrane potential. The hyperpolarizing actions of Y1 receptor agonists were abolished by application of the Y1 antagonist BIBP3226 (1 μm) confirming the selectivity of these analogs (Fig. 2D) (n = 5 for Pro34NPY plus Y1 antagonist; n = 6 for d-Arg25NPY plus BIBP3226). In contrast, both PYY3-36 and NPY13-36-mediated hyperpolarizations were not blocked by BIBP3226 (1 μm) in the ACSF (Fig. 2D), consistent with the idea that they were not mediated by postsynaptic Y1 receptor activation (change in membrane potential after PYY3-36 plus BIBP3226, 8.7 ± 1.7 mV, n = 7; hyperpolarization after NPY13-36 plus BIBP3226, 8.8 ± 2.6 mV, n = 6). In contrast, BIIE0246 (100 nm), a selective Y2 antagonist (Doods et al., 1999), eliminated 100 nm PYY3-36-induced hyperpolarization in POMC cells (shift in membrane potential in this condition, -0.2 ± 0.8 mV; n = 6; p > 0.05). Because possible indirect actions of Y2 agonists (PYY3-36 and NPY13-36) on network activity that could have affected POMC cell activity were eliminated under our experimental conditions (TTX, AP-5, CNQX, and BIC in the ACSF), the results presented here suggest that functional postsynaptic Y2 receptors exist in hypothalamic POMC neurons.
Mechanism of hyperpolarization
We further studied the ionic mechanisms underlying the hyperpolarizing PYY3-36 actions on hypothalamic POMC neurons. Voltage-clamp experiments showed that at -60 mV holding potentials, PYY3-36 induced a 10.1 ± 2.3 pA outward current (n = 7; p < 0.05) and when membrane potential was held at more negative potentials (-70 to -90 mV), the outward current became smaller or completely disappeared (data not shown). In addition, the whole-cell input resistance of POMC neurons decreased from 1.2 ± 0.9 to 0.7 ± 0.6 GΩ (n = 6; p < 0.05) in the presence of PYY3-36 leading to the hypothesis that PYY3-36 might activate a potassium conductance, possibly the G-protein-dependent inwardly rectifier subtype (Sun and Miller, 1999). To test this, we first evaluated the actions of PYY3-36 on POMC cells in which the membrane potential was maintained near -100 mV by constant injection of -40 to -60 pA through the recording pipette (Ki = 145 mm; Ko = 3.3 mm; the reversal potential for K+ predicted by Nernst equation under our experimental conditions is near -100 mV). In these conditions, PYY3-36 did not significantly alter the membrane potential of POMC cells (n = 7; change in membrane potential, 2.3 ± 1.4 mV; p > 0.05, not significant). These results further substantiate the idea that PYY3-36 modulates potassium currents in POMC neurons. We then delivered voltage ramps (from -140 to -10 mV for 800 ms) to the recorded neurons, and the current response before and during 100 nm PYY3-36 application was determined. As shown in Figure 3A, the application of PYY3-36 to the slice induced a robust inward current for potentials between -60 and -140 mV. This effect of PYY3-36 was accompanied by an increase in the whole-cell conductance (change in the slope of control and PYY conditions in Fig. 3A) during voltage-ramp protocols, which is consistent with channels opening. All of these experiments were performed in the presence of ionotropic glutamate and GABA receptor antagonists (AP-5/CNQX and BIC, respectively), TTX (0.5 μm), and CdCl2 (200 μm) to block voltage-activated sodium and calcium channels, respectively. With 15 mm external potassium, the net current induced by PYY3-36 reversed at approximately -64.2 ± 3.2 mV (n = 6) (Fig. 3B, gray trace). We then increased the external K+ from 3.3 to 15 mm and determined the reversal potential for the PYY3-36-induced current. With normal (3.3 mm) external K+, PYY3-36 induced a current that reversed at approximately -103.1 ± 3.4 mV (n = 5). This 38.9 mV shift in reversal potential observed after changing the extracellular K+ from 3.3 to 15 mm is similar to the value predicted by the Nernst equation for a selective K+ conductance and further supports the view that PYY3-36 inhibits POMC neurons by opening K+ channels in the postsynaptic cells. We also applied voltage steps to the recorded cells (n = 4) and evaluated the current response before and during application of 100 nm PYY3-36. Depolarizing voltage steps from -60 to -20 mV (15 mm external potassium concentration) revealed a small but consistent outward current induced by PYY3-36 (data not shown); when hyperpolarizing voltage steps from -60 to -120 mV were delivered, a robust PYY3-36-evoked inward current was evident (Fig. 3C, left).
Figure 3.

PYY3-36 activates GIRK-like currents and depresses whole-cell calcium currents in POMC neurons. A, Voltage-ramp protocols (from -140 to -10 mV for 800 ms; top) revealed that PYY3-36 activates an inwardly rectifying current reversing at approximately -60 mV in 15 mm external potassium. B, The PYY3-36-induced current (gray trace) was attenuated by 800 μm barium (black) in the ACSF, consistent with an inwardly rectifying subtype. C, Voltage steps (see protocol at bottom) showing the effect of PYY3-36 on the inward current response in a typical arcuate GFP-expressing neuron in the absence (left) and the presence (right) of 200 nm Tertiapin-Q. Without pretreatment with Tertiapin-Q, PYY3-36 activated an inward current that, in addition, was blocked by 100 μm barium in the external solution. In the presence of Tertiapin-Q, PYY3-36 failed to activate the inward current. These data suggest that PYY3-36 activates a GIRK-like current in GFP-containing cells. D, Bar graph summarizing the effect of high (800 μm) and low (100 μm) doses of external barium and Tertiapin-Q (200 nm) on the current induced by PYY3-36 after voltage steps from -60 to -120 mV during 200 ms. E, PYY3-36 also reduced POMC current responses to voltage ramps from -60 to +40 mV. F, Whole-cell calcium current evoked by voltage steps from -80 to 0 mV (top) before (left) and during (right) PYY3-36 application to POMC cells. PYY3-36 depressed voltage-dependent calcium currents in these hypothalamic neurons. G, Bar graph showing the mean depressing effect of PYY3-36 on calcium current.
Barium has been suggested to block inwardly rectifying potassium channels in other brain regions (Sodickson and Bean, 1996). The presence of 0.8 mm barium in the external solution eliminated the PYY3-36-evoked hyperpolarization in POMC cells under resting conditions (with no current injection through recording pipette) (n = 5; data not shown), suggesting that PYY3-36-evoked hyperpolarization in POMC neurons was attributable to the activation of an inward rectifier potassium channel. We also performed voltage-ramp experiments in the presence of 0.8 mm barium in the ACSF. PYY3-36 (100 nm)-induced currents in POMC cells were not seen under these conditions (Fig. 3B, black trace). Additional experiments were undertaken to determine the involvement of G-protein-gated inwardly rectifying K+ channel (GIRK) currents. External barium (100 μm) is reported to exert selective inhibitory actions on GIRK-type currents (Fernandez-Fernandez et al., 1999). In 100 μm barium, the current evoked by PYY3-36 (after hyperpolarizing voltage steps from -60 to -120 with 15 mm external potassium) (Fig. 3C) was strongly reduced (reduction, 92.7 ± 4.1%; n = 6) (Fig. 3C, left, D) further substantiating a role for GIRK conductances in inhibitory PYY3-36 actions on POMC neurons. The presence of GDP-β-S in the internal solution completely eliminated the PYY3-36-evoked current in POMC cells, similar to other neurons with GIRK responses (n = 6; data not shown) (Fu et al., 2004; van den Pol et al., 2004); in this and the next experiments, negative voltage steps from -60 to -120 during 200 ms were used to study potassium currents. Finally, we used tertiapin-Q, which selectively blocks native GIRK-type currents in neurons (Chen and Johnston, 2005). Tertiapin-Q (200 nm) depressed the PYY3-36-evoked current by 85.6 ± 6.6% (n = 5) (Fig. 3C, right, D). Together, our results suggest that PYY3-36 alters inwardly rectifying potassium currents in a G-protein activation-dependent manner (GIRK-type) in POMC neurons; activation of this current would inhibit POMC neurons.
PYY3-36 depresses calcium currents Voltage-dependent calcium channels might also be modulated by transmitters, including neuropeptides (Dolphin, 2003). We determined whether PYY3-36 can affect whole-cell calcium currents in hypothalamic POMC neurons with voltage ramp protocols (duration, 500 ms; from -60 to +40 mV) (Fig. 3E, top). In these experiments, we used CsMeSO4 in the internal solution and 40 mm TEA-Cl in the ASCF (by replacing equimolar concentrations of NaCl) to prevent the activation of voltage-dependent potassium currents. In addition, 0.5 μm TTX was bath applied to block voltage-activated sodium currents, and CaCl2 was replaced by BaCl2 in the external solution to increase the conductance of calcium channels and facilitate the detection of the current evoked by depolarization. The barium current response of POMC cells to voltage ramp commands was significantly depressed by 100 nm PYY3-36 (Fig. 3E) with a maximum effect near 0 mV (n = 4). Voltage steps from -80 to 0 mV for 300 ms also showed that PYY3-36 inhibited voltage-dependent calcium channels. The application of 100 nm PYY3-36 depressed the amplitude of barium current by 29.7 ± 3.4% (Fig. 3F,G). The mean PYY3-36 depressing effect on whole-cell barium currents is shown in the bar graph in Figure 3F. The effect of PYY3-36 was statistically significant (n = 5; p < 0.05) and reversible. The Y2 agonist NPY13-36 also reduced the amplitude of barium currents activated by voltage steps by 37.3 ± 3.5% (from -80 to 0 mV; n = 4; p < 0.05; data not shown). Together, our results suggest that Y2 receptor modulation by PYY3-36 and NPY13-36 inhibits voltage-dependent calcium channels in hypothalamic POMC neurons.
PYY3-36 depresses excitatory synaptic activity
Using voltage clamp (-60 mV holding potential) with BIC (30 μm) in the ACSF, fast synaptic inward currents were typically detected in POMC neurons (Fig. 4A) with a mean frequency of ∼2.5 ± 1.2 Hz (n = 12). When the cells were maintained at very negative holding potentials (-100 mV) excitatory synaptic current became larger, whereas more positive holding potentials (-20 mV) led to smaller synaptic events (data not shown). NMDA and AMPA receptor antagonists AP-5 (50 μm) and CNQX (10 μm) completely abolished all excitatory synaptic activity in these cells, similar to other hypothalamic regions (van den Pol et al., 1990), suggesting that glutamate is the primary excitatory neurotransmitter in POMC neurons (Fig. 4A, left). In current clamp, glutamate synaptic transmission blockade with AP-5 (50 μm) and CNQX (10 μm) induced a 31.2 ± 10.5% decrease in spike frequency, further substantiating a critical role for this fast-acting neurotransmitter in the intercellular signaling in the arcuate nucleus, including POMC neurons.
Figure 4.

PYY3-36 and NPY13-36, Y2 receptor agonists, attenuated POMC glutamate-mediated synaptic activity by presynaptic mechanisms. A, In voltage clamp (-60 mV holding potentials), EPSCs were detected as fast inward currents in POMC neurons. EPSCs were completely abolished after NMDA and AMPA receptor antagonist application (using 50 μm AP-5 and 10 μm CNQX, respectively), confirming their glutamatergic nature (left). PYY3-36 reduced EPSC frequency in POMC cells (right). B, The graph shows the time course of the inhibitory PYY3-36 (open circles) and NPY13-36 (filled circles) actions on the frequency of EPSCs in hypothalamic POMC neurons. Recovery was achieved after 10-20 min of peptide washout. C, Bar graph summarizing the depressing actions of PYY3-36 (left), NPY13-36 (middle), and NPY (right) on mEPSC frequency. D, Little change in mean amplitude of mEPSCs was observed after the application of PYY3-36 (top trace), NPY13-36 (middle traces), and NPY (bottom traces) as shown in these three typical hypothalamic POMC cells.
We then evaluated the actions of PYY3-36 and NPY13-36 on the glutamate inputs to POMC neurons. In these experiments, GABAA receptor-mediated synaptic transmission was eliminated by bath application of 30 μm BIC throughout the experiment. The application of PYY3-36 (100 nm) to the arcuate slices resulted in a consistent decrease (mean decrease in EPSC frequency after PYY3-36, 48.5 ± 2.1%) in the frequency of EPSCs in eight cells (Fig. 4A, representative traces, right) (p < 0.05). In Figure 4B (open circles), the time course of the mean PYY3-36 effect on the frequency of EPSCs is presented. The effect was long lasting, and recovery was observed after 10-20 min of peptide washout. Similarly to PYY3-36, the selective Y2 agonist NPY13-36 (100 nm) also depressed EPSC frequency in POMC neurons (Fig. 4B, filled circles) (n = 6; p < 0.05). To determine whether these Y2 agonist actions were attributable to presynaptic modulation of glutamate release onto POMC neurons, we studied miniature EPSCs (mEPSCs) in the presence of 0.5 μm TTX and 30 μm BIC in external solution. PYY3-36 decreased the frequency of mEPSCs by 33.4 ± 6.3% (Fig. 4C) (n = 7; p < 0.05); NPY13-36 reduced mEPSC frequency by 38.9 ± 9.5% (Fig. 4C) (n = 6; p < 0.05). Neither PYY3-36 nor NPY13-36 significantly affected the mean amplitude of mEPSCs in POMC neurons (Fig. 4D) (n = 5; p > 0.05). The mean amplitude of mEPSCs showed little change (9.1 ± 5.6 and 7.1 ± 3.2%, respectively) with PYY3-36 and NPY13-36. The cumulative probability of the mEPSC amplitude was not significantly altered by PYY3-36 (100 nm) or NPY13-36 (100 nm) (p > 0.05; Kolmogorov-Smirnov test; data not shown). These results are consistent with the view that PYY3-36 attenuates glutamate release onto POMC neurons via presynaptic Y2 receptors.
The effect of 100 nm PYY3-36 on GABAergic synaptic input to POMC neurons was also evaluated. These experiments were done with 50 μm AP-5 and 10 μm CNQX in the bath to block glutamate-mediated synaptic transmission. PYY3-36 significantly attenuated the frequency of GABAergic currents (BIC-sensitive) by 43.5 ± 6.9% compared with control pre-PYY3-36 conditions (n = 5; p < 0.05; data not shown). These results were consistent with previous findings (Batterham et al., 2002) and were not further studied.
A recent study suggested that NPY released by local axons might alter the activity of neurons producing POMC mainly by reducing their tonic inhibitory synaptic transmission (Cowley et al., 2001). In our experiments, spontaneous glutamatergic activity in POMC neurons was consistently found, and the application of NPY (100 nm) suppressed the frequency of EPSCs by 36.4 ± 6.7% (n = 5; p < 0.05; data not shown). These experiments were performed with 30 μm BIC in the bath. TTX (0.5 μm) was further added to the ASCF, and the effect of NPY on the mEPSCs was then studied. NPY (100 nm) reduced the mEPSC frequency by 30.4 ± 7.8% (Fig. 4C) (n = 5; p < 0.05). Neither the mean amplitude of mEPSC nor their cumulative distributions were significantly reduced by NPY (Fig. 4D, bottom traces, mean amplitude) (n = 5; p > 0.05). These results are consistent with the view that in addition to its effect on inhibitory transmission, NPY can depress the release of glutamate from presynaptic axons innervating POMC neurons. Furthermore, current-clamp experiments revealed an 87.3 ± 2.9% decrease in the frequency of POMC neuron action potentials accompanied by a -5.7 ± 1.5 mV hyperpolarization in membrane potential (n = 5; p < 0.05; data not shown) in the presence of NPY. Similar effects of NPY have been detected previously with a similar (Cowley et al., 2001) and a different GFP-POMC transgenic mouse (Roseberry et al., 2004). Together, our results suggest that, in general, NPY actions would lead to inhibition of POMC neurons in the hypothalamus.
PYY3-36 inhibits NPY neurons
In contrast to the anorexic POMC-containing cells in the arcuate nucleus, nearby neurons that produce NPY are thought to enhance food intake (Schwartz et al., 2000). Although it has been suggested that NPY cells might show inhibitory responses to Y2 agonists, consistent with histological findings of Y2 receptors in NPY cells (Broberger et al., 1997), identified NPY cells have not been tested (Batterham et al., 2002; Ghamari-Langroudi et al., 2005). To test this hypothesis, we locally applied PYY3-36 to arcuate NPY-producing neurons identified by selective GFP expression in transgenic mice (Pinto et al., 2004; Roseberry et al., 2004). PYY3-36 (100 nm) induced a 96.5 ± 1.7% reduction in spontaneous action potential firing and hyperpolarized arcuate NPY neurons by 9.4 ± 2.4 mV (Fig. 5A,B) (n = 6; p < 0.05). A lower PYY3-36 dose (10 nm) reduced spontaneous spike frequency by 63.6 ± 5.8% and changed the membrane potential by -4.1 ± 0.9 mV, consistent with a dose-dependent effect of this peptide on arcuate NPY cells (Fig. 5C) (n = 6; p < 0.05). PYY3-36 (1 nm) did not significantly alter spike frequency or membrane potential. The reduction in spike frequency evoked by 10 nm PYY3-36 was significantly greater in NPY cells (63% decrease) than in POMC cells (27% decrease) (Fig. 5C) (p < 0.05).
Figure 5.
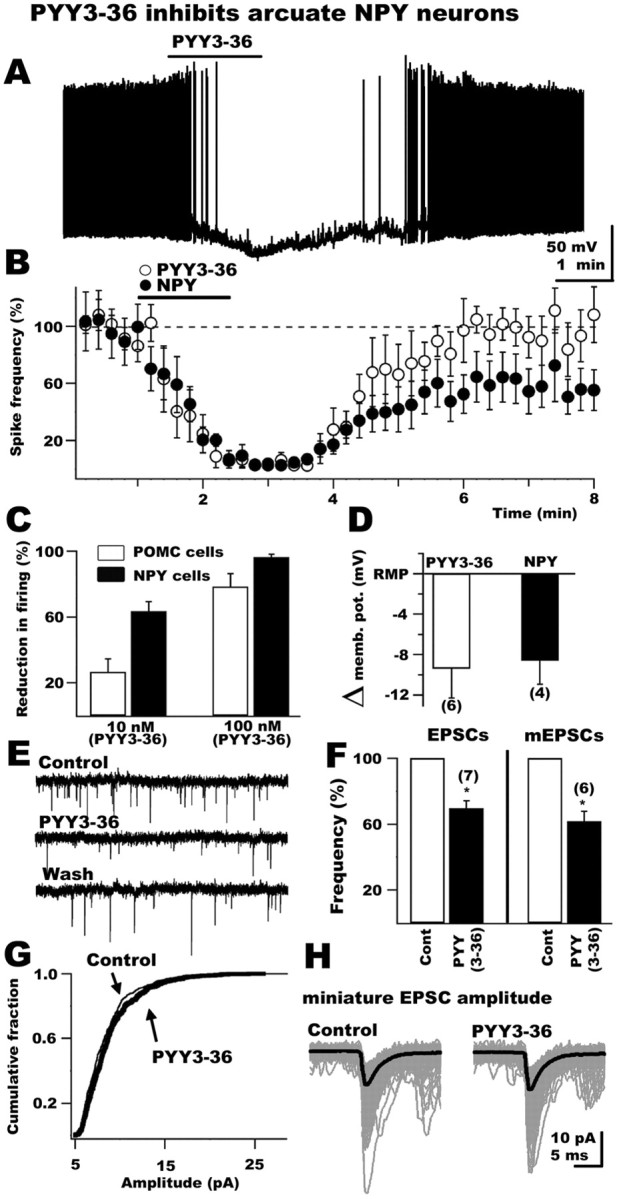
Inhibitory actions of PYY3-36 on identified arcuate NPY neurons. A, This representative recording shows the suppression in firing rate and membrane hyperpolarization of a GFP-expressing NPY neuron evoked by application of 100 nm PYY3-36 to the slice. B, Time course graph presenting the mean inhibitory actions of PYY3-36 (open circles) and NPY (filled circles) on NPY neurons. C, The bar graph compares the dose-dependent effects of PYY3-36 in POMC neurons (open bars) and NPY neurons (filled bars). D, This graph shows the direct hyperpolarizing action of PYY3-36 and NPY on GFP-expressing NPY neurons. memb. pot., Membrane potential. E, PYY3-36 reduced excitatory synaptic transmission in NPY neurons. F, Bar graph presenting the mean reduction in the frequency of both EPSCs and mEPSCs evoked by PYY3-36. G, H, Neither the cumulative distribution of mEPSC amplitude (G) nor the mean amplitude of miniature events (H) was altered by PYY3-36 consistent with a presynaptic action on glutamate release.
We then tested the effect of PYY3-36 on membrane potential in the presence of 0.5 μm TTX in the bath solution. Under this condition, 100 nm PYY3-36 still hyperpolarized NPY neurons, consistent with the presence of NPY receptors on the soma (Fig. 5D) (change in membrane potential after PYY3-36, -9.4 ± 2.9 mV; n = 6; p < 0.05). The Y1 agonist 100 nm [Pro34]NPY1-36 also hyperpolarized NPY neurons by 6.5 ± 0.4 mV (n = 5; p < 0.05), suggesting functional expression of Y1 receptors. Bath application of the Y2 selective antagonist BIIE0246 (100 nm) prevented the 100 nm PYY3-36-induced hyperpolarization (0.4 ± 2.2 mV change in membrane potential with BIIE0246 in the bath; n = 6; p > 0.05) but did not affect the negative shift in membrane potential evoked by [Pro34]NPY1-36. Pro34 NPY1-36 induced a 4.5 ± 0.6 mV hyperpolarization in the presence of BIIE0246 (n = 5; p < 0.05), consistent with the view that PYY3-36 inhibited NPY cells via postsynaptic Y2 receptor activation. Similar to POMC neurons, PYY3-36 reduced the input resistance of NPY-containing cells from 0.91 ± 0.04 to 0.56 ± 0.07 GΩ, a statistically significant change (n = 4; p < 0.05). PYY3-36 also activated GIRK currents and depressed voltage-dependent calcium currents in NPY neurons in experiments parallel to those described above for POMC cells, consistent with the view that similar postsynaptic mechanisms account for the inhibitory PYY3-36 effects on both arcuate NPY and POMC cells (data not shown).
In voltage clamp (-60 mV), PYY3-36 (100 nm) decreased the frequency of EPSCs by 30.4 ± 4.5% compared with preapplication control levels (Fig. 5E,F) (n = 7; p < 0.05), recorded in the presence of 30 μm BIC in the external solution to eliminate GABAergic transmission. These EPSCs were glutamatergic in nature, because they were completely eliminated by ionotropic glutamate NMDA- and AMPA-subtype receptor antagonists AP-5 (50 μm) and CNQX (10 μm), respectively (n = 6; data not shown). With TTX (0.5 μm) in the bath, 100 nm PYY3-36 decreased the frequency of miniature EPSCs by 38.2 ± 5.9% (Fig. 5F) (n = 6; p < 0.05). No effect of PYY3-36 on the mean mEPSC amplitude (Fig. 5H) (n = 6; p = 0.87) or the cumulative fraction (Fig. 5G) (n = 6; p > 0.01; Kolmogorov-Smirnov test) was detected. Together, these results indicate that PYY3-36 presynaptically reduces the release of glutamate from axon terminals innervating NPY neurons in the arcuate nucleus. Finally, we tested the actions of NPY on NPY neurons in the arcuate nucleus. NPY (100 nm) robustly depressed spontaneous spike frequency (by 96.3 ± 3.8%) and induced a 8.7 ± 1.8 mV hyperpolarization in NPY cells tested (n = 5; p < 0.05). These inhibitory actions of NPY persisted in the presence of 0.5 μm TTX (Fig. 5D) (hyperpolarization by NPY under this condition, 8.6 ± 2.3 mV; n = 4; p < 0.05), consistent with the view that they were spike-independent and attributable to the activation of postsynaptic NPY receptors in arcuate NPY neurons.
Because all peptidergic actions reported here were inhibitory in nature, we wanted to ensure that NPY neurons were capable of excitatory responses to peptide activation. To evaluate this, we applied the excitatory peptide hypocretin-2 (de Lecea et al., 1998) to arcuate NPY neurons. Hypocretin-2 (1 μm) reversibly increased NPY neuron spike frequency by 78.5 ± 8.4% (n = 6; p < 0.05), consistent with a previous study that showed hypocretin excitation of arcuate NPY neurons in the rat (van den Top et al., 2004). van den Top et al. (2004) found hypocretin-induced burst firing in putative rat NPY neurons; we found bursting in only one of six identified neurons in the mouse during hypocretin stimulation.
Discussion
In the present study, we used voltage- and current-clamp whole-cell recording to study the actions of PYY3-36 on identified GFP-expressing POMC or NPY neurons of the arcuate nucleus. In striking contrast to previous suggestions, PYY3-36 consistently and dose-dependently inhibited POMC neurons, reduced spike frequency or completely blocked spikes, and hyperpolarized the membrane potential. Multiple mechanisms underlay this inhibition, including a reduction in input resistance, activation of GIRK currents, attenuation of calcium currents, and a presynaptic depression of glutamate release onto the POMC cells. Thus, PYY3-36 would tend to depress the anorexigenic tone exerted by the POMC system. However, we also found PYY3-36 exerted similar inhibitory actions on NPY neurons; if PYY3-36 exerts an anorexic effect, it is most likely to do so by inhibiting the orexigenic NPY neuron.
Mechanisms of PYY inhibition
PYY3-36 consistently depressed the activity of both POMC and NPY cells by multiple direct mechanisms. PYY3-36 hyperpolarized these cell populations, leading to a reduction or absence of spikes. This was in part a result of activation of GIRK currents and also a result of the reduced input resistance, which would lead to a diminished driving force for excitatory input. GIRK currents are also activated by the GABAB receptor in POMC cells (Ibrahim et al., 2003). PYY3-36 reduced inward calcium currents; inhibition of calcium currents does not drastically affect POMC membrane potential, because PYY3-36 had minimal hyperpolarizing actions in the presence of external barium. PYY3-36 depression of calcium currents may result in an attenuation of those intracellular processes in which calcium plays a key second messenger role and may reduce calcium-dependent transmitter release. Along this line, PYY3-36 also depresses calcium currents in NPY neurons, which have been postulated to innervate nearby POMC cells (Cowley et al., 2001), further suggesting that PYY3-36 might reduce transmitter release from presynaptic NPY-containing axons onto POMC neurons.
Consistent with previous reports that the great majority of synaptic excitation in the arcuate nucleus is attributable to glutamate release (van den Pol et al., 1990; Belousov and van den Pol, 1997), blocking ionotropic glutamate receptors with AP-5 and CNQX blocked all excitatory synaptic input to both POMC and NPY cells, supporting the view (van den Pol, 2003) that the many remaining transmitters in the arcuate nucleus play primarily neuromodulatory functions. PYY3-36 reduced the excitatory synaptic input to POMC and NPY cells. In part, this was attributable to PYY3-36 actions on receptors that behaved like Y2 on glutamatergic axon terminals, because both PYY3-36 and the Y2 receptor agonist NPY13-36 reduced the frequency of miniature EPSCs with minimal effect on mEPSC amplitude. When the multiple mechanisms of inhibition described in the present study are compared with the sole possibility for excitation in POMC neurons (i.e., GABA disinhibition), PYY3-36 mediated inhibition greatly outweighs excitation.
It is not clear why our results on POMC neurons are different from those reported previously. There are several technical considerations that could account for some differences. We applied peptides by a flow pipe directed at the recorded cell, whereas the previous work used bath application. Flow pipes generally deliver the reagents to the recorded cell faster, and because the entire slice is not immersed in the reagents, washout is often faster and more complete. Fast application is less likely to be complicated by desensitization, which begins to occur even before a maximal response is reached. Bath application will presumably affect all cells in the slice, leading to modulation of all circuits. We used hypothalamic slices that are almost twice as thick as those used previously (300 vs 180 μm). A thick slice is less likely to damage the recorded cell and also less likely to disrupt axonal signaling from local neurons, particularly that on distal dendrites, which in some cases might be lost in thinner slices. For instance, we found more glutamatergic activity in POMC cells than reported previously, and this could be attributable to the sparing of a higher percentage of local excitatory inputs to the POMC cells; this possibility is consistent with the finding of asymmetrical synapses on POMC cells (Pinto et al., 2004) and with excitatory inputs from the lateral hypothalamus that terminate on arcuate neuron dendrites (Horvath et al., 1999). We also found a more negative resting membrane potential than reported previously [our study, RMP -53.2 mV; previous study (Cowley et al., 2001), -40 to -45 mV]. A more positive membrane potential may increase the driving force for GABA events, because it may move the potential away from the Cl- reversal potential but would also decrease it for glutamate synaptic events as the membrane potential moves toward the Na+ reversal potential.
NPY inhibition of POMC and NPY cells
In addition to studying the effects of PYY3-36 on POMC cells, we also examined the actions of the related peptide, NPY, which is synthesized and released by orexigenic neurons in the arcuate nucleus (McDonald and Koenig, 1993; Horvath et al., 1997) and by other neurons in the CNS that project to the hypothalamus (Everitt and Hokfelt, 1989). As reported previously (Cowley et al., 2001), NPY reduced the inhibitory synaptic tone to the POMC cells. This could be a result of presynaptic or postsynaptic actions on arcuate GABA neurons (Acuna-Goycolea et al., 2005). Although NPY-mediated attenuation of the inhibitory synaptic tone may lead to disinhibition, our finding that NPY also reduced excitatory glutamatergic tone, probably at Y2 receptors, might offset the attenuation of inhibitory synaptic tone. Some of the decrease in glutamate release may be from POMC cells, which are reported to use glutamate as a fast transmitter (Collin et al., 2003; Kiss et al., 2005) (but see Hentges et al., 2004). In addition, we found that NPY hyperpolarized POMC neurons in the presence of TTX, consistent with direct postsynaptic effects as documented previously (Cowley et al., 2001; Roseberry et al., 2004). POMC neurons were hyperpolarized by Y1 and Y2 agonists in a TTX-insensitive manner, suggesting that both receptor subtypes are involved in NPY inhibitory effects. The Y1 antagonist BIBP3226 has been shown to reduce NPY-induced hyperpolarization of POMC neurons (Roseberry et al., 2004), and we show here that Y2 receptors probably underline PYY3-36 postsynaptic actions in the same cells; because NPY can activate both Y1 and Y2 receptors, the relative contribution of these receptor subtypes to NPY inhibition merits additional study. Our results show little difference in the responses of POMC and NPY neurons to NPY receptor agonists.
NPY not only inhibits POMC cells but, as we show here, exerts a robust inhibitory action on nearby NPY cells, an action not reported previously. What might be the functional role of NPY inhibition of NPY cells? Perhaps the actions of NPY on CNS circuits involved in energy homeostasis is self-limiting, and once the orexigenic signal is sent, recurrent axon collaterals inhibit the additional activity of arcuate NPY neurons. This would be consistent with long-term actions of NPY and the very long-term actions of the peptide AgRP colocalized in NPY neurons that can enhance food intake for several days after a single intracerebral injection (Stanley and Leibowitz, 1985; Hagan et al., 2000). If this local inhibitory feedback on orexigenic neurons retards overeating, this may be a site of interest for drug actions that could potentially reduce obesity.
Relevance to feeding homeostasis
PYY3-36 was reported to reduce food intake by exciting POMC neurons. Our whole-cell recordings consistently showed that PYY3-36 reversibly inhibited POMC neurons in a dose-dependent manner, which would tend to be counter-anorexigenic. Consistent with our observations, a very recent paper similarly found that PYY3-36 reduced spike frequency in POMC cells (Ghamari-Langroudi et al., 2005). Our results are inconsistent with the view that PYY3-36 acting at POMC neurons would reduce feeding (Batterham et al., 2002). However, the PYY3-36-mediated inhibition of Y2 receptors on NPY neurons would much more likely explain anorexigenic actions of PYY3-36 and is also consistent with the absence of the anorectic effect of this peptide in Y2 knock-out mice (Batterham et al., 2002).
This leaves the question of why some laboratories find that PYY3-36 is anorexigenic (Batterham et al., 2002; Halatchev et al., 2004), whereas others find that it has no anorexigenic effect (Tschop et al., 2004). Our data offer a possible explanation. The dual inhibition of both POMC and NPY cells would tend to reduce the contribution of both of these arcuate neurons to the regulation of food intake. But other regions of the brain both within and outside the hypothalamus also participate in energy homeostasis, and these other brain regions may assume a greater contribution to energy balance during periods of highcirculating PYY3-36. Environmental stressors have been suggested to potentially block the anorectic actions of PYY3-36 (Batterham et al., 2004; Challis et al., 2004). Because NPY cells show a greater inhibition in response to PYY3-36 than POMC cells, PYY3-36 may reduce feeding initially by attenuation of the activity of NPY neurons, but with the combined inhibition of both NPY and POMC cells, environmental factors influencing other CNS circuits that modulate food intake may block the anorexigenic actions of the peptide.
Footnotes
This work was supported by National Institutes of Health Grants NS 34887, NS41454, and NS48476. We thank Dr. M. Low for kindly providing POMC-GFP mice, Drs. T. Horvath and J. Friedman for the NPY-GFP mice, and Vitaliy Rogulin for excellent help with our transgenic mouse colony.
Correspondence should be addressed to Anthony N. van den Pol, Department of Neurosurgery, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06520. E-mail: anthony.vandenpol@yale.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510510-10$15.00/0
References
- Acuna-Goycolea C, Li Y, van den Pol AN (2004) Group III metabotropic glutamate receptors maintain tonic inhibition of excitatory synaptic input to hypocretin/orexin neurons. J Neurosci 24: 3013-3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acuna-Goycolea C, Tamamaki N, Yanagawa Y, Obata K, van den Pol AN (2005) Mechanisms of neuropeptide Y, peptide YY, and pancreatic polypeptide inhibition of identified green fluorescent protein-expressing GABA neurons in the hypothalamic neuroendocrine arcuate nucleus. J Neurosci 25: 7406-7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batterham RL, Bloom SR (2003) The gut hormone peptide YY regulates appetite. Ann NY Acad Sci 994: 162-168. [DOI] [PubMed] [Google Scholar]
- Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, Cone RD, Bloom SR (2002) Gut hormone PYY3-36 physiologically inhibits food intake. Nature 418: 650-654. [DOI] [PubMed] [Google Scholar]
- Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, Cone RD, Bloom SR (2004) Physiology: does gut hormone PYY3-36 decrease food intake in rodents? Nature 430: 652-654. [Google Scholar]
- Bekkers JM, Stevens CF (1995) Quantal analysis of EPSCs recorded from small numbers of synapses in hippocampal cultures. J Neurophysiol 73: 1145-1156. [DOI] [PubMed] [Google Scholar]
- Belousov AB, van den Pol AN (1997) Local synaptic release of glutamate from neurons in the rat hypothalamic arcuate nucleus. J Physiol (Lond) 499: 747-761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broberger C, Landry M, Wong H, Walsh JN, Hokfelt T (1997) Subtypes Y1 and Y2 of the neuropeptide Y receptor are respectively expressed in proopiomelanocortin- and neuropeptide-Y-containing neurons of the rat hypothalamic arcuate nucleus. Neuroendocrinology 66: 393-408. [DOI] [PubMed] [Google Scholar]
- Challis BG, Coll AP, Yeo GS, Pinnock SB, Dickson SL, Thresher RR, Dixon J, Zahn D, Rochford JJ, White A, Oliver RL, Millington G, Aparicio SA, Colledge WH, Russ AP, Carlton MB, O'Rahilly S (2004) Mice lacking pro-opiomelanocortin are sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY(3-36). Proc Natl Acad Sci USA 101: 4695-4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Johnston D (2005) Constitutively active G-protein-gated inwardly rectifying K+ channels in dendrites of hippocampal CA1 pyramidal neurons. J Neurosci 25: 3787-3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin M, Backberg M, Ovesjo ML, Fisone G, Edwards RH, Fujiyama F, Meister B (2003) Plasma membrane and vesicular glutamate transporter mRNAs/proteins in hypothalamic neurons that regulate body weight. Eur J Neurosci 18: 1265-1278. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ (2001) Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411: 480-484. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Cone RD, Enriori P, Louiselle I, Williams SM, Evans AE (2003a) Electrophysiological actions of peripheral hormones on melanocortin neurons. Ann NY Acad Sci 994: 175-186. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, Garcia-Segura LM, Nillni EA, Mendez P, Low MJ, Sotonyi P, Friedman JM, Liu H, Pinto S, Colmers WF, Cone RD, Horvath TL (2003b) The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37: 649-661. [DOI] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett Jr FS, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG (1998) The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA 95: 322-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC (2003) G protein modulation of voltage-gated calcium channels. Pharmacol Rev 55: 607-627. [DOI] [PubMed] [Google Scholar]
- Doods H, Gaida W, Wieland HA, Dollinger H, Schnorrenberg G, Esser F, Engel W, Eberlein W, Rudolf K (1999) BIIE0246: a selective and high affinity neuropeptide Y Y(2) receptor antagonist. Eur J Pharmacol 384: 3-5. [DOI] [PubMed] [Google Scholar]
- Eggermann E, Bayer L, Serafin M, Saint-Mleux B, Bernheim L, Machard D, Jones BE, Muhlethaler M (2003) The wake-promoting hypocretin-orexin neurons are in an intrinsic state of membrane depolarization. J Neurosci 23: 1557-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmquist JK, Elias CF, Saper CB (1999) From lesions to leptin: hypothalamic control of food intake and body weight. Neuron 22: 221-232. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Hokfelt T (1989) The coexistence of neuropeptide Y with other peptides and amines in the central nervous system. In: Neuropeptide Y (Mutt V, Fuxe K, Hokfelt T, Lundberg J, eds), pp 61-72. New York: Raven.
- Fernandez-Fernandez JM, Wanaverbecq N, Halley P, Caulfield MP, Brown DA (1999) Selective activation of heterologously expressed G proteingated K+ channels by M2 muscarinic receptors in rat sympathetic neurones. J Physiol (Lond) 515: 631-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L-Y, Acuna-Goycolea C, van den Pol AN (2004) Neuropeptide Y inhibits hypocretin/orexin by multiple presynaptic and postsynaptic mechanisms: tonic depression of the hypothalamic arousal system. J Neurosci 24: 8741-8751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XB, van den Pol AN (1999) Neurotrophin-3 potentiates excitatory GABAergic synaptic transmission in cultured developing hypothalamic neurones of the rat. J Physiol (Lond) 518: 81-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghamari-Langroudi M, Colmers WF, Cone RD (2005) PYY3-36 inhibits the action potential firing activity of POMC neurons of the arcuate nucleus through postsynaptic Y2 receptors. Cell Metab 2: 191-199. [DOI] [PubMed] [Google Scholar]
- Grandt D, Schimiczek M, Beglinger C, Layer P, Goebell H, Eysselein VE, Reeve Jr JR (1994) Two molecular forms of peptide YY (PYY) are abundant in human blood: characterization of a radioimmunoassay recognizing PYY 1-36 and PYY 3-36. Regul Pept 51: 151-159. [DOI] [PubMed] [Google Scholar]
- Guo H, Castro PA, Palmiter RD, Baraban SC (2002) Y5 receptor mediate neuroprptide Y actions at excitatory synapses in area CA3 of the mouse hippocampus. J Neurophysiol 87: 558-566. [DOI] [PubMed] [Google Scholar]
- Hagan MM, Rushing PA, Pritchard LM, Schwartz MW, Strack AM, Van Der Ploeg LH, Woods SC, Seeley RJ (2000) Long-term orexigenic effects of AgRP-(83-132) involve mechanisms other than melanocortin receptor blockade. Am J Physiol Regul Integr Comp Physiol 279: R47-R52. [DOI] [PubMed] [Google Scholar]
- Halatchev IG, Ellacott KL, Fan W, Cone RD (2004) PeptideYY3-36 inhibits food intake in mice through a melanocortin-4 receptor-independent mechanism. Endocrinology 145: 2585-2590. [DOI] [PubMed] [Google Scholar]
- Hentges ST, Nishiyama M, Overstreet LS, Stenzel-Poore M, Williams JT, Low MJ (2004) GABA release from proopiomelanocortin neurons. J Neurosci 24: 1578-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath TL, Bechmann I, Naftolin F, Kalra SP, Leranth C (1997) Heterogeneity in the neuropeptide Y-containing neurons of the rat arcuate nucleus: GABAergic and non-GABAergic subpopulations. Brain Res 756: 283-286. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Diano S, van den Pol AN (1999) Synaptic interaction between hypocretin (orexin) and neuropeptide Y cells in the rodent and primate hypothalamus: a novel circuit implicated in metabolic and endocrine regulations. J Neurosci 19: 1072-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim N, Bosch MA, Smart JL, Qiu J, Rubinstein M, Ronnekleiv OK, Low MJ, Kelly MJ (2003) Hypothalamic proopiomelanocortin neurons are glucose responsive and express K(ATP) channels. Endocrinology 144: 1331-1340. [DOI] [PubMed] [Google Scholar]
- Jobst EE, Enriori PJ, Cowley MA (2004) The electrophysiology of feeding circuits. Trends Endocrinol Metab 15: 488-499. [DOI] [PubMed] [Google Scholar]
- Keire DA, Mannon P, Kobayashi M, Walsh JH, Solomon TE, Reeve Jr JR (2000) Primary structures of PYY, [Pro(34)]PYY, and PYY-(3-36) confer different conformations and receptor selectivity. Am J Physiol Gastrointest Liver Physiol 279: 126-131. [DOI] [PubMed] [Google Scholar]
- Kiss J, Csaba Z, Csaki A, Halasz B (2005) Glutamatergic innervation of neuropeptide Y and pro-opiomelanocortin-containing neurons in the hypothalamic arcuate nucleus of the rat. Eur J Neurosci 21: 2111-2119. [DOI] [PubMed] [Google Scholar]
- McDonald JK, Koenig JI (1993) Neuropeptide Y actions on reproductive and endocrine functions. In: The biology of neuropeptide Y and related peptides (Colmers WF, Wahlestedt C, eds), pp 419-456. Totowa, NJ: Humana.
- Mullins D, Kirby D, Hwa J, Guzzi M, Rivier J, Parker E (2001) Identification of potent and selective neuropeptide Y Y(1) receptor agonist with orexigenic activity in vivo. Mol Pharmacol 60: 534-540. [PubMed] [Google Scholar]
- Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL (2004) Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science 304: 110-115. [DOI] [PubMed] [Google Scholar]
- Potter EK, Fuhlendorff J, Schwartz TW (1991) [Pro34]neuropeptide Y selectively identifies postjunctional-mediated actions of neuropeptide Y in vivo in rats and dogs. Eur J Pharmacol 193: 15-19. [DOI] [PubMed] [Google Scholar]
- Rhim H, Kinney GA, Emmerson PJ, Miller RJ (1997) Regulation of neuro-transmission in the arcute nucleus of the rat by different neuropeptide Y receptors. J Neurosci 17: 2980-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseberry AG, Liu H, Jackson AC, Cai X, Friedman JM (2004) Neuropeptide Y-mediated inhibition of proopiomelanocortin neurons in the arcuate nucleus shows enhanced desensitization in ob/ob mice. Neuron 41: 711-722. [DOI] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Elmquist JK (2002) The need to feed: homeostatic and hedonic control of eating. Neuron 36: 199-211. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Morton GJ (2002) Obesity: keeping hunger at bay. Nature 418: 595-597. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte Jr D, Seeley RJ, Baskin DG (2000) Central nervous system control of food intake. Nature 404: 661-671. [DOI] [PubMed] [Google Scholar]
- Seeley RJ, Woods SC (2003) Monitoring of stored and available fuel by the CNS: implications for obesity. Nat Rev Neurosci 4: 901-909. [DOI] [PubMed] [Google Scholar]
- Sodickson DL, Bean BP (1996) GABAB receptor-activated inwardly rectifying potassium current in dissociated hippocampal CA3 neurons. J Neurosci 16: 6374-6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman BM, Flier JS (2001) Obesity and the regulation of energy balance. Cell 104: 531-543. [DOI] [PubMed] [Google Scholar]
- Stanley BG, Leibowitz SF (1985) Y injected in the paraventricular hypothalamus: a powerful stimulant of feeding behavior. Proc Natl Acad Sci USA 82: 3940-3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S, Wynne K, Bloom S (2004) Gastrointestinal satiety signals III. Glucagon-like peptide 1, oxyntomodulin, peptide YY, and pancreatic polypeptide. Am J Physiol Gastrointest Liver Physiol 286: 693-697. [DOI] [PubMed] [Google Scholar]
- Sun L, Miller R (1999) Multiple neuropeptide Y receptors regulate K+ and Ca2+ channels in acutely isolated neurons from the rat arcuate nucleus. J Neurophysiol 81: 1391-1403. [DOI] [PubMed] [Google Scholar]
- Tschop M, Castaneda TR, Joost HG, Thone-Reineke C, Ortmann S, Klaus S, Hagan MM, Chandler PC, Oswald KD, Benoit SC, Seeley RJ, Kinzig KP, Moran TH, Beck-sickinger AG, Koglin N, Rodgers RJ, Blundell JE, Ishii Y, Beattie AH, Holch P, et al. (2004) Physiology: does gut hormone PYY3-36 decrease food intake in rodents? Nature 430: 650-652. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Wuarin JP, Dudek FE (1990) Glutamate, the dominant excitatory transmitter in neuroendocrine regulation. Science 250: 1276-1278. [DOI] [PubMed] [Google Scholar]
- van den Pol AN (2003) Weighing the role of hypothalamic feeding neurotransmitters. Neuron 40: 1059-1061. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Acuna-Goycolea C, Clark KR, Ghosh PK (2004) Physiological properties of hypothalamic MCH neurons identified with selective expression of reporter gene after recombinant virus infection. Neuron 42: 635-652. [DOI] [PubMed] [Google Scholar]
- van den Top M, Lee K, Whyment AD, Blanks AM, Spanswick D (2004) Orexigen-sensitive NPY/AgRP pacemaker neurons in the hypothalamic arcuate nucleus. Nat Neurosci 7: 493-494. [DOI] [PubMed] [Google Scholar]
- Wisse BE, Schwartz MW (2001) Role of melanocortins in control of obesity. Lancet 358: 857-859. [DOI] [PubMed] [Google Scholar]